
IDS127 - Nouvelle réglementation Européenne et cartographie des réseaux biomédicaux
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- Mariana Alvarez : mariana.mtag@gmail.com
- Julien Charton : j-charton@hotmail.fr
- Jordy Ramos : j.ramos@cort37.fr
- Alexandra Laurent : alex.i.laurent@gmail.com
- Adébola Fatoke : fatokeadebola@gmail.com
Citation
A rappeler pour tout usage : Mariana Alvarez, Julien Charton, Jordy Ramos, Alexandra Laurent et Adebola Fatoke, "Nouvelle réglementation Européenne et cartographie des réseaux biomédicaux", Université technologique de Compiègne (France), Master Ingénierie de la santé, Mémoire de projet, httpps//travaux.master.utc.fr/, réf n° IDS127, https://doi.org/10.34746/tzhh-8778, janvier 2022, url directe : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids127
Résumé
Le développement informatique et technologique des dispositifs médicaux au sein des établissements de santé a contribué à l’amélioration des soins. Cependant, l’échange important de données par les dispositifs médicaux a complexifié la sécurité et la sureté des informations communiquées. Pour cette raison des réglementations ont vu le jour dans le but de mieux encadrer ces informations. C'est le cas du nouveau règlement européen 2017/745, qui a élargit la définition d’un dispositif médical et redéfinit les logiciels de santé comme étant des dispositifs médicaux à part entière. Cela induit une surveillance et une maintenance mais aussi une gestion des risques de ces nouveaux DM durant tout leur cycle de vie. De plus, la HAS a défini une nouvelle catégorie, les Dispositifs Médicaux Connectés (DMC), hiérarchisés en fonction de leur risque et autonomie vis à vis du patient.
Ces changements induisent des enjeux techniques, économiques ainsi que la redéfinition des responsabilités des différents acteurs concernés.
Les établissements de santé, comme le centre hospitalier WILLIAM MOREY de Chalon sur Saône sont donc impactés et se trouvent face à de nouvelles problématiques. En effet, le service biomédical et la DSI devront coopérer pour optimiser le traitement des patients et gérer au mieux l’utilisation et la sécurité des DATA à l’hôpital. Une cartographie détaillée des DM, DMC, flux de DATA et des acteurs responsables dans les différents services devient nécessaire, afin de faciliter la sureté, la disponibilité et l’accès des données aux personnels soignants et améliorer la qualité de soins du patient.
Ce Mémoire essaye d’apporter un éclaircissement sur la définition d’un DMC et la responsabilité des DATA au sein du bloc opératoire. En proposant une organisation plus efficace entre les services biomédicaux et informatique à l'avantage du patient. Il a été réalisé en collaboration avec le centre hospitalier WILLIAM MOREY de Chalon-sur-Saône, grâce aux informations fournies par le service biomédical et le service informatique sur le bloc opératoire.
Abstract
The technological development of medical devices in hospitals has contributed to the improvement of healthcare. However, the exchange of data by medical devices has made the security and protection of communicated information more complex. For this reason, regulations have emerged to better manage this information.
The new European regulation 2017/745 expanded the definition of medical devices and redefined healthcare software as medical devices (MD) in its own right. This converts the monitoring and maintenance as well as the risk management of these new MDs throughout their life cycle. Moreover, the Haute Autorité de santé (HAS), or French National Authority for Health, has defined a new class of Connected Medical Devices (CMD) hierarchized according to their risk and autonomy with respect to the patient. These changes induce technical and economic issues as well as the redefinition of the responsibilities of the different actors concerned. Hospitals, such as the WILLIAM MOREY hospital center at Chalon Sur Saône, are therefore affected and face new problems. Indeed, the biomedical service and the Information Technology (IT) department will have to cooperate to optimize the treatment of patients and better manage the use and security of DATA in the hospital. A detailed mapping of MDs, DMCs, DATA flows and the responsibility of the actors in the departments becomes necessary, to facilitate the safety, availability, and access of data to health records and improve the quality of patient care.
This thesis attempts to clarify the definition of a CMD and the responsibility of DATA within the operating room, by proposing a more efficient organization between biomedical and IT services to the benefit of the patient. This project has been carried out in collaboration with the WILLIAM MOREY hospital center in Chalon-Sur-saône, where the biomedical and IT department of the operating room provided the information used.
Remerciements
Avant de développer ce rapport, nous tenons à remercier toutes les personnes qui ont contribué à l’aboutissement de ce projet.
Tout d’abord nous remerçions nos deux suiveurs UTC, Isabelle CLAUDE et Jean-Matthieu PROT qui sont nos responsables de filière. Merci pour les conseils apportés à chaque jalon ainsi que l’aide donnée lorsque nous en avons eu besoin. Cela nous a permis d'être guidés tout au long de notre semestre.
Ensuite nous remercions Mr Mazille pour son regard critique, ses conseils et sa disponibilité. Étant ingénieur biomédical au centre hospitalier de Chalon sur saône, il a pu nous apporter les éléments concernant son expérience et nous mettre à disposition et en relation avec les professionnels qui répondaient à nos demande pour obtenir les informations.
Pour finir, nous remercions Mme KONIG Béatrice pour son regard critique sur notre bibliographique.
Liste des abréviations
- AFIB : Association Française des Ingénieurs Biomédicaux
- ANSM : Agence Nationale de la Sécurité du Médicaments et des Dispositifs Médicaux
- ANSSI : Agence Nationale de la Sécurité des Services d’Informations
- CE : Conformité Européenne
- CEI : Commission Electrotechnique Internationale
- DATA : Terme signifiant données Informatique
- DICOM : Digital Imaging and Communication in Medicine
- DM : Dispositif Médical
- DMC : Dispositif Médical Connecté
- DMDIV : Dispositif Médical de Diagnostic In Vitro
- DMIL : Dispositif Médical Intégrant du Logiciel
- DPI : Dossier Patient Informatisé
- DPO : Délégué à la Protection des Données
- DSI : Direction des Système Informatique
- EN : Norme Européenne
- GE : Général Électrique
- GAFA : Géant du Web
- GAM : Gestion Administrative du Malade
- HAD : Hospitalisation à Domicile
- HAS : Haute Autorité de Santé
- IADE : Infirmier Anesthésiste Diplômé d’Etat
- IBODE : Infirmier de Bloc Opératoire Diplômé d’Etat
- ISO : Organisme International de Normalisation
- IUD : Identifiant Unique du Dispositif
- IUP : Identifiant Unique du Patient
- ID : Identifiant du Dispositif
- IP : Identifiant de Production
- NIS : Network and Information System Security
- NF : Norme Française
- UE : Union Européenne
- ON : Organismes Notifiés
- OSE : Opérateurs de Services Essentiels
- RGPD : Règlement Général sur la Protection des Données
- RSSI : Responsable de la Sécurité des Systèmes d’Information
- SIH : Système d’Information Hospitalier
- RJ : Registered Jack ( prise jack en français)
- HL : Health Level
Téléchargements


Introduction
Depuis le 26 mai 2021, tous les acteurs et opérateurs économiques en lien avec les dispositifs médicaux doivent suivre le nouveau règlement européen 2017/745 [1].
Ce nouveau règlement élargit la définition du dispositif médical (DM). Le logiciel de santé devient lui aussi un DM et possède sa classification spécifique. Ce dernier doit répondre aux exigences mises en avant par le règlement tout au long de son cycle de vie. En ce qui concerne les dispositifs médicaux connectés (DMC), l’HAS a établi une procédure de classification et permet d’apporter un cadre réglementaire.
Plusieurs textes tel que le règlement 2016/679 (Règlement général de la Protection des Données) [2], la directive 2016/1148 [3], des guides réalisés par l’HAS [4] et l’ANSM [5] ainsi que des publications de l’AFIB [6] permettent de définir le logiciel de santé et de souligner l’importance de la sécurisation des données.
Ces Documents impliquent directement l'organisation et les responsabilités des différents acteurs de santé.
Les établissements de santé, comme le centre hospitalier de Chalon sur Saône WILLIAM MOREY se retrouvent face à la problématique suivante : Comment appliquer la nouvelle réglementation aux logiciels médicaux présents au Bloc opératoire.
L’objectif de ce mémoire est de considérer l’impact de ce nouveau règlement, pour permettre d’appréhender le développement informatique au sein de l’organisation Biomédical. La coordination des services informatiques et biomédicaux devient un élément essentiel pour un fonctionnement efficace. Un outil de cartographie répertoriant les différentes connexions entre équipements connectés et logiciels de santé entre Bloc opératoire et les structures informatiques, sera proposé et modifiable pour d’autres services.
I) Contexte
1. Définition : DM, logiciel de santé, DMC
Afin de comprendre les différences d’équipements présents au sein du bloc opératoire il est important de définir les termes suivants :
Les Dispositifs Médicaux est tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
— diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
— diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
— investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
— communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus, et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens [1].
Par exemple : table d’opération, défibrillateur, arceau amplificateur de brillance de bloc opératoire.
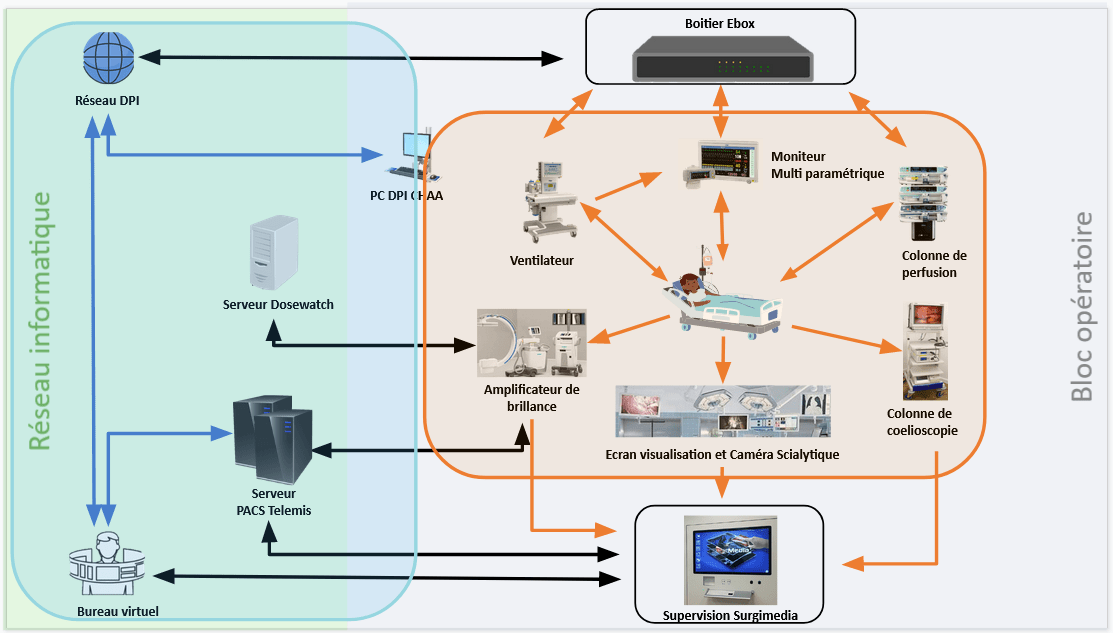
Le logiciel de santé est une solution numérique interprétable par un système informatique qui effectue une action ou une analyse d’information médicale à partir de données entrantes permettant de donner un résultat propre au bénéfice du patient [4]. (Il peut également être considéré comme un DM selon les critères du règlement Européen 2017/745).
Pour une application autre que médicale, un logiciel santé n’est pas considéré comme étant un DM.
Par exemple : Cpage (gestion parcours patient), logiciel de traitement d’image.
Un Dispositif Médical Connecté (DMC) est capable, outre sa fonction principale de DM, d’envoyer ou de recevoir des informations par l’intermédiaire d’un réseau de communication intégré (Câble, Wifi, Bluetooth, etc.). D’après l'HAS et la CNEDIMTS, un logiciel utilisé à des fins médicales ou "par le patient lui-même" et qui dispose d’une "fonction de télécommunication" est considéré comme un DMC [7]. Ces échanges et partages de données induisent une optimisation des soins.
Est considéré comme DMC tout dispositif répondant à la réglementation européenne 2017/745.
Par exemple, un logiciel de santé (Surgimedia -> supervision) connecté à un DM (amplification de brillance) est un DMC puisque celui-ci est destiné à des fins médicales et comporte une fonction de télécommunication. Cependant à ce jour, une montre connectée n’est pas considérée comme DMC mais plutôt comme un objet connecté de santé car celle-ci intègre un système de télécommunication mais fournit uniquement des données génériques sans fin médicale.
A contrario, le multiparamétrique est un DM capable d’être relié à d’autres dispositifs pour remonter des données. Dans ce cas, ils ne sont pas considérés comme des DMC.
Il existe une différence entre un DM, un logiciel de santé et un DMC explicité dans le tableau (tableau 1) suivant :
Tableau 1 : Récapitulatif des différences entre dispositif médical, logiciel santé et Dispositif médical connecté (Source : auteur inspiré par le nouveau règlement européen)
Dans ce contexte, il peut être mis en avant l'arrivée de nouvelles réglementations et des évolutions réglementaires induites par la numérisation de la santé. Il est essentiel de prendre en compte les risques liés à l’utilisation de ces logiciels et DMC (voir chapitre des risques). Cela induit la mise en place d’une classification de ces dispositifs. La classification se base sur des critères spécifiques à chaque type et utilisation médicale d'équipement.
Les critères sont régis par le nouveau règlement, à cela s'ajoutent les recommandations de l’HAS [4] et de l’ANSM [5] qui permet d'établir une évaluation clinique pour que les dispositifs puissent obtenir le marquage CE. Cela permet d’apporter un cadre réglementaire au Dispositif médical connecté ainsi qu'au logiciel, pour garantir une sécurité et une performance dans les soins procurés aux patients.
2. Risques liés aux DMC
Le risque est défini comme “Possibilité, probabilité d'un fait, d'un événement considéré comme un mal ou un dommage” selon Larousse.
Pour prévenir et gérer les risques, il faut considérer un événement potentiellement dangereux comme source de risque « que s’il est susceptible de porter atteinte à des enjeux humains, environnementaux, économiques (ou) culturels »[6].
A ce propos, il existe de nombreux risques liés au DMC et au DMIL. Les logiciels ainsi que les DMC produisent des données qui sont sensibles et qui représentent une valeur pour les piratages.(réf AFIB).
Cela touche le quotidien des professionnels et met en danger la prise en charge des patients de différentes façon [8] :
- Systèmes biomédicaux paralysés
- Plateaux techniques indisponibles
- Données de programmation des soins détruites
- Systèmes de messageries en panne
- Données de gestion et de ressources humaines perdues
- Données personnelles de santé usurpées.
Pour prévenir ou limiter les risques, les logiciels et les DMC doivent répondre aux exigences de sûreté et de performance. Ce qui permet d’induire une sécurisation des données médicales du patient produites par un dispositif (DATA) et de garantir son fonctionnement.
Répondre aux exigences en termes de sécurité, c’est protéger l'équipement et toutes les parties lui étant attachées des menaces extérieures qui pourraient altérer son bon fonctionnement.
Pour appliquer cela, il faut donc prévoir les menaces, qui peuvent être :
Lié aux services ou à l'établissement ce qui impacte :
- La santé des populations
- Les patients pris en charge dans les établissements
- Le secret médical
- La confidentialité des données
Lié à l’intégrité des équipements biomédicaux comme :
- La modification des données
- Les attaques informatiques
- Des failles structurales.
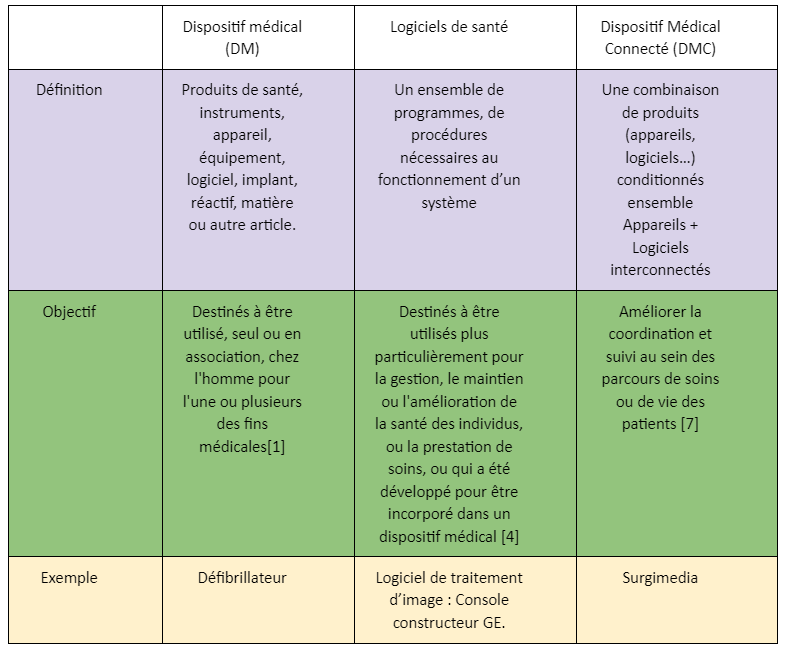
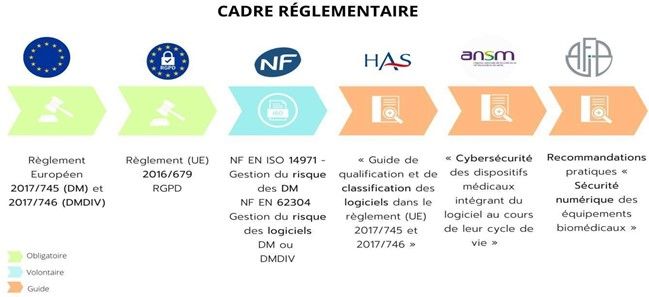
Tout cela induit la mise en place et la définition de critères de sécurité, divisés en plusieurs parties comme l'illustre la figure (figure 1) inspirée de l'ANSM ci-dessous [2] :
Figure 1 : Critères de sécurité (Sources auteurs, inspiré de l'ANSM)
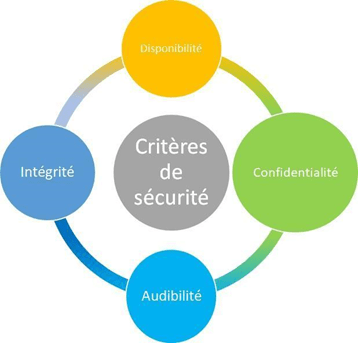
La confidentialité fait partie des éléments les plus délicats à protéger, puisqu'elle impacte directement la vie privée du patient et doit respecter le Règlement Général sur la Protection des Données (RGPD, Règlement (UE) 2016/679 relatif aux données personnelles) [2].
Il est important de prendre en compte que plusieurs acteurs ont des responsabilités et des actions à mener contre ces risques. Dans un premier temps, le fabricant doit évaluer son équipement et définir sa vulnérabilité en définissant un “niveau acceptable de risques” et en planifiant son impact.
Couplé au fabricant, dans les établissements de santé, le service Biomédical et l’informatique doivent conjointement assurer le rôle de prévention et de protection basé sur des réglementations et normes.
Par la mise en place de ces critères de sécurité, leur évaluation ainsi que leur suivi et la traçabilité de ceux-ci par les différents acteurs, permet de mettre en place les exigences de sûreté et d’assurer la protection du patient face aux risques.
3. La réglementation, les normes et les enjeux
Pour approfondir le cadre mis en avant dans la première partie, les obligations réglementaires présentes dans le cycle de vie des logiciels ainsi que des dispositifs médicaux (DM), mettent en relation différents règlements Européen et normes. À cela s'ajoutent des guides qui permettent de faciliter leurs applications. Ainsi, ceci permet aux acteurs principaux de respecter les exigences en vigueur pour les logiciels de santé qui ont pour spécificités une finalité médicale afin d'assurer la sécurité du patient.
Dans le contexte de notre projet, le règlement UE 2017/745 (C.II/ A. 16.1) qui s’applique aux dispositifs médicaux est la ligne directrice de toutes les exigences qui doivent être mises en place pour DM, logiciel et DMC. La réglementation ou la norme met en avant que les DM doivent être développés et fabriqués conformément à l'état de l'art qui repose sur les principes du cycle de vie, de gestion des risques, ainsi que de la sécurité de l'information, de la vérification et de la validation de sa conformité [1] . Ces critères obligatoires sont établis par le Conseil de l'Union Européenne et par sa Commission afin d’obtenir le marquage CE. Ce marquage autorise la libre circulation des dispositifs médicaux dans l’Union Européen et prouve sa conformité en termes de sécurité et de performance [1].
D’autres textes doivent être pris en compte tel que le Règlement 2016/679 – RGPD qui permet la gestion des risques liés aux logiciels. Il permet d'informer les patients et les professionnels sur leurs droits concernant la gestion des données personnelles et garantit la confidentialité des données. Pour cela, un Délégué à la Protection des Données (DPO) doit être nommé par l’établissement. Le DPO a la charge de l'organisation des actions à mener pour établir et mettre en œuvre les lignes directrices que permettent la mise en place de la RGPD des logiciels [2].
D'ailleurs, la Directive 2016/1148 NIS - Network and Information System Security - (FR arrêté du 14 septembre 2018). C'est un texte Européen qui structure les organismes permettant de lutter contre les cyberattaques et organise la protection des services essentiels des nations de façon homogène dans l’ensemble de l’UE. Il permet la mise en place d’une gouvernance par une stratégie nationale mis par l’Agence Nationale de la Sécurité des Systèmes d’Informations (ANSSI). Ce texte renforce la cybersécurité par des Opérateurs de Services Essentiels (OSE comme les CHU, centre de radiothérapie). La mise en place d’un OSE permet d’établir la sécurité numérique et recueillir les incidents par les déclarations. Cette directive permet aussi de renforcer la cybersécurité des Fabricants de Service Numérique (FSN) [3].
En plus des règlements, des directives, des normes et des guides sont établis par des organismes compétents comme l’AFNOR, l’HAS, l’ANSM et l’AFIB. Cela permet d’apporter un aspect opérationnel pour répondre aux exigences réglementaires que les fabricants et les établissements doivent mettre en place.
Le tableau(Tableau 2) ci-dessous met en avant les réglementations obligatoires concernant les logiciels, les normes qui peuvent leur être appliquées et les guides qui peuvent servir d’appuis pour les fabricants et les établissements de santé.
Tableau 2 : Obligations réglementaires et normative dans le cycle de vie des logiciels et DM (source auteur) [1]–[4], [6], [7], [9]–[22]
| Obligatoire | Volontaire | Guides |
| Règlement européen 2017/745 relatif aux DM | NF EN 62304 : Logiciels de santé | « Guide de qualification et de classification des logiciels dans le règlement (UE) 2017/745 – MDR et le règlement (UE) 2017/746 – IVDR » par la HAS |
| Règlement 2017/746 relatif aux DMDIV | Normes ISO/CEI 12207 et ISO/CEI 15288 : Cycle de vie des systèmes et du logiciel | Art L5311-1-18° du CSP : Les logiciels de gestion des laboratoires de biologie médicale |
| Règlement (UE) 2016/679 relatif aux données personnelles | NF EN ISO 13485 : Système de qualité d’un fabricant de DM ou DM DIV | « Guide sur les spécificités d’évaluation clinique d’un dispositif médical connecté (DMC) en vue de son accès au remboursement » par la HAS |
| Directive (UE) 2016/1148 relatif aux cybersécurité | NF EN ISO 14971 : Gestion du risque des DM | « Cybersécurité des dispositifs médicaux intégrant du logiciel au cours de leur cycle de vie » par l’ANSM |
| NF EN 62304 : Spécificité de la gestion du risque des logiciels DM ou DM DIV | Certification NF - Logiciel par l’AFNOR | |
| NF EN 62366 : Aptitude à l’utilisation des DM ou DM DIV | Recommandations pratiques « Sécurité numérique des équipements biomédicaux » par l’AFIB | |
| NF EN 60601-1-4 : Appareils électro- médicaux : systèmes électro-médicaux programmables | ||
| NF ISO/ IEC 25051 : 2014 Exigences de qualité de logiciel et évaluation par fabricants |
Tous ces textes permettent aux fabricants ainsi qu’aux acteurs économiques et aux établissements de fournir les éléments permettant de répondre aux exigences. Ce cadre réglementaire permet aussi de mettre en place une gestion des risques des données produites par ces équipements et de mettre en avant le rôle et les responsabilités de chaque acteur. Par cela, l'obtention de l’attestation individuelle de la certification de conformité (marquage CE) des Dispositifs est possible.
Le patient étant au centre de l’activité, les dispositifs sont des outils pour diagnostiquer, soigner, améliorer, et prévenir son état de santé. C’est pour cela qu’une réglementation a été établie, et sa mise en œuvre permet d’assurer une qualité et une sécurité des soins durant tout son parcours de santé.
En conclusion, les exigences réglementaires permettent de fournir les preuves qui établissent la sécurité et la performance des logiciels. Les normes permettent d’apporter un appui aux différents acteurs pour répondre à celles-ci. En plus de cela l’ANSM, l’HAS et l’AFIB apportent un éclairage par la publication de guides, de publications et la mise en place de suivi de gestion des risques. Toute cette organisation, comme le met en avant le schéma suivant (Figure 2), permet par son application de veiller à la performance dans le but d’assurer la sécurité auprès du patient.
Figure 2 : Schéma du cadre réglementaire des dispositifs médicaux aux logiciels de santé (Source auteur inspiré par l'ANSM)
II) Classification des DM
1. Classification selon la nouvelle réglementation 2017/745
La nouvelle réglementation européenne 2017/745, classe les DM selon quatre classes de risque : classe I, classe IIa, classe IIb et classe III. Cette classification prend en compte la durée d’utilisation (temporaire, court et long terme), le caractère potentiellement invasif et le type d’invasivité, la possibilité ou non de réutilisation, la visée thérapeutique ou diagnostique et la partie du corps concernée. Elle intervient dans le processus de marquage CE pour le fabricant et permet au service biomédical de déterminer le niveau de maintenance de son dispositif médical. La classification des DM du bloc opératoire est illustrée dans le tableau (Tableau 3) ci-dessous.
Tableau 3 : Classe des logiciels selon le règlement 2017/745

2. Classification selon la HAS
En plus de la classification de la réglementation 2017/745 [1], vu ci-dessus, la HAS propose une classification fonctionnelle basée sur 4 niveaux, allant de A à B, pour allier la santé et le développement numérique [4].
Le niveau A : Possibilité de personnalisation mais pas d’autonomie
Services supports aux patients, aux aidants ou aux professionnels dans le cadre de soins ou d’optimisation du parcours de soins ou la gestion médico/socio administrative sans action directe sur la santé des patients.
Le niveau B : Pas de personnalisation, ni d’autonomie
Information générale de l’utilisateur non personnalisée sur les conditions de vie, les règles hygiéno- diététiques, les pathologies/handicaps ou tout état de santé (au sens large du terme), les parcours de santé, de soins ou de vie, etc. Fournit également des supports ou outils de formation.
Le niveau C : Personnalisation, pas d’autonomie
Aide à la vie, à la prévention, au dépistage, au diagnostic, à l’observance, à la surveillance ou au traitement d’une pathologie, d’un état de santé ou dans le cadre d’une situation de handicap. Sans autonomie de la solution numérique dans la gestion de la décision thérapeutique.
Le niveau D : Personnalisation et autonomie
Gestion autonome de la décision après analyse des données et diagnostic afin d’ajuster automatiquement, le traitement à administrer, sans intervention humaine.
Ainsi, le logiciel du bloc opératoire comme "Cpage'' qui permet la gestion du parcours médical patient, est classé comme un logiciel de niveau A.
Les logiciels du bloc opératoire de Chalon-sur-Saône sont de niveau A. D’autres exemples sont présentés et classés sur la pyramide (Figure 3) ci-dessous :
Figure 3 : Pyramide des logiciels de bloc opératoire selon leur classification (Source auteurs inspiré de l’HAS)
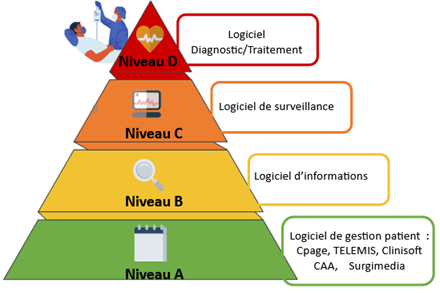
Pour conclure, l'application des deux classifications à TELEMIS (logiciel de gestion de PARCS) montre que le logiciel est de classe IIa et de niveau A. Ainsi la classification du règlement 2017/745 et celle proposée par l’HAS permettent de mettre en avant les exigences qui doivent être appliquées selon le type de logiciel utilisé. Ce qui permet la mise en place d’une classification de tous les logiciels utilisés à ce jour ainsi que la définition des rôles de chaque acteur. Ceci permet au patient de bénéficier d’un système de prise en charge sûr et efficace [4].
3. Responsabilités et impact des logiciels médicaux
Comme vu en amont, les logiciels de santé et les DM connaissent un développement technologique important. De plus en plus nombreux sur le marché et les plateaux techniques dans les filières de soins, ils dépendent d’une grande innovation technologique et digitale qu’il faut aujourd’hui contrôler pour maîtriser leur impact. Pour cela, il est important de définir les responsabilités des différents acteurs liées à ces DMC.
L’entrée en vigueur du règlement européen 2017/745 [1] a redéfinit la classification de ces logiciels de santé en DM ou DMC. Cette nouvelle réglementation renforce les exigences CE, ce qui induit un contrôle des nouveaux certificats mais également une vérification de l’usage clinique associé à ces certificats.
L'ANSM [5] et l’HAS [4] ont apporté un éclairage sur cette réglementation, comme vu précédemment, de manière à définir, pour les différents opérateurs, la qualification de ces produits comme DM et leur classification. Cette nouvelle réglementation s’applique sur l’ensemble du cycle de vie de ces logiciels de santé DM et DMC [17].
En ce qui concerne les exigences appliquées aux fabricants, il devra évaluer pour chaque solution numérique la fonction de la destination et les spécificités caractérisant la finalité médicale de son produit. Ainsi, si son logiciel dispose du statut de DM ou DM DIV, il devra définir sa classification selon les annexes VIII des règlements DM 2017/745 [1] et DMDIV 2017/746 [22]. Cela amènera à la reclassification de certains de ces logiciels, jusqu’à présent en classe I, vers une classe IIa, IIb voir III. Ils devront donc démontrer la conformité aux exigences essentielles applicables pour les DM ou DM DIV de cette classe. L'intervention d’ON sera ainsi nécessaire dans certains cas pour l'obtention du marquage CE [23]. Les incidents possibles seront également à prendre en compte par le fabricant (rappel, diffusion de nouvelles versions logiciels correctives). Cela passe par la mise en place d’une matériovigilance et d’un suivi post-commercial.
Le fabricant devra mettre en place une surveillance des produits après leur mise sur le marché et ce tout au long de leur cycle de vie. D'ailleurs, en fonction de l'intérêt clinique du DMC, le prix de remboursement voire le prix limite de vente de ces logiciels sera impacté [24]. Les DMC intègrent aussi une fonction de traitement des données personnelles. Le fabricant ou le distributeur devra donc joindre un dossier de déclaration indiquant que son produit suit la législation RGPD [2] pour pouvoir être remboursé.
Outre les fabricants, les établissements de santé vont également être impactés [25]. Une collaboration entre le service biomédical et le service informatique sera nécessaire pour veiller au bon fonctionnement des logiciels de santé DM et DMC. Le service biomédical devra donc définir à partir des documentations fabricant l’utilisation des logiciels de santé DM et des DMC mais également les personnes aptes à les utiliser (médecin, IBODE, technicien, pharmacien…). Du fait du passage de logiciels de santé à celui de DM ou DMC les maintenances et l’entretien de ses produits se feront en collaboration avec la DSI. Le service biomédical devra s’occuper de la partie matérielle alors que la DSI s’occupera des cyberattaques, des mises à jour ou encore des composants informatiques intégrant les logiciels de santé DM et DMC. Cette maintenance en partenariat permettra de prévenir les incidents de matériovigilance liés à l’utilisation de DMC. La figure suivante (Figure 4) montre cette coopération à mettre en place entre le service biomédical et la DSI pour gérer au mieux les DMC, dans le but de rendre le traitement du patient optimal.
Figure 4 : Coopération des acteurs pour les logiciels de santé DM et DMC (source auteur)
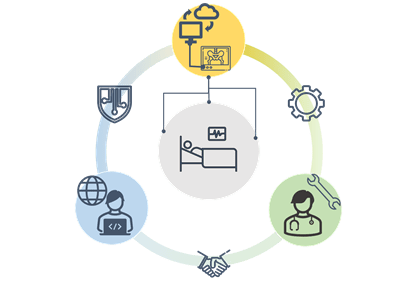
Il sera nécessaire d’avoir un suivi de l’ensemble des DMC de l’infrastructure pour prévenir les différents risques. Pour cela une documentation de l’infrastructure, des périphériques et outils connectés devra être faite dans le but d’avoir une surveillance de ces appareils dans l’établissement. Cette identification du matériel permettra au service biomédical, de gérer plus facilement les maintenances ainsi que les habilitations des intervenants et utilisateurs.
Le service biomédical, en partenariat avec la DSI, devra gérer les données des logiciels de santé DM et DMC de leur mise en service jusqu’à leur mise hors tension. Effectivement, à la différence d’un DM, un logiciel de santé DM et DMC stock et traite les données personnelles du patient qui devront être enregistrées et sécurisées avant son recyclage.
Le dernier chaînon de ce cycle impacté par la nouvelle réglementation est le patient. Assurément, le patient deviendra davantage acteur de son traitement en ayant accès à toutes les données de son dossier. Il devra alors bien comprendre les risques liés à ce type de produit et toute la législation qui lui permet d’être protégé en cas de défaillance du matériel ou d’une attaque de données. Mais pour cela il sera nécessaire que tous les opérateurs cités précédemment prennent à cœur leur rôle pour permettre au patient d’avoir une qualité de soin optimale.
Cette nouvelle réglementation permet ainsi d’attribuer les responsabilités nécessaires pour le bon fonctionnement et la sécurité d’utilisation des DM et DMC.
D’autres opérateurs des services des établissements de santé, tel que dans le bloc opératoire, vont voir leurs activités impactées. Au bloc opératoire le service biomédical est en contact étroit avec différents corps de métier pour veiller au bon fonctionnement des DMC et de leur utilisation lors des opérations. Le service biomédical doit donc prendre en compte les demandes et avis de chacun de ses acteurs (médecin, IBODE, cadre de santé, pharmacien…) [26].
III) Rôles et responsabilités
1. Processus général
La frontière entre l'ingénierie Biomédicale et l’Informatique devient plus mince au fil du temps. La gestion et la maîtrise des données patients est un domaine essentiel reliant ces services. Il est donc important de définir le périmètre d’action de chacun, pour qu’ils puissent travailler ensemble.
Les “responsabilités” étant toujours un sujet délicat au sein des établissements de santé, elles se sont accrues avec le développement rapide des Dispositifs Médicaux Connectés et des logiciels dans les services de soins.
Pour sécuriser les Données de santé, il faut une bonne coordination des services informatiques et biomédicaux. Permettant une efficacité lors des pannes et une amélioration du service vis à vis du patient.
Cependant les deux services n’ont pas tout à fait les mêmes objectifs, le biomédical ayant plus un but de fonctionnement et de moyen alors que l’informatique, la sécurité.
D’ou l’importance de mutualisation et coordination des services.
La cartographie des processus (Figure 5) de responsabilité, ci-dessous, permet d’avoir une vue d’ensemble de l'amélioration possible dans ce domaine.
Ils sont définis en 3 Processus : Management, Opérationnel et Support. Chacun de ces processus sera détaillé par la suite.
Figure 5 : Cartographie des Processus de responsabilité des données patient
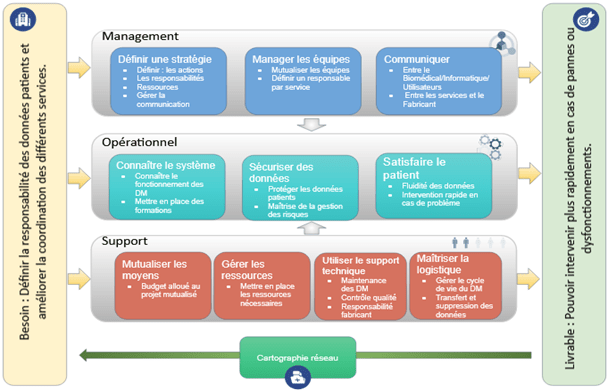
a. Processus : Management
Le but de ce processus est de gérer et mutualiser les ressources. Dans un premier temps, l’enjeu sera de définir les objectifs et actions à mener pour permettre une bonne communication entre les différents acteurs (Ingénieur biomédical, Service Informatique, Utilisateurs, Fabricants).
Un responsable de chaque secteur peut être désigné pour permettre un suivi rapide du projet, ou d’un système existant. De plus, la communication est un élément central. Définir un interlocuteur responsable pour chaque secteur permettra de réduire les acteurs et de gagner en efficacité.
b. Processus : Opérationnel
Le but de ce processus concerne la maîtrise, la sécurisation et la fluidité des données.
Pour bien définir un risque, il faut connaître le fonctionnement de son équipement. Le service biomédical conjointement au fabricant peuvent proposer, si besoin, des sessions de formations aux équipes du bloc opératoire, telles que les infirmières, les IBODE, les IADE, les chirurgiens…
Il est question ici du matériel commun au service de soins comme les respirateurs, pousse-seringues, équipement de monitorage connectés et bien d’autres. Cependant, le cœur du processus est la protection des DATA.
Pour cela il est important d’éviter des problèmes de failles de réseau, dû à des systèmes d’exploitation trop anciens, un manque de mises à jour ou des logiciels malware et ransomware. Ainsi que les faille d’ordre “humaines” (mail contenant un virus, disque dur externe vérolé)
La difficulté principale ici, est la protection de ces équipements et du réseau informatique de l’infrastructure, sans entraver la fluidité des DATA qui peuvent devenir des informations vitales. La Direction des services informatique a souvent une démarche de protection en verrouillant le réseau, au contraire de l'ingénierie biomédicale qui préfère la circulation des données.
La figure suivante (Figure 6) met en avant la relation entre la circulation des données souhaité par l'ingénierie biomédicale et sa sécurisation demandé par l’informatique :
Figure 6 : Balance entre la sécurité des données et sa circulation. (Source : auteurs inspirés du document AFIB “Groupe de travail AFIB 2019–2020 : sécurité numérique des équipements biomédicaux”[6])
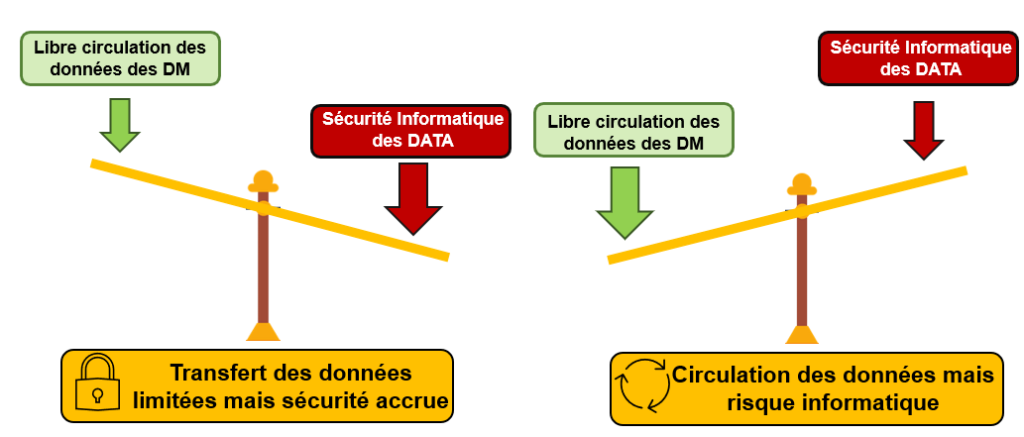
Il y a donc un compromis à effectuer entre sécurité et circulation des données, et en cas de dysfonctionnement, l’intervention doit être rapide et faite à la personne adéquate.
c. Processus : Support
Le but de ce processus est le support qui peut être apporté à ces objectifs. Le thème du budget devra être abordé. Un cas classique dans les institutions hospitalières est “d’acheter” en fonction des budgets attribués ou du budget restant. Par exemple, un équipement informatique peut être acheté par le service biomédical et vice versa en fonction des budgets restants, cela impacte directement la responsabilité des acteurs.
La proposition de mutualisation de certains financements pourrait être une alternative avantageuse. Parallèlement au financement, les équipes peuvent se mutualiser et les acteurs comme le fabricant sont impliqués comme support à cela. Il s’agit également ici de gérer le cycle de vie des dispositifs de santé, par le transfert ou l’effacement des données patient vers un équipement plus récent, si ce dernier est destiné au rebut.
Cette cartographie (figure 5) s’appuie sur des propositions faites dans le document “Groupe de travail AFIB 2019–2020 : sécurité numérique des équipements biomédicaux “ [6]
Beaucoup des éléments abordés sont déjà mis en place dans les établissements de soins. Cet outil a pour but d’avoir une vue d’ensemble, pour proposer un outil concret comme une cartographie du réseau. Cependant il sera difficile de délimiter le périmètre d'intervention de chaque acteur, mais il sera possible d’apporter un éclaircissement sur le rôle du service biomédical de Chalon-sur-Saône, grâce aux connaissances que nous a apportées le technicien sur le dit service.
2. Analyses du parcours de la DATA présente dans le parcours de soins.
L’évolution de la technologie médicale a considérablement augmenté la quantité d’informations du système sanitaire. Il est nécessaire de connaître la provenance et la destination de ces données.
La connaissance du parcours des données au sein de l’hôpital permet d’identifier le personnel compétent pour la résolution des problèmes et de sécuriser les données en bloquant tout échange au bon moment. Pour suivre cette DATA, il est indispensable d’avoir une cartographie du réseau dans l’infrastructure concernée. Ainsi, il est primordial d’identifier les DMC qui communiquent entre eux ou avec des logiciels à l’intérieur et à l’extérieur de la salle ainsi que les serveurs.
Des informations transitent de la préadmission jusqu’à la salle de réveil. Il faut définir comment celles-ci naviguent entre les différents services de l'établissement pour en assurer une sécurité et une sûreté.
La figure suivante (Figure 7) présente les différentes étapes du parcours de soin du patient :
Figure 7 : Schéma illustratif du parcours de soin du patient (Source : auteur)
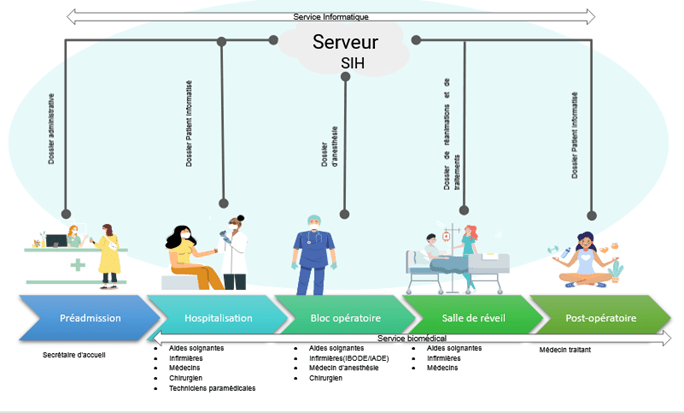
Pour commencer le patient est accueilli, son dossier administratif est créé et tracé par une secrétaire d'accueil dans un logiciel permettant de mettre en place la Gestion Administrative du Malade (GAM) comme Cpage [27]. Ce logiciel alimente le système d’information de l’établissement (SIH), il permet de créer un dossier numérique qui a pour objectif de garantir une continuité de la gestion administrative (IUP, identité, facturation…) au patient.
A la suite de son accueil, le patient va rejoindre un service d'hospitalisation (ambulatoire ou spécifique). A cette étape un dossier de soin va être créé et institué dans le SIH [28] par un logiciel métier comme Cristal [29] ou Easily qui permettra de partager entre les différents acteurs de soins (aides-soignantes, infirmières, médecins, chirurgien, techniciens paramédicales…) d'accéder au Dossier Patient Informatisé. Ce dossier conserve et sécurise les informations de santé concernant le suivi, les traitements, les résultats d’examens, les allergies… et permet un échange entre les différents professionnels. Ceci permet une coordination et une continuité des soins proposés au patient. La mise en place d’un DPI lui permet aussi d’avoir toutes les informations concernant son hospitalisation (transparence des soins).
Ensuite le patient va être pris en charge pour aller au bloc opératoire. Le DPI sera complété par les acteurs de soins puis il sera ajouté le dossier d'anesthésie. Le dossier d’anesthésie est alimenté par le logiciel métier des anesthésies comme clinisoft CCA. Le DPI est alimenté par les DATA venant des DM, logiciels et des DMC utilisés au bloc opératoire pour réaliser l’acte comme TELEMIS, DOSEWATCH, et Surgimedia. Cela entraîne un flux important de données, ce qui induit la mise en place d’une organisation, une sécurisation ainsi que d’une répartition des rôles et des responsabilités. La partie suivante présentera une cartographie mettant en avant cette organisation.
Après le bloc opératoire, le patient va passer en salle de réveil, à cette étape le DPI est complété par les données de réanimations, de traitements post opératoire…
Pour finir le patient retourne à domicile, son GAM va être clôturé par la facturation et son DPI va être transmis à son médecin traitant. Cela permettra de poursuivre les soins même hors de l’établissement, comme l'Hospitalisation à Domicile (HAD) qui du coup induit un échange de DATA entre le patient et le médecin par des DMC. Toutes ces données seront conservées dans le SIH de l’établissement. Cela permet de poursuivre les soins mais aussi de mettre en place une sécurité et une transparence sur les actes médicaux réalisés ainsi que les données personnelles de monitorage et de traitement du patient.
Nous constatons que la sécurité des DATA est primordiale à chaque étape de la prise en charge du patient qui entre pour un acte chirurgical. L'organisation du flux de données vers le SIH est primordiale car ceci permettra de définir les acteurs ainsi que les rôles et responsabilités qui leur sont liés. Grâce à ces informations, le maintien des performances et de la sécurité de ces équipements sera possible (soignants, paramédicaux, service informatique, service biomédicale...), leurs rôles ainsi que leur responsabilité pour permettre le maintien de la performance de ces équipements informatiques. Mais aussi garantir une continuité, une transparence et une sécurité des soins au patient. Pour cela la législation impose un cadre et des organismes comme l’ANSM, l’HAS et l’AFIB apporte des guides, des publications pour aider l'établissement à mettre en place cette organisation et déterminer le rôle de chacun [4]–[6].
IV) Cartographie
Pour suivre et sécuriser les informations utiles au bloc opératoire, les techniciens et ingénieurs biomédicaux ont une image mentale du réseau du bloc opératoire. Chaque cartographie mentale peut être légèrement différente entre les acteurs du fait de la pluridisciplinarité d’un tel service. Pour pallier ce problème qui peut entraîner des risques majeurs du fait ne pas avoir d’outils adaptés, mais aussi pour aider à la compréhension et définir les responsabilités des acteurs, il est essentiel de faire une cartographie physique du réseau tenu à jour à chaque implémentation de nouveaux appareils ou de nouvelles connexions.
Cette cartographie aura pour objectif de définir la responsabilité des utilisateurs des DMC et logiciels connectés. En effet, les utilisateurs de tels dispositifs doivent être conscients des risques qui y sont associés mais aussi savoir la responsabilité qui leur incombe dans l’utilisation des données fournies. Avec cette cartographie, ils pourront plus facilement suivre le parcours des données et contacter la personne adéquate en cas de dysfonctionnement.
Ainsi, les utilisateurs feront appel à la personne compétente pour résoudre le problème rencontré. Par exemple, si le même problème se présente dans 2 salles de bloc opératoire avec un réseau identique, il sera plus facile de diagnostiquer le problème. Dans un tel cas, il pourrait s’agir d’un problème de transfert de données vers le serveur, il faudra alors contacter la DSI. Si le problème a lieu uniquement dans une salle, alors l’utilisateur du DMC appellera le service biomédical pour constater si le problème vient du DM, du logiciel de santé ou d’un problème de réseau. Ainsi le service biomédical pourra aisément définir d’où vient le dysfonctionnement et intervenir rapidement. En effet, la localisation du problème est alors plus aisée pour le service biomédical et peut ainsi intervenir plus rapidement pour éviter de ralentir la prise en charge du patient au bloc opératoire.
Le dernier intérêt, et non des moindres, d’une cartographie détaillée d’un service de bloc opératoire est l’intégration du type de DATA créé et transmis. Le format de l’information permet de relever sa criticité [6] et ainsi prévoir une gestion de risque efficace en cas de dysfonctionnement ou de cybercriminalité. Le codage de la DATA est important car il va permettre de stocker des informations du patient et du traitement nécessaire au bon déroulement de la prise en charge du patient tout en garantissant une protection de ces données [2].
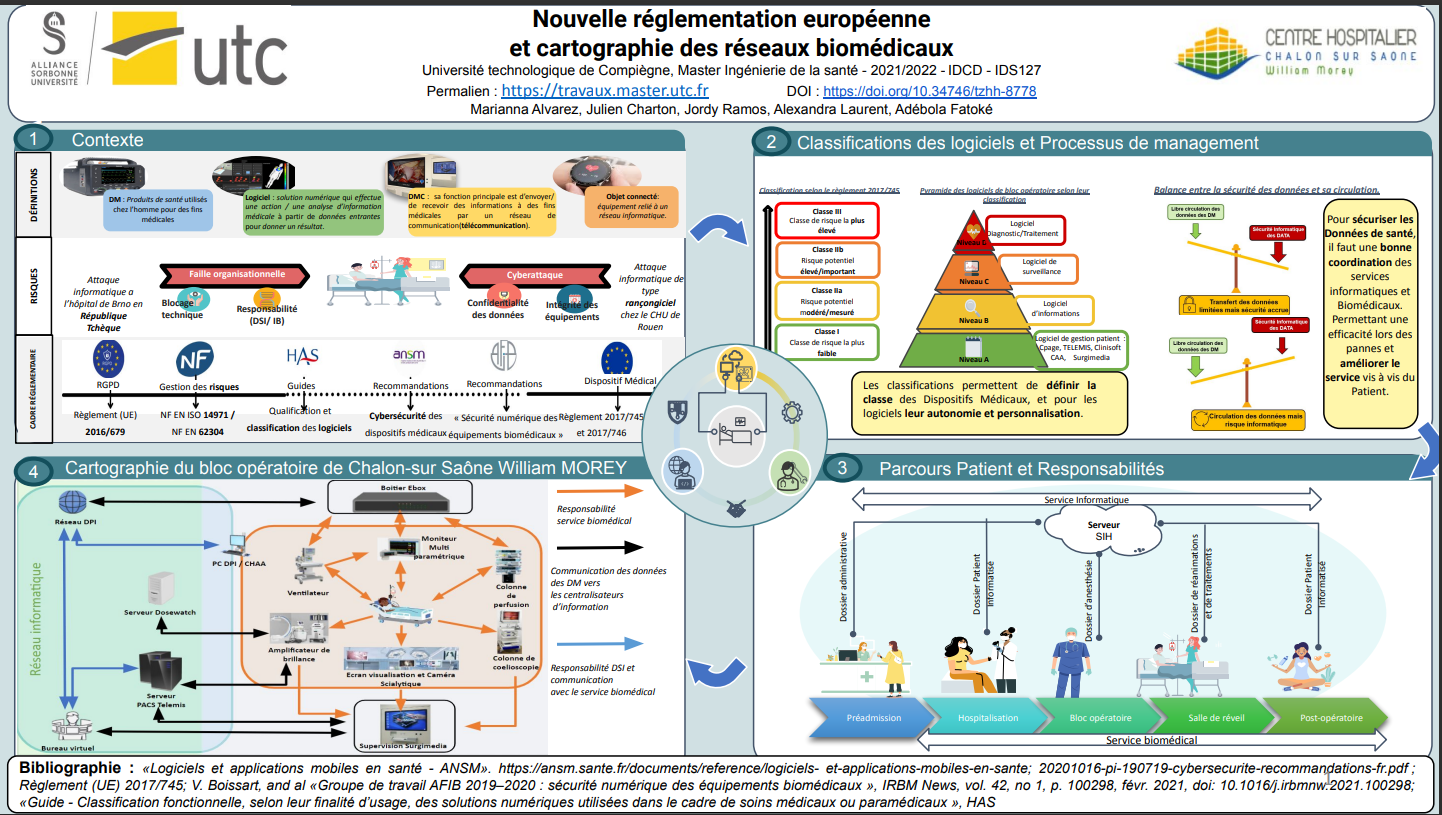
La cartographie suivante (Figure 8) est propre au bloc opératoire de Chalon-Sur-Saône, les flux de DATA entre chaque système y sont représentés [2], [6].
Figure 8 : cartographie du bloc opératoire de Chalon-Sur-Saône WILLIAM MOREY
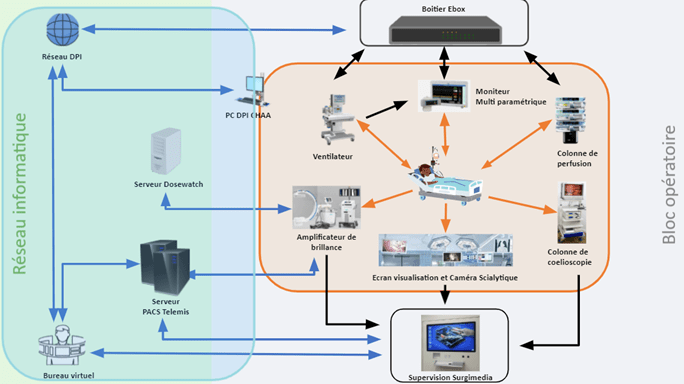
a. Interaction DM-patient
Au bloc opératoire les dispositifs médicaux connectés au patient collectent les informations sur les paramètres physiologiques qui permettent une analyse et adaptation par le personnel médical. C’est le cas du ventilateur et de la cœlioscopie sur le schéma ci-dessus (cadre orange).
Le ventilateur récupère les informations sur le volume courant, volume minute, la pression expiratoire positive (PEP) et le débit d’insufflation. Le clinicien interviendra sur ces paramètres en fonction des besoins du patient. La coelioscopie permet d’enregistrer les images et/ou vidéo pour un diagnostic ou un traitement ultérieur. Ces informations récupérées par les DM sont gérées par le service biomédical, garant des dispositifs médicaux dans l’hôpital.
b. Echange de données entre DMC et serveurs
Les informations vitales recueillies par les DM sont envoyées par câbles RJ45 à travers les prises sur les serveurs ou par relation de DICOMISATION (Dosewatch). Par exemple, le ventilateur sur le schéma ci-dessus est relié au boîtier ebox par câble série pour les échanges de données vitales. Quant à la cœlioscopie, elle transfère les images et/ou vidéo par câble vidéo (HDMI/SDI/s-Vidéo) avec le DMC SURGIMEDIA, qui permet la Dicomisation des données. Ces informations étant en provenance des DM entrent donc dans le champ de compétences du service biomédical, en revanche les logiciels auxquels ils sont connectés sont du ressort de la DSI (Serveurs (PACS, DPI…). A ce niveau un partenariat entre service biomédical et DSI est fortement recommandé pour faciliter la disponibilité et l’accès des données aux personnels soignants.
c. Transmission des données du serveur sur le réseau
Les serveurs convertissent dans leur ensemble les données issues des DMC en format HL7. Le format HL7 est une norme internationale qui permet les échanges informatisés de données cliniques, financières et administratives entre systèmes d'information hospitaliers. Grâce à cette opération, les données sont envoyées sur le réseau principal de l’établissement (Figure 9). Ce qui permet au personnel soignant de consulter le dossier patient dans n’importe quel service. Ainsi par l’intermédiaire du sous répartiteur, le boîtier Ebox envoie les données vitales en format HL7 sur le réseau DPI par fibre. Le réseau DPI, serveur TELEMIS et SURGIMEDIA sont reliés à leur tour par fibre au bureau virtuel qui n’est rien d’autre qu’un ordinateur permettant l’accès sur toutes les autres interfaces tel que le SURGIMEDIA, le serveur TELEMIS et DOSEWATCH. Ces informations sont gérées par le service informatique, qui garantit la sûreté des données et le fonctionnement des différents services à l’aide d’un serveur Back-up, permettant de prévenir un problème lié au réseau via une duplication des données. Ces informations envoyées sur le réseau sont gérées par le service informatique.
Figure 9 : Transmission des données (Source Auteurs)
Conclusion
Par l'application de la nouvelle réglementation Européenne 2017/745 et 2017/746, les dispositifs médicaux font face à de nouvelles exigences. Cela concerne notamment les logiciels de santé. Tout cela, impact l’implantation, l’utilisation, l’organisation et la redéfinition des responsabilités au sein des établissements de santé.
Dans ce mémoire, Il a pu être établi les éléments qui posent le contexte avec la définition des dispositifs médicaux et des logiciels de santé. Ainsi que l’impact des logiciels et des DMC au sein du système de santé et de la prise en charge du patient.
A la suite il a été mis en avant la vision réglementaire et normative qui a permis de mettre en évidence des exigences et une gestion des risques pour apporter de la sécurité, une performance des logiciels et des flux de DATA pour garantir au patient des soins de qualité et un parcours adapté à son besoin.
Une mise en relation entre contexte réglementaire et le bloc opératoire a pu être établie. Cela a permis de réaliser une cartographie des logiciels et des flux de DATA présents au sein du bloc opératoire qui permettra de définir le rôle, les acteurs et leurs responsables. Il est difficile de répartir des rôles distincts entre le service informatique et le service biomédical.
A la suite d’entretien avec les différents acteurs de Chalon-sur-Saône WILLIAM MOREY et selon la politique de l’établissement et son organisation historique, la mutualisation des deux services ou la mise en place d’une organisation permettant de les mettre en lien n’est pas la solution la plus optimale pour répondre aux exigences garantissant une sécurité et une performance aux patients.
Dans ce cas, il peut être envisager les solutions suivantes :
- Créer une division informatique biomédicale au sein du service biomédical.
- Développer les compétences des techniciens.
- Créer un groupe de travail DSI/Biomédical où mutualiser les deux services mais sous certaines conditions (accord et volonté de la direction, des services et d’un gain de productivité).
Références bibliographiques
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017.
[2] Commission Européenne, « Règlement (UE) 2016/679 », ed. ec, www.ec.europa.eu, Règlement, mai 2016. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/reglementation/rg-679-2016/FR154349?rechercheID=3309049&searchIndex=4&activeTab=reglementations
[3] « Directive 2016/1148/CE mesures destinées à assurer un niveau élevé commun de sécurité des réseaux et des systèmes d’information. », ed. ec, www.ec.europa.eu, juill. 2016.
[4] HAS, « Guide - Classification fonctionnelle, selon leur finalité d’usage, des solutions numériques utilisées dans le cadre de soins médicaux ou paramédicaux », HAS - Haute autorité de santé, févr. 17, 2021. https://www.has-sante.fr/jcms/p_3238360/fr/classification-fonctionnelle-selon-leur-finalite-d-usage-des-solutions-numeriques-utilisees-dans-le-cadre-de-soins-medicaux-ou-paramedicaux
[5] ANSM, « Logiciels et applications mobiles en santé », www.ansm.sante.fr, janv. 29, 2021. https://ansm.sante.fr/documents/reference/logiciels-et-applications-mobiles-en-sante (consulté le oct. 18, 2021).
[6] V. Boissart et al., « Groupe de travail AFIB 2019–2020 : sécurité numérique des équipements biomédicaux », IRBM News, vol. 42, no 1, p. 100298, févr. 2021, doi : 10.1016/j.irbmnw.2021.100298.
[7] HAS, « Guide sur les spécificités d’évaluation clinique d’un dispositif médical connecté (DMC) en vue de son accés au remboursement », HAS, Guide pratique 1, janv. 2019.
[8] ANSM, « L’ANSM lance une consultation publique sur un projet de recommandations pour la cybersécurité des dispositifs médicaux », ansm.sante.fr, oct. 16, 2020. https://ansm.sante.fr/actualites/lansm-lance-une-consultation-publique-sur-un-projet-de-recommandations-pour-la-cybersecurite-des-dispositifs-medicaux (consulté le oct. 18, 2021).
[9] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux », Journal officiel de l’Union européenne, Document 31993L0042, Journal officiel n° L 169 du 12/07/1993 p. 0001-0043.
[10] « Directive 95/46/CE (annulé et remplacé par règlement Européen 679/2016) », ed. ec, www.ec.europa.eu, Directive.
[11] « Code de la santé publique », Ed. Legifrance, Paris, Version consolidée au 16 août 2020.
[12] G. Farges et al., Guide des bonnes pratiques de l’ingénierie biomédicale en établissement de santé, Les Pratiques de la Performance. Paris : Editions Lexitis, www.lespratiquesdelaperformance.fr, 2011. Consulté le : déc. 28, 2012.
[13] PGSSI-S, « Guide pratique - Régles pour les dispositifs connectés d’un systéme d’Information de santé », Ministère des affaires sociales et de la santé, Guide pratique V1, nov. 2013.
[14] « Norme ISO/IEC/IEEE 12207-2:2020 - Ingénierie des systèmes et du logiciel - Processus du cycle de vie du logiciel - Partie 2 : Relation et correspondance entre l’ISO/IEC/IEEE 12207:2017 et l’ISO/IEC 12207:2008 », Ed. Afnor, Paris, www.afnor.org, oct. 01, 2020. Consulté le : sept. 23, 2021.
[15] « Norme ISO/IEC/IEEE 15288:2015 - Ingénierie des systèmes et du logiciel - Processus du cycle de vie du système », Ed. Afnor, Paris, www.afnor.org, mai 15, 2015. Consulté le : sept. 23, 2021.
[16] « Norme NF EN 60601-1-8/A11 - Appareils électromédicaux - Partie 1-8 : exigences générales pour la sécurité de base et les performances essentielles - Norme collatérale : exigences générales, essais et guide pour les systèmes d’alarme des appareils et des systèmes électromédicaux », Ed. Afnor, Paris, www.afnor.org, avr. 21, 2018.
[17] « Norme NF EN 62366-1/A1 : 2020 », Ed. Afnor, Paris, www.afnor.org, aout 2020.
[18] « Norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », Ed. Afnor, Paris, www.afnor.org, avr. 30, 2016.
[19] « Norme NF EN ISO 14971 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux », Ed. Afnor, Paris, www.afnor.org, déc. 18, 2019.
[20] « Norme NF ISO/IEC 25051 : 2014 - Ingénierie du logiciel - Exigences de qualité pour le logiciel et évaluation (SQuaRE) - Exigences de qualité pour les progiciels et instructions d’essai », Ed. Afnor, Paris, www.afnor.org, sept. 06, 2014. Consulté le : oct. 18, 2021.
[21] « Norme PR NF EN 62304 - Logiciels de santé - Processus du cycle de vie du logiciel - Projet de révision de la norme », ed. Afnor, Paris, www.afnor.fr, mars 12, 2021.
[22] « Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017.
[23] ANSM, « Exemples de logiciels et applications mobiles illustrant le positionnement réglementaire - ANSM », www.ansm.sante.fr, janv. 29, 2021. https://ansm.sante.fr/documents/reference/exemples-de-logiciels-et-applications-mobiles-illustrant-le-positionnement-reglementaire (consulté le oct. 18, 2021).
[24] HAS, « Évaluer les dispositifs médicaux connectés, y compris ceux faisant appel à l’intelligence artificielle », www.has-sante.fr, févr. 19, 2019. https://www.has-sante.fr/jcms/c_2905546/fr/evaluer-les-dispositifs-medicaux-connectes-y-compris-ceux-faisant-appel-a-l-intelligence-artificielle (consulté le oct. 18, 2021).
[25] Kevin Gutierrez, « Vers une nouvelle transition dans les dispositifs médicaux – Règlement (UE) 2017/745 », www.sante-digitale.fr, nov. 15, 2019. https://sante-digitale.fr/vers-une-nouvelle-transition-dans-les-dispositifs-medicaux-reglement-ue-2017745/ (consulté le oct. 05, 2021).
[26] ANAP - Appui Santé & Médico-Social, « Bloc opératoire - Optimiser les interfaces avec la pharmacie », anap.fr, janv. 06, 2016. https://ressources.anap.fr/bloc-operatoire/publication/1480-optimiser-les-interfaces-avec-la-pharmacie (consulté le oct. 18, 2021).
[27] GIP - CPage, « cpage », www.cpage.fr, 2018. https://www.cpage.fr/offre-globale.html (consulté le nov. 05, 2021).
[28] « Loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé (1) », ed.legifrance, Paris, legifrance.fr, 2002‑303, févr. 2002. Consulté le : nov. 05, 2021.
[29] DCI - Cristal - Net, « DPI - Modules et fonctionnalités », Cristal. https://www.dcicristalnet.com/produit/modules-et-fonctionnalites (consulté le nov. 05, 2021).
Annexe
1. Annexe 1 : Mode d’emplois de la cartographie
Mode d’emplois de la cartographie :
Ce document a pour objectif de vous guider et vous présenter le fonctionnement de la cartographie interactive du réseau.
La cartographie a pour objectif de définir les connexions entre les DM/DMC et logiciels connectés au sein d’un service de soin. Cela permettra le maintien efficace de la performance et la sécurité pour les patients.
Les utilisateurs de tels dispositifs doivent être conscients des risques qui y sont associés mais aussi savoir la responsabilité qui leur incombe dans l’utilisation des données fournies. Avec cette cartographie, il est plus facile de suivre le parcours des données et contacter la personne adéquate en cas de dysfonctionnement.
Pour ce travail, la cartographie a été réalisée pour un bloc opératoire de l'hôpital de Chalon sur Saône, mais elle peut être modifiée et mise à jour pour tout autre service de soins.
Notice d’utilisation de la cartographie :
Accueil avec les légendes, symbolique des icônes

Il est présenté les acteurs, les types d’information et de connexion utilisés pour la transmission des données.
2. Annexe 2 : La cartographie dans son ensemble
Cette interface donne une vue globale des éléments constitutifs de la cartographie. Pour des informations sur les interactions DM-PATIENT, il faut cliquer sur le patient. Dans cette rubrique est schématisé le type de DATA collecté par le DM. Cliquez sur l'icône "flèche" pour retourner à la page précédente ou sur l'icône "maison" pour retourner à la cartographie principale.