
IDS081 - Gestion de l'évaluation biologique des dispositifs médicaux selon la norme NF EN ISO 10993-1:2018
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

Imane ESSAAID 
Kawtar IDIHYA 
Woodeline
PIERRE-LOUIS
Saryane WAOUSSI
Contacts
- Imane ESSAAID : imaneessaaid@hotmail.com
- Kawtar IDIHYA : idihyakawtar@gmail.com
- Woodeline PIERRE-LOUIS : pwoodeline@gmail.com
- Saryane WAOUSSI : saryanewaoussi@gmail.com
Résumé
Les normes de la série 10993 ont pour but d'assurer la sécurité des utilisateurs face aux risques biologiques (cancérogénicité, toxicité…) associés à l'emploi des dispositifs médicaux. Ce mémoire a pour but d'aider toute personne désirant appréhender les risques biologiques pouvant se produire de la conception des dispositifs médicaux à leur mise sur le marché.
Ce projet s’appuie sur la Norme ISO NF EN 10993-1 : 2018 : Évaluation biologique des dispositifs médicaux, Partie 1 : Évaluation et essais au sein d'un processus de gestion du risque. Il définit l’ensemble des données relatives à l’évaluation biologique, détaille ses différentes étapes ainsi que les tests de biocompatibilité réalisés sur les dispositifs médicaux selon leurs classes, et leur contact direct ou indirect avec le corps du patient.
Une analyse détaillée de la norme a permis de mettre à disposition de tous, des outils facilitant la prise en main rapide et efficace de ce référentiel. Ces outils comprennent une cartographie interactive détaillant les étapes à réaliser ainsi qu'un outil d'autodiagnostic permettant de se situer dans l'avancement de la procédure.
Une liste des documents à fournir en vue d'une preuve de conformité vis-à-vis des exigences de cette norme, du Règlement Européen 2017/745, ainsi que les étapes de l’évaluation biologique sont également disponibles dans ces outils.
Mots clés : Dispositifs médicaux, Evaluation biologique, Norme ISO NF EN 10993-1, Biocompatibilité.
Abstract
The purpose of the 10993 series of standards is to ensure the safety of users with respect to the risks associated with the use of medical devices. The purpose of this brief is therefore to help anyone who wishes to understand the biological risks that can occur from the design of medical devices to their marketing.
This project is based on ISO NF EN 10993-1:2018 Biological evaluation of medical devices, Part 1 : Evaluation and testing within a risk management process. It defines all the data related to the biological evaluation, details its different steps and the biocompatibility tests performed on medical devices according to their classes ; in direct or indirect contact with the patient's body.
A detailed analysis of the standard has made it possible to provide everyone with tools to facilitate a quick and efficient handling of the standard. These tools include an interactive map detailing the steps to be taken and a self-diagnosis tool to help the user find his or her way through the procedure.
Keywords : Medical devices, Biological evaluation, ISO NF EN
10993-1, Biocompatibility.
Glossaire
Evaluation biologique : Activité de vérification de la conception qui est faite dans un contexte de processus de gestion du risque plus larges.
Biocompatibilité : Capacité d’un dispositif médical ou d’un matériau à produire une réponse hôte appropriée dans une application spécifique.
Risque biologique : Combinaison de la probabilité de dommage pour la santé survenant suite à des réactions négatives associées à des interactions avec un dispositif médical ou un matériau et de la gravité de ce dommage.
Analyse du risque : Utilisation des informations disponibles pour identifier les phénomènes dangereux et estimer le risque.
Matériau : Polymère naturel ou synthétique, métal ou alliage, céramique ou bien composite, y compris les tissus rendus non viables, utilisé comme dispositif médical ou partie de dispositif médical.
Implant : Dispositif médical destiné à être introduit entièrement dans le corps humain ou à remplacer une surface épithéliale ou la surface de l’œil au moyen d’une intervention clinique et qui est destiné à rester en place après l’intervention.
Sécurité biologique : Absence de tout risque biologique inacceptable dans le contexte de l’utilisation prévue.
Toxique : Pouvant entraîner une réponse biologique négative
Contact direct : C’est le fait pour un dispositif d’entrer en contact direct avec la peau ou les tissus de l’organisme.
Contact indirect : Dispositif médical ou composant de dispositif médical par lequel passe un fluide ou un gaz, avant que le fluide ou le gaz entre en contact physique avec un tissu de l’organisme (dans ce cas, le dispositif médical ou le composant de dispositif médical lui-même n’entre pas en contact physique avec le tissu de l’organisme).
Contact unique : Un même dispositif utilisé seul ou plusieurs de ces mêmes dispositifs utilisés pendant un temps de contact unique et non interrompu.
Contact multiple : Un dispositif devant être changé à plusieurs reprises en fin de vie, dont les temps de contact de son utilisation s’additionnent.
Contact répété : Un dispositif utilisé de façon interrompue et pendant différents temps de contact qui s’additionnent.
Exposition limitée (A) : Dispositifs dont la somme cumulée de durée de contact unique, multiple ou répété est inférieure à 24 heures.
Exposition limitée (A) avec contact transitoire : Dispositifs en contact sur une durée très brève avec le corps humain.
Exposition prolongée (B) : Dispositifs dont la somme cumulée de durée de contact unique, multiple ou répété à long terme est susceptible de dépasser 24 heures, tout en restant inférieure à 30 jours.
Contact permanent (C ) : Dispositifs dont la somme cumulée de durée de contact unique, multiple ou répété à long terme dépasse 30 jours.
Cartographie : Outil d’aide à la compréhension d’un objet de l’étude.
Outil d’autodiagnostic : Interface interactive permettant l’auto-évaluation rapide par rapport à un référentiel.
Sources :
« Guide GMED-Evaluation biologique des dispositifs médicaux selon la norme 10993-1 ». sept. 2020, Consulté le : sept. 27, 2020. [En ligne].
« Norme ISO 10993-1:2018(fr), Évaluation biologique des dispositifs médicaux —Partie 1 : Évaluation et essais au sein d’un processus de gestion du risque »,Ed. Afnor, Paris, www.afnor.org, août. 01, 2018.
Téléchargements

Mémoire d'Intelligence Méthodologique : Gestion de l'évaluation biologique des dispositifs médicaux selon la norme NF EN ISO 10993-1

Outil d'aide à l compréhension de la norme
Mémoire complet :
Gestion de l'évaluation biologique des dispositifs médicaux selon la norme NF EN ISO 10993-1:2018
Introduction
La norme ISO 10993-1 :2018, Évaluation biologique des dispositifs médicaux, Partie 1 : Évaluation et essais au sein d'un processus de gestion du risque, a été créée par le comité technique ISO/TC 194. Cette norme s’adresse aux fabricants afin que l’évaluation biologique de leurs dispositifs médicaux soit correctement effectuée [1]. Elle a pour objectif principal de protéger les utilisateurs des risques biologiques associés à l’usage des différents dispositifs. Pour ce faire, il est nécessaire de recueillir les informations relatives aux matériaux utilisés lors de la conception de ces derniers.
Il s’agit de la première partie d’une série de 20 normes harmonisées, toutes portant sur l’évaluation biologique et basées sur les textes des directives UE 93/42/CEE et UE 90/385/CEE.
Révisée puis publiée en Août 2018, l’ISO 10993-1(2018) n’apporte pas de changements majeurs à la version précédente. A celles déjà existantes dans la version de 2010, elle ajoute certaines définitions importantes pour une meilleure compréhension des exigences réglementaires. Contrairement à la version précédente, elle n’est pas encore harmonisée, ni selon le règlement 2017/745, ni selon la directive 93/42/CEE. Elle éclaircit néanmoins un certain nombre de points, comme la caractérisation chimique des matériaux impérative dans l’évaluation des dispositifs médicaux [2].
Le guide « Use of International Standard ISO 10993-1, "Biological evaluation of medical devices - Part 1 : Evaluation and testing within a risk management process" a été rédigé en Septembre 2020. Il remplace le « Blue Book Memorandum #G95-1 » rédigé par le bureau d’évaluation des dispositifs (Office of Device Evaluation) en 1995. Dans ce guide, de structure similaire à la norme, sont renseignées les recommandations de la FDA (Food Drug Administration) concernant l’évaluation biologique des dispositifs médicaux. En effet, les matériaux utilisés lors de la conception des dispositifs médicaux peuvent induire des effets indésirables sur le patient. Ces effets sont la conséquence de l’incompatibilité desdits matériaux avec le corps humain. Les recommandations de la FDA permettent donc aux industries de déterminer les tests de biocompatibilité nécessaires pour les dispositifs médicaux en contact direct ou indirect avec le corps.
Il est important de noter que, par rapport à la norme, certaines modifications ont été apportées dans ce guide, notamment dans les annexes. Le nombre et le type de tests à réaliser diffèrent et dans la plupart des cas, un plus grand nombre de tests est exigé par la règlementation américaine [3].
L’augmentation des exigences règlementaires dans le monde des dispositifs médicaux traduit la nécessité d’avoir un système d’évaluation biologique robuste. Bien assimiler la complexité des exigences essentielles est donc primordial pour les fabricants de dispositifs médicaux. La question qui se pose est alors la suivante : Comment optimiser l’évaluation biologique des dispositifs médicaux selon la norme ISO 10993-1 :2018 au sein d’une entreprise ?
Afin de répondre à cette problématique, il est important dans un premier temps, de se familiariser avec la norme ISO NF EN 10993-1 : 2018, son contenu, son domaine d’application, et les spécificités de la version 2018. Une fois ce premier objectif réalisé, il sera possible de comprendre les enjeux associés à l’évaluation biologique. Un guide a été élaboré afin d’aider les fabricants à mettre sur pied des dispositifs conformes aux normes en vigueur, et ainsi faciliter l’obtention du marquage CE [4].
1 L’évaluation biologique : Généralités et objectifs
1.1 La sécurité du patient, au cœur de l’évaluation biologique
1.1.1 Les incidents liés à la biocompatibilité
De 2008 à 2014, environ 1 000 000 de prothèses de hanches ont été posées en France [5]. Plusieurs types de prothèses de hanche existent, et parmi elles se trouvent les prothèses totales dites « métal – métal ». L’usure et la corrosion libèrent de petites particules de cobalt en plus ou moins grande quantité dans l’organisme.
Après ce relargage, le cobalt peut atteindre des organes vitaux tels que le foie et les reins. La toxicité locale du cobalt peut entraîner une hypersensibilité ou une tumeur bénigne [6].
Des études, menées aux Etats-Unis par un chirurgien orthopédique ont montré que le cobalt dans le sang, diffusé suite à la pose d’une prothèse de hanche, pouvait avoir une incidence directe sur l’état neurologique du patient, et ceci, après avoir lui-même subi de tels effets secondaires [7].
D’autre part, en 2010, le scandale PIP (Poly Implant Prothèse) a éclaté. L’industrie française découvre alors qu’une entreprise de prothèses mammaires, remplissait ses implants avec du silicone industriel frauduleux. Il a été mis en évidence que les documents fournis par l’entreprise concernant la biocompatibilité de leurs matériaux n’ont pas été vérifiés, mettant alors la vie de dizaines de milliers de femmes en danger [8].
En 2019, l'ANSM décide de retirer du marché les implants mammaires en polyuréthane et en silicone. En effet, le port de ces implants, dits macrotexturés peuvent créer une inflammation chronique qui déclenche l’apparition d’une maladie nommée le Lymphomes Anaplasique à Grandes Cellules (LAGC).
Ces différents incidents poussent à s’interroger sur les enjeux relatifs à l’évaluation biologique des dispositifs médicaux.
1.1.2 Les enjeux humains et économiques liés à cette évaluation
Les exemples précédents mettent en évidence que l’objectif principal de l’évaluation biologique est d’assurer la protection des patients en contact avec les dispositifs médicaux. Pour la sécurité des utilisateurs, il convient de faire une étude qui permettra d’anticiper les risques auxquels ils sont exposés afin de prévoir les alternatives associées. Cette étude permet non seulement d’améliorer la qualité des dispositifs, mais aussi la productivité de l’entreprise fabricante. Ainsi, les utilisateurs auront l’assurance de la performance des équipements d’une part, et d’autre part, cette évaluation accompagnera les entreprises biomédicales tout au long du cycle de vie du DM.
Une durée de contact prolongée et l’implantation d’un dispositif peuvent entrainer un processus plus long et plus détaillé du fait des séquelles que ce dernier est susceptible d’engendrer, ce qui peut s’avérer être un challenge pour les fabricants de dispositifs médicaux.
Il est également intéressant de se pencher sur les coûts financiers associés à cette procédure. Une entreprise de petite envergure qui vient de se lancer dans la conception d’un dispositif médical (ex : Prothèse mammaire, Implants cardiaques…) peut se trouver dans l’incapacité de dépenser une grande somme d’argent, d’autant plus si le nombre de tests à effectuer est important. Certains professionnels interrogés, responsables de l’évaluation biologique au sein de leur entreprise, ont confirmé, que cette problématique était une question majeure lors de ce processus.
Les conséquences sur la santé et les moyens financiers engagés dans cette procédure impliquent d’adopter la bonne stratégie afin de ne pas connaître de trop grandes pertes. Il est donc important de correctement réaliser les tests de biocompatibilité tout au long du cycle de vie du dispositif médical. Cela permettra alors de mesurer l’impact de chacun des constituants, en fonction de leur nature, de leur durée de contact avec le corps humain, et ainsi d’évaluer le risque biologique pour la santé du patient.
1.2 Importance de la biocompatibilité lors de l’évaluation biologique
1.2.1 La biocompatibilité, un élément essentiel tout au long du cycle de vie du DM
Selon le Règlement Européen 2017/745 un dispositif médical est défini comme : « tout instrument, appareil, équipement, logiciel, implant, réactif ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l’homme pour l’une ou plusieurs des fins médicales précises suivantes : diagnostic, prévention, contrôle, prévision, pronostic, traitement ou atténuation d’une maladie […] et dont l’action principale voulue dans ou sur le corps humain n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens.. » [9].
Les matériaux utilisés lors de la fabrication des dispositifs médicaux sont variés :
- Les alliages métalliques : Titane et alliage de titane, Alliage de cobalt…
- Les céramiques : Alumine, Zircone, Hydroxyapatite (HAP)….
- Les polymères ou plastiques
- Les composites
- Les matériaux d’origine naturelle (animale ou végétale)
- Les verres (etc…)
Ces matériaux sont utilisés pour leurs propriétés mécaniques de résistance à la corrosion et leur biocompatibilité. Cette dernière est définie comme « l’aptitude d’un dispositif à être en contact avec un système vivant, sans produire d’effet indésirable inacceptable » [10].
Une étude de biocompatibilité a deux objectifs principaux d’une part, prouver l'absence vraisemblable d'effet délétère du matériau ou dispositif considéré, et d'autre part, d’accumuler des données prédictives du comportement in vivo du matériau ou dispositif. Dans cette stratégie, il faut tenir compte non seulement des caractéristiques et des propriétés (physiques, chimiques, mécaniques et morphologiques), mais aussi de la tolérance de ces matériaux vis-à-vis des contraintes qui leur sont appliquées [11]. Cette notion de biocompatibilité est au cœur de l’évaluation biologique comme décrite dans la norme ISO NF EN 10993-1.
Dans cette dernière un tableau d’aide à la décision permet de déterminer les essais de biocompatibilité nécessaires pour un dispositif médical qui dépend :
- Du type de contact avec le corps humain.
- De la durée de contact avec le corps humain :
- A-Limitée (⩽ 24h)
- B-Prolongée (> 24 h à 30 jours)
- C-Permanente (> 30 jours)
- De la biocompatibilité des matières premières.
- Du procédé de fabrication.
Les essais de biocompatibilité sont à réaliser sur le produit fini qui doit avoir subi l’ensemble des étapes du procédé de fabrication, de la réception des matières premières à l’emballage final et à la stérilisation, si le produit est destiné à être vendu stérile. Il est nécessaire que les échantillons ou prototypes testés soient parfaitement similaires au dispositif médical qui va être commercialisé.
Ces essais doivent être réalisés par :
- Un laboratoire accrédité pour ces essais (accréditation Cofrac en France, par exemple)
- Un laboratoire reconnu par l’organisme notifié
- Ou l’organisme notifié lui-même
Les rapports d’essais sont généralement joints au dossier de marquage CE en annexe tandis que le dossier lui-même comprend :
- Le résumé du protocole
- Les critères d’acceptation
- Les résultats obtenus
- La conclusion sur la biocompatibilité du produit.
Il est fortement recommandé de rédiger tous les rapports des tests en anglais pour faciliter l'accès aux marchés étrangers après l'obtention du marquage CE [13].
Pour entamer cette évaluation biologique, il est important de suivre les exigences des référentiels associés à cette évaluation.
1.2.2 Référentiels liés à l’évaluation biologique : La série de normes NF EN ISO 10993
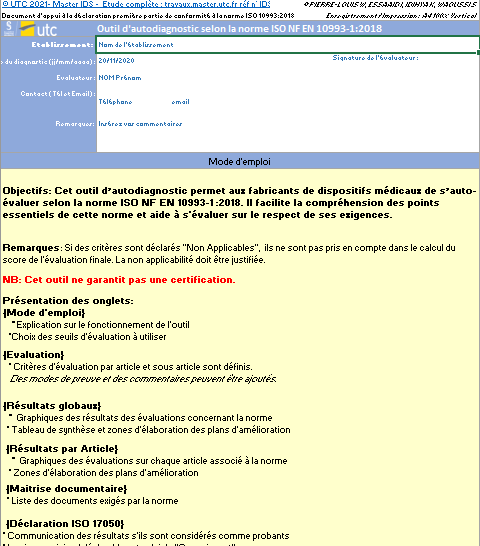
Figure 1 : Importance de l'évaluation biologique et risques liés aux dispositifs médicaux (Source : Auteurs)
Lors de leurs utilisations, les dispositifs médicaux peuvent induire des effets indésirables comme ceux cités précédemment (1.1.1). Les normes de la série ISO NF EN 10993 sont présentes afin d’éviter que des évènements similaires ne se produisent (Figure 1).
Celles-ci offrent un ensemble d’outils à la fois stratégiques, techniques et scientifiques permettant aux fabricants de s’assurer de l’absence de tout risque biologique pour le patient et/ou l’utilisateur d’un dispositif médical [12].
L’évaluation biologique se base sur des éléments et procédures qui évaluent les avantages, les inconvénients et la pertinence : des propriétés physiques et chimiques des différents matériaux des dispositifs médicaux, de tout historique d'usage clinique ou toutes données d'exposition humaine, de toutes données de sécurité biologique sur le dispositif et les matériaux de composition et enfin sur des modes opératoires d'essai.
Le Règlement européen 2017/745 relatif aux dispositifs médicaux traite aussi de l’évaluation biologique et décrit le processus au cours duquel les fabricants doivent identifier, estimer et évaluer les risques biologiques, mais également évaluer le rapport bénéfices/risques global afin de répondre aux exigences réglementaires [13]. Il est également important de définir les responsables de la sécurité biologique des dispositifs médicaux au sein de chaque organisme.
En effet, les techniques d’évaluation biologique sont définies et font partie d’une démarche globale de sécurité biologique qui, elle-même, est intégrée au plan de gestion de risque conformément à l’ISO 14971:2007 [14]. Le plan de gestion de risque est un processus continu qui met en jeu l’identification des dangers biologiques, l’estimation des risques biologiques associés et la détermination de leur acceptabilité. Il doit obligatoirement être mis en œuvre pour l’obtention du marquage CE. Cette étude requiert plusieurs étapes et différents tests doivent être réalisés.
1.2.3 Les étapes de l’évaluation biologique selon la norme NF EN ISO10993-1
Un guide, rédigé par le GMED, a été réalisé pour permettre aux fabricants d’éradiquer ou de réduire au maximum les risques biologiques [16]. Il a pour but d’accompagner les fabricants dans l’évaluation biologique de leur dispositif médical.
Il détaille de manière générale, et selon la norme ISO NF EN 10993-1, les différentes étapes à réaliser [14] :
- Identification des normes caractéristiques pour l'évaluation biologique des dispositifs médicaux :
Cette étape consiste à identifier les normes spécifiques et applicables à chaque dispositif médical et dont le niveau d’exigence est le plus élevé.
- Elaboration, assemblage et conception du dispositif médical :
Il faut ensuite détailler la description du fonctionnement des dispositifs médicaux, leur classe, l’identification de tous les éléments qui les constituent, leur composition chimique, et la spécification du rôle de chaque composant constituant le dispositif médical. Il faut également identifier les étapes de fabrication et leur durée de vie.
- Classification du dispositif médical :
La spécification de la nature (sans contact, au contact d’une surface, communiquant avec l’extérieur, implantable) et de la durée de contact (exposition limitée, prolongée, avec contact transitoire, ou contact permanent) du dispositif médical avec le corps du patient sont à prendre en compte.
- Identification des complications biologiques :
Il faut définir tous les paramètres à prendre en compte lors de l’évaluation, en se basant sur la composition du dispositif médical, sa catégorisation et son utilisation.
- Informations physiques et chimiques pour le rapport des complications biologiques :
Il est nécessaire d’avoir une analyse la plus détaillée possible. En effet, la réalisation d’une liste complète des informations toxicologiques qualitatives et quantitatives du dispositif permet un processus d’évaluation plus simple et plus rapide. Il est nécessaire de mettre l’accent sur cet aspect car les compositions chimiques initiales du dispositif lors de sa fabrication ne sont généralement plus les mêmes en fin de conception.
- Rapport complet :
Si les données précédentes ne sont pas suffisantes pour répondre aux exigences listées à l’étape 4, des tests additionnels sont à prévoir afin de prouver la non-satisfaction des résultats obtenus. Cependant, si les résultats des tests de sécurité biologique issus de ces recherches sont satisfaisants, une analyse globale des résultats peut être effectuée [15].
- Etude complète des résultats :
Elle permet de confirmer la sécurité du dispositif médical à mettre sur le marché. Il est recommandé au fabricant de faire une veille sur les résultats des essais biologiques.
2 Des outils pour mieux appréhender cette norme
2.1 Evaluation des besoins des utilisateurs
Afin de mieux répondre aux attentes des destinataires de ce projet, un sondage en ligne, à destination des entreprises fabricantes de dispositifs médicaux, a été réalisé. Les réponses obtenues ont permis d’identifier les éléments essentiels à intégrer dans les outils réalisés. Parmi ces recommandations, il est notamment ressorti la nécessité d’expliciter les spécificités de la version 2018 de la norme NF EN ISO 10993-1.
Une autre approche, consistait à entrer en contact avec des entreprises fabricantes de dispositifs médicaux. Les questions posées ont permis d’en savoir un peu plus sur ces entreprises et sur leurs méthodes d’application de cette norme.
La plupart des entreprises contactées avaient déjà réalisé les tests préconisés dans la norme (toxicité, cancérogénicité, cytotoxicité…) par l’intermédiaire de laboratoires sous-traitants. Certaines entreprises ont également bien voulu partager leurs données concernant les coûts associés à ces tests. Cela a permis d’évaluer l’investissement financier conséquent lié à ce processus et l’importance d’effectuer les bons tests au bon moment afin de ne pas avoir de pertes économiques.
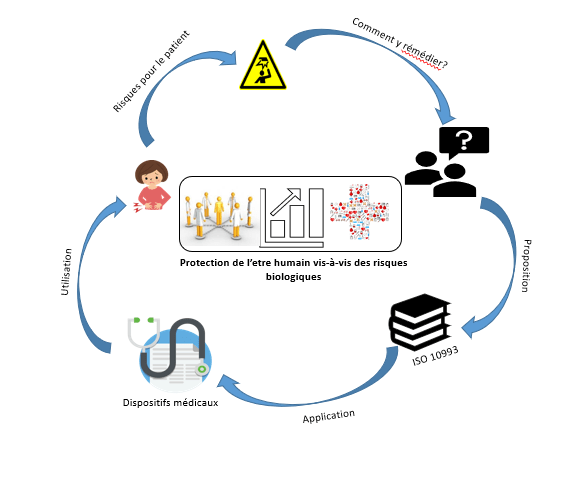
À l’issue de ces entretiens, l’avis de ces entreprises en ce qui concerne l’utilisation d’un guide pour l’évaluation biologique de leurs dispositifs médicaux a été le suivant (Figure 2) :
Figure 2 : Statistiques - Pertinence du projet (Source : Auteurs)
Ces résultats attestent bien qu’un guide à destination des néophytes peut être utile et c’est en se basant sur les informations recueillies que des outils d’appropriation de la norme ont été élaboré.
2.2 Outil d’aide à la compréhension de la norme
Figure 3 : Interface d’accueil – Cartographie (Source : Auteurs)

Les informations fournies par les entreprises ont permis de créer une cartographie adaptée, facile de prise en main et utile (Figure 3). Cet outil, réalisé à l’aide du logiciel PowerPoint, permet à toute personne voulant en savoir plus sur la norme, d’accéder rapidement aux informations dont elle a besoin pour entamer un processus d’évaluation biologique.
a. Mode d’emploi
Au niveau de l’interface d’accueil (Figure 3), un mode d’emploi est accessible via simple clic (Figure 4). Dans celui-ci, est détaillé le mode de fonctionnement de l’outil, l’intérêt de chaque section et les résultats fournis par ce dernier :
Figure 4 : Mode d'emploi – Cartographie (Source : Auteurs)

b. Guide express
Figure 5 : Mode d'emploi – Cartographie (Source : Auteurs)

Cette interface se divise en 4 parties (Figure 5) :
-L’aide à la compréhension de la norme selon trois niveaux de profondeur :
- Un premier par article
- Un par sous-article
- Un par sous-partie des sous-articles (si existants)
Cette partie présente avec des termes simples et compréhensibles de tous, les exigences principales de la norme. Il est également possible de trouver la liste de tous les documents à fournir en vue d’une preuve de conformité selon l’ISO NF EN 10993-1.Le deuxième onglet, La série 10993, présente la norme dans sa série. Les autres normes sont explicitées et leur intérêt dans l’évaluation biologique est développé.
- Le deuxième onglet, La série 10993, présente la norme dans sa série. Les autres normes sont explicitées et leur intérêt dans l’évaluation biologique est développé.
-Le troisième onglet concerne les différentes étapes à réaliser dans le cadre de l’évaluation biologique.
L’onglet suivant, Guides Officiels, concerne les guides pouvant aider à l’évaluation biologique, en France et à l’international.
c. Informations sur les tests
Dans cette partie, chacun des tests présentés dans la norme est défini et des informations supplémentaires sont ajoutées (Figure 6).
Figure 6 : Informations sur les différents tests– Cartographie (Source : Auteurs)

d. Questionnaire : Quel tests pour mon DM ?
Afin que les fabricants de dispositifs médicaux se repèrent facilement sur les tests à réaliser pour leur matériel, un questionnaire rapide, basé sur l’Annexe A de la norme est mis à leur disposition (Figure 7). Des informations sur les tests requis par la Food Drug Administration ainsi que les tests supplémentaires requis dans la version 2018 de la norme y sont renseignés.
Figure 7 : Quel test pour DM ? – Cartographie (Source : Auteurs)

Pour compléter cet outil et afin de se situer vis-à-vis des exigences listées dans cette cartographie, un outil d’autodiagnostic a été développé.
2.3 Outil d’auto-évaluation vis-à-vis de la norme NF EN ISO 10993-1 :2018
L’outil d’autodiagnostic de la norme ISO NF EN 10993-1 :2018 est destiné aux fabricants de dispositifs médicaux souhaitant s’autoévaluer, et avoir une vue globale sur leur conformité aux exigences de cette norme. En effet, il facilite la compréhension des points essentiels et les aide à bien respecter les étapes de cette évaluation.
Cet outil a été réalisé sous format Microsoft Excel. Il comprend 6 feuilles contenant les onglets suivants (Figure 8) :
Figure 8 : Onglets de l’outil d’autodiagnostic (Source : Auteurs)

- Mode d’emploi : Dans cet onglet est détaillé le mode de fonctionnement de cet outil ainsi que le paramétrage des seuils d’évaluation (Figure 9).
Figure 9 : Onglet : {Mode d'emploi} (Source : Auteurs)
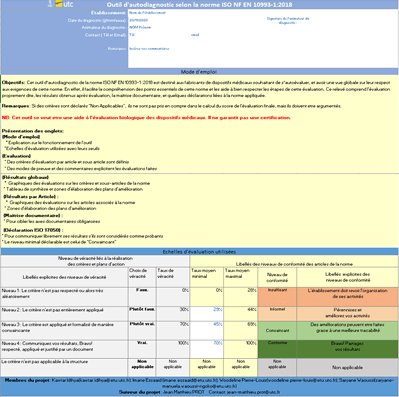
Les onglets comportent des en-têtes permettant de renseigner les données relatives à la procédure d’évaluation : identité de l’utilisateur, de l’organisme ainsi que la date.
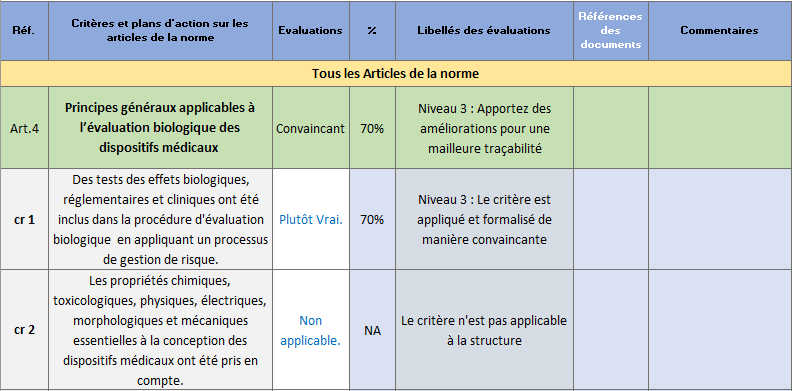
- Evaluation : dans cet onglet, sont listés 100 critères permettant de s’autoévaluer vis-à-vis des exigences relatives à l’évaluation biologique des dispositifs médicaux. Ces critères ont été déterminés grâce à la réalisation d’une analyse opérationnelle de la norme (Figure 10).
L’utilisateur peut donc choisir entre les niveaux de véracité suivants :
Vrai : Le critère est respecté, appliqué et justifié par un document.
Plutôt Vrai : Le critère est respecté.
Faux : Le critère n'est pas respecté.
Plutôt Faux : Le critère n’est pas complètement appliqué.
Non applicable : Critère non applicable.
Figure 10 : Onglet : {Evaluation} (Source : Auteurs)
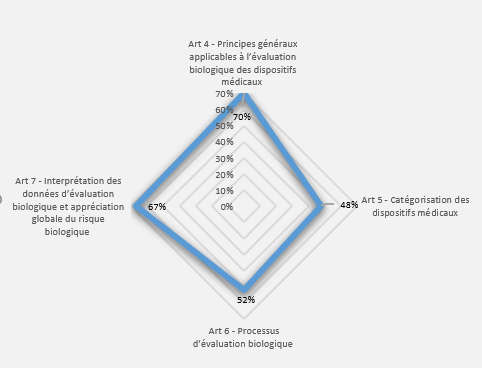
- Résultats globaux : Cet onglet présente les résultats de l’évaluation sous forme de graphes radar (Figure 11).
Figure 11 : Graphe radar – Onglet {Résultats globaux} (Source : Auteurs)
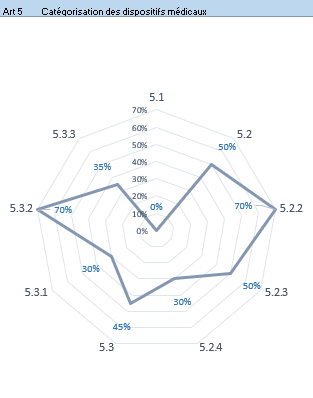
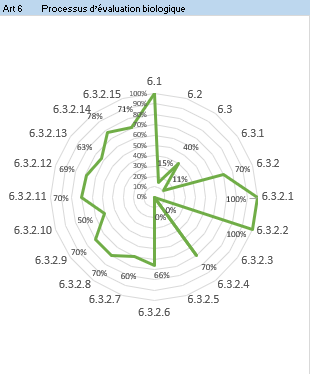
- Résultats par article : cet onglet présente les résultats de l’évaluation pour chaque article sous forme de graphe radar (Figure 12). Cela permet à l‘utilisateur de visualiser les articles pour lesquels il y a une non-conformité et d’établir un plan d’action.
Figure 12 : Articles 5 et 6 – Onglet : {Résultats par articles} (Source/ Auteurs)
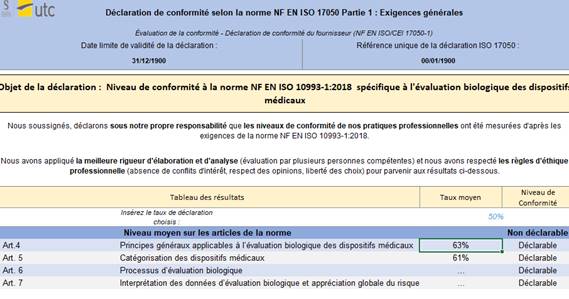
- Déclaration selon l’ISO 17050 : Il s’agit d’un document que remplit l’utilisateur afin de communiquer sa conformité aux exigences réglementaires de la norme ISO NF EN 10993-1 :2018. Cette déclaration ne fait pas office de preuve de conformité (Figure 13).
Figure 13 : Onglet : {Déclaration} (Source : Auteurs)
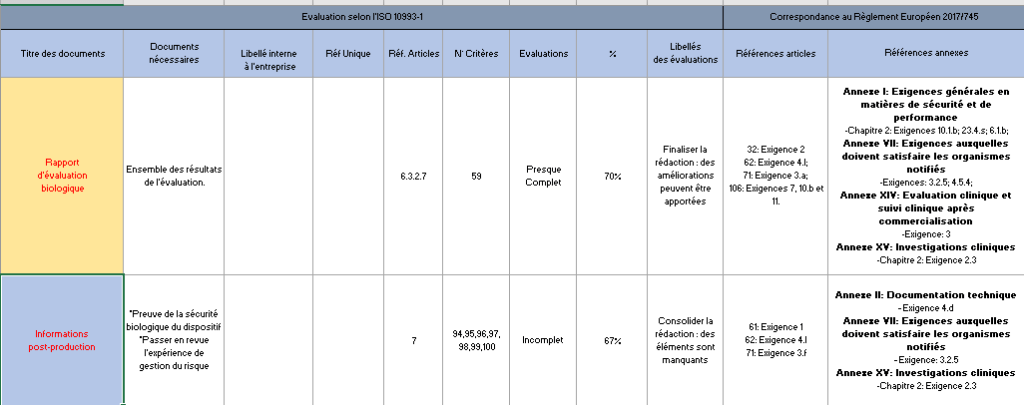
- Maîtrise documentaire : L’onglet maîtrise documentaire donne un aperçu des documents qui doivent être inclus dans le dossier d’évaluation biologique. Il permet de situer son avancement grâce à un graphique et fait également référence aux exigences du Règlement 2017/745 (Figure 14) :
Figure 14 : Tableau récapitulatif – Onglet : {Maîtrise documentaire} (Source : Auteurs)
Conclusion
L'évaluation des interactions possibles et des effets secondaires potentiels des dispositifs médicaux mis en contact avec le corps humain est exigée par les autorités réglementaires.
Les normes de la série ISO 10993 sont présentes afin d’éviter que les risques biologiques ne se produisent. Ils ont pour but de protéger l’être humain contre les risques associés à l’usage des différents dispositifs médicaux.
Les deux outils qui constituent cette aide opérationnelle sont destinés à l’utilisation des fabricants afin de faciliter leur compréhension et l’application de la version 2018 de l’ISO NF EN 10993-1 relative à l’évaluation biologique. Ils ont pour but d’apporter une solution simple d’utilisation afin de comprendre cette norme et de réduire le risque pour le fabricant de faillir à la satisfaction des exigences réglementaires. Cela permet également de garantir la sécurité du patient vis-à-vis des risques biologiques associés à l’usage des différents dispositifs médicaux.