IDS117 - Impacts des nouveaux règlements européens (2017/745 et 2017/746) sur l'ingénierie biomédicale hospitalière
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs




Contacts
- BOUDET Lincey : lincey.boudet22@gmail.com
- EL KHOURY Sylvia : selkhoury29@gmail.com
- MAROT Camille : camille.marot1998@gmail.com
- WAXWEILER Maëva : maeva.waxweiler@sfr.fr
Citation
Lincey BOUDET, Sylvia EL-KHOURY, Camille MAROT et Maeva WAXWEILER, "Impacts des règlements Européens 2017/745 et 2017/746 sur l'ingénierie biomédicale en établissement de santé", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS), Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS117, janvier 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids117/ , DOI : https://doi.org/10.34746/zjxj-x676
Résumé
Avant 2021, certains scandales liés aux dispositifs médicaux ont remis en cause les directives européennes 93/42, 90/385 et 90/79, vis-à-vis de la sécurité du patient. Suite à cela, ces directives ont été remplacées par de nouveaux règlements européens : 2017/745 relatif aux dispositifs médicaux et 2017/746 relatif aux dispositifs de diagnostic in vitro. L’enjeu majeur de ces règlements concerne ainsi la sécurité du patient, en veillant à valoriser les preuves et démonstrations du bénéfice clinique de chaque dispositif médical mis sur le marché.
Les fabricants de dispositifs médicaux sont directement concernés par ces règlements. Cependant, les exploitants, plus particulièrement l’ingénierie biomédicale en établissement de santé, sont eux aussi impactés par cette nouvelle réglementation qui bouleverse en réalité leurs différentes missions et responsabilités.
Il est essentiel de notifier que les dispositifs médicaux possèdent un cycle de vie qui se décompose en deux cycles : un cycle de vie propre à sa fabrication et un cycle de vie propre à son exploitation dans un établissement de santé. Ensemble les fabricants et les exploitants sont donc unis pour la maîtrise de la sécurité du patient tout au long du cycle de vie des dispositifs médicaux. Il est donc essentiel que ces deux acteurs prennent bien conscience des nouvelles exigences imposées par ces nouveaux règlements.
Abstract
Before 2021, multiple European council directives such as the 93/42, 90/385, and 90/79 directives were revisited in regards to patient safety due to multiple scandals concerning medical devices in the EU. Hence, these directives were replaced by new European guidelines including the 2017/745 EU regulation, linked to medical devices, and the 2017/746 instructions, regarding in vitro diagnostic devices. The main goal is the safety of the patient, while still highlighting the clinical benefits of each medical product put on the market.
These new regulations primarily affect medical device manufacturers. Nevertheless, consumers are also impacted by these changes, especially in the field of biomedical engineering within the health sector. These adjustments might alter their mission and responsibilities.
It is important to note that medical devices possess a specific lifecycle. The latter can be subdivided into two additional cycles. The first refers to its production. Whereas the other describes its use in the health sector. The manufacturers and consumers are both accountable for the patient’s safety along the comprehensive lifecycle of a device. Therefore, we can conclude that both actors should, in good conscience, abide by these new regulations.
Téléchargements

Remerciements
Dans un premier temps, nous tenons à remercier l’ensemble de l’équipe pédagogique de l’Université de Technologie de Compiègne et plus particulièrement Monsieur Gilbert Farges pour avoir suivi le projet. Tout au long du projet, ses conseils, retours et sa bienveillance ont été primordiale.
Nous remercions également Madame Valérie Boissard, responsable de la cellule d’ingénierie biomédicale au Centre Hospitalier de Luxembourg, que nous avons eu l’opportunité de rencontrer via des sessions de visioconférence. Son implication durant les différentes phases du projet à été indispensable à l’aboutissement de celui-ci. Nous souhaitons remercier Monsieur Alessio Del Mastro, responsable biomédical au Centre Hospitalier de Compiègne-Noyon, Monsieur Alexandre Jaborska, ingénieur biomédical au CHU d’Amiens pour leurs retours au cours du projet. Ils ont aussi pris le temps de répondre à nos mails et nos sollicitations
De plus, nous souhaitons remercier tous les acteurs biomédicaux ayant répondus au sondage élaboré par notre équipe projet. Les résultats du sondage nous ont permis de rendre notre projet plus concret et plus enrichissant.
Enfin, nous souhaitons remercier nos camarades de classe dont Monsieur Mickaël Bourjac, Monsieur Julien Charton, Monsieur Lucas Zugaj ainsi que Monsieur Laurent Blanpain qui nous ont partagé leurs expériences ainsi que leurs contacts pour nous aider à mener à bien notre projet.
Abréviations
- ANSM : L'Agence nationale de sécurité du médicament et des produits de santé
- BPAC : Bonne Pratique d'Activités Connexes
- COFRAC : Comité français d'accréditation
- DM : Dispositif Médical
- GMAO : Gestion de la Maintenance Assistée par Ordinateur
- GHT : Groupement Hospitalier de Territoire
- HAS : Haute Autorité de Santé
- ON : Organisme Notifié
- SBM : Service d’ingénierie biomédicale
- SNITEM : Syndicat National de l'Industrie des Technologies Médicales
- UNCAM : Union Nationale des Caisses d’Assurance Maladie
Mémoire complet
Impacts des nouveaux règlements européens (2017/745 et 2017/746) sur l'ingénierie biomédicale hospitalière
Introduction
Les dispositifs médicaux sont des équipements réglementés utilisés dans le domaine de la santé, qui ont pour objectif principal d’améliorer la qualité de vie des patients, jusqu’à même de sauver des vies. Ils ont la particularité d’être très diversifiés et d’être ainsi répartis en quatre classes selon le niveau de risque qui leur est attribué. Ce sont donc des équipements qui jouent un rôle particulièrement important dans le domaine de la santé et pour preuve, c’est un secteur qui représente un chiffre d’affaires s’élevant à près de 30 milliards d’euros selon le SNITEM durant l’année 2019 en France [1]. De plus, plusieurs autres acteurs entrent en jeu au cours du cycle de vie des dispositifs médicaux, comme l’ingénierie biomédicale. En effet, l’ingénierie biomédicale est l’exploitant de ces dispositifs. Ainsi, la responsabilité de l’ingénieur biomédical est engagée pour garantir la sécurité du personnel soignant et du patient.
C’est notamment pour toutes ces raisons, que la commission européenne établit une réglementation très stricte pour assurer la sécurité de leurs usagers et la qualité de ces dispositifs. Cette réglementation européenne, se chargeant d’encadrer la commercialisation de ces produits, est ainsi en constante évolution et exige une démonstration favorable et optimale d’un rapport bénéfice/risque en termes d’exigences de sécurité et de performances [2].
Cette évolution a conduit ainsi à la mise en place de deux nouveaux règlements européens 2017/745 et 2017/746 visant à renforcer de manière significative les prérequis nécessaires à l’obtention du marquage CE, tout en mettant en œuvre des outils pour assurer une traçabilité et une transparence notables. Ce changement actuel de réglementation définit de nombreux acteurs économiques, tels que le fabricant, le mandataire, l’importateur ou encore le distributeur, tous partie prenante dans la mise sur le marché du dispositif médical [3].
Chacun de ces acteurs présentent désormais des responsabilités et obligations parfaitement établies et inscrites dans ce nouveau règlement.
Cette mise en application va nécessiter un enjeu considérable en termes d’adaptation pour chacun de ces acteurs, et pour toutes les entreprises de manière générale. Cependant, en parallèle de ces acteurs, et du système de santé français et européen, les institutions hospitalières restent avant tout l’utilisateur final de ces dispositifs médicaux. Leurs responsabilités sont fortement impactées vis-à -vis de l’exploitation de ces équipements. Ainsi, en lien avec l’article de madame Valérie Boissart (Ingénieur biomédical du Centre Hospitalier de Luxembourg), il est donc important de s’intéresser ici aux exigences de ces règlements qui peuvent impacter à la fois le fabricant et l'exploitant ?
Ce mémoire de projet est rédigé afin d’aider l’ingénierie biomédical dans les établissements de santé responsable de la gestion des équipements médicaux à adopter les bonnes actions afin de respecter les deux nouvelles règlementations 2017/745 et 2017/746.
I. Ingénierie biomédicale et réglementation au service des patients
A. Ingénierie biomédicale
1. Fonction de l'ingénierie biomédicale
La fonction de l’ingénierie biomédicale est l'expertise ; une expertise qui est technologique, technique et physique dans le domaine de la santé, appliquée à la gestion des dispositifs médicaux (DM).
Ces expertises sont appliquées sur les technologies des dispositifs médicaux au bénéfice de leur qualité et de leur sécurité. Tout ceci, afin de prodiguer une qualité et une sécurité des soins délivrés aux patients par des soignants qui utilisent ces DM. L’ingénierie biomédicale est donc au service de la sécurité du patient et du personnel soignant et médical.
En outre, le responsable biomédicale (ingénieur biomédical) a une fonction qui est transversale. En effet, il est le lien entre l'ensemble des utilisateurs de dispositifs médicaux, que ce soit les professionnels de santé, la direction de l’institution, les techniciens ainsi que les fournisseurs.
Afin de parvenir à la fonction de l’ingénierie biomédicale, il existe plusieurs organisations différentes dans les établissements de santé.
2. Organisation de l'ingénierie biomédicale
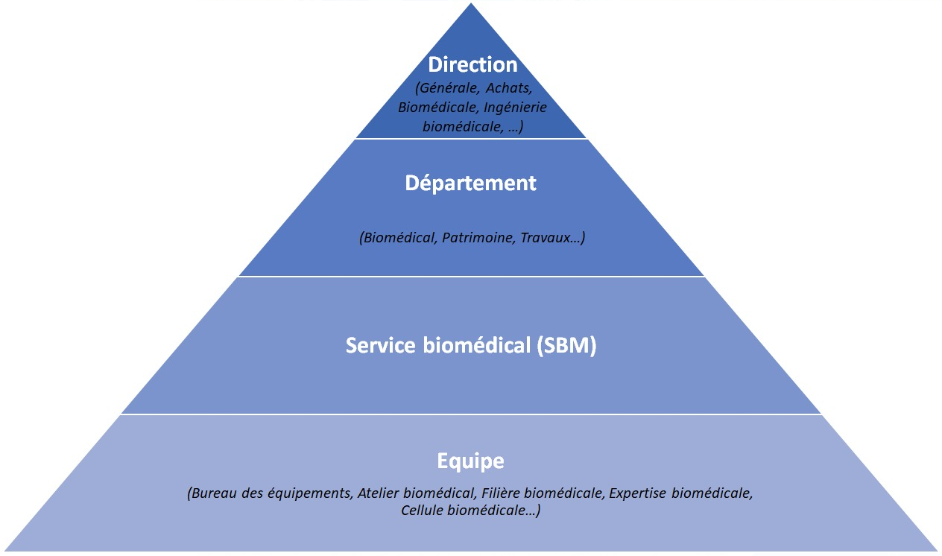
Il existe différents types de positions, organisation hiérarchiques et appellations qui visent à atteindre la même fonction (Figure 1) [4] :
- Service biomédical (SBM) : Le service biomédical représente une entité à part entière dans un centre hospitalier. Le service biomédical met en œuvre les moyens humains et matériels afin d’assurer la disponibilité et le fonctionnement optimal des équipements biomédicaux au sein de l’établissement, tout en respectant les conditions exigées par la réglementation européenne.
- Département : Il existe différents départements tels que Biomédical, Patrimoine, Travaux…
- Direction : Il existe différentes directions telles que Générale, Achats, Biomédicale, Ingénierie biomédicale, Travaux, … Pour ce qui est de la direction Biomédicale, elle existe lorsque le département biomédical est au sein d’un GHT par exemple.
- Structures transversales : Bureau des équipements, Atelier biomédical, Filière biomédicale, Expertise biomédicale, Cellule biomédicale…
Figure 1 : Positions hiérarchiques et appellations associées à la fonction de l'ingénierie biomédicale (source : auteurs)
La fonction de l’ingénierie biomédicale dans l’organisation de santé est principalement transversale et pluridisciplinaire. En effet, elle possède diverses missions telles que l’achat, la maintenance, l’analyse des besoins … Ceci éclaircit le fait que la fonction de l’ingénierie biomédicale possède des rattachements hiérarchiques divers dans les différents organigrammes des établissements de santé [5].
De plus, il ne faut pas oublier l’ingénierie biomédicale au service de l’industrie. En effet, les ingénieurs biomédicaux sont également présents dans les entreprises de l'industrie biomédicale. Leur qualité d'expert permet aux entreprises d’améliorer la qualité des soins et de répondre aux besoins du secteur de la santé, notamment en termes d’innovations médicales.
3. Les différentes missions de l'ingénierie biomédicale
Les missions de l’ingénierie biomédicale servent à contribuer à la sécurité du patient et du personnel médico-soignant par la qualité et la disponibilité des DM et donc la qualité et l’efficacité des services de soins.
En effet, le service biomédical est chargé de la gestion du parc d’équipements médicaux, soit l’inventaire, l’étiquetage et le suivi du dispositif de son arrivée dans l’établissement jusqu'à sa réforme. La gestion du parc inclut également les maintenances préventives et curatives des équipements. Enfin, la matériovigilance qui représente le suivi des incidents ou risque d’incidents afin de mettre en place des actions préventives et/ou correctives constitue aussi l’une des principales missions du service biomédical [6].
Tout cela est généralement géré depuis la GMAO afin d’assurer la traçabilité des équipements (Gestion de la Maintenance Assistée par Ordinateur). En d’autres termes, le service biomédical est un acteur majeur dans la partie de l’exploitation du cycle de vie des dispositifs médicaux au sein d’un établissement de santé, et doit répondre à différentes missions pour permettre une bonne gestion de l’ensemble des dispositifs médicaux. Il constitue un véritable prestataire de services. Les moyens humains du service sont généralement fonction de la taille de l’établissement. L’équipe biomédicale étant composée de techniciens biomédicaux et d'ingénieurs ou responsables biomédicaux.
Les missions de l’ingénierie biomédicale sont donc de différents types (figure 2) : principales, managériales, spécifiques, et transversales [6], [7].
Figure 2 : Listes des missions de l'ingénierie biomédicale (source : auteurs d'après [6], [7] )

B. Réglementations nationales et internationales
1. Différence entre directives et règlements
En Europe, les règlements et les directives sont d’application obligatoires. Cependant, il existe une différence entre les directives et les règlements en termes d’application de ceux-ci. En effet, les règlements, comme le règlement 2017/745, sont applicables tels qu’ils sont rédigés alors que les directives peuvent être transcrites avant d’être appliquées. C’est-à-dire que celles-ci sont à traduire selon chaque membre de l’Union Européenne, ce qui signifie que chaque pays a la possibilité d’interpréter ces directives différemment selon le contexte national. Elles ne connaissent pas de principe d’harmonisation strict. En ce qui concerne les normes, elles sont d’applications volontaires.
2. La fin des directives 93/42, 90/385, 98/79 et réglementation en vigueur
Avant 2017, plusieurs directives étaient mises en place afin d’assurer la conformité aux exigences européennes des dispositifs médicaux (DM), des dispositifs médicaux implantables et actifs (DMIA) et des dispositifs médicaux de diagnostic in vitro (DMDIV). Si les équipements médicaux sont conformes à ces directives, un marquage CE leur est attribué afin de permettre leur mise sur le marché européen. Les directives concernées sont les suivantes (figure 3) [8]:
Figure 3 : Les directives relatives aux dispositifs médicaux (source : auteurs)
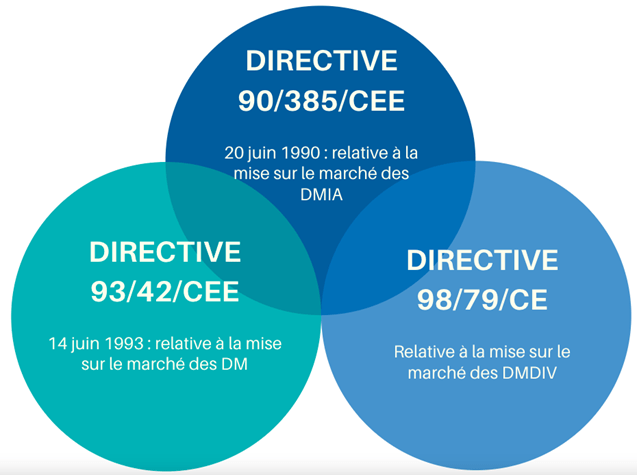
Cependant, depuis le 26 mai 2021, ces directives sont devenues obsolètes suite à la mise en place et l'entrée en vigueur des nouveaux règlements européens.
En 2017, deux règlements ont été élaborés par la commission européenne : le règlement 2017/745 et le règlement 2017/746. Le règlement 2017/745, visant à remplacer la directive 90/385/CEE et la directive 93/421/CEE. Quant au règlement 2017/746, il vise à remplacer la directive 98/79/CE. Depuis le 26 mai 2021, le règlement 2017/745 est entré en application.
Et c’est seulement à partir de 2022 que le règlement 2017/746 entrera en application. Il est bon de rappeler qu’en raison de la crise sanitaire de la Covid-19, l’application de ces règlements a été retardée d’une année.
Cependant, c’est à partir de mai 2025, que tous les dispositifs médicaux devront être conformes aux nouveaux règlements pour être commercialisés (figure 4).
Figure 4 : Calendrier d'application des règlements 2017/745 et 2017/746 (source : auteurs)
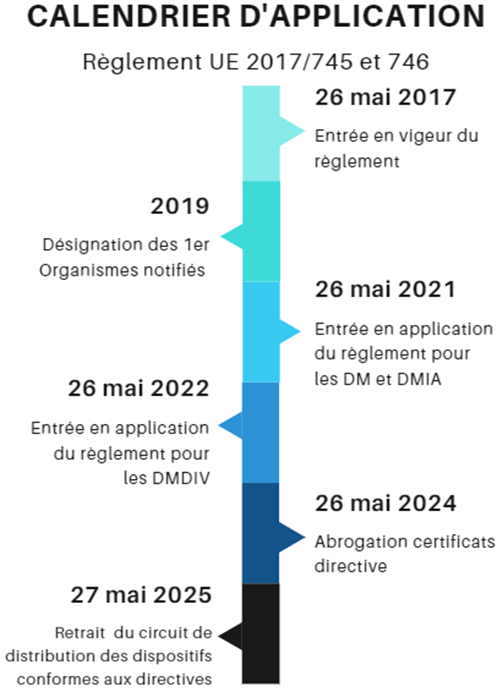
Dans le règlement 2017/745, de nombreuses exigences ont évolué ainsi le champ d’application a été élargi. Ces évolutions permettent de réglementer les étapes du cycle de vie du dispositif médical impactant majoritairement le fabricant.
En France, la réglementation en vigueur depuis l’application du nouveau règlement a changé, voici une liste non exhaustive des textes de lois constituant la réglementation Française [9]:
- Règlement (UE) 2017/745 du 5 avril 2017 relatif aux dispositifs médicaux
- Décret 2018-1186 du 19 décembre 2018 : relatif à la maintenance des Défibrillateurs Automatique Externe (DAE)
- Loi 2018-527 du 28 juin 2018 relative au défibrillateur cardiaque
- Arrêté du 12 Avril 2017 relatif aux lits médicaux
- Arrêté du 3 mars 2003 relatif aux contrôles qualité
- Décret n°2001-1154 du 5 décembre 2001 concernant l’obligation de maintenance et de contrôle qualité des dispositifs médicaux.
- Loi n° 98-535 du 2 juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits à l’homme
- Décret du 17 janvier 1996 relatif à la matériovigilance des dispositifs médicaux
- Arrêté du 3 octobre 1995 relative aux modalités d’utilisation et de contrôle des dispositifs médicaux
- Décret du 16 mars 1995 relatif aux dispositifs médicaux
- Décret 94-1050 du 5 décembre 1994 relatif à la « Sécurité anesthésie ».
3. Comparaison entre le règlement 2017/745 et le règlement 2017/746 et leurs points clés
Ces deux nouveaux règlements européens se chargent alors de faire évoluer le cadre réglementaire en imposant divers changements, qui ont pour objectif principal d’augmenter la qualité et la sécurité de ces dispositifs médicaux au regard des patients.
Le règlement européen 2017/745 est relatif et encadre les dispositifs médicaux, tandis que le règlement 2017/746 se charge des dispositifs médicaux de diagnostic in vitro (DMDIV).
Pour mettre en œuvre cet objectif, les règlements visent principalement à renforcer la démonstration de la sécurité des produits en choisissant d’implémenter de nouvelles règles notamment dans la désignation, l’organisation et la surveillance des organismes notifiés. Mais aussi dans l’évaluation clinique, la traçabilité et la surveillance après commercialisation. Ces deux règlements sont très similaires, mais quelques différences restent cependant notables [10], [11]. (Voir annexe n°1 présentant le tableau 3 de comparaison entre le règlement européen 2017/745 et 746 et leurs points clés).
C. La sécurité du patient et des professionnels de santé : un enjeu majeur
1. Pourquoi cette nouvelle réglementation ?
Le secteur des dispositifs médicaux est un secteur particulièrement réglementé et soumis à de nombreuses exigences concernant leur mise sur le marché. Les dispositifs médicaux sont régis par des directives, depuis maintenant plus de 25 ans. Cependant, comme expliqué précédemment, chaque pays a la possibilité d’interpréter ces directives différemment selon leur langue natale, elles ne connaissent pas de principe d’harmonisation. Or, le secteur des dispositifs médicaux connaît depuis de nombreuses années, certains scandales qui accusent ces directives de ne plus assurer la sécurité des patients et la qualité de ces dispositifs [3].
Par conséquent, la mise en place de ce nouveau règlement vise principalement à unifier et harmoniser l’ensemble des exigences du dispositif médical sous un seul et même règlement. Ces nouveaux
règlements européens 2017/745 et 746 représentent un enjeu majeur pour renforcer la sécurité des patients vis-à-vis de l’utilisation de ces dispositifs médicaux.
Comme cité précédemment, ces principales modifications précisent ainsi les prérequis que nécessitent le marquage CE, sans compter une précision des outils de traçabilité et une transparence désormais plus présente pour les patients, qui de nos jours veulent être de véritables acteurs de leur santé.
2. Le scandale des prothèses PIP
Parmi ces scandales, on peut citer le cas des prothèses mammaires Poly Implant Prothèse (PIP). En effet, ce scandale a dévoilé au grand jour une fraude de la société PIP, qui utilisait un gel de silicone différent de celui qui était indiqué dans le dossier de conception. Et pour lequel les analyses physico-chimiques ont prouvé sa non-conformité puisque le silicone industriel était responsable notamment d’une inflammation des nœuds lymphatiques, une fois celui-ci répandu dans l’organisme. Cette fraude a permis de soulever un problème majeur concernant le non-respect des exigences et des directives qui peut donc avoir un impact considérable en termes de santé publique [12].
3. Les changements côté patient en raison de l'application des nouveaux règlements européens
De nombreux changements côté patient sont donc à prévoir après l’application de ces nouveaux règlements (figure 5) :
Figure 5 : Les principaux changements pour le patient dus aux nouveaux règlements européens (source : auteurs)
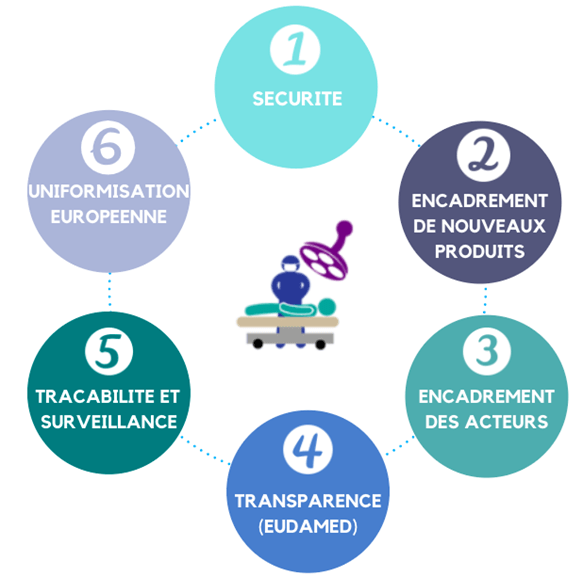
- Concernant la sécurité, le règlement prévoit augmenter le niveau de sécurité en valorisant les preuves et les démonstrations du bénéfice clinique des dispositifs médicaux mis sur le marché afin d’assurer une vérification de la sécurité et de la performance des produits plus importante [13].
- L’encadrement des nouveaux produits s’est également élargi aux produits n’ayant pas de finalité médicale mais qui présentent tout de même des risques similaires, comme le cas des lentilles de contact non correctrices [13].
- En plus de l’encadrement des produits, le nouveau règlement prévoit également un encadrement des acteurs en précisant davantage les obligations de chacun pour garantir une conformité indiscutable avec les règles de sécurité [13].
- La transparence représente aussi un véritable changement dans l’évolution de la réglementation, en mettant en œuvre des notices, carte d’implants, et une base donnée (EUDAMED), permettant de rendre les informations relatives aux dispositifs médicaux directement accessibles aux patients, aux organismes de réglementation, aux opérateurs économiques, et aux prestataires de soins de santé de l’Union Européenne [11], [13].
- De plus, pour assurer une traçabilité et une surveillance optimale des dispositifs médicaux, un identifiant unique a été attribué pour chaque produit. Ce nouvel outil facilite largement le suivi des dispositifs médicaux tout le long de leur cycle de vie [13].
- L’uniformisation européenne est aussi un changement majeur, puisque cela va permettre d’appliquer les mêmes règles dans toute l’Union Européenne pour tous les patients [13].
II. Fabricants, exploitants : unis pour la maîtrise de la sécurité tout au long du cycle de vie du dispositif médical
La mise en place des nouveaux règlements européens concernant les dispositifs médicaux précise les rôles et les obligations des différentes parties prenantes concernées par la nouvelle réglementation. Il est donc intéressant de mener une réflexion structurée vis-à-vis de l’impact de celle-ci.
A. L'impact du règlement sur le cycle de vie d'un dispositif médical
1. Définition d'un dispositif médical (DM)
La nouvelle réglementation 2017/745 a modifié la définition des DM. Elle inclut en plus des instruments cités dans la directive 93/42/CEE les implants [14], les réactifs et tous les équipements médicaux permettant de prédire ou de pronostiquer une maladie et de faire de l’investigation pour une fonction anatomique. Les produits qui servent à communiquer des informations à travers un examen in vitro d’échantillons provenant du corps et ceux destinés à la stérilisation des dispositifs sont considérés comme des DM ( Voir annexe n°2 ).
De plus, la réglementation 2017/745 s’applique également pour les dispositifs non médicaux (d’après l’article 1 paragraphe 2), qui sont par exemple : les lentilles non correctrices, les appareils d’épilation à lumière pulsée intense, les équipements utilisés pour la liposuccion etc... Ces dispositifs cités dans l’annexe XVI malgré qu’ils n’aient pas une visée principalement médicale, ils présentent des risques importants c’est pourquoi il est nécessaire de durcir les exigences qui les concernent pour assurer la sûreté et la fiabilité de leur utilisation [15].
Le changement de cette définition et l’inclusion de dispositifs non médicaux obligent les acteurs qui orbitent autour de ces produits à revoir le statut des dispositifs déjà sur le marché et ceux en cours de développement [16].
Ces acteurs sont essentiellement l’exploitant et le fabricant. De plus, les nouvelles exigences apportées par la réglementation 2017/745, impactent plusieurs étapes des cycles de vie des DM, cela induit des modifications au niveau de la responsabilité des acteurs [10].
2. Les cycles de vie du DM
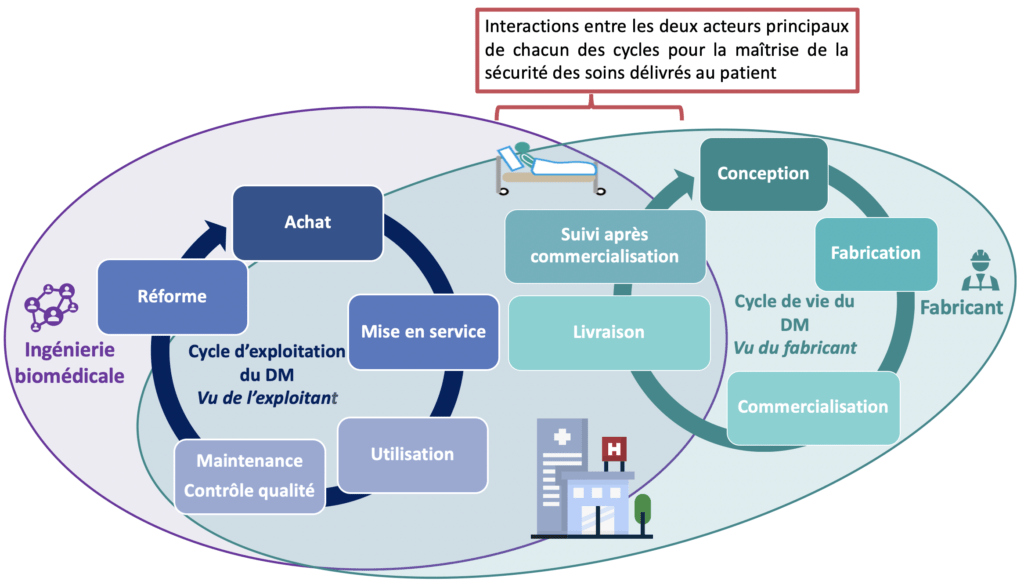
Un cycle de vie est la succession d’étapes que traverse un produit tout au long de sa durée de vie. Les dispositifs médicaux possèdent deux cycles de vie différents. Il possède un cycle de vie qui correspond au moment où ils sont exploités et donc il implique les exploitants du DM et un second cycle de vie qui renferme les étapes de fabrication et de mise sur le marché du DM. Ce dernier dépend essentiellement du fabricant. Dans chacun de ces cycles les rôles et les responsabilités des acteurs respectifs sont spécifiés. Cependant, certaines étapes de ces cycles de vie sont communes aux deux acteurs.
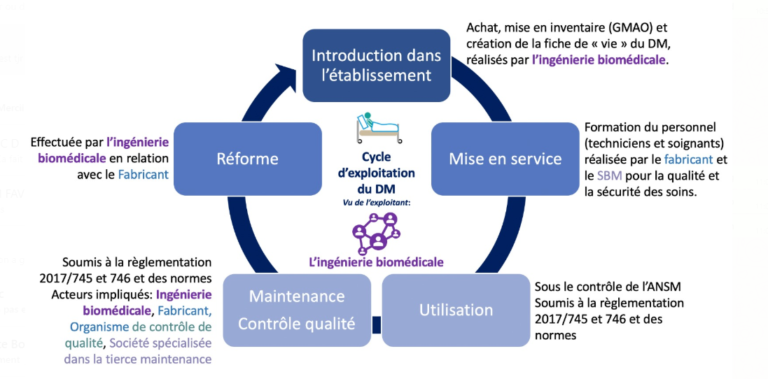
a. Le cycle d'exploitation du DM vu de l'exploitant
Ce cycle concerne l’exploitant qui est le service d’ingénierie biomédicale. Un de ses rôles est de mettre en place un plan et de gérer les équipements médicaux de l’établissement de santé auquel il appartient. Une fois que l’ingénierie biomédicale a retenu l’offre proposée par un des fabricants, le dispositif médical est introduit au sein de l’établissement : il est enregistré dans la GMAO, de cette manière il fait partie de l’inventaire de l’établissement de santé. Ensuite, une formation est effectuée par le fabricant retenu et le service d’ingénierie biomédicale au personnel soignant et aux techniciens. Après la mise en service, le DM peut être utilisé tout en déclarant la matériovigilance auprès de l’ANSM et en effectuant son contrôle de qualité. Ce contrôle de qualité consiste à évaluer les maintiens des performances définis par le fabricant ou ceux établis par le directeur de l’ANSM. Il se fait en interne par l’exploitant ou par un prestataire, et en externe par un organisme de contrôle de qualité externe accrédité par le COFRAC et agréé par l’ANSM. Dans les deux cas, il est sous la responsabilité de l’exploitant du DM [17].
En plus du contrôle de qualité, le service d’ingénierie biomédicale doit effectuer régulièrement la maintenance du DM. Cette maintenance est réalisée par les exploitants ou le fournisseur du dispositif ou une société spécialisée dans la tierce maintenance. Elle renferme toutes les activités ayant pour but de maintenir ou de restaurer l’état du DM afin de préserver la sûreté de son fonctionnement [18]. Finalement, la dernière étape du cycle d’exploitation du DM est sa réforme. Il ne faut pas oublier que les patients sont au centre du cycle d’exploitation des DM, l’objectif principal est d'assurer une bonne prise en charge des patients tout en utilisant les DM. Dans la figure 6 ci-dessous, le cycle complet d’exploitation du DM avec les différents acteurs est présenté.
Figure 6 : Schéma du cycle d’exploitation du DM vu de l’ingénierie biomédical montrant les différents acteurs (source : auteurs)
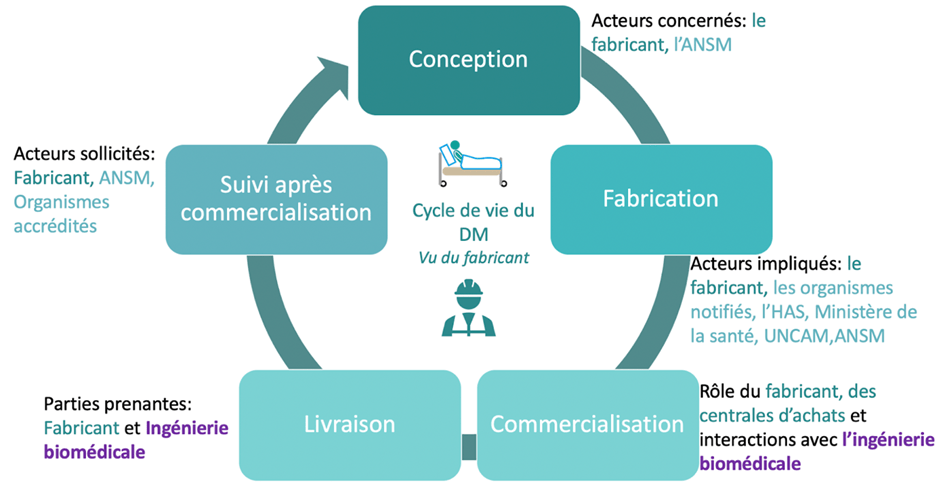
b. Le cycle de vie du DM vu du fabricant
Ce sont les fabricants des dispositifs médicaux qui sont impliqués dans ce cycle de vie. Il débute par la conception du DM qui se fait après avoir effectué une étude de marché et de faisabilité et obtenu l’accord de l’ANSM.
Ensuite, la fabrication commence durant cette procédure le fabricant doit obtenir le marquage CE et il peut demander l’évaluation de la prise en charge de son DM afin qu’il soit remboursé. Pour obtenir le marquage CE il doit selon la classe de son DM faire appel ou non à un organisme notifié.
Par la suite, le DM peut être commercialisé à travers les centrales d’achats ou vendu directement aux établissements de santé cela dépend de leurs statuts s’ils sont des établissements de santé privés ou publics. Après avoir livré l’équipement, le rôle du fabricant ne s’achève pas, il doit faire le suivi après commercialisation.
Pour cela, il doit effectuer des audits et maintenir le marquage CE. Le règlement 2017/745 impose au fabricant de fournir un PSUR (rapport périodique actualisé de sécurité) pour toutes les classes sauf la classe I, pour cette dernière un rapport de surveillance après commercialisation est suffisant. Il est donc nécessaire pour le fabricant de mettre en place un plan de surveillance après commercialisation pour collecter toutes les informations et par la suite les analyser. Le fabricant agit également en faveur de la sécurité du patient et des usagers du DM. Le schéma suivant reprend les grandes étapes du cycle de vie du DM vu du fabricant (figure 7) , [19].
Figure 7 : Schéma du cycle de vie du DM vu du fabricant avec les acteurs impliqués dans les différentes étapes (source : auteurs)
3. Les interactions fabricant et exploitant dans la maitrise de la sécurité
Comme montré précédemment, il existe deux cycles de vie pour les DM, un qui montre l’exploitation de ce dernier et l’autre sa fabrication et sa mise sur le marché. Mais tous les deux ont doivent assurer la sécurité du patient et des utilisateurs du DM. Cet objectif commun a été renforcée par la nouvelle réglementation 2017/745 notamment dans l’annexe I intitulé « Exigences générales en matière de sécurité et de performances ».
Dans cette annexe, les exigences que doit effectuer le fabricant pour atteindre cet objectif sont bien explicitées. Le service d’ingénierie biomédicale n’est pas mentionné mais la sécurité du patient et des utilisateurs du DM est une de ces préoccupations majeures. Il assure cette sécurité en formant les usagers du DM, en établissant des contrôles de qualité et de maintenance et en déclarant les matériovigilances.
Dans toutes ces étapes, le service d’ingénierie biomédicale interagit avec le fabricant pour aider l’exploitant à assurer la sécurité des patients et des utilisateurs du DM. De ce fait, comme il existe des interactions entre le fabricant et l’exploitant pour la maîtrise de la sécurité, la conclusion est que le service d’ingénierie biomédicale doit revoir ces responsabilités suite à la mise en place des exigences de la réglementations 2017/745 qui concernent principalement le fabricant.
La figure 8 met en avant les interactions entre le fabricant et l’exploitant qui sont unis pour la maitrise de la sécurité du patient et des utilisateurs du DM :
Figure 8 : Schéma récapitulatif du cycle de vie et du cycle d’exploitation des DM montrant les interactions entre les deux acteurs (source : auteurs)
B. Responsabilité du fabricant
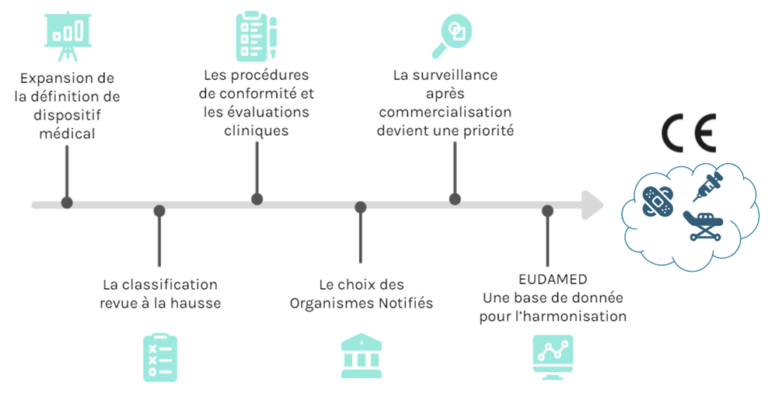
Les responsabilités du fabricant ont été grandement impactées par les nouveaux règlements 2017/745 et 2017/746. En effet, les fabricants de dispositifs médicaux sont largement et directement impactés par l’entrée en vigueur de ces règlements. Ces règlements font évoluer la réglementation au niveau de toutes les étapes de la mise sur le marché des dispositifs médicaux, dont le champ d’application a été élargi. Dans cette partie, seront explicités les grands changements pour le fabricant apportés par ces règlements.
1. Renforcement de la réglementation avant la mise sur le marché : classification, nouvelles procédures d'évaluations et organismes notifiés
a. La classification des dispositifs médicaux à la hausse
Selon le règlement 2017/745, la classification est également renforcée. En effet, certains dispositifs appartenant à la classe I passe en classe IIa, comme les logiciels médicaux. Ainsi que les dispositifs médicaux implantables actifs de classe IIb qui passe en classe III avec le règlement.
Dans le cadre du renforcement de la classification, les fabricants devront redéfinir la classe de leurs dispositifs et suivre la nouvelle classification pour les produits en cours de développement [20].
b. De nouvelles procédures d'évaluations de conformité et d'évaluations cliniques
Les procédures d’évaluations de conformité correspondent à la conduite réglementaire du fabricant afin d’évaluer la conformité de son produit avant la mise sur le marché. Dans le règlement 2017/745, ces procédures sont modifiées et renforcées afin d’être spécifiques aux caractéristiques des dispositifs médicaux, plus particulièrement selon leurs classes. Dans le cadre du renforcement des procédures d’évaluations de conformité, les fabricants devront choisir la procédure adaptée à leur dispositif selon sa classe.
Les évaluations cliniques correspondent à l’étude des données cliniques dans l’objectif d’affirmer que le produit respecte les exigences réglementaires. Ces évaluations ont lieu avant la mise sur le marché mais également pour la surveillance du dispositif après sa commercialisation [20]. Ces évaluations cliniques ont été renforcées par le règlement, en augmentant le niveau de preuves demandé au fabricant. Enfin selon le règlement, les dispositifs implantables doivent obligatoirement suivre des investigations cliniques par le fabricant. Dans le cadre des évaluations cliniques, les fabricants doivent respecter les nouvelles procédures citées précédemment afin d’assurer la sécurité et la surveillance des dispositifs [1].
Ainsi les fabricants devront mettre à jour leur SMQ (Système de Management Qualité) avec les nouvelles procédures. De plus, ils sont désormais dans l’obligation de désigner une personne en qualité de responsable règlementaire [19].
c. Le choix des Organismes Notifiés (ON)
Les organismes notifiés, désignés par un Etat membre de l’Union Européenne, représentent une organisation indépendante qui s’avère être responsable de l’évaluation de la conformité des dispositifs médicaux, afin de vérifier si le produit peut être mis sur le marché. Ils ont donc un rôle primordial dans la certification des dispositifs.
La mise en place de cette nouvelle réglementation européenne a induit des changements majeurs au niveau des organismes notifiés (voir annexe n°3), en choisissant de renforcer les critères de désignation et de surveillance de ces organismes, par la mise en place d’une procédure spécifique. Par ailleurs, ces organismes seront aussi dans l’obligation de consulter un groupe d'experts indépendant pour certains dispositifs à haut risque, avant de prendre la décision de certifier le produit.
Avant l’entrée en vigueur des nouveaux règlements européens 2017/745 et 746, les organismes notifiés étaient plus nombreux, il en existait plus de cinquante capables de certifier selon la directive 93/42/CEE et une vingtaine selon la directive 98/79/CE relative aux dispositifs médicaux de diagnostic in vitro. Contre six seulement aujourd’hui au titre du règlement DIV [21]. Au vu de cette nouvelle réglementation, on a donc constaté une nette diminution de ces organismes en raison de ces nouveaux critères renforcés rendant alors ces organismes plus difficiles d’accès. Cela entraîne le retardement de l’évaluation ou de la réévaluation de conformité de certains nouveaux dispositifs médicaux au regard du règlement européen pour 2021. Ce phénomène a alors pour conséquence de retarder le marquage CE, et ainsi la mise sur le marché.
Ces changements, impactant les organismes notifiés, entraînent alors des conséquences sur les fabricants qui devront impérativement anticiper les démarches afin de ne pas être impactés par le retard engendré par le changement de réglementation.
2. La surveillance après commercialisation devient une priorité : traçabilité, plan de suivi clinique et base de données EUDAMED
a. La traçabilité et le plan de suivi clinique sont des moyens pour assurer la surveillance après commercialisation
Cette nouvelle réglementation implique beaucoup de changements concernant la surveillance après commercialisation.
Dans un premier temps, chaque dispositif doit être identifié grâce à un identifiant unique, ce qui permettra un suivi plus efficace des dispositifs sur le marché. Dans un second temps, le règlement impose un plan de suivi clinique après commercialisation, impliquant : un calendrier des activités pour le suivi clinique après commercialisation, d’attester de la sécurité et des performances du dispositif, et identifier toutes mauvaises utilisations.
De plus, après l’application du plan de suivi, un rapport périodique de sécurité doit être réalisé pour les classes IIa, IIb et III ainsi qu’un rapport non périodique pour les classes I [19].
b. L’harmonisation grâce à une base de données : EUDAMED
EUDAMED est une base de données européenne pour les dispositifs médicaux où les fabricants doivent s’identifier et déclarer les dispositifs médicaux sur le marché. EUDAMED résume les informations concernant les dispositifs : l’enregistrement des acteurs, l’identifiant unique des dispositifs, les organismes notifiés, les rapports d’investigations cliniques, et enfin la surveillance du marché.
Cette base de données est mise en place afin de renforcer l’harmonisation entre les pays membres et la transparence en promouvant l’accès des informations au public et aux professionnels. Il est donc indispensable pour les fabricants de s’enregistrer sur EUDAMED et de fournir les informations nécessaires [22].
En conclusion sur les changements qui concernent les fabricants, il est important pour eux de distinguer ces changements et d’anticiper leur application. En effet, cela va impliquer plusieurs acteurs, comme les organismes notifiés, qui ne seront pas forcément prêts (figure 9).
Figure 9 : Les grands changements apportés par le règlement concernant le fabricant (source : auteurs)
3. L'impact de ces changements sur le fabricant et son implication
La nouvelle réglementation impact largement le fabricant, comme vu précédemment. De plus, dans les textes les fabricants sont cités et bien définis, ce qui n’est pas le cas de tous les acteurs impactés par ce règlement. Avant la mise en application du règlement en 2019, une enquête a été réalisée afin de prendre conscience de l’état d’avancement pour la transition vers le règlement.
Les résultats démontrent que la répartition par état de conformité au 26 mai 2020 montre un pic autour de 60-80%. De plus, 15% des fabricants seront prêts à 100% au 26 mai 2020. Cela malgré les difficultés rencontrées qui sont mises en avant par l’enquête [23].
Cela prouve bien que les fabricants ont conscience de l’impact du règlement et qu’ils agissaient déjà avant l’application du règlement pour la conformité de leurs produits et afin de respecter les nouvelles obligations dont le non-respect pourrait conduire au retrait de la certification CE (Article 13).
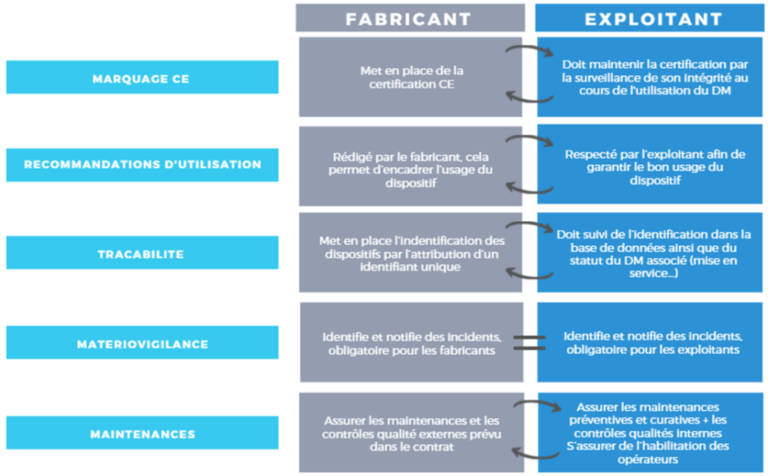
Le marché du dispositif médical implique de nombreux acteurs, comme les fabricants qui agissent selon les textes réglementaires. Cependant, l’ingénierie biomédicale entre en jeu dans l’exploitation des dispositifs, ainsi il existe des interactions entre les acteurs qu’il est nécessaire de mettre en évidence. Le fabricant est dans l’obligation réglementaire de produire des dispositifs qui possèdent la certification CE, cependant lors de l’exploitation, l’exploitant qui correspond à l’ingénierie biomédicale doit garantir le maintien de la certification CE. En effet, tout au long de l’utilisation du matériel, la responsabilité de l’ingénieur biomédical est engagée pour garantir la sécurité du personnel soignant et du patient. En d’autres termes, il faut savoir que durant l’utilisation du dispositif dans un établissement de santé, celui-ci peut perdre sa certification CE, et c’est l’ingénieur biomédical qui est garant de la conformité du dispositif médical au règlement européen. Afin de mieux comprendre la mise en cause de la responsabilité de l’ingénierie biomédicale, il est possible de mettre en évidence les relations entre les responsabilités du fabricant et celles de l’exploitant (voir figure 10).
Figure 10 : Mise en évidence des relations entre les responsabilités du fabricant et de l’exploitant (Source : auteurs)
C. Responsabilité de l'exploitant : l'ingénierie biomédicale
1. La responsabilité de l'exploitant est une source d'incompréhension
La responsabilité de l’ingénierie biomédicale française est de mettre en place une politique de gestion des DM, étendue dans plusieurs activités, en passant par l’introduction des nouveaux DM dans l’établissement, leurs mises en services, leurs utilisations, les différentes maintenances, les contrôles qualités réglementaires et la réforme. Tout ceci forme le cycle de vie d’exploitation du DM qui est soumis à la nouvelle réglementation (figure 6).
Leur responsabilité, par rapport à la nouvelle réglementation européenne, est souvent source d’incompréhension. En effet, celle-ci en elle-même ne s’adresse pas directement à l’exploitant mais plutôt au fabricant. C’est pour cela que de nombreux ingénieurs biomédicaux ne se sentent pas concernés par ces nouveaux règlements. Suite à plusieurs entretiens avec différents ingénieurs biomédicaux, il en est ressorti que certains d’entre eux affirment que ces réglementations ont un impact très faible sur leurs services.
Cependant ces nouveaux règlements portent sur l’attribution de la certification CE d’un dispositif médical avant sa mise sur le marché et défini par conséquent tous les éléments qui établissent la perte de cette certification CE. Le risque est réel et survient principalement après la mise sur le marché, chez l’exploitant. L’ingénieur biomédical, acteur principal après la mise sur le marché, est par conséquent au cœur de cette responsabilité de garantir cette conformité CE jamais dénaturée. De ce fait, les nouvelles réglementations ont donc un impact sur l’ingénierie biomédicale même si certains exploitants n’en n’ont pas encore conscience.
Elle a donc pour principale responsabilité de désigner formellement les personnes chargées de veiller au respect de la réglementation et ainsi surveiller la conformité du marquage CE.
Outre la désignation d’une personne responsable de ces réglementations, il est également important de comprendre que la surveillance va bien au-delà d’un suivi de ces nouveaux certificats CE, puisque cela consiste aussi en la mise en place d’une organisation nouvelle concernant la gestion des dispositifs médicaux tout au long de leur cycle de vie : de l’acquisition, à son installation, en passant par la maintenance, la matériovigilance et la mise au rebut.
2. Impact des nouvelles exigences européennes sur l'ingénierie biomédicale
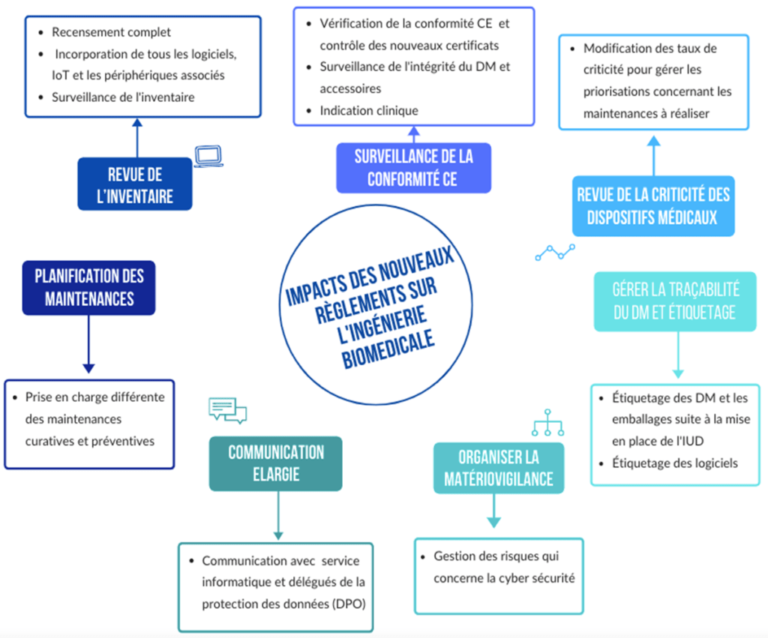
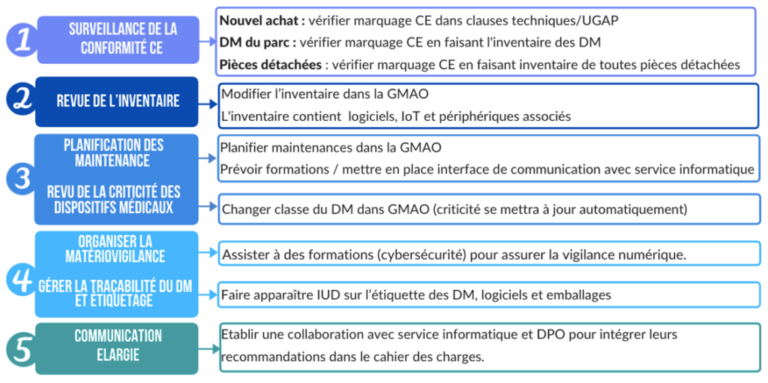
a. Revue de l’inventaire : un recensement complet de tous les éléments du DM
L’inventaire constitue un élément essentiel dans la gestion du parc d’équipements. Il doit être tenu et actualisé en permanence pour pouvoir refléter la réalité de la situation actuelle de l’établissement de santé. Cet inventaire se doit de répondre à différents objectifs (figure 11) :
- Il doit fournir une évaluation technique des dispositifs médicaux disponibles (il précise le type, la quantité disponible et l’état de fonctionnement de celui-ci)
- Il doit planifier le programme des maintenances préventives et curatives à réaliser sur chaque dispositif : il constitue donc un véritable outil de gestion
- Il peut répertorier des informations financières vis-à-vis des évaluations économiques et budgétaires
- Il assure aussi un rôle d’historique du matériel, comme : les dossiers, les manuels de fonctionnement et maintenance, les procédures d’essai et d’assurance, les inventaires des accessoires, des consommables ou des pièces de rechange
Figure 11 : Les objectifs de l’inventaire au sein de l’ingénierie biomédicale (source : auteurs)

Cependant, avec l’application de cette nouvelle réglementation, les inventaires doivent être revus et modifiés pour plusieurs raisons.
En premier lieu, la définition d’un dispositif médical a changé, comme expliqué précédemment. Désormais, la définition s’est élargie à certains produits n’ayant pas vraiment de destination médicale mais avec des risques similaires. De plus, les logiciels ainsi que les IoT (Internet of Things) font également partie intégrante de la définition d’un dispositif médical. Avec ces ajouts, il est donc absolument nécessaire d’effectuer un recensement complet de tous les éléments des dispositifs médicaux. L’inventaire doit ainsi incorporer à sa base de données tous les logiciels, IoT et les périphériques associés [24].
Parmi les changements importants, on compte aussi la modification de la classification des DM qui doit nécessairement aussi aboutir à une modification de l’inventaire.
Par conséquent, la surveillance de cet inventaire devient donc, de ce fait, une tâche importante et quotidienne pour garantir un suivi et des maintenances optimales qui doivent veiller à vérifier si les « nouvelles pièces de réparation », encore sous la directive 93/42, garantissent l’intégrité du dispositif médical selon les nouveaux règlements européens [24].
b. Surveillance de la conformité CE et usage clinique vérifié
C’est assez pernicieux d’avoir un règlement qui aujourd’hui touche dans sa définition l’essentiel du métier de la responsabilité de l’exploitant du DM tel qu’un médecin, un hôpital. Un médecin doit prodiguer des soins, un diagnostic, une thérapie, et pour cela il doit absolument avoir à disposition des DM qui soient performants, sécurisés, et qui garantissent que le diagnostic puisse être juste. Par définition le seul texte concernant le DM va être la certification CE. La définition du marquage CE énonce donc qu’un objet destiné à diagnostiquer, soigner un patient s’appelle dispositif médical. En ce sens, tout objet nommé “dispositif médical” doit donc posséder le marquage CE et doit donc être conforme à sa définition et à son usage. En effet, si les nouvelles réglementations sont à destination principalement des fabricants et des organismes notifiés pour la certification CE des DM, elle a aussi un impact important sur les établissements hospitaliers, utilisateurs finaux de dispositifs médicaux, étant donné qu’ils ont une responsabilité concernant l’utilisation de ces DM qui doivent être en accord avec la définition qui en est faite. De ce fait, la responsabilité des exploitants (utilisateur du DM) est donc de toujours utiliser du matériel qui est marqué CE pour les missions qui lui sont confiées, c'est-à-dire soigner le patient en toute sécurité [24].
La responsabilité de l’ingénierie biomédicale par rapport à ces nouveaux règlements est donc de veiller à ce que la certification CE de l’objet, pendant tout son cycle de vie, reste garantie lors de son utilisation. Il est donc important, pour l’ingénierie biomédicale, de faire en sorte qu'à aucun moment la certification CE soit perdue autour de l’objet. Pour cela, elle doit vérifier la conformité du DM au marquage CE, faire un contrôle des nouveaux certificats, surveiller l’intégrité des DM après leur mise en service, vérifier si leurs accessoires et leurs consommables sont autorisés et vérifier que l’indication clinique soit associée au certificat CE [24].
Lors du passage actuel entre l’ancienne certification et la nouvelle réglementation européenne il est dit qu'au-delà de la date fatidique les équipements devront avoir un nouveau certificat CE. Cela amène à soulever de nombreuses questions telles que :
- Quelle personne dans l’organisation hospitalière sera responsable de s’assurer que les DM sont conformes à la réglementation ?
- Comment les DM nouvellement définis seront-ils identifiés et répertoriés ? Et dans ce sens quels seront les impacts sur la prise en charge biomédicale de ces dispositifs médicaux ?
Ces questions impliquent toutes, l'obligation pour chaque organisation hospitalière de définir clairement les responsabilités de l’ingénierie biomédicale conformément à ces nouvelles réglementations, et de fournir suffisamment de personnes qualifiées afin que les services biomédicaux effectuent une gestion efficace des DM [24].
Par conséquent, l'intégrité des DM est l'objectif primordial à garantir dans les établissements hospitaliers pour maintenir leur certification CE, qui est de la responsabilité des fabricants mais aussi indirectement celle de l’exploitant (l’ingénierie biomédicale).
En outre, il est important de notifier que dans un établissement hospitalier, ce sont les pharmaciens qui possèdent les compétences requises afin de s’occuper de la surveillance de la conformité CE des dispositifs médicaux implantables actifs (DMIA) et les ingénieurs biomédicaux, quant à eux, possèdent les compétences requises afin de s’occuper de la surveillance de la conformité CE des dispositifs médicaux [24].
c. Revue de la criticité des dispositifs médicaux
La criticité d’un dispositif médical représente aussi bien la quantité de risques que peut engendrer l’utilisation de ce dispositif selon des conditions d’utilisation particulières, que les risques encourus par l’absence de ce dispositif. Celle-ci se définit aussi par la classification du dispositif médical et le risque que l’ingénierie biomédicale peut associer à ce DM, notamment son risque de perte du marquage CE, expliqué dans la partie ci-dessous [24].
Pour définir cette criticité, les ingénieurs biomédicaux vont donc être amenés à se poser la question suivante : quels seront les dispositifs médicaux dans l’inventaire qui ont le risque le plus important de perdre leur certification CE ?
En effet, cette criticité s’est vue être modifiée suite à la mise en place de la nouvelle réglementation puisque la classification des dispositifs médicaux a été modifiée. Par conséquent, selon cette nouvelle classification, l'ingénierie biomédicale est tenue de modifier ces taux de criticité qui s’avèrent très importants pour pouvoir gérer les priorisations des interventions des techniciens biomédicaux concernant les maintenances à réaliser [24].
d. Gérer la traçabilité des dispositifs médicaux et l’étiquetage
La traçabilité qui représente aussi un enjeu majeur de la nouvelle réglementation, met en place des codes d'identification unique (IUD) qui doivent apparaître sur l’étiquette des dispositifs médicaux et sur tous les emballages. Sans compter que l’ajout des logiciels dans la définition d’un dispositif médical, nécessite aussi un identifiant unique et un étiquetage qui doit être indiqué sur le support physique du logiciel mais aussi au niveau du système logiciel [3].
Ainsi, l’ingénierie biomédicale se voit dans l’obligation de revoir tout leur système d'étiquetage afin de garantir le respect de cette nouvelle réglementation vis-à-vis de la traçabilité tout au long du cycle de vie du dispositif médical. Ces données sont ensuite collectées et publiées dans la base de données EUDAMED pour assurer une transparence optimale [24].
e. Organiser la matériovigilance
La matériovigilance a pour but de surveiller les incidents, les défauts de conception qui sont susceptibles d'entraîner la mort ou de graves dégradations de l'état de santé d'un patient, d'un personnel soignant ou d'un tiers. Elle consiste à enregistrer, signaler, évaluer et exploiter des alertes afin de faire de la prévention. La matériovigilance a donc pour mission, de façon préventive et corrective, d’empêcher des incidents et des risques d’incidents graves impliquant des équipements médicaux [24].
De plus il existe deux types d’alertes, une ascendante qui provient des responsables locaux, des professionnels de santé et les tiers vers l’ANSM, et une descendante qui provient de l’ANSM vers les responsables locaux, les professionnels de santé et les utilisateurs [24].
Suite à ces nouvelles règlementations, l’ingénieur biomédical doit ainsi acquérir une nouvelle compétence dans la gestion des risques puisque l’intégration des logiciels dans la définition d’un dispositif médical impose des connaissances et une surveillance dans le domaine de la cyber sécurité. Cette compétence est une obligation de savoir qui fait désormais partie de la mission de l'ingénierie biomédicale pour s'assurer que les équipements médicaux sont conformes à la réglementation européenne en vigueur. Cette nouvelle tâche enrichit l’ampleur de la matériovigilance dont les ingénieurs biomédicaux sont déjà responsables [24].
f. Planification des maintenances
Tout dispositif médical nécessite une prise en charge et une surveillance continue pour garantir une sécurité optimale. En effet, avant la mise sur le marché de ces dispositifs médicaux, les fabricants sont tenus de respecter certaines exigences réglementaires en établissant une procédure de conformité qui va devoir désormais répondre aux règlements européens. Les fabricants doivent donc spécifier les exigences de conformité de leur dispositif médical, et en ce sens, il est ensuite de la responsabilité de l'ingénierie biomédicale de programmer un planning de maintenances pour chaque dispositif, selon sa classe et sa criticité, pour maintenir les performances d'origine des équipements médicaux [24].
Suite à la nouvelle classification donnée par les nouvelles réglementations, la prise en charge des maintenances curatives et préventives ainsi que les règles de sécurité associées, doivent être modifiées et adaptées puisque la classification des dispositifs médicaux a été revue à la hausse.
Par conséquent, certains dispositifs médicaux doivent simplement répondre à une maintenance plus régulière, tandis que pour certains dispositifs médicaux, nouvellement ajoutés dans la classification, il est essentiel de prévoir une organisation complètement nouvelle concernant leurs maintenances [24].
De plus, il se pose également la question de la conformité et habilitations des intervenants concernant les maintenances à réaliser, notamment à propos de la partie informatique pour gérer les logiciels nouvellement intégrés aux dispositifs médicaux [24].
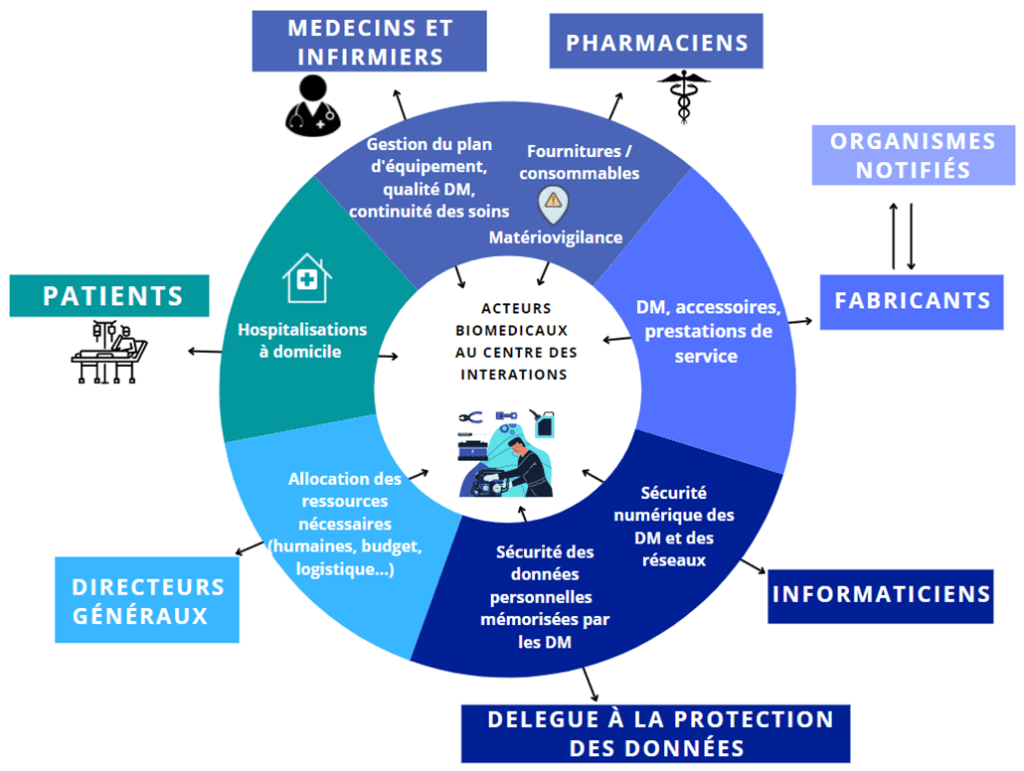
g. Une communication élargie
Un autre impact majeur de ces nouvelles réglementations va finalement concerner les acteurs qui vont graviter autour de l’ingénierie biomédicale. En effet, l’ingénieur biomédical possède un rôle central et se charge de faire le lien entre tous les utilisateurs, tant le corps médical que les professionnels paramédicaux, la direction, les techniciens mais aussi les fournisseurs [24].
Ce rôle et cette communication vont finalement être élargis pour pouvoir formaliser des collaborations et mettre en place un dialogue avec les différents services de l’hôpital et les différents partenaires (figure 12).
Suite à l’intégration des logiciels dans la définition d’un dispositif médical, la communication avec le service informatique ou encore avec les délégués de la protection des données (DPO) est donc primordiale. Dans ce sens, l’ingénieur biomédical se voit dans l’obligation d’élargir son champ de communication pour pouvoir veiller au respect et à la bonne organisation de l’ingénierie biomédicale selon les nouveaux règlements européens [24].
Figure 12 : Interactions entre les différents acteurs impliqués dans l'utilisation des DM (source : auteurs)
Voici un schéma récapitulatif de l’impact des nouveaux règlements européens 2017/745 et 2017/746 sur l'ingénierie biomédicale (figure 13) :
Figure 13 : Récapitulatif de l’impact des nouveaux règlements européens 2017/745 et 2017/746 sur l'ingénierie biomédicale (source : auteurs)
3. Risque pour l'ingénieur biomédical
Les risques de poursuites judiciaires étant élevés et pouvant être préjudiciables à l'organisation, la surveillance de la conformité aux réglementations relatives aux dispositifs médicaux est devenue encore plus importante. En effet, pour rappel, l’ingénierie biomédicale doit garantir, à tout moment du cycle de vie d’un dispositif médical, que celui-ci est conforme à la certification CE. En ce sens, le risque majeur pour l’ingénierie biomédicale est la perte de la certification CE. De ce fait, l’exploitant devra faire attention à ne pas dénaturer le certificat CE d'origine du dispositif médical qui est sous sa surveillance. Cette dénaturation se traduit, par exemple, par un oubli de vérification ou le non-respect des recommandations du fabricant par l’achat d’accessoires qui ne sont pas compatibles avec la certification CE. Ce qui conduirait donc à une perte du marquage CE du dispositif, amenant l’exploitant à utiliser un équipement qui perdra son usage défini par la certification CE, mettant en danger les patients et le personnel, et devenant ainsi un incident de matériovigilance. Le signalement des différents incidents aux autorités sanitaires est l'un des principaux processus de gestion des risques et est obligatoire dans tous les États membres européens. Avec l'introduction de nouvelles réglementations, cette gestion des risques est désormais étendue à la sécurité des réseaux, nommée cyber sécurité, autour des DM [24].
De plus, comme dit précédemment, la nouvelle réglementation a induit une nette diminution des organismes notifiés et donc le retard du marquage CE et ainsi la mise sur le marché. Le retard est donc finalement susceptible d’engendrer une perturbation de l’ingénierie biomédicale qui ne pourrait pas exploiter ces dispositifs médicaux [24].
Il est donc nécessaire pour l’ingénieur biomédical et son équipe de prévoir et anticiper des mesures afin de pouvoir prévenir une pénurie et faire en sorte de fournir des DM conformes à la nouvelle réglementation [24].
Pour cela, il serait intéressant d’utiliser un outil qui permettrait de mettre en avant des points de vérification, qui serait des points de risques de perte de la certification CE.
III. Proposition d'actions pour respecter les exigences européennes
A. Sondage auprès de l'ingénierie biomédicale
Les nouvelles réglementations européennes 2017/745 et 2017/746 concernent principalement les fabricants des dispositifs médicaux. Les dernières exigences imposées à ces acteurs ont pour objectif principal de renforcer la sécurité des DM vis-à-vis des patients et des usagers. Mais les fabricants ne sont pas les seuls à œuvrer pour garantir cette sécurité, les exploitants des DM qui sont le service d’ingénierie biomédicale partagent également le même objectif. D’ailleurs, ils sont tous les deux unis pour la maîtrise de la sécurité des patients et du personnel soignant. Comme ces réglementations ne s’adressent pas directement aux exploitants, ils ont donc du mal à comprendre comment ils en sont concernés.
Après avoir identifié les impacts de ces réglementations sur le service d’ingénierie biomédicale dans les parties précédentes, il semble judicieux de les présenter à ces acteurs. Pour cela, un sondage renfermant différentes questions concernant chacun des 7 impacts relevés, a été transmis à des centaines de professionnels biomédicaux en établissement de santé en France. Les réponses obtenues permettront de confirmer ou infirmer les impacts déterminés précédemment.
Grâce à ce sondage, 51 personnes ayant tous des métiers biomédicaux différents ont participé. Les professions des répondants sont réparties de cette manière : 66,7% sont des ingénieurs biomédicaux, 25,5% sont des techniciens biomédicaux, 2% sont des directeurs biomédicaux, et le reste sont des consultants en santé ou des responsables qualité.
A la suite de ce sondage, chacune de ces réponses ont été analysées pour pouvoir présenter tout d’abord les impacts prioritaires validés par les répondants et ensuite établir une proposition de processus que devra entreprendre le service d’ingénierie biomédicale pour respecter les exigences réglementaires.
B. Validation des impacts prioritaires (sondage)
L’objectif du sondage est de faire valider les impacts par les professionnels biomédicaux eux-mêmes. Les résultats du sondage sont exploités dans cette partie, afin de mettre en avant la pertinence des impacts relevés précédemment, selon les acteurs biomédicaux. Voici le tableau 1 récapitulatif reprenant l’ordre de pertinence des impacts ainsi que l’interprétation des résultats.
Tableau 1 : Pertinence des impacts et interprétation des résultats du sondage (source : auteurs)



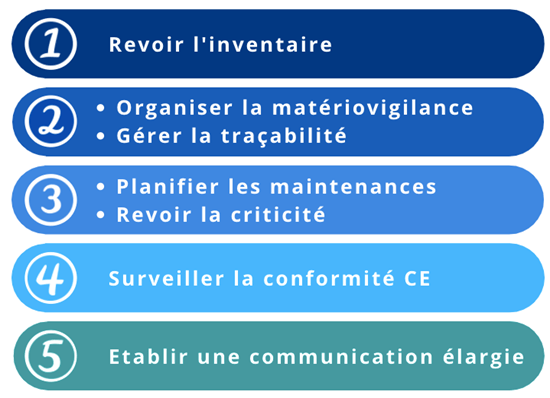
L’étude des résultats du sondage permet de rendre compte de l’avis des professionnels et ainsi de proposer des moyens adaptés à leurs besoins. Cette étude a démontré que les différents impacts relevés sont pertinents, ils sont désormais classés par ordre de priorité selon l’avis de la communauté biomédicale (figure 14). De plus, des résultats similaires ont été révélés pour certains impacts, notamment pour l’organisation de la matériovigilance et la gestion de la traçabilité ainsi que pour la planification des maintenances et la revue de la criticité, ce qui semble très pertinent comme résultats. En effet, on peut faire un lien cohérent entre ses différents impacts, l’organisation de la matériovigilance et la gestion de la traçabilité sont liées à la gestion du risque et la planification des maintenances et la revue de la criticité sont liées à la priorisation de la gestion des dispositifs médicaux.
Figure 14 : Classification des impacts selon les résultats du sondage (source : auteurs)
Les résultats obtenus par le sondage sont très révélateurs de la situation actuelle. Il est bon de notifier que les acteurs biomédicaux ont pleinement conscience de l’impact de ces nouveaux règlements sur leurs missions quotidiennes. Cependant, les nouvelles missions permettant de répondre aux exigences réglementaires ne sont pas définies clairement sous forme actions ou autres moyens. Ainsi, les impacts sont réels mais les professionnels ne sont pas en capacité de mettre en application des moyens pour répondre aux exigences réglementaires.
La classification montre que les trois premiers impacts (les plus pertinents), soit l’inventaire, la gestion du risque et la priorisation des tâches, ont un impact direct sur les tâches quotidiennes de l’ingénieur biomédical. Alors que la classification des impacts suivants (moins pertinents), soit la surveillance du marquage CE et la communication élargie, montre que ce sont des impacts qui ne concernent pas les ingénieurs biomédicaux selon les résultats. Cependant, la conformité CE est la mission principale que les ingénieurs biomédicaux doivent respecter, et cela ne fait pas uniquement référence à la responsabilité du fabricant. Ce qui affirme le manque de prise de conscience sur la transition entre nouvelle et ancienne réglementation, soit l'incompatibilité des pièces sous la nouvelle réglementation pour des dispositifs achetés sous l’ancienne réglementation. En ce qui concerne la communication élargie, l’actualité montre une technologie omniprésente sur les dispositifs. Ainsi, l’ingénieur biomédical étant responsable de l’ensemble des dispositifs, il doit s’assurer de la conformité intégrale, incluant les réseaux numériques.
Pour conclure cette partie, les résultats confirment la position actuelle de l’ingénierie biomédicale face aux nouvelles règlementations. Afin de guider les acteurs biomédicaux à la compréhension des impacts, et de leurs responsabilités dans la garantie de la conformité CE, un processus proposé dans ce rapport avec une classification correspondant aux exigences du règlement.
C. Proposition de processus pour l'ingénierie biomédicale
Suite à la validation de ces différents impacts par l’ingénierie biomédicale, il s’agit alors de proposer des pistes d'activités nécessaires dans le but de préparer une BPAC éventuelle et de faciliter le respect et la mise en œuvre de ces deux nouveaux règlements européens (tableau 2 et figure 15).
Tableau 2 : Présentation d'un processus d'activités pour l'ingénierie biomédicale pour respecter les nouveaux règlements (source : auteurs)
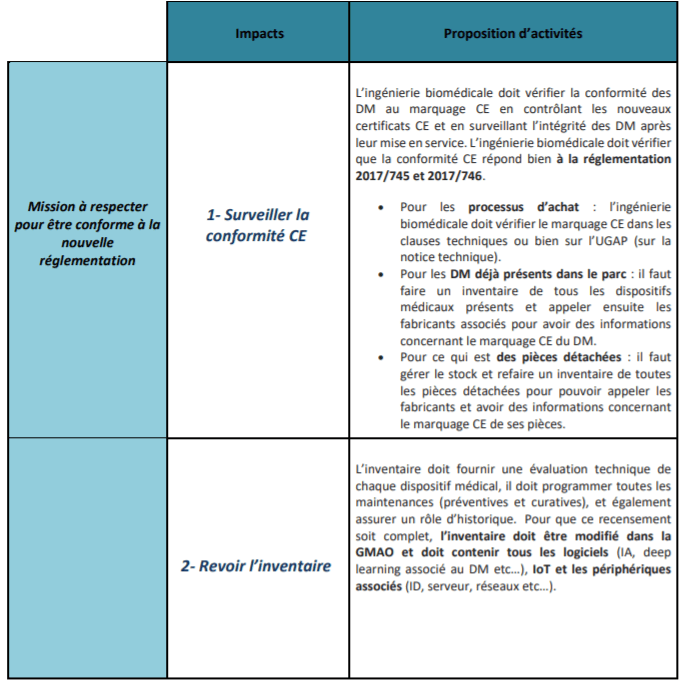
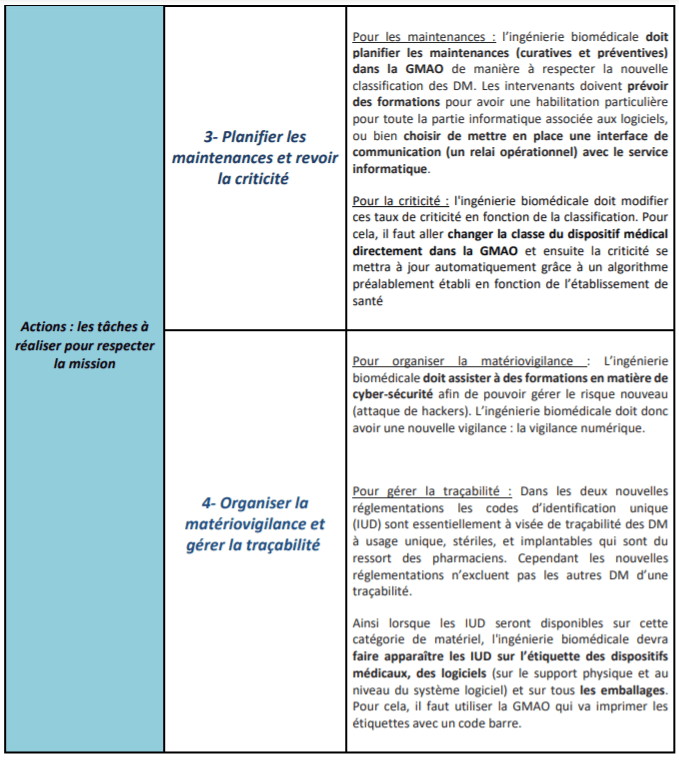

Figure 15 : Récapitulatif des activités nécessaires pour le respect des règlements européens (source : auteurs)
Conclusion générale
Ces règlements européens ont donc pour finalité principale de garantir la conformité des dispositifs médicaux afin d’assurer une sécurité exemplaire vis-à-vis du patient. Ces nouvelles exigences ne sont pas sans impact tant du côté des fabricants, qui se retrouvent les premiers impactés, qu'au niveau de l'ingénierie biomédicale qui retrouve finalement son quotidien bouleversé par la mise en place d’une nouvelle organisation.
A partir du sondage, les différents impacts qui ont été relevés précédemment, ont été exposés aux professionnels de santé qui n’ont pas conscience de l’impact que possèdent ces réglementations sur leurs activités. Ces derniers les ont approuvés et les ont classés par ordre de priorisation. Des suggestions de processus permettant d'être conforme aux exigences des nouvelles réglementations ont été proposées au service d’ingénierie biomédicale. Il serait intéressant par la suite d’établir un guide des bonnes pratiques avec son outil de diagnostic ou de management qui aiderait l’ingénierie biomédicale à vérifier leur niveau de conformité vis -à -vis des règlements 2017/745 et 2017/746.
Références bibliographiques
[1] « Panorama et analyse qualitative de la filière industrielle des dispositifs médicaux en France », 2019. Consulté le : oct. 10, 2021. [En ligne]. Disponible sur : https://www.snitem.fr/wp-content/uploads/2020/01/Snitem-Panorama-chiffre-des-DM-2019.pdf
[2] C. Collignon, R. Aubourg, P.-E. De Joannis, et al. , « Parcours du dispositif médical en France », Haute Autorité de Santé, oct. 09, 2020. https://www.has-sante.fr/jcms/p_3213810/fr/parcours-du-dispositif-medical-en-france (consulté le nov. 14, 2021).
[3] C. Mangeol, « Enjeux et exigences de la nouvelle réglementation européenne des dispositifs médicaux », Thèse de doctorat en pharmacie, Université d’Aix-Marseille, Faculté de pharmacie de Marseille, 2019. Consulté le : oct. 10, 2021. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-02140163
[4] Y. Ouedraogo, « Gestion de la maintenance biomédicale », Université de Technologie de Compiègne, 2016. Consulté le : oct. 10, 2021. [En ligne]. Disponible sur : http://www.utc.fr/tsibh/public/3abih/16/stage/ouedraogo/index.html#IIPresentation_du_service_biomedical
[5] G. Farges, I. Claude, J. M. Prot, et al. , « Benchmark des services biomédicaux : vision médiane et diversité de la maintenance hospitalière… », IRBM News, vol. 40, no 5, p. 100200, oct. 2019, doi : https://doi.org/10.1016/j.irbmnw.2019.07.001.
[6] M. Decouvelaere, « Valoriser le métier d’ingénieur biomédical : des fiches pratiques pour décrire concrètement ses missions et ses actions », IRBM News, vol. 36, Issue 3, p. 57‑106, juin 2015, doi : https://doi.org/10.1016/j.irbmnw.2015.02.001.
[7] A. Paquet, K. Sivakumar, et G. Farges, « Nouvelle bonne pratique d’activités connexes. BPAC 6 : ingénierie biomédicale au sein d’un groupement hospitalier de territoire en France. Partie 1 : enjeux et élaboration », IRBM News, vol. 40, no 5, p. 1‑4, oct. 2019, doi : https://doi.org/10.1016/j.irbmnw.2019.07.002.
[8] « DM et DMIA - Principaux textes législatifs et réglementaires », ANSM, Agence nationale de sécurité du médicament et des produits de santé, janv. 29, 2021. https://ansm.sante.fr/documents/reference/reglementation-relative-aux-dispositifs-medicaux-dm-et-aux-dispositifs-medicaux-de-diagnostic-in-vitro-dmdiv/dm-et-dmia-principaux-textes-legislatifs-et-reglementaires (consulté le oct. 08, 2021).
[9] C. Mesnil, « Principaux textes et décrets », BioMesnil Médical, mai 2016. Consulté le : nov. 04, 2021. [En ligne]. Disponible sur : https://www.biomesnil.com/principaux-textes/
[10] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. Consulté le : nov. 04, 2021. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra
[11] « Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. Consulté le : nov. 04, 2021. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/746/oj/fra
[12] « Prothèses mammaires PIP : chronologie d’un scandale », Le Monde.fr, janv. 18, 2012. Consulté le : oct. 10, 2021. [En ligne]. Disponible sur : https://www.lemonde.fr/societe/article/2012/01/18/les-grandes-dates-du-scandale-des-implants-pip_1625045_3224.html
[13] « Ce que change la nouvelle réglementation pour les patients et les professionnels de santé », SNITEM, Syndicat National de l’industrie des Technologies Médicales, mai 26, 2021. https://www.snitem.fr/ce-que-change-la-nouvelle-reglementation-pour-les-patients-et-les-professionnels-de-sante/ (consulté le nov. 04, 2021).
[14] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, Document 31993L0042, Journal officiel n° L 169 du 12/07/1993 p. 0001-0043, juill. 1993. Consulté le : nov. 14, 2021. [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1993/42/oj/fra
[15] G. Promé, « Règlement 2017/745 - Annexe XVI - dispositifs non médicaux », Blog Qualitiso, août 22, 2016. https://www.qualitiso.com/dispositifs-esthetique-reglement-dispositifs-medicaux/ (consulté le nov. 04, 2021).
[16] G. Promé, « Règlement (UE) 2017/745 : guide pour les fabricants », Blog Qualitiso, mai 14, 2017. https://www.qualitiso.com/reglement-ue-2017-745-guide/(consulté le nov. 04, 2021).
[17] « Maintenance et contrôle qualité des dispositifs médicaux », ANSM, Agence nationale de sécurité du médicament et des produits de santé, févr. 16, 2021. https://ansm.sante.fr/documents/reference/maintenance-et-controle-qualite-des-dispositifs-medicaux (consulté le nov. 04, 2021).
[18] « Obligation de maintenance », ANSM, Agence nationale de sécurité du médicament et des produits de santé, févr. 16, 2021. https://ansm.sante.fr/documents/reference/maintenance-et-controle-qualite-des-dispositifs-medicaux/obligation-de-maintenance (consulté le nov. 04, 2021).
[19] A. Groell, ZKEIK Hajar, et BENACEUR Kheira, « Surveillance après commercialisation des dispositifs médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositif Médical et Affaires Règlementaires (DMAR), Mémoire de projet, réf n° IDS005, https://doi.org/10.34746/nq28-4w94, janv. 2019. Consulté le : nov. 04, 2021. [En ligne]. Disponible sur : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids005/
[20] G. Promé, « Évaluation clinique des Dispositifs Médicaux », Qualitiso, janv. 05, 2019. https://www.qualitiso.com/evaluation-clinique-dispositif-medical/ (consulté le nov. 04, 2021).
[21] « Questions et réponses sur la mise en œuvre progressive du nouveau règlement relatif aux dispositifs médicaux de diagnostic in vitro », European Commission, oct. 14, 2021. https://ec.europa.eu/commission/presscorner/detail/fr/qanda_21_5210 (consulté le nov. 04, 2021).
[22] « EUDAMED - European Database on Medical Devices », European Commission. https://ec.europa.eu/tools/eudamed/#/screen/home (consulté le nov. 04, 2021).
[23] G. Promé, « Résultats de l’enquête « Règlement (UE) 2017/745 & Fabricants » », Blog Qualitiso, oct. 11, 2019. https://www.qualitiso.com/resultats-enquete-reglement-2017-745-et-fabricants/#Resultats_generaux (consulté le nov. 04, 2021).
[24] V. Boissart, « Impacts du nouveau règlement 2017/745 sur la gestion biomédicale des dispositifs médicaux », IRBM News, vol. 42, Issue 2, p. 100300, avr. 2021, doi : https://www.sciencedirect.com/science/article/pii/S1959756821000043?via%3Dihub.
Annexes
Annexe n° 1 : Le tableau de comparaison des deux règlements européens
Tableau 3 : Comparaison des deux règlements européens (source : auteurs)

Annexe n°2 : La définition d'un dispositif médical selon la nouvelle réglementation
Selon le règlement 2017/745, un dispositif médical aux fins du présent règlement, on entend par « dispositif médical », tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
- diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
- investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
- communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens.
Les produits ci-après sont également réputés être des dispositifs médicaux :
- les dispositifs destinés à la maîtrise de la conception ou à l'assistance à celle-ci,
- les produits spécifiquement destinés au nettoyage, à la désinfection ou à la stérilisation humain, y compris les dons d'organes, de sang et de tissus, et dont l'action principale voulu des dispositifs visés à l'article 1er, paragraphe 4, et de ceux visés au premier alinéa du présent point;”
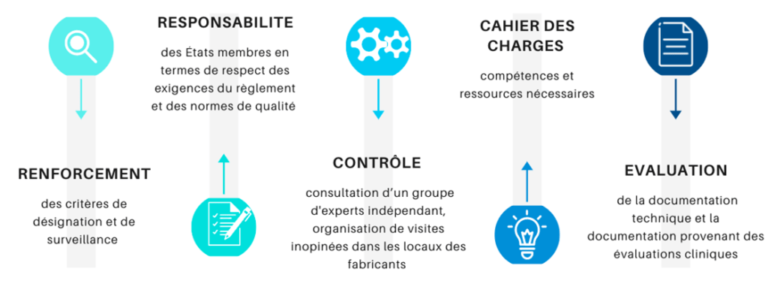
Annexe n° 3 : Les principes attendus de la nouvelle réglementation européenne sur les organismes notifiés
La mise en place de cette nouvelle réglementation européenne a induit des changements majeurs au niveau des organismes notifiés, en choisissant de renforcer les critères de désignation et de surveillance de ces organismes, par la mise en place d’une procédure spécifique. En effet, en plus de devoir organiser des visites, spontanées ou non, dans les locaux des fabricants ; ces organismes devront également répondre à un cahier des charges particulier dans lequel il devra y figurer les compétences et les ressources nécessaires. Par ailleurs, ces organismes seront aussi dans l’obligation de consulter un groupe d'experts indépendant pour certains dispositifs à haut risque, avant de prendre la décision de certifier ou non le produit, dans le but de prendre des décisions mûrement réfléchies en préservant la sécurité et la qualité des dispositifs innovants. Bien sûr, ce renforcement de procédure ne concerne pas seulement la désignation initiale puisque une surveillance accrue et une réévaluation de ces organismes aura lieu périodiquement, en évaluant la documentation technique et la documentation provenant des évaluations cliniques. Toutes ces évaluations seront menées par l’ANSM (Autorité Nationale de la Sécurité des Médicaments) en collaboration avec une équipe européenne d’évaluation (figure 16).
Figure 16 : Les principes attendues de la nouvelle réglementation vis-à-vis des organismes notifiés (source : auteurs)