
IDS079 - Management de la qualité des dispositifs médicaux selon l'ISO 13485 : 2016 et son amendement A1
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

FOSSO MATCHINDE Megane Shandy 
WAOUSSI NGOKO Saryane Manuela
Contacts
Citation
A rappeler pour tout usage : FOSSO MATCHINDE Megane Shandy, WAOUSSI NGOKO Saryane Manuela « Management de la qualité des dispositifs médicaux selon l'ISO 13485 : 2016 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids079
Résumé
La venue du nouveau règlement européen crée un déséquilibre dans toutes les entreprises biomédicales. Il vise à insister sur la sécurité du patient et à renforcer davantage les actes posés à l’endroit de celui-ci. Ceci inclue une obtention plus difficile du marquage CE. En effet, les fabricants sont désormais appelés à se conformer selon les exigences des nouvelles règlementations 2017/745 et 2017/746. Ce changement brusque oblige tous les fabricants des dispositifs médicaux à faire face à de nouvelles difficultés pour pouvoir commercialiser leurs produits. Le présent document a été établi pour identifier tout d’abord les acteurs concernés par le nouveau règlement, ensuite aider ces derniers à mettre un accent sur l’efficacité et l’efficience de leur système de management de la qualité selon la norme NF EN ISO 13485 : 2016. La charge accrue du travail, la non-qualité et le manque de moyens financiers sont des éléments qui pourraient freiner les fabricants dans ce processus. Malgré les enjeux associés à ce nouveau départ, il reste possible si toutes les entreprises s’impliquent dans cette démarche. De plus, les outils développés et mis au service de ces entreprises seront aussi des plus-values pour ce changement.
Abstract
The coming of the new European regulation creates an unbalance in all biomedical companies. It aims to insist on patient safety and to further strengthen the actions taken towards the patient. This includes making it more difficult to obtain the CE mark. Indeed, manufacturers are now required to comply with the requirements of the new regulations 2017/745 and 2017/746. This abrupt change forces all medical device manufacturers to face new challenges in order to be able to market their products. This report has been prepared to first identify the stakeholders affected by the new regulations and then to help them focus on the effectiveness and efficiency of their quality management system according to the ISO 13485 standard. Increased workload, non-quality and lack of financial means are elements that could hinder manufacturers in this process. Despite the challenges associated with this new start, it is still possible if all companies get involved. Moreover, the tools developed and put at the service of these companies will also be an added value for this change.
Téléchargements
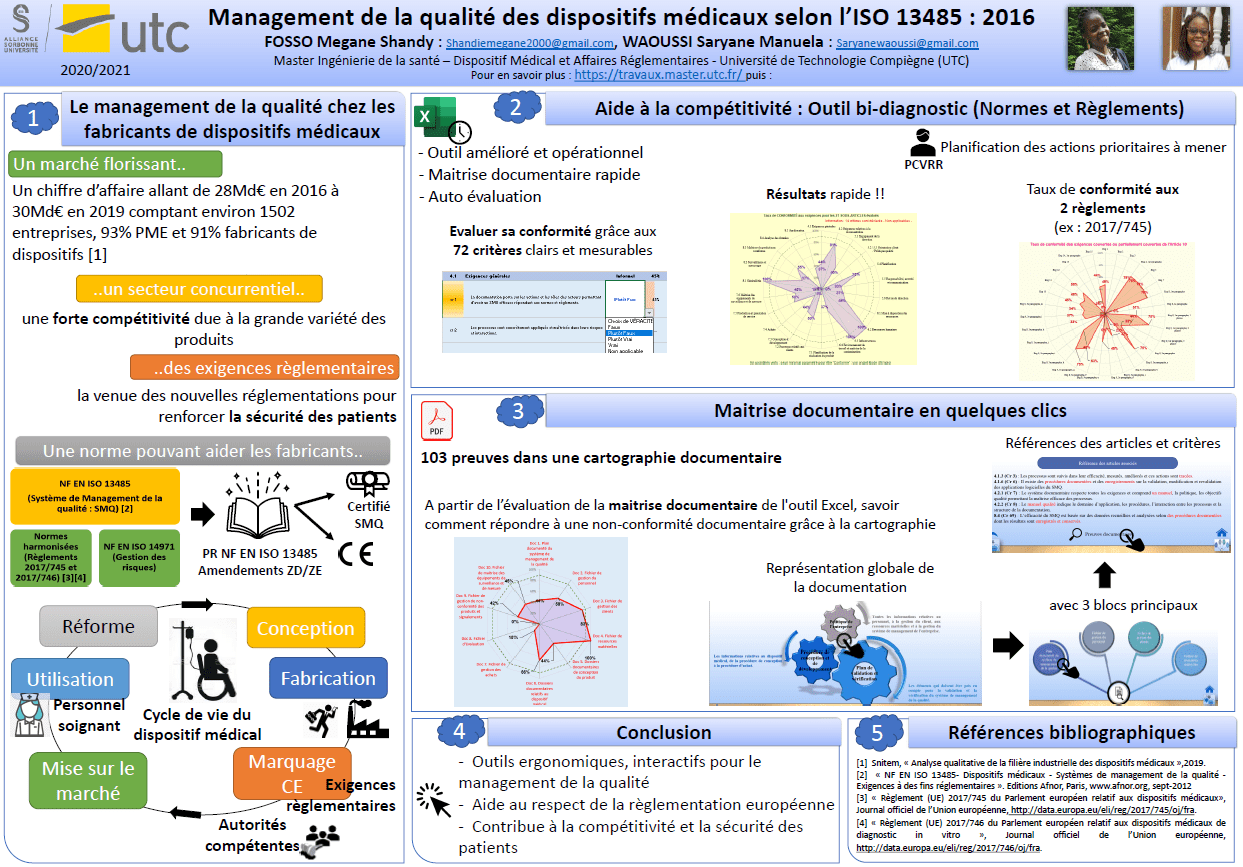
Poster du mémoire

Mémoire d'Intelligence Méthodologique : Management de la qualité des dispositifs médicaux selon l'ISO 13485 : 2016

Cartographie documentaire selon la norme ISO 13485 : 2016

Vidéo de présentation de l'utilisation de la cartographie documentaire
Outil d'autodiagnostic selon la norme ISO 13485 : 2016
Mémoire complet : Management de la qualité des dispositifs médicaux selon l'ISO 13485 : 2016
Introduction
L’industrie des dispositifs médicaux entre dans une ère très spécifique, par de très fortes exigences réglementaires qui ont été mis en place dans le but de renforcer la sécurité des patients. Ces exigences renforcent également la compétitivité du secteur des dispositifs médicaux due à la grande variété de produits. Bien que ces exigences soient très lourdes pour les fabricants, ralentissant légèrement les délais de mise sur le marché des dispositifs médicaux et mobilisant une quantité de ressources, leurs avantages sont plus importants.
Pour se commercialiser sur le marché européen, un dispositif médical doit obligatoirement obtenir un marquage CE. C’est une certification garantissant la satisfaction des dispositifs médicaux aux exigences essentielles des directives ou réglementations en termes de performances et de sécurité des utilisateurs.
Dans le but d’être compétitif sur le marché des dispositifs médicaux, l’exercice de la qualité par la mise en place d’un système de management de la qualité est primordial. Cette démarche qualité vise à guider et à harmoniser les activités d’une entreprise, à satisfaire aux exigences des clients de manière permanente.
Alors la norme NF EN ISO 13485 : 2016 « Dispositifs médicaux – Systèmes de management de la qualité – Exigences à des fins réglementaires » permet non seulement aux entreprises de mettre en œuvre un bon système de management de la qualité mais, aussi d’avoir un gain en temps pour la mise sur le marché de leurs dispositifs médicaux, en facilitant l’obtention du marquage CE [1].
Avec les annexes qui y sont associées, le respect de cette norme permettra aux entreprises de s’ouvrir au marché européen en offrant une présomption de conformité aux exigences réglementaires qui visent à la sécurité des patients.
Ce mémoire a pour objectif d’étudier le management de la qualité des dispositifs médicaux selon la norme ISO 13485 : 2016 et son premier amendement (A1) [2] pour l’élaboration d’un outil d’autodiagnostic qui permettra l’évaluation du respect aux exigences générales de la norme et, au respect des exigences générales des nouvelles réglementations 2017/745 relatif aux dispositifs médicaux et dispositifs médicaux implantables actifs [3] et 2017/746 relatif aux dispositifs médicaux de diagnostic in vitro [4].
I. L’impact de la nouvelle réglementation européenne sur le système de management de la qualité des entreprises biomédicales
1. Le marché des dispositifs médicaux
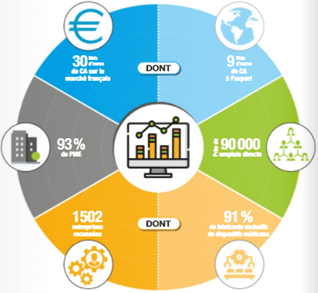
En 2019, le chiffre d’affaire du marché français est estimé à 30M€, dont 9M€ à l’exportation. Comparé à celui de 2016 (environ 28M€), on peut dire que le secteur français des dispositifs médicaux est fleurissant et se porte bien [5]. Il est illustré à la figure ci-dessous (Figure 1).
D’après le syndicat national de l’industrie des technologies médicales (SNITEM), environ 1502 entreprises sont comptées dont 93% sont des Petites et Moyennes entreprises (PME) pour le marché des dispositifs et 91% sont des entreprises de fabrication de dispositifs médicaux[5]. Ce nombre a augmenté de 200 nouvelles entreprises depuis les trois dernières années (environ 13% de ces structures sont des startups). Près de 2/3 des entreprises ont allouées 6% de leurs chiffres d’affaires au volet R&D, plus de 60% de ces entreprises ont une activité de production et plus de 80% ont une activité commerciale[5].
Figure 1 : Représentation du marché des dispositifs médicaux [5]
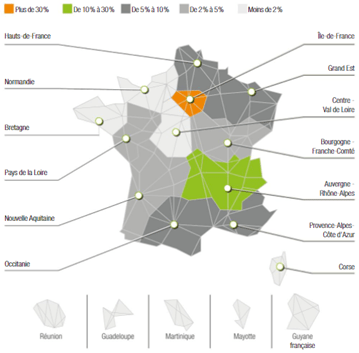
Le secteur des dispositifs médicaux génère environ 90 000 emplois directs, 27% sont des entreprises d’origines étrangères dont 57% sont d’origines européennes. Des entreprises de ce secteur sont implantées sur tout le territoire français, néanmoins, certaines régions regroupent un nombre important que d’autres[5], comme le montre la (Figure 2).
Figure 2 : Répartition géographique des entreprises de dispositifs médicaux sur le territoire français [5]
2. Scandales et incidents liés aux dispositifs médicaux
Ces dix dernières années ont été scandaleuses pour le secteur des dispositifs médicaux où plusieurs incidents ont été enregistrés, mettant en danger la sécurité du patient à l’utilisation des dispositifs médicaux tant à l’échelle nationale qu’internationale.
Aux Etats – unis, d’après le Consortium international des journalistes d’investigation (ICIJ), une enquête appelée « Implants Files » a été réalisée dévoilant 10 implants causant le plus d’incidents. D’après cette enquête, 5,4 millions d’incidents ont été recensé en dix ans causant ainsi 82 000 morts, 1,7millions de blessés et le reste dû aux défaillances [6].
En France, 30 000 femmes et environ 1million dans d’autres pays, se sont fait implanter des prothèses mammaires de la société Poly Implant Prothèse (PIP) qui étaient potentiellement défectueuses [7]. En 2010, l’Agence Française de sécurité sanitaire des produits de santé (Afssaps) a annoncé le retrait des implants mammaires PIP sur le marché, à cause de l’utilisation anormal d’un gel différent de celui qu’ils ont déclaré avant la commercialisation [8]. Le premier signalement a été en 2011 à Marseille par le décès d’une femme ayant développé un cancer après la rupture de ses prothèses [8].
Ces scandales m’étant en danger la vie des patients ont montré la nécessité de renforcer la règlementation. C’est pour cette raison qu’ont été mises en place les nouvelles réglementations (2017/745 et 2017/746) d’application obligatoire abrogeant les directives.
3. Généralités et application de l’ISO 13485
a. Historique et exigences
Née de la conférence des organisations nationales de normalisation, la norme ISO a été créée en 1946 [9], se diversifiant en différentes normes parmi lesquelles l’ISO 13485. Cette dernière quant à elle s’est développée selon le canevas de la (Figure 3) :
Figure 3 : Historique de création de la norme ISO 13485 (Source : Auteurs)
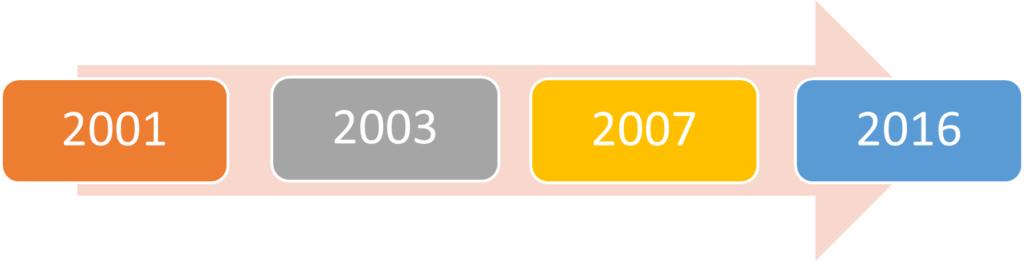
La version 2016 est la cinquième (dernière) édition de l’ISO 13485 basée sur l’ISO 9001 : 2008 montrant l’aptitude de toute entreprise à démontrer sa conformité aux exigences spécifiées d’un système de management de la qualité efficace [10].
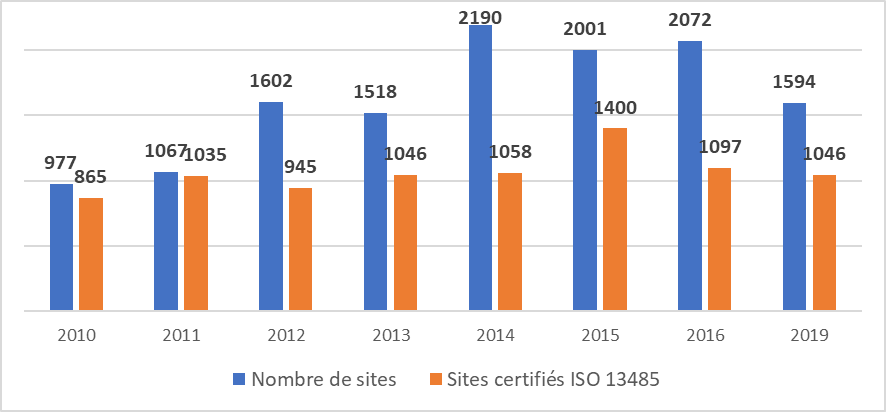
Aujourd’hui, en France, il existe plus de 1046 entreprises ou organismes certifiés ISO 13485 [11]. Ce nombre est en constante amélioration, comme le montre la (Figure 4).
Figure 4 : Nombre de certification ISO 13485 délivrés en France (Source : Auteurs d’après [11])
La norme ISO 13485 énonce les exigences liées au système de management de la qualité des entreprises fabricantes des dispositifs médicaux. Elle s’assure de la conformité des dispositifs médicaux aux exigences des clients, mais fournit aussi une approche internationale. Son domaine d’application concerne principalement les structures de production et de fabrication des dispositifs médicaux ; elle peut aussi être utilisée par des organismes de certification pendant le processus d’audit.
Cette évolution de la version 2016 est donc observée au niveau de deux éléments principaux : L’application des exigences réglementaires sur un système de management de la qualité, ainsi que sur la gestion des risques selon la norme NF EN ISO 14971 [10]. Cette approche sur les risques s’étend aux dispositions du système de management de la qualité au niveau des différentes étapes de la conception et de la fabrication du produit.
b. La sécurité du patient, au cœur du processus de management de qualité des dispositifs médicaux
La (Figure 5) ci-dessus identifie les acteurs liés au cycle de vie d’un dispositif médical.
Figure 5 : Acteurs du cycle de vie des dispositifs médicaux (Source : Auteurs)
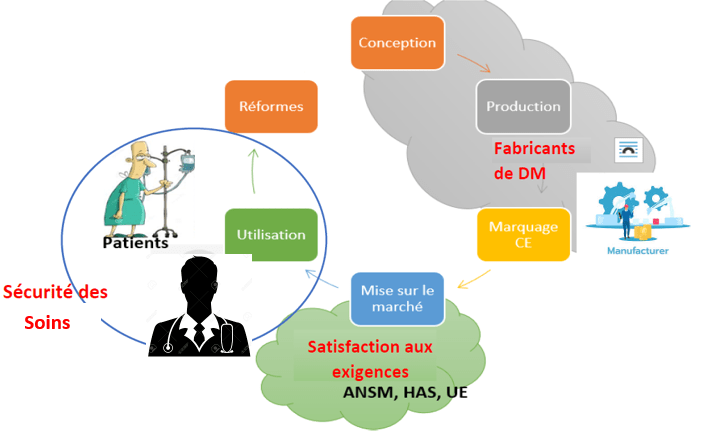
Les organismes qui veillent à la sécurité des patients sont : l’Agence Nationale de sécurité du Médicament (ANSM), la Haute Autorité de Santé (HAS), l’Union Européenne (UE). Les fabricants étant les acteurs principaux de ce cycle, doivent donc travailler en respectant les exigences des règlementations en vigueur liées à la conception des dispositifs médicaux, en fonction des recommandations des organismes précédents.
Le rôle du fabricant est primordial dans le cycle de vie du dispositif médical car il doit prouver la conformité de ses dispositifs médicaux aux organismes en charge de ceux-ci. Parce que les défauts de conception existent et ont toujours des conséquences, tant humaines que matérielles, l’application des exigences de l’ISO 13485 permettra aux entreprises « fabricants » la clarification et la responsabilité de chaque membre[12].
Lors de la conception de son dispositif en se conformant aux règlementations, le fabricant saura également gagner la confiance et la fidélité des clients [13].
4. Nouvelle règlementation et marquage CE
Avec la venue de la nouvelle réglementation, les exigences réglementaires sont devenues beaucoup plus strictes et le temps d’obtention du marquage CE relativement plus long. L’application du PR NF EN ISO 13485/A1 permettra donc aux entreprises « fabricants » de dispositifs médicaux de répondre aux exigences des nouvelles règlementations européennes, afin d’obtenir un marquage CE rapide de leurs dispositifs et un gain de temps pour rapidement les mettre sur le marché.
Comme le montre la (Figure 6), cette norme est dont dite « harmonisée » : grâce à l’ajout des annexes ZD et ZE donnant une correspondance des exigences des règlements 2017/745 et 2017/746 et la norme ISO 13485 : 2016, permettant aux fabricants de dispositifs médicaux d’avoir une présomption de conformité partielle aux exigences essentielles de règlementation visant à la sécurité des patients.
Figure 6 : ISO 13485 : Norme harmonisée (Source : Auteur d'après [9])
![Zone de Texte: Figure 6 : ISO 13485 : Norme harmonisée (Source : Auteur d'après [9])](https://travaux.master.utc.fr/wp-content/uploads/sites/16/2020/12/ids079-mim-fig06-1024x505.png)
5. Les enjeux socio-économiques pour les fabricants des dispositifs médicaux
La non qualité est un élément que plusieurs fabricants sous-estiment [14]. Elle est généralement observée lors du non-respect des systèmes de management de la qualité par certains fabricants, entrainant notamment des pénalités, une reprise tardive des activités, des pertes en ressources matérielles, humaines et énergie. En revanche en respectant les exigences de la norme, le fabricant pourrait ne pas se retrouver dans ces dépenses.
La compétitivité de l’entreprise est également mise au défi. Pour une bonne compétitivité, les entreprises peuvent agir sur le rapport bénéfices/risques ; Mais également, elles peuvent aussi agir sur la certification d’un système de management qualité, car de nos jours c’est un argument commercial non négligeable, puisqu’il rassure le client sur le niveau qualité et donc le sérieux de l’entreprise [13]. En effet, toutes les entreprises « fabricants » des dispositifs médicaux sont en perpétuelle concurrence, encore plus celles dont les types de dispositifs fabriqués sont les mêmes. Le défi consistera donc pour chacune de ces entreprises d’être performantes, et donc toujours à la recherche de l’amélioration, afin de défier ses semblables sur le marché.
Le manque de moyens financiers [15] : Certains fabricants se voient peu dépenser de l’argent pour la mise en place d’un système de management. Généralement, c’est le cas des Start-up qui n’ont pas à leur début assez de moyens pour se permettre cela, et ne sont pas encore prêts à s’endetter auprès des banques.
La charge de travail accrue : En effet, le système de management peut varier d’un qualiticien à un autre. Le personnel aura donc tendance à trouver que la charge de travail a considérablement /ou pas augmenté pour certains cas, et donc qu’il dépense beaucoup plus en énergie. De ce problème découle le besoin de temps tant pour appliquer le processus de la démarche que pour se relaxer. Il est aussi possible de noter l’impression de tourner en rond, la durée des réunions, les nombreuses documentations, la fatigue du personnel [16].
En conclusion, si la norme NF EN ISO 13485 est appliquée dans une entreprise, celle-ci aurait la possibilité d’améliorer les processus et l’efficacité [17] permettant d’assurer la sécurité des patients tout au long du cycle de vie du dispositif médical.
II. Solutions proposées pour la mise en place d’un système de management de la qualité
Pour aider les entreprises souhaitant se faire certifier ISO 13485, des solutions ont été mis en place pour permettre à ces entreprises d’assurer leurs conformités aux exigences notamment :
1- Vidéo d’appropriation de la norme
Une vidéo a été développée en 2016 par la société DM EXPERT (disponible sur YouTube) [18]. C’est une vidéo instructive d’aide à l’appropriation de la norme ISO 13485. Elle fait une présentation beaucoup plus opérationnelle, synthétique d’une durée d’environ 15min, facilement utilisable par les PME. Chaque élément essentiel des articles 4 à 8 est étudié et expliqué.
2- Guide de l’Organisation internationale de Normalisation
Un manuel pratique intitulé « ISO 13485 : 2016 – Dispositifs Médicaux – Un guide pratique » pour tous les organismes souhaitant mettre en œuvre un système de management de la qualité conformément à la norme ISO 13485 [19]. Ce guide a été élaboré par l'Organisme internationale de Normalisation, disponible uniquement sous une version Anglaise, permettant l’appropriation de la norme. Il contient environ 200 pages, ce qui rend énorme le temps de compréhension. Pour les TPE et PME, il sera important d’avoir une aide supplémentaire, rapide et simple pour la mise en place du système du management de la qualité.
3- Applications ou Logiciels
Ces moyens ont été développés par des cabinets de conseil en système de management de la qualité pour aider leurs clients à gérer au mieux leurs systèmes de management de la qualité et à garantir leurs conformités à la norme. C’est le cas des entreprises Iterop[20] et Apsalys (MasterControl)[21].
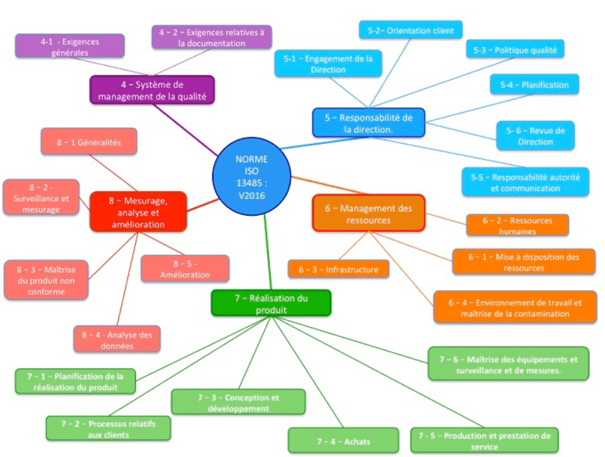
4- Cartographie des processus
Une cartographie des processus qui propose une appropriation visuelle et interactive de la norme comme illustrée à la (Figure 7). Elle se présente de deux façons :
- Approche par articles et sous-articles : Cette approche présente toutes les 227 exigences de la normes [22].
- Approche par processus : Cette approche présente tous les 72 processus (approche plus rapide, synthétisée) [22].
Figure 7 : Cartographie des processus de la norme ISO 13485 : 2016 [22]
5- Outil d’autodiagnostic
C’est un outil sous format Excel permettant l’évaluation rapide des entreprises de leur niveau de conformité dans le but de faire des améliorations et se préparer à la certification. Il regroupe toutes les exigences de la norme et donne une représentation graphique des résultats de l’évaluation [22].
C’est vers cette lancée que des anciens étudiants de l’UTC, ont effectués des projets auxquels ils ont inclus chacun un outil d’autodiagnostic et/ou une cartographie des processus permettant l’évaluation du niveau de conformité [22] [23] [24].
III. Outils pouvant aider à la compétitivité des fabricants
L’application de l’Analyse Normative Opérationnelle (méthode de synthétisation) a permis de synthétiser la norme en transformant les exigences complexes en objectifs clairs et mesurables. Ces derniers ont été utilisés pour le développement de la cartographie documentaire et l’amélioration de l’outil. La cartographie des processus ayant déjà été élaborée et 100% opérationnelle [22], une cartographie documentaire a donc été élaborée.
1- Cartographie documentaire de la norme ISO 13485 : 2016
Grâce à la norme PR NF EN ISO 13485/A1, il a été permis d’effectuer une correspondance entre la norme NF EN ISO 13485 : 2016 et les règlements européens 2017/745 et 2017/746 axée sur les obligations générales du fabricant mentionnés à l’article 10, les exigences générales du Chapitre I Annexe I et les exigences relatives à l’évaluation de conformité des Annexes IX et XI.
Cette correspondance a permis de ressortir les références des articles et les preuves documentaires de la norme NF EN ISO 13485 : 2016 qui correspondent aux règlementations européennes 2017/745 et 2017/746 qui ont été intégrées à une cartographie documentaire. Ainsi, les entreprises pourront satisfaire aux exigences de ladite norme ainsi que la satisfaction partielle ou totale aux exigences des règlements de manière aisée et plus rapide. Cela en s’appropriant des preuves documentaires correspondant aux différents critères de l’outil développé et des articles de la norme NF EN ISO 13485 : 2016.
Pour sa réalisation, le logiciel PowerPoint a été utilisé pour sa facilité d’utilisation et aussi parce qu’il démontre clairement l’interactivité entre les différents documents et les articles associés. La page d’accueil de la cartographie est présentée à la (Figure 8) :
Figure 8 : Page d'accueil de la cartographie (Source : Auteurs)

Comme présenté, elle contient trois sections :
a. Mode d’emploi
Le mode d’emploi présente l’objectif de cette cartographie et donne des indications sur son utilisation (Figure 9). Il est mieux explicité dans le Mode d'emploi multimédia.
Figure 9 : Mode d'emploi _ Boutons de navigation (Sources : Auteurs)

b. Commencez la manipulation de l’outil
C’est à ce niveau que commence la manipulation proprement dite. Cette partie démontre une possibilité d’organisation d’une entreprise qui applique un système de management de la qualité, ainsi que tous les critères correspondant à l’outil et références des articles correspondant à la documentation de la norme NF EN ISO 13485 :2016. Les trois (03) principaux axes de cette section sont les suivants (Figure 10) :
Figure 10 : Principaux axes de la manipulation de l'outil (Source : Auteurs)
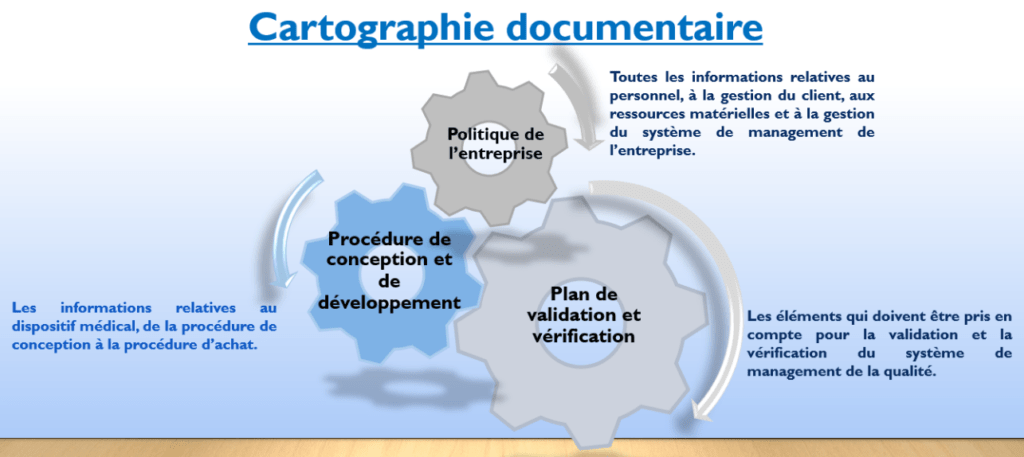
Politique de l’entreprise
Chaque entreprise l’établit en fonction de son organisation. Elle comprend toutes les informations relatives au personnel, aux clients et aux ressources comme l’indique la (Figure 11) :
Figure 11 : Sections de la politique de l'entreprise (Source : Auteurs)

- Plan documenté du système de management de la qualité : Il comprend les étapes de planification du système de management de la qualité, ainsi que les références des articles et critères correspondants.
- Fichier de gestion du personnel : C’est un fichier relatif à la gestion du personnel. Il comprend donc toutes les informations des employés de la structure et les axes d’amélioration de ces derniers.
- Fichier de gestion des clients : Il comprend toutes les exigences des clients, ainsi que les alternatives prévues pour satisfaire ces derniers.
- Fichier de ressources matérielles : Il comprend les éléments nécessaires pour la conception et/ou la maintenance du dispositif médical.
Procédure de conception et de développement
Cette partie comprend les informations relatives au dispositif médical, de la procédure de conception à la procédure d’achat. Subdivisée en trois (03) sous parties, elle se présente comme suit (Figure 12) :
Figure 12 : Procédure de conception et de développement (Source : Auteurs)
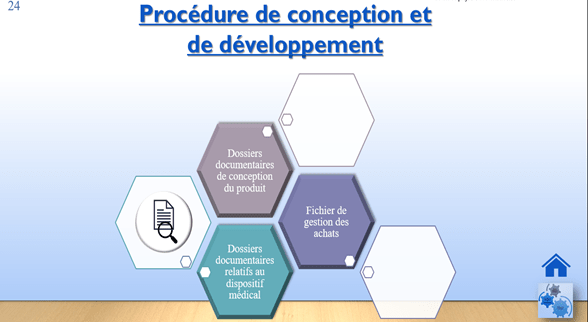
- Dossier de conception du produit : prenant en compte les risques que peuvent subir tout dispositif médical pendant ou après la conception. Les étapes de cette conception y sont donc mentionnées, afin de s’assurer que la personne en charge de la conception respecte toutes les exigences des articles mentionnés.
- Document relatif au dispositif médical : L’organisme devra mentionner toutes les informations d’ordre technique des dispositifs médicaux conçus. Ainsi, il sera plus facile de tracer ces éléments en cas de dommages causés par le produit.
- Fichier de gestion des achats : Il comprend les informations relatives aux achats effectuées et aux fournisseurs concernés.
Plan de validation et vérification
Après conception des dispositifs, ces derniers doivent être vérifiés dans le but de s’assurer qu’une fois en contact avec le patient, il ne causera pas de dommages. Ce plan est structuré selon trois (03) axes comme le montre la figure suivante (Figure 13) :
Figure 13 : Plan de validation et vérification (Source : Auteurs)

- Fichier d’évaluation : Il comprend toutes les informations détaillées du dispositif médical après conception. Notamment des informations liées aux audits et à la conformité du produit aux exigences.
- Fichier de gestion de non-conformité des produits et signalement : En fonction du fichier d’évaluation établi, les non-conformités présentes sur le dispositif devront être identifiées, ainsi que les alternatives prévues pour améliorer le produit.
- Fichier de maitrise des équipements de surveillance et de mesure : Il doit comprendre toute les informations relatives aux équipements de surveillance et de mesure. Pour les appareils de ce type possédant des logiciels, l’organisme devra fournir une preuve de la maitrise de ces logiciels.
c. Bibliographie
Cette section comprend toutes les informations, sites et liens utilisés pour la réalisation de la cartographie documentaire. Dans ce cas il a été mentionné la référence normative ainsi que le lien de l’outil d’autodiagnostic de ce projet.
2- Outil bi-diagnostic : Etat de la norme NF EN ISO 13485 et des règlements européens
Cet outil est l’élément principal qui permettra au fabricant d’effectuer son évaluation de conformité à la norme NF EN ISO 13485 : 2016 et d’établir des axes d’amélioration. Il est l’amélioration d’un outil qui avait déjà été mis en place par des anciens étudiants [22]. En prenant compte des apports de la nouvelle réglementation, l’outil permet également d’avoir une estimation du respect aux règlements 2017/745 et 2017/746 en fonction des exigences qui sont couvertes partiellement ou totalement par la norme (en fonction de l’amendement A1 de la norme ISO 13485). Les onglets présents dans l’outil sont listés suivant la (Figure 14).
Figure 14 : Les onglets composants l'outil d'autodiagnostic (Source : Auteurs)
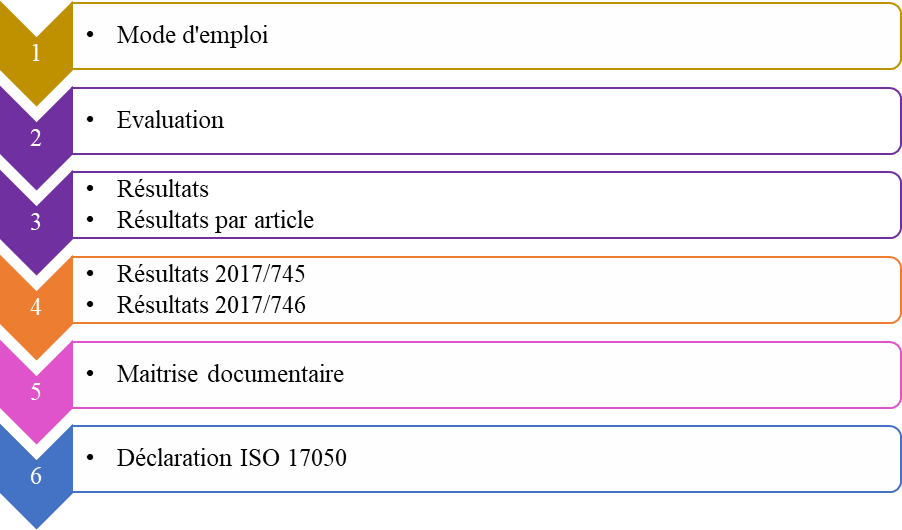
a. Un mode d’emploi
C’est l’onglet qui donne le mode de fonctionnement et les paramétrages de l’outil. Sur cet onglet, seules les cases en fond blanc et police bleu sont modifiables, présentées comme sur la (Figure 15) (standard défini dans le but de facilement capter l’attention des utilisateurs).
Tous les onglets comprennent des en-têtes permettant d’y figurer les métadonnées qui sont modifiables uniquement sur les onglets « Mode d’emploi » et « Evaluation ». Ces métadonnées font partir de la procédure d’évaluation : nom de l’établissement, identité et contact des responsables et participants, date, etc…
Figure 15 : En-tête de l’onglet_ Mode d'emploi (Source : Auteurs)
Il est également possible d’ajuster le taux moyen minimal de conformité en fonction de l’objectif de conformité visé par l’entreprise. Chaque critère est évalué suivant un taux de véracité allant de « 100% », « 75% », « 45% » à « 0% », chacun correspondant au choix de véracité « Vrai », « Plutôt vrai », « Plutôt faux » et « Faux ». Si le choix de véracité est « Non applicable », alors ce critère ne sera pas pris en compte lors du calcul des résultats de l’évaluation et il est impératif de donner le justificatif de la non-applicabilité du critère.
Il existe également un code de couleur correspondant aux critères associés à un mode de preuve de conformité documentaire. L’échelle d’évaluation utilisée est représentée comme sur la (Figure 16).
Figure 16 : Echelle d'évaluation (Source : Auteurs)

b. Evaluation
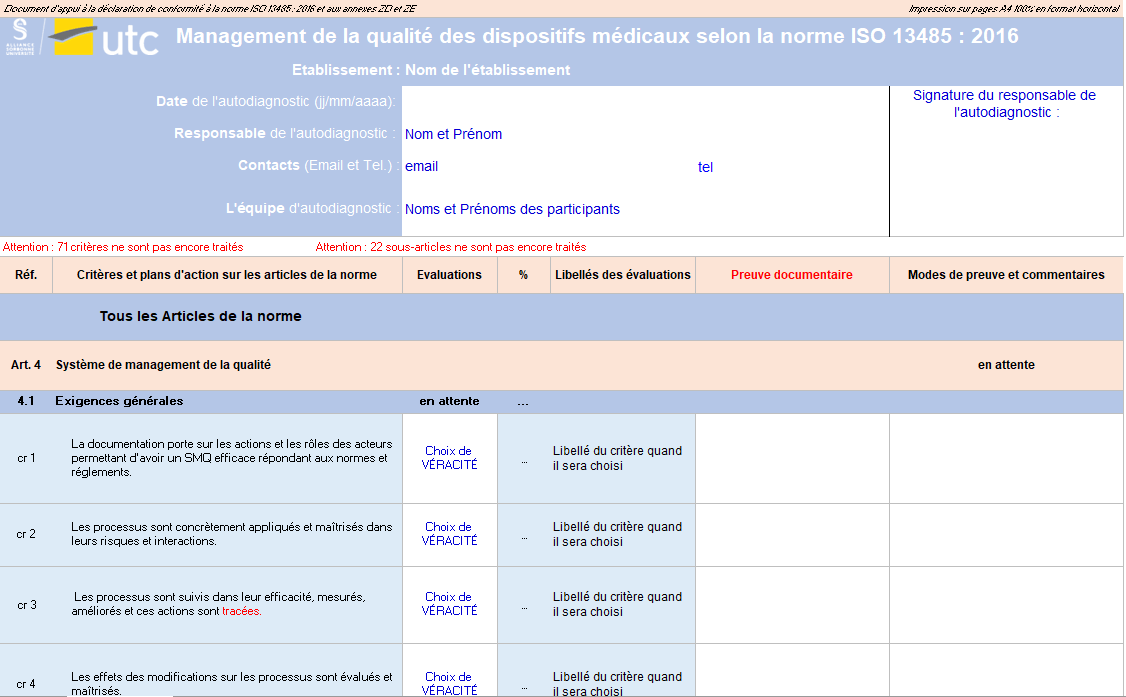
L’évaluation est basée sur 72 critères, qui sont des substituts des exigences essentielles établies dans la norme. Certains critères contiennent des mots écrits en rouge pour attirer l’attention des utilisateurs sur l’importance de celui - ci car il exige la présence d’une documentation. Une fois un critère évalué, il est impératif d’inscrire un commentaire et si possible une preuve de satisfaction sur lequel le fabricant pourra prouver sa conformité.
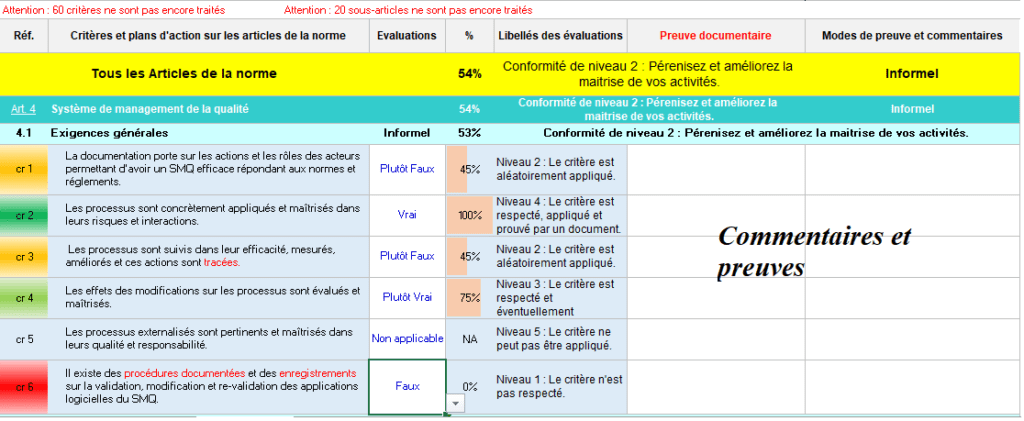
Au cours de l’évaluation, il est mentionné en rouge le nombre de critères et de sous-articles qui n’ont pas été évalués comme indiqué sur la (Figure 17). Cela a été mis en place dans le but de permettre à l’utilisateur de facilement se repérer lors de l’évaluation et de pouvoir se mettre à ligne si un critère a été oublié.
Figure 17 : Onglet _ Evaluation : Début de l’article 4 (Source : Auteurs)
c. Résultats et Résultats par article
Ces deux onglets permettent de donner plusieurs visualisations graphiques des résultats de l’évaluation. Ce qui a pour but d’avoir une représentation rapide sur l’avancement des différents critères et de faire une comparaison de résultats avec le diagnostic précédent.
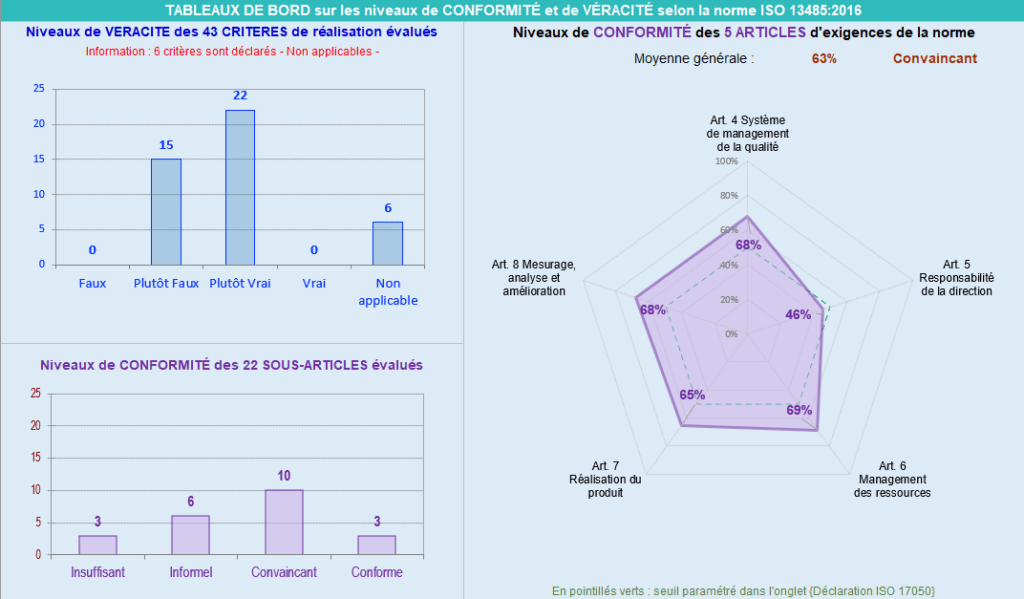
L’onglet « Résultat » donne la représentation de la synthèse globale de l’évaluation des critères, la représentation du niveau de véracité des critères évalués et le niveau de conformité des sous articles évalués représentés par la (Figure 18). Cette représentation donne également le nombre de critères déclarés « Non applicables ».
Figure 18 : Onglet _ Résultats : Niveaux de conformité et de véracité (Source : Auteurs)
Cet onglet présente également un graphe visualisant le bilan global du niveau de respect de conformité aux différents sous-articles de la norme comme présenté à la (Figure 19). Il est également possible pour l’utilisateur d’effectuer des commentaires sur les résultats et de préconiser des plans d’actions crédibles de façon prioritaire pour les graphes présentant des lacunes pour l’amélioration du système du management de la qualité.
Figure 19 : Onglet _ Résultats : Bilan global (Sources : Auteurs)
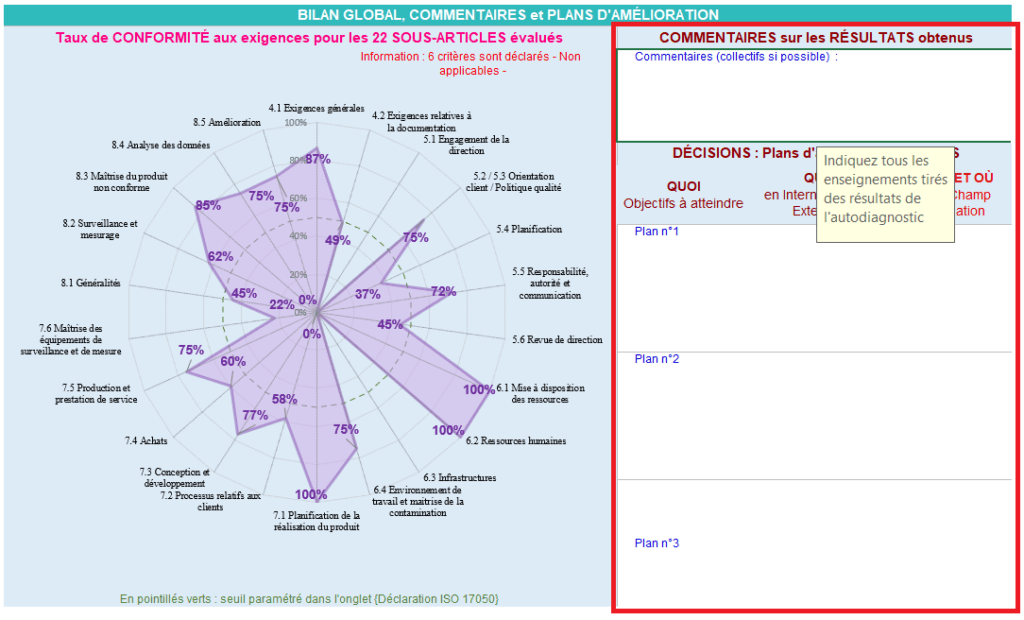
Les pointillés verts ici représentent le seuil minimal pour être conforme paramétré par l’utilisateur depuis l’onglet « Mode d’emploi »
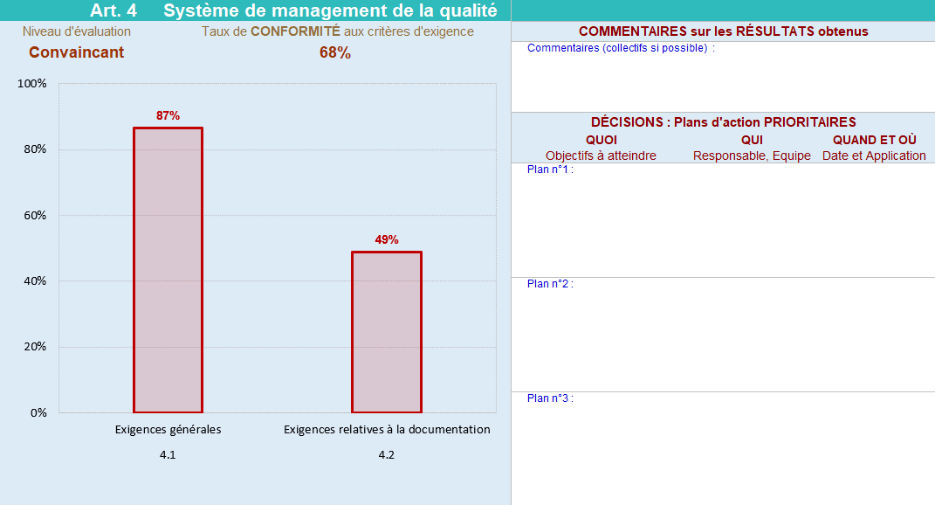
L’onglet « Résultats par article » a le même principe que l’onglet « Résultats » ; mais il est beaucoup plus explicite dans la mesure où il donne des représentations détaillées de chaque article par sous – article comme visualisé à la (Figure 20) qui est la représentation des sous - articles 4.1 et 4.2 de l’article 4 de la norme ISO 13485.
Figure 20 : Onglet _ Résultats par article : Représentation de l'article 4 (Source : Auteurs)
d. Résultats 2017/745 et 2017/746
Ces onglets sont des principales améliorations apportées à l’ancienne version de l’outil. Ils donnent la représentation de l’estimation des niveaux de respect des critères associés aux règlements 2017/745 et 2017/746 vis-à-vis de la norme ISO 13485.
Ils s’illustrent de la manière suivante (Figure 21) :
Figure 21 : Onglet _ Résultats 2017/745 : Article 10 (Source : Auteurs)

Ces onglets ont été établit sur la base de la norme PR NF EN ISO 13485/A1. Comme l’indique cette norme, elle permet de faire la correspondance entre les critères de la norme NF EN ISO 13485 et les exigences des règlements européens 2017/745 et 2017/746. Cette correspondance est axée sur les obligations générales du fabricant mentionnées à l’article 10, les exigences générales du Chapitre I Annexe I et les exigences relatives à l’évaluation de conformité des Annexes IX et XI (règlements 2017/745 et 2017/746).
Comme pour les onglets « Résultats et Résultats par article », ces onglets permettent de donner la visualisation graphique de la correspondance règlementaire qui se fera automatiquement lorsque l’utilisateur sera entrain de remplir l’onglet « Evaluation ».
Certaines exigences réglementaires faisant intervenir plusieurs critères de la norme, une moyenne de ces critères a été faite pour l’élaboration des représentations graphiques.
Il est également important de savoir que ces représentations ont été faite sur la base des exigences réglementaires totalement ou partiellement couvertes par la norme ; ce qui signifie que si l’utilisateur respecte à 100% les exigences de la norme, cela ne signifie pas qu’il respecte également à 100% les exigences règlementaires.
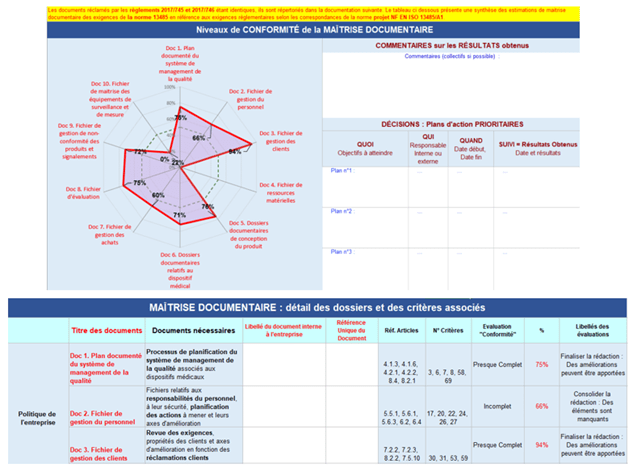
e. Maitrise documentaire
Cet onglet permet de faire un bilan documentaire et donne une visualisation de l’état de conformité des preuves documentaires qu’exige la norme afin de satisfaire aux exigences essentielles de celle-ci comme présenté à la figure 20. Accompagnée de la cartographie documentaire, son utilisation sera très importante aux entreprises souhaitant mettre en place un système de management de la qualité.
Comme pour les onglets « Résultats 2017/745 et 2017/746 », l’élaboration de cet onglet a été basée sur la correspondance entre la norme ISO 13485 et les règlements européens 2017/745 et 2017/746. Après avoir effectué la correspondance entre les exigences règlementaires, il a été constaté que la documentation utilisée pour satisfaire aux exigences de la norme ISO 13485, est identique à celle utilisée pour la satisfaction aux exigences règlementaires concernées par la norme PR NF EN ISO 13485/A1 (Article 10, Annexe I Chapitre I, Annexe IX et XI des règlements).
C’est pour cette raison que cet onglet permet également le suivi documentaire correspondant à la réglementation européenne (Figure 22).
Figure 22 : Onglet _ Maitrise documentaire (Source : Auteurs)
f. Déclaration ISO 17050
Cet onglet permet de faire une auto - déclaration en fonction des résultats obtenus lors de l’évaluation. Cette déclaration est une preuve d’identification (en cas de conformité satisfaisante aux critères d’évaluation) du niveau de respect aux exigences de la norme ISO 13485 : 2016 avant toute certification par un organisme d’accréditation.
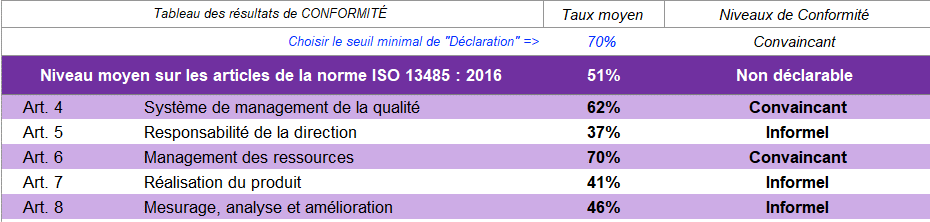
C’est le taux moyen qui est la base (élément important) pour effectuer cette déclaration. Il est choisi par l’utilisateur en fonction de l’objectif de conformité de l’entreprise. Ce taux varie entre plusieurs valeurs tel que : « 50% », « 60% », « 70% », « 80% » et « 90% ».
Cet onglet est également important dans la mesure où il permet de savoir à quel niveau se situe l’entreprise avant d’effectuer un audit. En effet, si le niveau moyen sur les articles de la norme est supérieur au taux moyen fixé alors l’entreprise peut effectuer l’audit. Au cas contraire, il serait judicieux pour l’utilisateur de vérifier dans le tableau, lequel des articles doit être amélioré pour avoir un bon niveau de conformité pour permettre d’effectuer l’audit (Figure 23).
Figure 23 : Onglet _ Déclaration ISO 17050 (Source : Auteurs)
Conclusion
L’utilisation des dispositifs médicaux non conformes a créé de nombreux décès. Pour éviter que ces évènements ne se reproduisent, le nouveau règlement a été établi. Il met donc un accent sur la sécurité du patient et de ce fait implique la complexité d’obtention du marquage CE par des exigences plus strictes. Le principal défi pour les entreprises fabricantes est la compétitivité avec leurs concurrents sur le marché des dispositifs médicaux. Certaines entreprises sont encore dans l’incapacité de se mettre au pas du fait du manque de moyens financiers, et d’autres encore du fait de la non-implication de tous les membres de l’entreprise.
Dans le souci d’accompagner les entreprises vers cette nouvelle démarche, un outil d’autodiagnostic a été établi. Il permettra aux fabricants des dispositifs médicaux de vérifier leur niveau de respect des exigences de la norme ISO 13485, en parallèle avec celles des nouvelles règlementations. Une synthèse des documents liés aux articles de l’ISO 13485 : 2016 a également été élaborée, dans le but de faciliter la lecture de la norme.
Les correspondances entre la norme NF EN ISO 13485 : 2016 et les exigences de la nouvelle règlementation par la norme l'application de la norme PR NF EN ISO 13485/A1 ont été au centre de ce projet. Les outils développés seront mis au service des fabricants afin que ces derniers puissent évaluer la conformité de leur système de management de la qualité selon les exigences de la norme NF EN ISO 13485 : 2016 en fonction des modifications des nouveaux règlements 2017/745 et 2017/746 ; et également d’avoir une satisfaction partielle ou totale à certaines exigences de ces derniers.