IDS147 - Mise en place d'un système de management de la qualité selon l'ISO 13485 v2016 : zoom sur la validation des applications logicielles
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteure

Contact
- Christina Roxanne GBELAY : cgbelay@laposte.net
Citation
A rappeler pour tout usage : Christina Roxanne GBELAY, « Mise en place d'un système de management de la qualité selon l'ISO 13485v2016 : Zoom sur la Validation des Applications Logicielles », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Stage, https://travaux.master.utc.fr/, réf n° IDS147, septembre 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids147/
Résumé
Le 26 mai 2021 marque un tournant pour les professionnels travaillant dans le secteur des dispositifs médicaux. En effet, cette date correspond à l’entrée en application de la nouvelle réglementation autour des Dispositifs Médicaux (DM) et des Dispositifs Médicaux de Diagnostic In Vitro (DMDIV) : Les règlements 2017/745 et 2017/746.
Cette nouvelle règlementation, visant à harmoniser et à renforcer les pratiques de commercialisation des dispositifs médicaux sur le marché européen, apporte avec elle de nouvelles dispositions, pour assurer la qualité, la sécurité et la performance des dispositifs médicaux [1].
Parmi ces dispositions, la mise en place d’un Système de gestion de la qualité renforcé, pour les industriels du dispositif médical, devient une exigence indispensable.
Pour cette mise en place une norme harmonisée spécifique aux industriels du DM existe : L’ISO 13485 : 2016.
La validation des applications logicielles est une exigence de cette norme touchant de près les industriels du DM. Cette exigence représente encore aujourd’hui, près de 6 ans après son apparition, une source importante de non-conformité, lors des audits de certification ISO 13485 : 2016. Cette exigence est cependant incontournable pour la majorité des entreprises de dispositifs médicaux, car il est très rare à l’heure actuelle, de trouver une entreprise qui n’utilise pas de logiciel dans le soutien de ses activités [2].
Mots-clés : Dispositif médical - Système de Management de la qualité - Validation des applications Logicielles du SMQ - ISO 13485
Abstract
May 26, 2021 marks a turning point for professionals working in the medical device sector. Indeed, this date corresponds to the entry into application of the new regulation around Medical Devices (DM): Regulation 2017/745 (MDR).
This new regulation, aimed at harmonizing and strengthening marketing practices for medical devices on the European market, brings with it new provisions to ensure the quality, safety, and performance of medical devices [1].
Among these provisions, the implementation of a reinforced Quality Management System for medical device manufacturers is becoming an essential requirement. For this implementation, a harmonized standard specific to medical devices industrials exists : ISO 13485 : 2016.
The validation of software for medical devices quality system is a requirement of this standard that closely affects medical devices industrials. This requirement remains today, almost 6 years after its appearance, a major source of non-compliance during ISO 13485 : 2016 certification audits. This requirement is however unavoidable for most medical device companies, because it is very rare at present, to find a company that does not use software in support of its activities [2].
Keywords : Medical Device – Quality Management system - Validation of software for QMS - ISO 13485
Téléchargements

Remerciements
Je tiens tout premièrement à remercier l’ensemble de l’équipe pédagogique du master IDS, qui nous a délivré des enseignements de qualité, et qui nous a mis en interaction avec de nombreux professionnels de notre domaine.
Merci à Mme Claude, à M. Prot et à M. Farges, pour l’ensemble des efforts qu’ils mettent en œuvre pour nous apporter une formation qualitative, merci à eux pour leur accompagnement, leur écoute attentive et leur soutien.
Un remerciement particulier à Monsieur Prot, qui a été mon interlocuteur principal, en tant que tuteur d’université, et qui m’a délivré de précieux conseils tout au long de mes 2 années d’études.
Je tiens également à remercier l’ensemble des enseignants que j’ai eu lors de ma formation en licence professionnelle biomédical, et plus particulièrement Mme Mondolini, M. Azan, M. Dubayle, Mme Lénat et M. Tome. En effet, ces derniers m'ont fait découvrir en profondeur le domaine du dispositif médical, et les perspectives professionnelles de ce dernier, c’est également en partie grâce à leurs encouragements, au travers de recommandation, que j’ai eu l’opportunité d’intégrer cette formation.
Mille mercis à Lida Farah, ma tutrice d’apprentissage, de m’avoir prise en tant qu’Apprentie Chargée Affaires Règlementaires et Qualité. J’ai eu l’opportunité de découvrir une personne professionnelle et pleine de bienveillance. Cette alternance fut un réel challenge, mais j’ai pu compter sur son écoute, et je ressors énormément grandit de cette expérience dans mon domaine, tant sur le plan professionnel que sur le plan humain. Merci également à l’ensemble de l’équipe de ZOS IS pour leur accueil.
Pour terminer, je tiens à remercier les personnes qui comptent le plus dans ma vie : L’ensemble de ma famille et mes amis.
Merci à ma mère de m’avoir encouragé tout au long de ma vie et de m’avoir supporté, ainsi qu’à mon père de m’avoir orienté et d’être resté à mon écoute, même avec des milliers de kilomètres nous séparant.
Merci également à Nolwenn G., d’avoir toujours cru en moi, de m’avoir motivé et apporté son soutien inconditionnel, et à Emmanuel C. de m'avoir épaulé durant ces deux années de master, de m’avoir réconforté dans les moments durs et d’être l’une de mes sources motrices les plus importantes.
Glossaire et abréviations
- Norme harmonisée : Norme donnant une présomption de conformité à un texte règlementaire
- Règlements : Actes législatifs devant être mis en œuvre dans leur intégralité sans transposition et sans interprétation. Il s’applique en tant que tel à tous les états concernés.
- Directive : Acte législatif fixant des objectifs et devant être transposé dans chaque pays concerné et pour lequel une liberté d’interprétation est possible.
- DM : Dispositif Médical
- DMDIV : Dispositif Médical de Diagnostic In Vitro
- SMQ : Système de Management de la Qualité
- TPE : Très Petite Entreprise
- MDR : Médical Device Regulation (Règlement 2017/745)
- ISO : Organisation Internationale de normalisation
- NF : Norme Française
- EN : Norme Européenne
- SOUP : Software Of Unknow Pedigree
Mémoire complet :
Mise en place d’un Système de Management de la Qualité, selon l’ISO 13485v2016 : Zoom sur la Validation des Applications Logicielles
Introduction
Les logiciels de santé sont des technologies numériques visant à gérer, à améliorer et à maintenir la santé des individus ou des prestations de soins [3].
Avec l’entrée en vigueur de la nouvelle règlementation autour des dispositifs médicaux (Règlement 2017/745), de nombreux logiciels passent de la catégorie de « simple » logiciel de santé à dispositif médical et ce passage s’accompagne d’un cadre juridique spécifique et beaucoup plus pointilleux que celui encadrant les logiciels de santé.
L’enjeu est crucial dans le secteur de la santé, car les logiciels régissent de plus en plus les activités en santé, d’autant plus que la plupart des sociétés ayant développé un logiciel de santé qui devient dispositif médical, mesurent rarement l’ampleur des exigences et démarches accompagnant la commercialisation d’un dispositif médical [4][5][6][7].
Ainsi, deux cas de figure peuvent se présenter pour ces sociétés :
Soit, elles n’ont pas pu ou su marquer leur dispositif médical logiciel sous les directives, avant la date de mise en application du règlement (26 mai 2021), dans ce cas elles ne disposent pas de la période dérogatoire accordée aux dispositifs marqués CE sous directive, et doivent retirer leur logiciel du marché européen jusqu’à obtenir le marquage CE.
Soit, elles ont pu marquer leur dispositif médical logiciel sous directive, avant la date de mise en application du règlement, elles disposent alors d’une période dite “période dérogatoire” jusqu’au 27 mai 2024, leur permettant de continuer à commercialiser leur logiciel sur le marché européen (sous réserve d’être conforme aux dispositions transitoires de l’article 120 du règlement).
Mais, si elles souhaitent maintenir leur logiciel dispositif médical sur le marché européen après le 27 mai 2024, ces dernières doivent rapidement réaliser la transition vers le règlement européen, car au-delà de cette date, les certificats de conformité obtenus sous directive sont rendus caduques, engendrant un retrait immédiat de ces DM [1].
Dans le cadre de ma dernière année de Master, j’ai eu l’opportunité de travailler au sein d’une entreprise se trouvant dans le 2ème cas de figure et devant donc effectuer la transition vers le règlement 2017/745.
Mon mémoire de fin d’alternance vient éclairer sur la mise en place d’un SMQ dans ce type de structure, pour laquelle un système de management de la qualité n’est pas encore implémenté et se concentrera plus particulièrement sur une exigence spécifique de la norme 13485v2016 : Le point 4.1.6 concernant la « Validation des applications logicielles ».
Chapitre 1 : ZOS Information System : De Logiciel de santé à Dispositif Médical Logiciel
Présentation de ZOS IS
Histoire de ZOS IS : L'association d'un directeur à son prestataire indépendant
ZOS Information System est une entreprise créée en 2018 par M. Souhail Bou Khaled, le dirigeant actuel de l’entreprise. C’est une entreprise encore jeune qui est née d’une collaboration pérenne, de plus de 10 ans, entre un client (Mr Bou Khaled), et son prestataire indépendant.
Le prestataire indépendant a donc commencé à travailler en collaboration avec Mr Bou Khaled en free-lance, ZOS IS n’était pas encore créée.
Le dirigeant actuel de ZOS IS était alors déjà à la tête de plusieurs entreprises du réseau de sociétés “Elia médical”, Prestataires de Santé à Domicile (PSAD) spécialisés dans l’assistance respiratoire à domicile.
Au début de cette collaboration, le prestataire avait été missionné pour mettre en place une interface (type « site web ») permettant au PSAD Elia-Médical, de gérer les rendez-vous des patients dont ils sont en charge. Le PSAD étant chargé de la livraison, l’installation, le suivi et également la maintenance du matériel médical et des traitements associés [8].
Au fur et à mesure du temps, les missions confiées au prestataire indépendant sur l’interface ont évolué et se sont diversifiées, et plusieurs fonctionnalités ont vu le jour sur l’interface initialement demandée. La collaboration de service initiale a alors naturellement abouti à une collaboration plus encadrée, grâce à la création de ZOS IS par le client et l’embauche du prestataire en tant que premier salarié de la société nouvellement fondée.
C’est à la suite de cela que le produit de ZOS IS a vu le jour, I-GEIA est née. D’abord catégorisé en tant que logiciel de santé, il a ensuite été requalifié en tant que dispositif médical.
À noter que le nom I-GEIA vient de la mythologie grecque, et plus précisément de la déesse HYGIE, déesse de la santé, de la propreté et également de l'hygiène [9].
Organisation de ZOS IS
Zos Information System est actuellement une TPE (Très Petite Entreprise), qui se revendique en tant que société spécialisée dans le développement de solutions numériques sur mesure (logiciels, applications web et mobile), en associant innovation et appareils connectés. Elle s’est développée petit à petit, d’année en année et continue encore à se développer. Actuellement, elle compte 9 salariés stables et 2 étudiants en apprentissage [10].
Trois services peuvent être identifiés au sein de la société :
- Le service recherche et développement (R&D) : C’est le service en charge du développement et de la maintenance du produit. Il est composé d’une équipe de 4 développeurs, encadrée par un Responsable technique et créatif. Un apprenti développeur a également intégré ZOS IS au cours de l’année 2021-2022 et poursuivra normalement son alternance durant l’année 2022-2023.
- Le service support : Ce service est constitué d’une personne chargée du support technique, qui s’occupe de la gestion de l’ensemble des retours client et utilisateur, et d’une Chargée de communication et de marketing qui gère la mise en place d’une stratégie de communication à la fois en interne et également en externe pour faire connaitre les services offerts par l’entreprise, fidéliser sa clientèle, attirer de nouveaux clients et ainsi étendre son marché.
- Le service affaires réglementaires et qualité : Ce service nouvellement créé est en charge d’assurer la conformité de ZOS IS à l’ensemble des exigences qui lui sont applicables, de par la nature du produit qu’il commercialise (un dispositif médical). Il compte pour l’instant une Responsable Qualité et Affaires règlementaires, qui a été accompagné par moi-même, en tant qu’apprentie Chargée affaires réglementaires et qualité.
L’organigramme suivant (figure 1) représente l’organisation hiérarchique de ZOS IS en date actuelle :
Figure 1 : Organigramme de ZOS IS. Source : Auteure
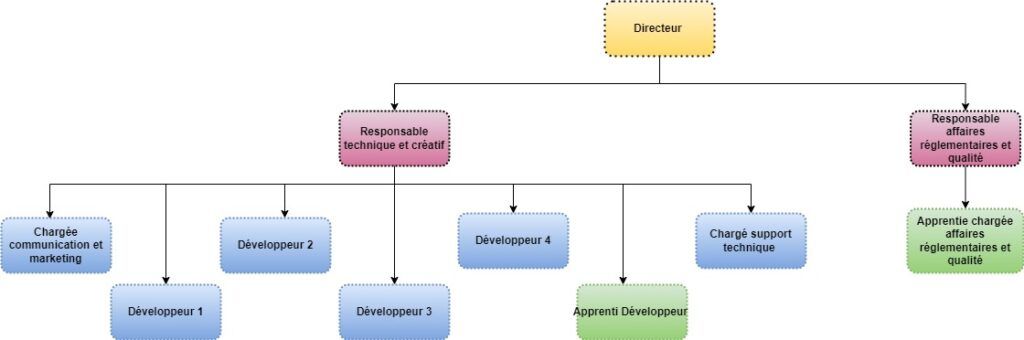
L’ensemble des salariés sont répartis dans deux locaux différents. Un local se situe à Boulogne-Billancourt et regroupe le service R&D et le service support. Le service affaires réglementaires et qualité est quant à lui délocalisé aux Loges-en-Josas, pour des raisons de limitation en termes d’espace dans les locaux de Boulogne-Billancourt.
Les clients de ZOS IS sont les sociétés du réseau d’Elia médical ainsi que d’Univ’air médical, également Prestataires de Santé à Domicile spécialisés dans l’assistance respiratoire à domicile [8].
Présentation du produit commercialisé par ZOS IS
I-GEIA : Vers une télésurveillance ultra-performante des patients en assistance respiratoire
ZOS IS se concentre actuellement sur un unique produit, le dispositif médical I-GEIA. L’équipe étant relativement petite, comparée à la charge de travail, il est actuellement difficile pour l’entreprise d’élargir son portefeuille produit.
I-GEIA était donc à la base un “simple” logiciel de santé, mais les demandes récurrentes d’amélioration venant des clients et utilisateurs ont conduit à l’ajout de fonctionnalité faisant basculer le logiciel de santé dans la catégorie bien particulière des dispositifs médicaux, soumettant ainsi le logiciel aux obligations établis dans le règlement 2017/745.
I-GEIA est une plateforme informatique permettant le suivi des personnes en assistance respiratoire qui font de l’apnée du sommeil.
L’apnée du sommeil, ou syndrome d’apnées-hypopnées obstructives du sommeil, est une pathologie qui se traduit par des arrêts respiratoires répétés et incontrôlés, qui conduisent à des micro-réveils constants du patient, qui n’en a pourtant pas conscience.
Le nombre d’individus souffrant de ce syndrome est en augmentation au fur et à mesure de l’amélioration des techniques de diagnostic, et est à l’origine de nombreux troubles tels que les ronflements, la fatigue chronique, la somnolence diurne (en pleine journée), les troubles cardiovasculaires, voir même le décès du patient etc.
Aussi, un suivi régulier et précis de ces patients est nécessaire pour limiter ces troubles [11], [12].
Ainsi, le logiciel i-GEIA permet aux Prestataires de Santé à Domicile, spécialisés dans l’assistance respiratoire, et aux médecins prescripteurs d’accéder d’une manière numérique aux données des patients appareillés d’un dispositif respiratoire. Il permet de stocker les dossiers patients pour fournir un suivi de l’historique de l’appareillage et des interventions effectuées au domicile du patient.
Pour les patients équipés d’appareils connectés (ventilation assistée ou appareil à pression positive continue), le logiciel recueille les données d’observances des serveurs des fabricants par une interface de programmation d’application (API) et les transforme en graphiques pour faciliter la lecture. Le logiciel génère ainsi une liste d’alertes médicales selon un algorithme et des critères définis.
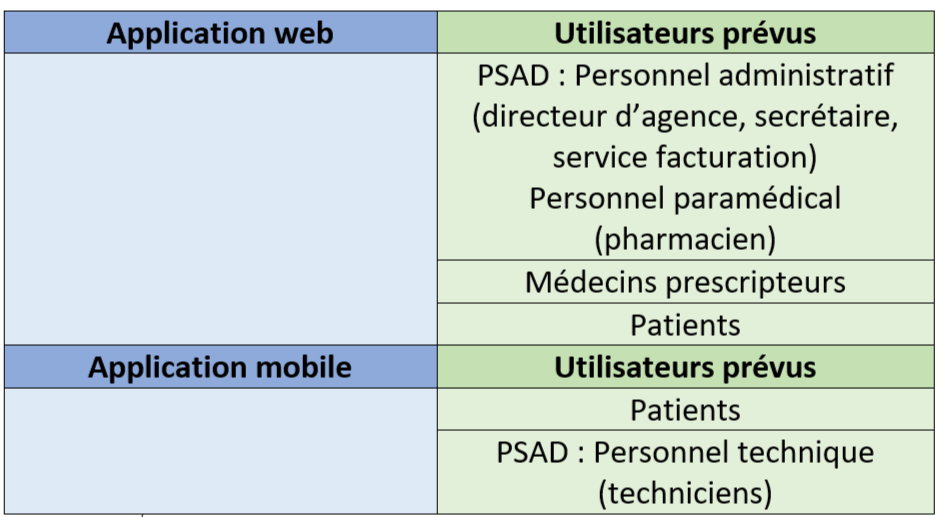
Le logiciel comprend plusieurs fonctionnalités et existe sous quatre configurations :
- I-GEIA pour le PSAD est sous le support d’une application web : Le PSAD représente l’entité en charge du matériel médical, de son installation ainsi que de sa maintenance. Il constitue l’intermédiaire privilégié entre le patient, chez qui le matériel est installé, et le médecin prenant en charge ce patient (médecin ORL, médecin généraliste, cardiologue, neurologue …).
- I-GEIA pour les techniciens est sous le support d’une application mobile : Les techniciens font partie du personnel du PSAD et sont notamment en charge de l’installation et des visites chez les différents patients. L’application leur permet de saisir les données lors de leurs visites aux domiciles des patients.
- I-GEIA pour les médecins est sous le support d’une application web : Le médecin est la personne qui suit le patient et qui lui a prescrit la machine médicale. Le logiciel permet au médecin prescripteur de suivre les données d’observance de ses patients. Les données d’observance sont :
- L’index apnée/hypopnées : il représente les apnées non corrigées par l’appareil
- Les fuites aux masques : elles peuvent être le signe d’un problème d’adaptation du masque à la morphologie faciale du patient.
- La durée d’utilisation de l’appareil par le patient : qui permet de constater la bonne utilisation ou non par le patient de son traitement.
- La pression efficace
Il permet également de visualiser les jours où une alerte médiale a été détectée chez un patient, ainsi que de visualiser les visites effectuées par le PSAD chez le patient (à la suite d’une alerte ou lors d’une visite de suivi habituelle). Ces différents indicateurs vont donc donner différentes indications permettant d’améliorer la prise en charge du patient.
- I-GEIA pour les patients : I-GEIA patient peut être présent sous le support à la fois d’une application web et d’une application mobile. L’application permet à chaque patient d’être un acteur central de sa santé. En effet, le patient peut suivre quotidienne et facilement plusieurs de ces paramètres, mais aussi contacter, au travers de l’application, son médecin ou le PSAD pour toute interrogation.
Le tableau ci-dessous (tableau 1) présente un résumé des différentes plateformes et des utilisateurs associés :
Tableau 1 : Récapitulatif des configurations de i-GEIA. Source : Auteure
I-GEIA se développe de jour en jour et les demandes d’améliorations en termes de modification ou d’ajout de fonctionnalité ne cesse d’augmenter.
Le logiciel tend par ailleurs à implémenter une fonctionnalité d’aide à la décision, permettant au médecin, au travers de réponse à diverses questions et d’un bilan final, de prendre des décisions concernant la modification du traitement du patient.
I-GEIA sur le marché français
La réalisation d’une analyse concurrentielle du marché auquel se confronte I-GEIA, a permis d’identifier 2 concurrents principaux :
- Adel santé par Datamedcare
Adel santé est une plateforme de télé-suivi qui collecte et agrège des données de santé provenant de différents types de dispositif médical, pour les mettre à la disposition des différents acteurs du parcours de soin. Adel santé gère comme I-GEIA des patients atteints de pathologies respiratoires [13].
- MUST-G5 (Orthop) par Must Informatique
MUST-G5 est un outil informatique dédié au PSAD permettant de gérer leur intervention (planification...), les rapports d’intervention ainsi que la facturation SESAM-Vitale et SCOR (l’envoi de feuilles de soins sous format électronique à l’assurance maladie et aux organismes d’assurance maladie complémentaire). MUST-G5 gère comme I-GEIA des patients atteints de pathologies respiratoires [14].
D’autres concurrents indirects peuvent être également cités tel que Resmed avec leur logiciel AirView ou Phillips respironix dream station, mais ces derniers mettent en place ce type d’outil directement intégrer à leur propre appareil respiratoire qu’ils fabriquent [15], [16].
Peu d’informations sont trouvables sur ces diverses technologies pour permettre d’effectuer un comparatif significatif et pertinent.
ZOS IS : Une entreprise jeune et prometteuse avec une organisation interne à développer
Figure 2 : SWOT de ZOS IS. Source : Auteure
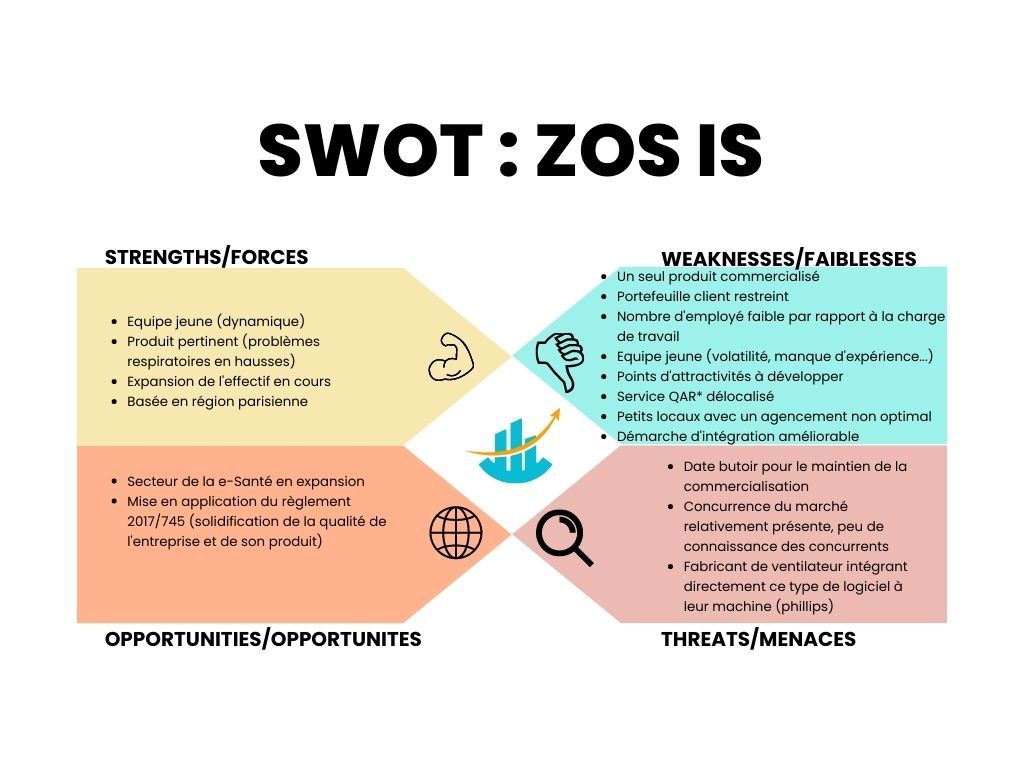
Le SWOT ou MOFF en français est une analyse globale d’une entreprise, permettant d’évaluer ses forces et faiblesses ainsi que d’identifier les opportunités pouvant s’offrir à elle et les menaces auxquelles elle pourrait être confrontée.
L’analyse de l’entreprise par le SWOT (figure 2) montre que les forces et donc points forts de l’entreprise à développer réside dans ses ressources humaines, qui sont des salariés jeunes et dynamique [17]. La pertinence de leur produit est également un atout important (pathologies respiratoires en hausses d’années en années) [18].
Les faiblesses de l’entreprise et donc les points d’amélioration pour lesquels cette dernière doit porter une attention particulière, afin de rester performante sur son marché sont notamment :
- Réfléchir sur l’éventualité d’étendre son portefeuille produit afin que son chiffre d'affaires ne soit plus dépendant d’un seul et unique produit
- Trouver des locaux, aussi bien localisé que ceux de Boulogne, suffisamment grand pour accueillir l’ensemble des services et suffisamment agencé pour permettre une meilleure cohésion de groupe
- Amélioration de l’attractivité de la société et de sa politique d’intégration des salariés pour limiter le turn-over et favoriser une atmosphère de travail plus stimulante
- Réfléchir sur une stratégie permettant d’étendre son portefeuille client
Plusieurs opportunités s’offrent également à l’entreprise, telles que l’expansion de la e-Santé, offrant une réflexion sur la possibilité de concevoir un nouveau produit entrant dans le cadre de la e-santé, mais également le nouveau cadre juridique s’imposant à l’entreprise, qui va permettre d’améliorer de façon notable la qualité des pratiques en interne, mais également l’image renvoyée par la société.
L’entreprise devra faire également attention à certains points externes à son organisation, qui pourraient l’impacter négativement :
- La date butoir (27 mai 2024) à laquelle les dispositifs médicaux marqué CE sous directives ne pourront plus être commercialisés, si ces derniers n’ont pas obtenu le marquage CE sous le règlement 2017/745. ZOS IS doit donc impérativement réaliser sa transition « directive -> règlement » et obtenir son marquage CE, si elle souhaite maintenir son DM sur le marché
- Une veille concurrentielle doit également être effectué afin d’assurer la plus-value de son outil par rapport aux outils similaires se développant
ZOS IS est une société en plein essor dont les perspectives de développement sont multiples et doivent passer par une amélioration de son organisation interne. Une amélioration du suivi et des besoins des salariés pourrait permettre un meilleur épanouissement de ses derniers et une amélioration de leur efficacité et efficience. L’entreprise à un large panel de clientèle potentiel auquel il serait intéressant qu’elle réussisse à s’ouvrir, le désir de recrutement de nouveau salarié tend à se concrétiser et permettrait également d’augmenter les performances de l’entreprise.
Chapitre 2 : Vision macro de la mise en place d'un Système de Management de la Qualité selon l'ISO 13485 : 2016
Présentation de la norme et des points principaux
La nouvelle règlementation autour des dispositifs médicaux explicite la nécessité de mettre en place un système de gestion de la qualité pour tout fabricant de dispositifs médicaux. Ce système de gestion de la qualité doit garantir la conformité au règlement.
Le règlement nous donne le « Quoi », c’est-à-dire « quoi faire », mais ne nous donne pas le « comment », c’est-à-dire le “comment faire”. Pour savoir « comment faire » et donc comment appliquer les exigences pour être conforme à la règlementation, l’utilisation des normes harmonisées est une solution pertinente.
ZOS IS développe un dispositif médical logiciel, ainsi la mise en place d’un système de gestion de la qualité est obligatoire. Une norme harmonisée existe, permettant de donner la présomption de conformité à certains points exigés par le règlement, concernant le système de gestion de la qualité : C’est la norme ISO 13485 : 2016 “Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires”.
L’ISO 13485 est la norme par excellence utilisée dans le domaine du dispositif médical pour mettre en place un système de management de la qualité, permettant de démontrer l’« aptitude [des entreprises du DM] à fournir régulièrement des dispositifs médicaux et des services associés conformes aux exigences des clients et aux exigences réglementaires applicables » [19].
En suivant cette norme, ZOS IS pourra se conformer en partie aux exigences demandées par le règlement pour la mise en place d’un système de gestion de la qualité. « En partie » signifie que l’application complète de cette norme ne garantis pas la conformité totale à l’exigence du règlement. Pour déterminer les parties couvertes, partiellement couvertes ou non couvertes par la norme, il existe le référentiel NF EN ISO 13485/A11 : 2021, qui vient expliciter le degré de couverture, au travers des annexes Z [20], [21].
ZOS IS étant dans un premier temps une société développant un produit de type logiciel de santé, la mise en place d’un SMQ et donc l’instauration d’une politique et d’une culture qualité n’était ni une obligation, ni une préoccupation première de la société. De ce fait, ZOS IS, lors du début de l’alternance, n’avait pas encore débutée sa démarche de mise en place d’un SMQ. Il existait donc très peu de documentation pouvant servir de base à l’implémentation d’un SMQ.
Une première étape était donc d’étudier la norme ISO 13485 : 2016, afin d’identifier l’ensemble des exigences de cette norme et de déterminer quelles étaient celles applicables à l’entreprise et à son produit (Dispositif Médical actif de classe IIa).
En effet, il existe des parties de cette norme qui ne s’applique pas à toute entreprise et à tout dispositif.
Par exemple l’exigence 7.5.2 “Propreté du produit” n’est pas applicable à ZOS IS de par la nature de son dispositif (logiciel en tant que dispositif médical) ce dernier ne peut pas être soumis à des phénomènes de contamination biologique, car il n’est pas en contact direct de l’humain, ou encore le point 7.5.5 qui s’adresse à des dispositifs médicaux spécifiques, les dispositifs médicaux stériles [19].
Une fois cette analyse de la norme effectuée et cette détermination des exigences applicables, qui ont été agencées sous forme de tableau, afin de faciliter la visibilité de l’ensemble des exigences (Annexe 1), la mise en application de ces dernières a été planifiée et réalisée.
L’ensemble des exigences de la norme s’articulent autour de 5 points :
A) La mise en place d’un système de management de la qualité
Point centrale de la norme, la mise en place d’un SMQ s’axe sur l’approche processus. L’approche processus permets à un organisme de structurer l’ensemble de ses activités tout au long du cycle de vie de ses DM.
Pour mettre en place l’approche processus, l’organisme doit d’abord identifier l’ensemble des processus qui interviennent au sein de sa structure.
Un processus représente une activité ou un ensemble d’activités, réalisée(s) dans l’entreprise, qui transforment des éléments d’entrée en éléments de sortie.
Par exemple, si l’on prend un exemple de la vie quotidienne et que l’on considère donc l’activité « Nourrir sa famille », comme un processus alors l’élément d’entrée pourra être « La famille est affamée » et l’élément de sortie « La famille est nourrie ».
Un exemple de processus interne à l’entreprise qui a pu être identifié au sein de ZOS IS est le sous-processus « Traiter les réclamations », l’élément d’entrée étant « Réclamations des utilisateurs » et les éléments de sortie étant « Modification du logiciel » et « Notifications (fiches d’avertissements) ».
Les processus peuvent être regroupés/subdivisés en 3 macro-processus (ensemble cohérent permettant de rassembler plusieurs processus ou sous processus en grand domaine) :
- Les processus de management/pilote : Ils représentent les processus qui viennent piloter la démarche qualité et en assurer son amélioration continue.
- Les processus de réalisation : Ils représentent les processus centraux de l’entreprise. En effet, ce sont les processus qui vont contribuer directement à la réalisation de(s) élément(s) de sortie (service ou produit) offert(s) par l’entreprise (de l’identification du besoin client jusqu’à sa satisfaction).
- Les processus support : Ils représentent les processus qui vont participer au bon déroulement de l’ensemble des autres processus. En effet, ils vont fournir aux autres processus les ressources nécessaires pour leur fonctionnement correct (matérielles et immatérielles) [22].
Une fois les processus identifiés, il faut les organiser dans la logique entreprise. Pour cela, chaque processus peut être classé dans un des 3 macro-processus précédemment cités.
Il est à noter que la classification des processus dans ces trois macro-processus peut varier d’une entreprise à une autre. Par exemple : le processus “maintenance informatique” en fonction de sa destination (destination de client finaux ou à destination de service de production interne) fera soit partie du macro-processus réalisation, car il contribuera directement à la réalisation du service ou du produit, ou bien il fera partie du macro-processus support, car il interviendra en tant qu’aide au maintien des différentes activités. Dans le cas de l’entreprise ZOS IS par exemple, nous avons positionné le processus maintenance (informatique), dans le processus de réalisation, car la maintenance de notre logiciel dispositif médical participe directement à la réalisation de notre produit.
Les processus ainsi regroupés en macro-processus, une cartographie des processus peut être réalisée.
La cartographie des processus est une représentation visuelle permettant de mettre en évidence les interactions entre les processus, de façon simplifiée et illustrée, et donc plus communicatif pour l’ensemble du personnel.
Le choix de la représentation de la cartographie (figure 3) est laissé à la libre appréciation de la personne en charge de sa réalisation, cependant les cartographies processus sont généralement représentées avec une structure de ce type :
Figure 3 Exemples de cartographie de processus. Sources : Stratégik, Qualitexpert, Advaloris
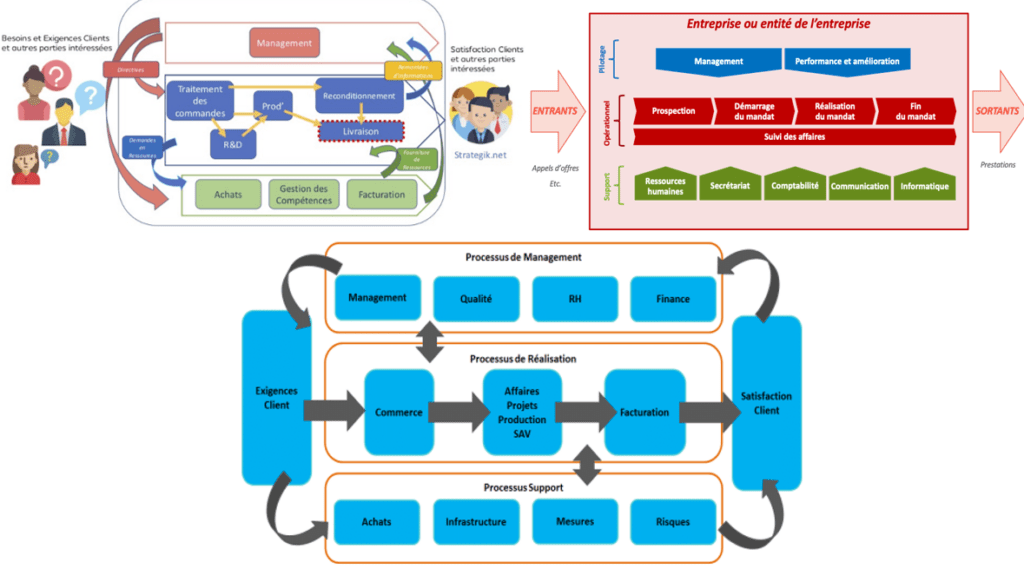
Enfin, chaque processus au sein de la structure doit être maitrisé, et leur maitrise passe par une approche fondée sur les risques (analyse des risques des différents processus afin d’identifier les activités critiques et de mettre en place, si nécessaire, des actions proportionnées) [23].
B) La responsabilité de la direction
Point non-négligeable, la responsabilité de la direction dans la norme est clairement explicitée.
La norme indique que la direction doit faire preuve d’engagement dans la démarche de mise en place d’un SMQ. Cet engagement passe notamment au travers de l’établissement d’une politique qualité, par la direction, au travers d’une planification d’objectifs qualités.
Cette politique qualité décrit les axes stratégiques de l’entreprises, le(s) but(s), l(es) objectif(s) de l’entreprise et la direction dans laquelle cette dernière souhaite aller. Elle est essentielle et doit être connue de l’ensemble des salariés, car elle vise à diriger l’entreprise et à lui permettre, au travers de l’atteinte des objectifs, d’améliorer continuellement son activité. Elle est également un outil de management non-négligeable, si elle est utilisée à bon escient, en fédérant les collaborateurs autour des axes stratégiques communs de l’entreprise.
La direction devra également sensibiliser ses salariés en communiquant régulièrement et dès que nécessaire sur la qualité (et notamment la politique et les objectifs qualité).
Souvent négligée par les entreprises, la responsabilité de la direction est pourtant le point moteur de la mise en place efficace d’un système de management de la qualité performant.
En effet, l’attribution des ressources est dépendante de la direction, une direction qui ne mettra pas en place les ressources nécessaires à l’avancé et à l’efficacité de la mise en place du SMQ, contribuera à un ralentissement considérable de sa bonne mise en place, et pourra même conduire à des non-conformités (SMQ mis en place mais non-conforme). Sans l’appui de la direction les ressources nécessaires sont très limitées pour la mise en place d’un SMQ performant.
C) Le management des ressources
Lié comme décrit précédemment à la direction, le management des ressources au sein de l’organisme est fondamental.
En effet, il faut s’assurer que l’ensemble des ressources nécessaires sont, tout au long du cycle de vie du dispositif médical, disponibles et exploitables (humaines, matérielles...).
Ces ressources vont permettent notamment de satisfaire et ainsi répondre aux exigences règlementaires applicables, aux exigences en termes d’infrastructure, en termes d’environnement de travail et en termes de compétence des acteurs de la qualité.
La norme ISO 13485 : 2016 exige ainsi que l’organisme soit en capacité de fournir les ressources adéquates à la mise en place et au maintien du système de management de la qualité, et également capable de fournir des preuves de la qualification du personnel clé dans le SMQ.
D) La réalisation du produit
Dans le cadre de l’ISO 13485 le produit est un dispositif médical.
La norme centre la réalisation du produit sur la maitrise des risques. Le DM doit ainsi être conçu, développé et produit en minimisant au maximum les risques liés à la conception, à l’aptitude à l’utilisation, à la production, à l’achat de composant ou de matière première, au transport etc...
L’organisme doit pouvoir fournir les preuves de ses activités de gestion des risques en tenant notamment un registre.
Lors de la réalisation du produit la norme exigence également que plusieurs facteurs soient pris en compte tel que l’environnement de travail, le contrôle de la contamination (non applicable dans le cas de ZOS IS, comme vu précédemment), l’infrastructure de l’entreprise, la manipulation du produit (non applicable à ZOS IS), le stockage adéquat du produit (non applicable à ZOS IS), les méthodes de distribution, la traçabilité du produit.
E) Le mesurage, l’analyse et l’amélioration
Enfin, une fois que l’ensemble des exigences de la norme sont appliquées au sein de l’organisme, la bonne application de la norme ne s’arrête pas là.
En effet, il ne suffit pas d’appliquer les exigences, il faut s’assurer que ces exigences soient toujours exécutées et conformes dans le temps.
Ce dernier point implémente cette nécessité. En effet, la norme ISO 13485 exige que les produits, les processus et les services soient surveiller et mesurer au regard des objectifs prédéfinis, des exigences et des activités planifiés, pour rendre compte des résultats.
L’organisme doit réaliser et mettre en œuvre des procédures qualité, ou des méthodes pour s’assurer que le produit répond ou dépasse toutes les exigences du client et/ou de la réglementation. De plus, des données doivent être collectées et analysées afin de s’assurer de l’efficacité du SMQ mis en place. Les données devront prendre en compte les informations recueillies à partir des audits et de la mesure des processus (au travers d’indicateur) et du produit ainsi que des retours des clients et des fournisseurs [24][25][26].
Pour répondre à l’ensemble des points précités, il est important de se positionner sur l’état d’avancement de l’application des exigences. L’outil d’autodiagnostique de l’ISO 13485 : 2016 réalisé par des étudiants de l’UTC, a été effectué après quelques mois passés en entreprise (figure 4).
Cet outil a pu montrer que nous avions progresser dans la mise en place du SMQ, mais que nous avions encore une quantité importante d’exigence à mettre en place, et notamment au niveau de l’article 8 “Mesurage, analyse et amélioration” et de l’article 5 “Responsabilité de la direction”.
Figure 4 Autodiagnostique de ZOS IS sur sa conformité à l'ISO 13485v2016 après quelques mois passés en entreprise
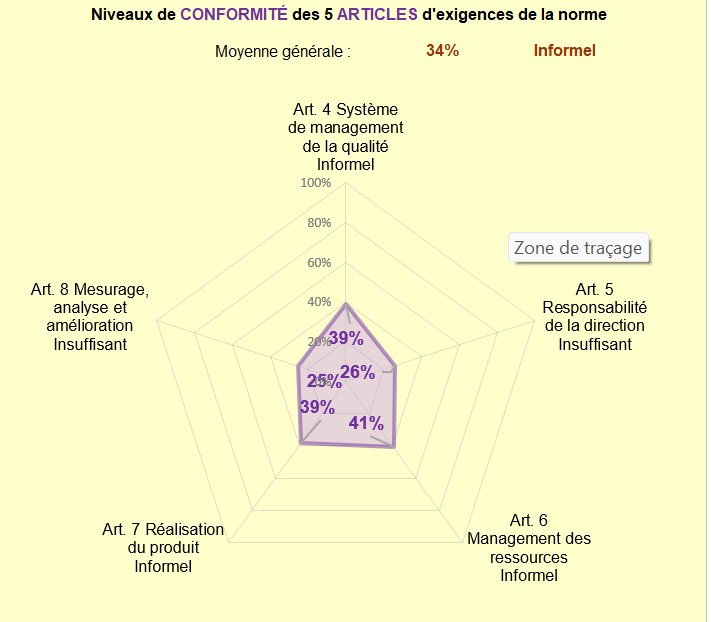
Aussi, autour de ces points s’articule une exigence fondamentale : la mise en place d’un système documentaire.
Aide à la rédaction du système documentaire du SMQ
La mise en place d’un système documentaire est l’une des exigences qui a été principalement mise en place lors de l’alternance. Le système documentaire est l’un des éléments centraux de la norme. Il représente l’ensemble des documents que l’entreprise doit réaliser et qui serviront de preuve de la bonne mise en place du SMQ.
Le système documentaire demandé par la norme est relativement imposant, en effet, en une norme on décompte 103 preuves documentaires évoquées (obligatoires pour la plupart, et conditionnelles pour certaines). Parmi elles, on retrouve 30 procédures, 48 enregistrements, et 25 documents autres [27].
Les procédures représentent une façon spécifiée d’effectuer une activité (et donc un processus), elles consistent en un descriptif organisationnel et détaillé, permettant de mener à la réalisation du processus/de l’activité associé.
Ainsi, si une procédure n’est pas suivie, il est fort probable que les données de sorties du processus ne soient pas conformes aux exigences attendues [22]. Parmi les procédures exigées par la norme, qui ont pu être réalisées au sein de l’entreprise ZOS IS, on retrouve :
- La procédure « Revue de direction »
- La procédure « Traitement des réclamations »
- La procédure « diffusion des fiches d’avertissement »
- La procédure « Gestion des ressources humaines »
Une procédure est donc très souvent associée à un processus, la norme exige que des processus existent pour plusieurs activités. Pour garder une preuve, les processus peuvent être documentés sous forme de fiche de processus.
Parmi les processus exigés par la norme et généralement présents chez tout fabricant de dispositifs médicaux, on peut retrouver :
- Le processus « gestion des retours d’informations »
- Le processus « gestion des réclamations »
- Le processus « maitrise des non-conformités »
- Le processus « Notification aux autorités compétentes »
- Le processus « Gestion des actions correctives et préventives »
Les enregistrements représentent des preuves de la bonne exécution d’une activité exigée par la norme. Par exemple, la norme exige que l’organisme s’assure que son personnel possède les compétences nécessaires, lorsque que le travail qu’il réalise peut avoir une incidence sur la qualité, et que la preuve de ses compétences et également de la bonne réalisation de ses formations soit conservée sous forme d’un enregistrement.
Les 25 autres documents exigés par la norme représentent des documents annexes aux procédures et aux enregistrements, tels que le manuel qualité, le dossier du dispositif médical, ou encore le dossier de conception.
Pour réaliser l’ensemble des documents exigés, la méthodologie suivante a été appliquée :
En premier lieu, il faut s’assurer d’avoir correctement saisi ce qui est exigé en termes de contenu dans le document, la forme peut également avoir une importance en fonction du document, mais est généralement laissé à la libre appréciation du rédacteur.
Une première recherche est réalisée pour déterminer l’utilité et le but du document, et ainsi avoir une idée globale de ce qui pourrait être attendu par ce document.
Ensuite, la recherche de différents modèles accessibles, pour avoir une base sur la structure du document et notamment sur le contenu pouvant être attendu par ce dernier, est réalisée (cette étape n’est pas toujours possible, car certains documents n’existent pas en libre accès).
Enfin, pour les documents qui concernent directement ou indirectement des membres du personnel, tels que les procédures, une interrogation a été menée pour obtenir des informations sur l’état actuel des choses. Par exemple, pour les procédures déjà réalisées en interne de façon informelle, le personnel jouant un rôle dans la procédure a été interrogé afin de se renseigner sur leur façon actuelle de procéder.
Par la suite, en comparant ce qui était fait à ce qui pouvait être attendu, des documents ont pu être réalisés, en prenant en compte les pratiques internes (afin de ne pas bouleverser les membres du personnel et conserver ce qui était déjà correctement réalisé en interne), mais également en vérifiant si tout ce qui était attendu était rempli, et si cela n’était pas le cas les informations manquantes étaient rajoutés aux informations collectées.
Une fois les documents réalisés, ces derniers ont été présentés à la Responsable Qualité et Affaires Règlementaires afin qu’elle apporte les corrections nécessaires.
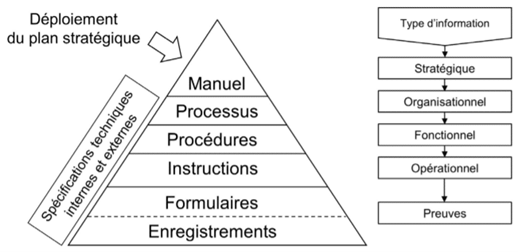
Le système documentaire du SMQ peut être structuré sous forme pyramidale (figure 5), du déploiement de la stratégie de l’entreprise au travers de la politique qualité, à la réalité opérationnelle du terrain.
Figure 5 Exemple du système documentaire du SMQ organisé sous forme pyramidale. Source : QualitExpert
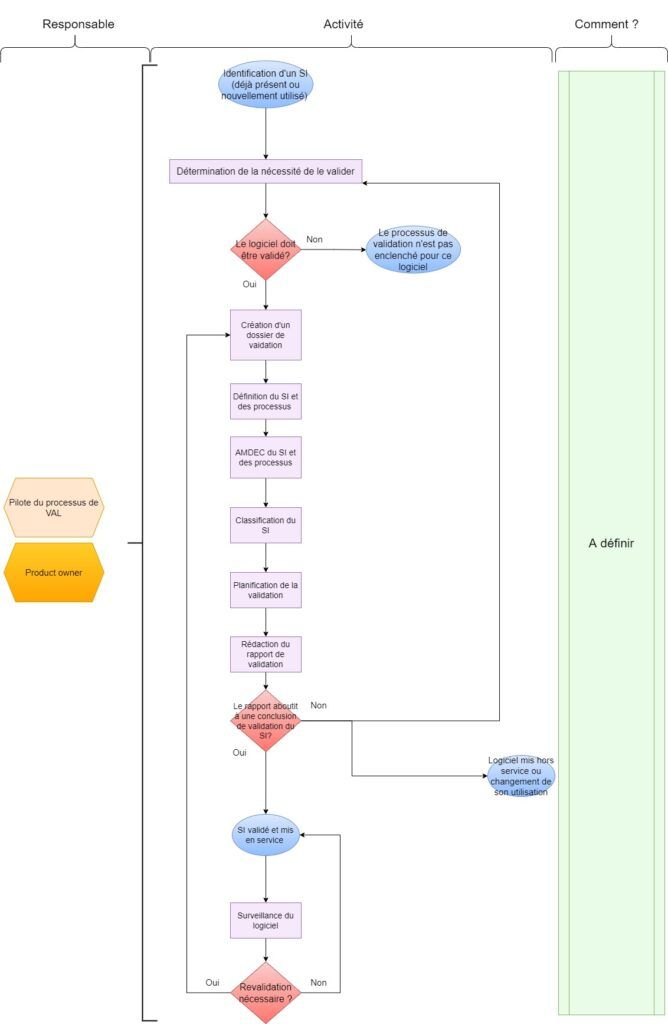
Chapitre 3 : Validation des applications logicielles
Présentation de la validation des applications logicielles : Une exigence cruciale, bien souvent négligée
La validation des applications logicielles est une des exigences nouvellement apportées par la révision de l’ISO 13485 : 2003. En effet, l’ISO 13485 : 2016, évoque à plusieurs reprises la nécessité de valider les « applications logicielles » :
- Au point 4.1.6 la norme exige que « L’organisme documente des procédures pour la validation des applications logicielles utilisées dans le système de management de la qualité. Ces applications logicielles doivent être validées avant leur première utilisation et, lorsque approprié, après la modification de ce logiciel ou de son application »
- Au point 7.5.6 elle évoque que « L’organisme doit documenter des procédures pour la validation des applications logicielles utilisées en production et dans le cadre des prestations de service. Ces applications logicielles doivent être validées avant leur première utilisation et, lorsque approprié, après la modification de ce logiciel ou de son application »
- Enfin, au point 7.6 elle mentionne que « L’organisme doit documenter des procédures pour la validation des applications logicielles utilisées pour la surveillance et la mesure des exigences. Ces applications logicielles doivent être validées avant leur première utilisation et, lorsque approprié, après la modification de ce logiciel ou de son application »
Aussi, les fabricants rencontrent encore des difficultés avec cette thématique, car cette exigence est souvent source de non-conformité lors des audits de certification ISO 13485 : 2016. En effet, bien souvent, tous les logiciels devant être validés au sein des organismes ne le sont pas. Ce fait, s’explique principalement par le manque d’information existant sur cette exigence, la difficulté pour certaines entreprises de recenser tous leurs logiciels, de comprendre l’impact potentiel de ces logiciels, mais également par la chronophagie potentielle de la mise en place de cette exigence.
Mais qu’entend exactement la norme par la terminologie « validation des applications logicielles » ?
Les applications logicielles représentent les systèmes informatisés/logiciels utilisés en entreprise.
Le processus de validation des applications logicielles selon l’ISO 13485 : 2016, revient à confirmer que ces logiciels, pouvant impacter la sécurité ou la qualité des produits de l’entreprise (ici les dispositifs médicaux), fonctionnent correctement, au regard de leur finalité d'utilisation.
D’après l’ISO 13485 elle représente « toutes activités permettant d’établir un niveau de confiance pour assurer que le logiciel est bien approprié à son utilisation prévue, est digne de confiance, mais également fiable ».
Ainsi, le but premier de cette exigence est d’éviter les non-conformités, en réduisant les risques d’incidents liés aux applications logicielles utilisées dans le cadre du SMQ, qui peuvent impacter les DM.
L’objectif final du processus de validation des applications logicielles consistera à avoir la capacité de fournir des preuves documentées de la fiabilité, la sécurité et la performance des logiciels utilisés dans les activités du SMQ.
Une fois qu’on a saisi à quoi correspondait cette exigence, il faut déterminer comme l’appliquer. En effet, la norme 13495 : 2016 ne fournit pas de méthodologie précise pour réaliser cette validation. Cependant, des référentiels annexes fournissent des indications pour réaliser correctement cette validation.
Parmi ces référentiels, on retrouve principalement des référentiels internationaux tels que l’AAMI TIR36 : 2007 “Validation of software for regulated processes”, le FDA Guidance “General principles of software validation”, mais également le document non normatif ISO/TR 80002-2 “Logiciels de dispositifs médicaux - Partie 2 : Validation des logiciels pour les systèmes de qualité des dispositifs médicaux”, document disponible uniquement en anglais et principalement utilisé dans le cadre de l’alternance pour mettre en place cette exigence.
Pour résumer, ces différents référentiels fournissent plusieurs activités et outils, à exécuter au cours du cycle de vie du logiciel à valider, en fonction de différents paramètres. Ces activités et outils vont viser à garantir que le logiciel répond à ses exigences et à sa destination, pour ainsi permettre de réduire les risques qui lui sont associés et à renforcer la confiance dans le logiciel [19][28][29][30][31].
Méthodologie pour réaliser la validation des applications logicielles
Généralités
La norme ISO 13485 : 2016, n’apporte pas de méthodologie précise sur la réalisation de la validation des applications logicielles, cependant les rédacteurs de celle-ci, sûrement conscients de la chronophagie potentielle de cette tâche, ont émis un point important devant être mis en avant et même en premier lieu de l’application de cette exigence : La proportionnalité des efforts par rapport au risque du logiciel, et donc l’adaptation de la quantité de travail à fournir en fonction du risque logiciel.
En effet, la norme soutient que “L’approche spécifique et les activités associées à la validation et la revalidation du logiciel doivent être proportionnées au risque associé à son utilisation”, cette formule est fondamentale et doit être bien prise en compte par les fabricants de DM, car elle permettra d’axer les efforts de façon ciblée et ainsi de ne pas dépenser du temps et de l’énergie dans la validation de logiciel à risque faible.
Le guide ISO/TR 80002-2 parle également du recours à la « pensée critique », tout au long du processus de validation, qui fait directement référence à la nécessité d’avoir une réflexion approfondie quant à la nécessité de réaliser telle ou telle activité pour la validation (moins le risque sera élevé, plus la rigueur et les efforts de validation devront être faibles). Au-delà de l’analyse du risque du logiciel, la norme évoque le besoin d’analyser les risques de défaillances des processus qui vont être contrôlés ou en partie contrôlés par le logiciel.
Avant de débuter l’exposition de la méthodologie mis en place au sein de ZOS IS pour valider ses applications logicielles (grandement basée sur l’ISO/TR 80002-2), le processus global de validation utilisé est représenté ci-dessous (figure 6), c’est le fil directeur qui sera utilisé pour décrire la méthode de validation :
Figure 6 Principales activités de validation en fonction des phases du cycle de vie du logiciel. Source : Figure 2 ISO/TR 80002-2
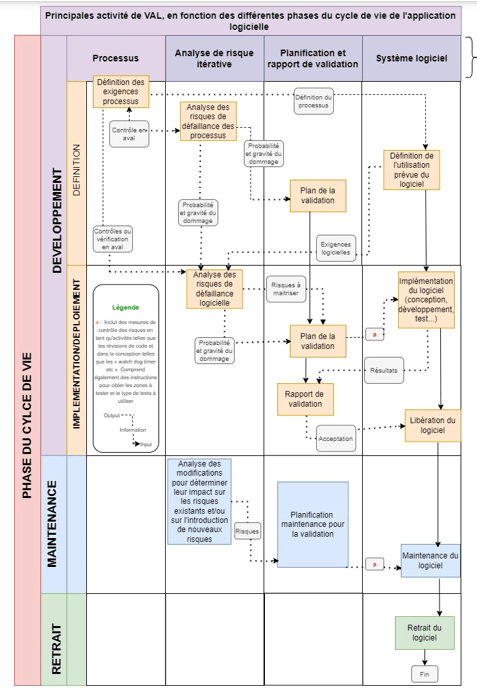
Comme le montre cette représentation issue de l’ISO/TR 80002-2, le cycle de vie d’une application logicielle peut se décliner en trois phases :
- La phase de développement : phase au cours de laquelle le besoin logiciel nait, et conduit à l’étude du besoin utilisateur ainsi qu’à la conception et la mise en œuvre du logiciel. C’est également la phase où l’on est censé déterminer le besoin de validation
- La phase de maintenance : phase au cours de laquelle le logiciel est censé avoir été validé et est mis en service, et doit être régulièrement surveillé au regard des modifications apportées ou des bugs pouvant survenir
- Et la phase de retrait : phase au cours de laquelle le logiciel n’est plus utilisé au sein de l’entreprise et doit donc être retiré des serveurs de l’entreprise
Au cours de ces phases, les besoins de validation peuvent varier, et les activités et outils utilisés pour la validation vont également évoluer.
Une procédure doit être réalisée avant d’entreprendre la validation des applications logicielles, cette procédure doit définir les différentes étapes de la validation, mais également explicité l’effort de validation par rapport au risque en indiquant, en fonction du niveau de risque déterminé, les activités et livrables requis.
Dans un premier temps, l’action qui a été réalisée pour débuter la validation a été l’étude du guide ISO/TR 80002-2, son décryptage, sa compréhension et une réorganisation des informations fournis afin de conserver les éléments apparaissant les plus pertinents. Une fois l’étude de ce guide terminé et la clarification des éléments apportés par ce dernier, la démarche de validation des applications logicielles, présentes au sein de ZOS IS, a pu débuter.
Inventoriât des systèmes informatisés et détermination de l'applicabilité de la validation
Préambule : l’exigence de validation des applications logicielles de l’ISO 13485 : 2016 exige que les « applications logicielles [soient] validées avant leur première utilisation ». De ce fait, avant d’être utilisé en entreprise tout logiciel devant être validé doit l’être, sous peine d’être non conforme à la norme. En réalité, cela est très rarement possible notamment en raison de l’apparition tardive de l’exigence. Par exemple, dans le cas des structures comme ZOS IS, qui n’étaient pas à l’origine soumise à la règlementation des dispositifs médicaux, les logiciels utilisés n’ont pas pu être validés avant leur première utilisation.
En l’état, ce point n’est pas dramatique car l’avantage de ces logiciels est qu’il y a déjà, rétrospectivement, des informations sur son utilisation, sur des dysfonctionnements rencontrés, des retours utilisateurs, du personnel formé etc qui vont être des données d’entrée à la validation et vont permettre généralement d’écourter une partie du travail à réaliser.
Cependant, tout nouveau logiciel intégré à l’entreprise doit être en temps normal validé, si l’exigence lui est applicable.
La première étape de la validation logicielle est celle visant à identifier l’ensemble des systèmes informatisés utilisés au sein de l’entreprise, et donc à les inventorier. Cette première étape est décisive et peut aboutir à un listing conséquent. L’inventaire des SI doit être effectué avec une attention particulière et de façon précautionneuse en explorant et interrogeant tous les services de l’entreprise. En effet, certains logiciels peuvent avoir été mis en place par certains services, pour faciliter leur travail, sans que la personne en charge du listing n’ait été tenue au courant, et ces derniers peuvent nécessiter d’être validés.
Au sein de ZOS IS, l’inventoriât a permis d’identifier 6 logiciels qui pourraient entrer dans le champ d’application de la validation des applications logicielles.
A noter que les SOUP (Logiciels tiers utilisés, mais dont aucune preuve des performances est accessible), les dispositifs médicaux logiciels, et les logiciels utilisés en tant que composant, partie ou accessoire d’un DM ne sont pas concerné par la présente liste.
Une fois l’inventoriât réalisé le fabricant doit déterminer si les logiciels identifiés doivent réellement subir une validation au sens que l’entend l’ISO 13485 : 2016.
Pour cela, le fabricant doit axer sa réflexion sur plusieurs éléments en se posant les questions suivantes pour chaque logiciel listé :
- Le logiciel automatise-t-il ou exécute-t-il une activité requise par les exigences réglementaires ? Pour cette question, il faut se concentrer en particulier sur les exigences relatives aux systèmes de gestion de la qualité des dispositifs médicaux et celles du règlement 2017/745.
Cela peut par exemple concerner des SI agissant au niveau de la capture de signatures et/ou d'enregistrements électroniques, le maintien de la traçabilité des produits, l'exécution et la capture des résultats des tests, la maintenance des journaux de données tels que les CAPA (actions correctives et préventives), les non-conformités, les plaintes etc
- Une défaillance ou des défauts cachés du logiciel peuvent-ils affecter la sécurité ou la qualité du SMQ ou du Dispositif ?Il faut se demander si, lorsque le logiciel à une défaillance ou un dysfonctionnement et ne peut plus être utilisé comme habituellement, cela peut conduire à un impact sur la qualité et la sécurité du SMQ et du dispositif. Cette question sera développée de façon approfondie au travers d’une analyse de risque.
Par exemple : Si un logiciel d’étiquetage, dont la fonction est d’imprimer directement sur le conditionnement d’un DM, l’UDI, le numéro de lot, les dates (expiration ect) ou encore la référence, subit une défaillance (erreur dans l’impression des informations précitées), l’impact sur la sécurité du patient peut être désastreux, en fonction du DM utilisé (par exemple dans le cas d’un cathéter pour lequel l’étiquetage contiendrait une erreur dans la taille de ce dernier, le chirurgien en lisant l’étiquetage imprimer va se tromper et prendre une mauvaise taille, ce qui peut conduire à la mort du patient)
Le logiciel est-il utilisé dans le SMQ ? Pour le système documentaire du SMQ ?
Le logiciel est-il utilisé en production et dans le cadre des prestations de services, d’une manière qui affecte la capacité du produit à se conformer à des exigences spécifiées ?
Dans le cas où au moins une des réponses est positive à au moins une de ces questions, le logiciel nécessite une validation. Dans certains cas, une justification peut être apportée pour exclure le logiciel de cette obligation de validation.
Les SI suivants entrent généralement dans le cadre de la validation (liste non exhaustive) :
- Logiciel sur équipement de production
- Logiciel sur équipement de test
- ERP (Enterprise Ressource Planning) => gros logiciel avec plusieurs modules et recoupé dans plusieurs services
- Logiciel de gestion des documents, et des enregistrements
- Logiciel de gestion des réclamations, des CAPA
- Logiciel de gestion du SAV (similaire à de la production)
- Logiciel de gestion des équipements, des dispositifs de mesure
- Logiciel de suivi de données cliniques
- Logiciel d’étiquetage (rentre dans les équipements de production)
- Logiciel de suivi des infrastructures
- Feuilles Excel contenant des macros ou des formules complexes sur les activités précitées
A contrario ce type de SI ne rentre pas dans le cadre de la validation (liste non exhaustive) :
- Logiciel financier ou administratif qui n’a pas d’impact sur le SMQ
- Microsoft office (sauf macro), la FDA a déclarée qu’on considérait qu’il y avait des millions d’utilisateurs depuis des milliers d’années et que donc c’était validé
- Logiciel de mailing, sauf si dans les mailings il y a des enregistrements qualité (dans le cas où l’on à paramétré le mail pour faire des choses concernant la qualité)
- Les systèmes d’exploitation
Dans le cas de ZOS IS, trois SI listés ont été identifiés comment devant être soumis à une validation, car ils interviennent à la fois dans la conformité du produit, dans la traçabilité, et dans la gestion du système documentaire. Un dossier de validation a alors été créé pour chacun de ces logiciels.
Définition des systèmes informatisés et des processus associés
Lorsque la détermination de l’ensemble des SI devant être soumis à la validation selon l’ISO 13485 : 2016 a été réalisée, l’étape de définition rentre en jeu.
Cette deuxième étape doit également être faite de façon rigoureuse, elle consiste à définir de façon relativement précise le logiciel.
Pour définir correctement le logiciel, plusieurs éléments doivent être déterminés en collaboration directe avec les utilisateurs du logiciel :
- Le(s) fonctionnalité(s) du logiciel
- Le(s) exigence(s) des utilisateurs (spécification des besoins utilisateurs), l’utilisation du logiciel et le contexte d’utilisation
- Les spécifications des besoins utilisateurs sont importantes et doivent être définies de la façon la plus exhaustive possible avec les utilisateurs directs du logiciel. Cette spécification interviendra par la suite dans l’analyse des risques.
- L’utilisation du logiciel doit être correctement identifiée, car le logiciel est validé sur la base de son utilisation prévue, et donc si cette utilisation est amenée à être modifiée au cours du temps, cela pourra avoir un impact sur l’état validé du logiciel
- Le contexte d’utilisation aide à l’identification de l’utilisation logiciel et peut passer par la réponse aux questions suivantes :

- Les spécifications de la conception fonctionnelle : ces spécifications sont à définir essentiellement dans le cas où le logiciel est conçu totalement, ou en partie, en interne de l’organisme
- Les éléments de sortie attendus du logiciel peuvent également être documentés (par exemple : le logiciel doit fournir des enregistrements de conformité du produit, des résultats de calcul intervenant dans la conformité du produit ou encore le résultat des indicateurs qualités de l’entreprise)
L’ensemble de ces éléments vont servir d’éléments d’entrée pour réaliser l’analyse des risques et ainsi pouvoir définir l’effort de validation à appliquer pour chaque SI. Ils doivent être formalisés et donc documentés.
Également, les processus contrôlés ou en partie contrôlés par le logiciel devront être définis.
La définition d’un seul logiciel a pour l’instant pu être documentée au sein de la structure, mais ce dernier est à améliorer en incluant tous les utilisateurs directs.
Analyse des risques (de défaillances)
Cette troisième étape est cruciale et doit être l’un des éléments minimums de la validation des applications logicielles à réaliser, avant un audit de certification ISO 13485 : 2016, si l’on souhaite éviter des non-conformités sur cette exigence.
En effet, l’analyse de risque est obligatoire pour tout système informatisé devant être soumis à validation. Il est donc fondamental que l’entreprise ait entrepris une démarche d’analyse des risques pour l’ensemble des logiciels à valider, lui permettant ainsi de prioriser la validation de ses logiciels. Ainsi, dans le cas où les logiciels à valider n’auraient pas pu l’être avant l’audit de certification, l’entreprise pourra justifier cela en déclarant que, de par la priorisation des logiciels à valider, elle s’est concentrée en premier lieu sur ceux avec le risque le plus élevé (sous-entendu que les logiciels à haut risque sont validés lors de l’audit).
Pour réaliser son analyse des risques l’entreprise peut réutiliser sa procédure de gestion des risques (normalement présente dans toute entreprise de DM) ou elle peut se baser sur d’autres méthodes d’analyse, telles que l’AMDEC (Analyse des modes de Défaillance, de leurs Effets et de leur Criticité).
L’AMDEC est une méthode de gestion des risques permettant d’évaluer les modes de défaillances potentielles d’un processus, d’un produit, ou d’une organisation.
L’analyse des risques réalisée pour chaque logicielle est effectuée pour évaluer les conséquences d’une défaillance logicielle sur :
- la conformité du produit
- le patient
- le système management de la qualité
- la réglementation
- la qualité des documents et des enregistrements
Elle peut ainsi être réalisée en recherchant les séquences d’événements suivants (sans s’y limiter) :
- Pour les logiciels utilisés sur ou avec des équipements en production :
- La conséquence sur l’équipement qu’il contrôle ou supporte,
- La conséquence sur la conformité du produit,
- La conséquence sur le patient
- Pour les logiciels utilisés pour l’entretien :
- La conséquence sur le service qu’il contrôle ou supporte,
- La conséquence sur la conformité du produit ou du service,
- La conséquence sur le patient,
- Pour les logiciels utilisés pour traiter des données de qualité ou réglementaires :
- La conséquence sur la qualité des documents ou des enregistrements,
- La conséquence sur la conformité du produit,
- La conséquence sur le patient,
- Pour tous les logiciels :
- La conséquence d’une faille de sécurité ou d’une attaque,
- La conséquence sur la qualité des documents ou des enregistrements,
- La conséquence sur la conformité du produit,
- La conséquence sur le patient,
Le choix de la méthode de réalisation de l’analyse des risques au sein de ZOS IS s’est basé sur la méthode AMDEC, la procédure de gestion de risque n’a pas été utilisée car cette dernière était uniquement adaptée à l’analyse des risques liée au produit de l’entreprise (le logiciel I-GEIA).
L’analyse des risques de défaillance des processus contrôlés ou en partie contrôlés par le logiciel est réalisée.
Cette méthode est particulièrement adaptée car elle prend directement en compte l’aspect du risque lié aux défaillances possibles. Elle a été réalisée pour le logiciel ayant été définie comme évoqué dans la partie précédente et a abouti à la conclusion que le logiciel avait un niveau de risque mineur.
Etablissement d'un plan de validation
C’est lors de cette étape que la formule de la norme 13485 : 2016 « L’approche spécifique et les activités associées à la validation et la revalidation du logiciel doivent être proportionnées au risque associé à son utilisation » intervient et prend tout son sens.
Lorsque l’analyse des risques a été réalisée et les niveaux de risques des différents logiciels a validé ont été définis et a permis de prioriser les logiciels, il faut effectuer la planification de la validation au travers de la réalisation d’un plan de validation (également appelé parfois « Protocole de Validation » ou « Validation Master Plan » pour « Plan Maitre de Validation »).
Le plan de validation est le document qui permettra d’établir les activités et outils choisis pour poursuivre la validation des systèmes informatisés, il décrit également les tâches de vérifications appropriées.
Aussi, plus le plan fera intervenir des outils et activités, plus le niveau d’effort pour la validation et la rigueur exigé pour la documentation associée sera important. Et cette détermination se base principalement sur les analyses des risques de défaillance réalisée pour les SI, mais le guide ISO/TR 80002-2 indique également que l’analyse des risques des défaillances du processus associé viendra conditionner la rigueur pour la documentation.
Cette activité de planification aboutit à un plan documenté qui décrit et fournit :
- Les responsabilités (qui est en charge de réaliser quoi)
- Les choix effectués en termes d’activités/d’outils : c’est-à-dire les décisions, les choix pris d’activité et d’outil (une liste non exhaustive des activités/outils est disponible dans la 80002-2)
- Les raisons des choix effectués : les facteurs de décision ayant abouti à ces choix d’outil et d’activité (basé sur le risque)
- Des preuves objectives de l'application de la pensée critique au processus de planification de la validation.
- Les documents de sortie attendus
Les activités principales retrouvées pour valider les logiciels sont les suivantes :
La Qualification de Conception (QC) :
Elle concerne essentiellement les logiciels qui ont été développés totalement, ou en parties, en interne de l’entreprise (peut être adapté à des logiciels acheté et hautement configurable, dans ce cas l’éditeur doit pouvoir fournir la documentation nécessaire à cette qualification).
Pour les logiciels conçus en interne, une procédure de conception doit normalement exister, et les documents suivants réalisés et ajoutés à la QC :
- Des revues de conception (vérification et validation)
- Les spécifications du logiciel
- Des tests du développement logiciel
En fonction du risque du logiciel les documents suivants pourront alimenter la QC :
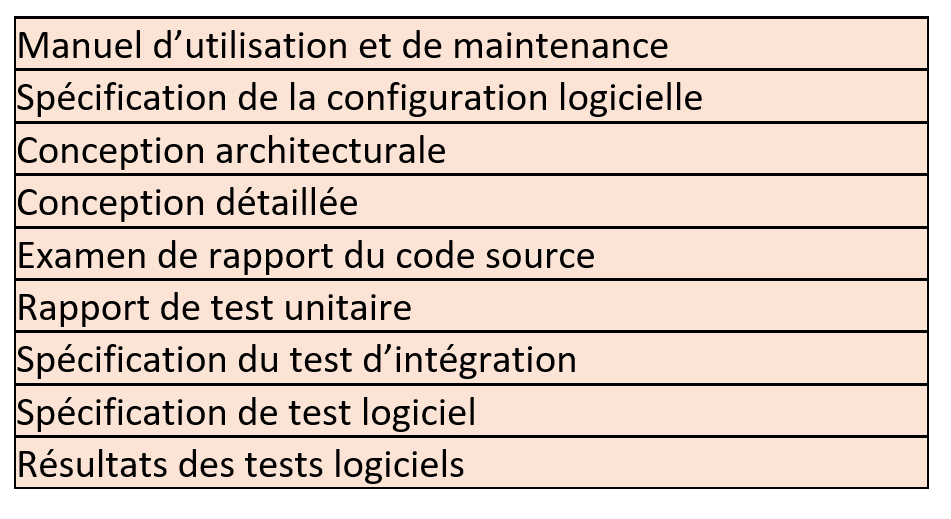
La Qualification d’Installation (QI) :
Elle vise à s’assurer que le système informatisé est correctement installé et dans le bon environnement. Elle concerne essentiellement les logiciels nécessitant une installation spécifique.
Cette qualification permet de tester l’installation du software mais également du hardware (réseau, accès, connections). En effet, il peut arriver que des prérequis techniques (par exemple en termes de débit, de réseau etc) à l’installation d’un logiciel soient nécessaires, il faut donc déterminer si l’infrastructure de l’entreprise répond bien à ces prérequis.
Lors de cette qualification, les éléments suivants pourront également être vérifiés :
- Si des contrats de maintenance existent avec l’éditeur et auquel cas si une maintenance effective existe.
- Vérifier que les paramétrages sont bons et que tout est correctement paramétré
- Vérifier que toute la documentation utile du logiciel (notice d’utilisation etc) est présente et à jour (conforme à la version utilisée…)
- Vérifier que les formations du personnel sont à jour (il est pertinent d’avoir un utilisateur clé qui sera le référent d’un SI)
- Tester la migration des données, elle doit être documentée pour avoir la preuve qu’elle a été faite correctement
Il est aussi important que l’entreprise rédige des documents qui déroulent le fonctionnement du logiciel (des instructions du logiciel, elles ne doivent pas être forcément très détaillées, mais contenir au moins les bases d’utilisation et les instructions des fonctionnalités critiques pour expliquer comment se servir du logiciel)
Aussi, en fonction du risque du logiciel les documents suivants pourront alimenter la QI :

La Qualification Opérationnelle (QO) :
Elle a pour but d’assurer que chaque fonction du système informatisé fonctionne comme prévu et ainsi de vérifier que le logiciel fonctionne selon l’emploi prévu et les besoins utilisateur.
Il faut donc se poser la question « Est-ce que le logiciel fonctionne spécifiquement à ceux que l’on souhaite »
Pour réaliser cette validation, il est possible de se baser sur la QC de l’éditeur, si ce dernier a fourni un certain nombre de tests, qui devront être réeffectués (car l’environnement de l’entreprise peut être différent de l’environnement initial de test : le nombre de connexion, le type d’utilisateur etc.).
Il faut également tester le fonctionnement du logiciel dans un mode dégradé et tester prioritairement les éléments à risque : Par exemple, si un champ à risque existe dans lequel il faut remplir un numéro de lot et qu’il est implémenté pour recueillir 14 digit, il faut regarder ce qu’il se passe lorsque l’on va mettre un nombre de digit différent à 14 (16 ou 9 par exemple) pour voir comment le logiciel réagis (déclenchement d’un signal d’erreur ou d’alertes ? ou le logiciel ne réagit pas et dans ce cas il y a un problème et une action doit être entreprise).
Il faut tester que les fonctions de sécurité marchent (alerte qui se déclenche, ou qui doivent apparaitre), tester les sauvegardes/restauration (si on fait exprès de perdre les données, est-ce qu’on les récupère bien ?), les fonctions de maintenance (marchent-elles ? par exemple si possibilité d’avoir accès à distance au logiciel pour dépanner. Est-ce que cet accès fonctionne ?)
A noter que les protocoles de QO ne sont pas forcément testés par une seule personne mais par différentes personnes compétentes (les utilisateurs), et une personne doit être en charge de récupérer et compiler tous les documents. En fonction du risque du logiciel les documents suivants pourront aussi alimenter la QO :

La Qualification de Performance (QP) :
Elle vise à garantir que le système informatisé fonctionne comme prévu dans son environnement cible et avec les utilisateurs cible. Elle permet donc de qualifier le logiciel dans son environnement réel, dit « de routine » (soit on la réalise sur la vraie plateforme, soit sur une plateforme de test, mais dans un contexte de routine réel).
Avec la QP, on va beaucoup plus loin, elle est plutôt exigée pour des logiciels à haut risque car la QI et la QO sécurise déjà beaucoup le logiciel, puisque normalement en les réalisant il est démontré qu’il n’y a pas de gros bugs, que tout est bien installé, et que tout le monde est correctement formé.
Cependant, la QP permet de faire des scénarios d’utilisation complets, ce qui est beaucoup plus lourd en termes de travail à fournir, mais permet de voir les performances du logiciel.
Pour réaliser les tests, il faut :
- Des testeurs indépendants, celui qui développe ne peut pas être celui qui teste. Il faut une stratégie pour savoir qui va tester car c’est une activité très chronophage => personne dédiée (spécifiquement embauchée pour réaliser les tests) ou personne interne (effectuant déjà d’autres activités), cette dernière option peut s’avérer compliqué dans le cas où la charge de travail est déjà importante. Mais en TPE comme chez ZOS IS, il est compliqué d’embauché, il faut donc essayer de réduire le nombre de logiciels à valider et avoir réellement une stratégie décroissante en fonction du risque.
- Avoir un environnement représentatif (données réalistes, mais sans risque de corrompre les données de l’entreprise), nécessité de dupliquer l’environnement
- Former les testeurs à la documentation, il faut documenter les tests
Dans le cas où des problèmes, des bugs ou encore appelé des déviations surviendraient au cours des tests, les actions suivantes devraient être menées :
- Enregistrer les déviations, faire une analyse de cause, et définir une action corrective si nécessaire (il serait pertinent d’expliquer dans la procédure de validation ce qui serait un problème mineur, pour lequel on ne fait pas d’action, et ce qui sera un problème critique où une action directe doit être menée)
- Se poser la question si on refait des tests après la mise en place des actions (tests complémentaires)
- Si on fait des tests de régression, si l’on refait toute la QO, toute la validation, ou juste quelques tests dits de « débug »
A noter qu’il existe différents niveaux de criticité d’un bug (par exemple : mineur, significative, majeur, critique et catastrophique). Ce qui engendre qu’il y a possibilité de déclarer par exemple que tous les bugs mineurs n’ont pas besoin d’être corrigé (mais cela doit être défini dans un document).
En fonction du risque du logiciel les documents suivants pourront alimenter la QP :

Exécution du plan de validation et rédaction du rapport de validation
Une fois effectué, le plan de validation doit être appliqué, les activités réalisées et les outils utilisés.
La réalisation du plan de validation va aboutir à plusieurs résultats qui devront être recensés et étudiés.
C’est l’étape 6 de rédaction du rapport de validation.
En plus de compiler tous les résultats de données, ce rapport devra explicitement conclure sur l’état validé du logiciel, et le fait donc qu’il puisse être utilisé.
Dans le cas où des résultats de test ne correspondant pas aux attendus auraient été obtenus, plusieurs possibilités s’offrent telles que :
- L’ouverture de non-conformité et la réalisation des actions pour se rendre conforme
- L’acceptation du logiciel en l’état, dans le cas où le résultat non attendu est justifié ou argumenté
Il est très important de bien conclure à l’issue de ce rapport et de statué sur le fait que le logiciel peut être utilisé en toute sécurité ou que les risques associés à ce logiciel sont maitrisés. Bien souvent les entreprises oublient cette conclusion dans leur rapport de validation.
En résumé, le rapport de validation final doit porter sur les points suivants :
- Un résumé global du projet de validation
- Une liste de tous les livrables générés
- Tout écart par rapport à la procédure de validation des applications logicielles
- Une conclusion indiquant si le système informatisé est validé ou non
A noter : un rapport de qualification peut être réalisé en amont du rapport final de validation. Ce rapport devra synthétiser l'ensemble des phases de qualifications, avec leurs résultats, les bugs ou déviations apparues, les actions correctives ou préventives menées et les modifications si applicables. Il pourra également inclure une conclusion préalable par rapport à la qualification.
Surveillance des changements et revalidation
Une fois les logiciels validés, la validation des applications logicielles ne s’arrête pas là.
La 7ème et dernière étape de la validation consiste à s’assurer que les logiciels validés, restent dans un état validé tout au long de leur utilisation, en s’adaptant, en gérant et en contrôlant divers types de changements.
Différentes raisons existent pour lesquelles le logiciel changerait après sa mise en service. Parmi les types de modifications de maintenance les plus courants, on peut retrouver :
- Les modifications de maintenance corrective (utilisées pour corriger les erreurs et les défauts du logiciel)
- Les modifications de maintenance préventive (utilisées pour améliorer les performances, la maintenabilité ou d'autres attributs du logiciel)
- La maintenance adaptative effectuée pour mettre à jour l'environnement opérationnel du logiciel (par exemple, modifications du système d'exploitation, du matériel du système ou d'autres applications avec lesquelles le logiciel s'interface).
Toute modification apportée à un SI validé doit être réalisée de manière contrôlée conformément à une procédure (la procédure « contrôle des modifications » doit être définie dans une entreprise de DM).
Ainsi, il est nécessaire de définir un intervalle minimum au cours duquel, les logiciels validés seront (re) analysés, afin de déterminer si des changements ont pu conduire à une modification de leur état validé (intervalle à définir dans la procédure de validation).
Lors d’une modification logicielle, une analyse d’impact doit être réalisée quant à son effet sur :
- L'utilisation prévue
- Le risque de défaillance
- L’introduction de nouveaux risques
- Les mesures de contrôle des risques existantes
- Toute fonctionnalité du logiciel lui-même
Les modifications pouvant aboutir à la nécessité d’une revalidation peuvent être :
- Modification du processus dans lequel le logiciel est utilisé
- Lorsque le processus entièrement ou partiellement contrôlé par le logiciel change, une analyse d'impact doit être réalisé (impact sur les mesures de contrôle des risques, l’utilisation prévue...)
- Changement de sa configuration
- Changement de son niveau de risque
- Changement dans l’utilisation prévue du logiciel
- S'il y a un changement significatif dans l'utilisation prévue du logiciel, il doit être validé pour la nouvelle utilisation prévue ou la nouvelle utilisation doit cesser (dans ce cas, une évaluation des risques est nécessaire pour s'assurer qu'aucun risque n'a été introduit pendant la période d'utilisation non autorisée)
Un plan de maintenance doit être réalisé pour définir quelles sont les activités opérationnelles réalisables sans que la validation du logiciel soit affectée et quelles modifications nécessitent des efforts de validation et donc une revalidation.
Chaque opérateur du logiciel doit être formé à reconnaitre la différence entre les activités opérationnelles normales et tout changement nécessitant potentiellement une (re)validation.
Il peut être également pertinent que le “Plan de validation”, indique les éléments conduisant à une revalidation d’un système informatisé et décrit les actions et activités nécessaires à cette revalidation.
Une fois cette dernière étape implémentée, la validation des applications logicielles au sein d’une entreprise est en théorie conforme.
Un résumé global sous forme de logigramme de la démarche de validation des applications logicielles est disponible en annexe 2 [19][28][29][30][31].
Chapitre 4 : Bilan personnel de mon expérience d'apprentie
Evolution personnelle au sein de ZOS IS
Lors de l’intégration du Master Ingénierie de la Santé parcours Dispositifs Médicaux et Affaires Règlementaires, mon projet professionnel s’est rapidement tourné vers le métier de Chargé des Affaires Réglementaires des dispositifs médicaux.
Le métier de Chargé des Affaires Règlementaires des DM englobe toutes les activités permettant de garantir que les dispositifs médicaux sont, tout au long de leur cycle de vie, conforment à la règlementation qui leur est applicable.
Le 30 août 2021, j’intégrais l’entreprise ZOS Information System, non pas en tant qu’apprentie Chargée des Affaires Règlementaires des DM, mais en tant qu’apprentie Chargée des Affaires Réglementaires et de la Qualité des DM.
Le métier de Chargé Affaires Règlementaires et Qualité des DM, regroupe l’aspect règlementaire, comprenant les éléments cités précédemment et l’aspect qualité englobant les activités permettant d’assurer le bon fonctionnement et le bon suivi du système de management de la qualité.
Ces deux aspects dans le secteur des DM sont très étroitement liés et peuvent se confondre contrairement à d’autres secteurs ou les deux aspects se différencient de façon beaucoup plus marquée (dans le secteur pharmaceutique par exemple). C’est pourquoi, l’ambivalence du poste ne m’a pas bloqué pour saisir cette opportunité d’alternance.
L’alternance était pour moi la modalité d’étude la plus pertinente, car elle me permettait de mettre directement en application les savoirs qui m’ont été délivrés lors de mes années d’étude, et ainsi de mieux les intégrer et d’acquérir plus facilement et plus rapidement les savoir-faire associés.
Aussi, mon début d’alternance n’a pas été sans difficulté, en effet plusieurs facteurs sont entrés en jeu dans cette complexité, tels que :
- La taille de l’entreprise : ZOS IS est une TPE, de ce fait le nombre de salariés est restreint, et comme dans de nombreuses TPE les frontières en termes de travail ne sont pas toujours définies, ce qui conduit très souvent à porter plusieurs casquettes. Ainsi, le service qualité et affaires règlementaires, à mon arrivé, ne comptait qu’une personne, la Responsable qualité et affaires réglementaires, ce qui pouvait représenter un frein pour une progression efficiente, compte tenu de la quantité de travail et de l’ensemble des démarches devant être réalisée pour obtenir le marquage CE dans les délais impartis.
- L’expérience de la Responsable qualité et affaires réglementaires : Bien que cette dernière ait pu suivre une formation certifiante, la responsable QAR n’avait pas encore acquis un recul suffisamment solide en termes d’expérience professionnel dans le domaine, lui permettant d’avoir des connaissances sur les attentes (notamment des organismes notifiés) mais également d’avoir un regard plus construit et clair sur la ligne directrice à suivre.
Ainsi, malgré un démarrage difficile, l’autonomie et la diversité des tâches confiées, m’ont permis d’avoir une réelle vision globale sur la grande majorité des activités du chargé ARQ et ainsi de travailler sur différents aspects du domaine, me permettant d’acquérir et d’approfondir rapidement diverses compétences. Ce fut donc une alternance qui s’est révélée très challengeante et formatrice.
J’ai en effet pu, lors de cette expérience, approfondir l’étude de plusieurs référentiels incontournable dans le secteur du DM, ce qui me confère à l’heure actuelle une maitrise importante de plusieurs de ces référentiels :
- Le règlement 2017/745, texte législatif qui indique les différentes exigences permettant de mettre sur le marché européen un dispositif médical, et les diverses obligations qui incombent aux différents acteurs de l’industrie du DM
- L’ISO 13485 : 2016, portant sur la mise en place d’un système de management de la qualité chez les industriels du DM
- L’ISO 14971 : 2019, sur l’application de la gestion des risques aux dispositifs médicaux, et le guide associé FD CEN ISO/TR 24917 qui donne des recommandations sur l’application de l’ISO 14971
- La NF EN 62366-1 : 2015, sur l’application de l’ingénierie de l’aptitude à l’utilisation aux dispositifs médicaux, processus permettant d’évaluer et de réduire les risques associés à une utilisation normale d’un DM
- La NF EN 62304 : 2006, qui vient définir les exigences en ce qui concerne le cycle de vie des dispositifs médicaux logiciels
- L’ISO/TR 80002-2 : 2017, qui est un guide portant sur la validation des applications logicielles du SMQ
Grâce au guide ISO/TR 80002-2 j’ai pu apprendre à maitriser plusieurs aspects de l’exigence de validation des applications logicielles.
J’ai également pu participer à la gestion des risques, et ainsi mieux maitriser les livrables associés et développer ma réflexion sur l’Identification et l’analyse des risques, leur estimation, leur évaluation et leur maitrise. Mon apprentissage sur la NF EN 62366 m’a également apporté des éléments sur la gestion des risques, car cette dernière développe des éléments permettant de prévenir les risques dus aux erreurs d’utilisation d’un DM. J’ai ainsi pu débuter la rédaction du dossier d’ingénierie d’aptitude à l’utilisation du DM logiciel I-GEIA, et notamment les parties concernant la spécification d’utilisation d’I-GEIA (partie très étroitement liée à certains attendus du dossier technique du DM).
J’ai pu développer mes capacités rédactionnelles, mais également de synthèse, en apprenant la rédaction des procédures, des fiches processus et de divers autres documents.
Enfin, cette autonomie m’a appris à prendre des initiatives, à être force de proposition, mais également à réussir à communiquer de façon adaptée et diplomate avec l’ensemble des acteurs impliqués dans la démarche qualité. J’ai ainsi pu participer à la sensibilisation du personnel à la qualité au travers de différentes présentations. Pour résumer mon évolution en termes de compétence, j’ai réalisé un graphique radar (figure 7), qui illustre mon regard personnel sur ma progression globale au travers de cette alternance :
Figure 7 Graphique de l'évolution de mes compétences à la suite de mon alternance. Source : Auteure
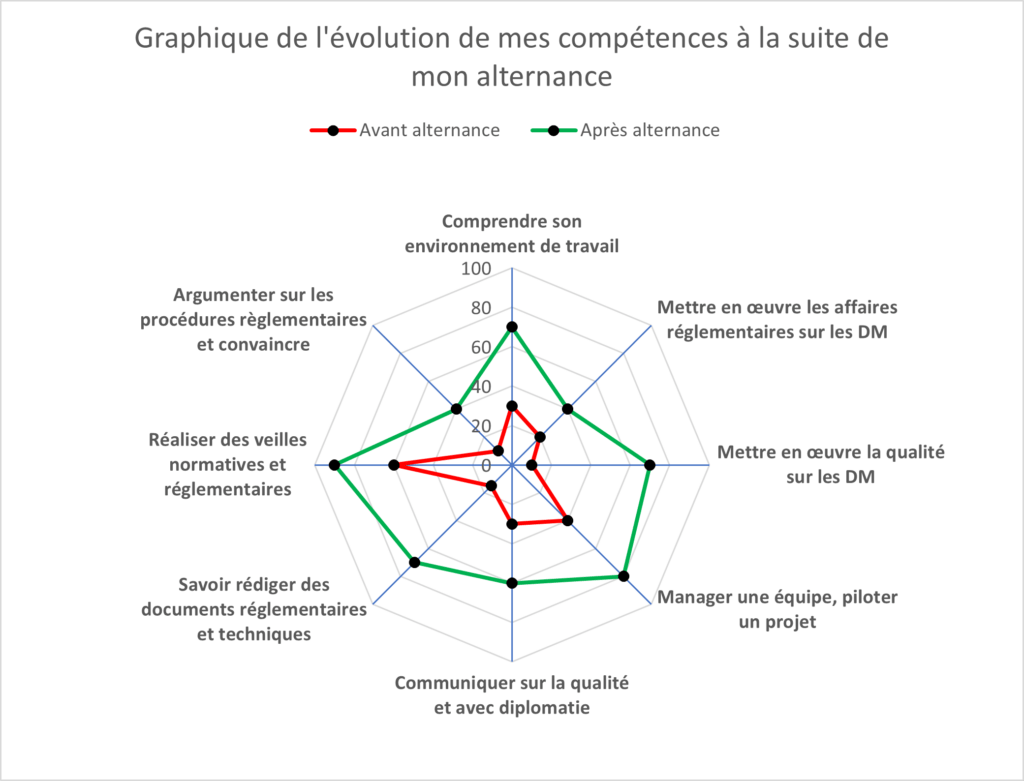
Mon bilan global de cette première réelle expérience dans mon domaine a donc été positif et très enrichissant.
Il me reste encore à développer plus en profondeur certains éléments, notamment concernant la gestion de risque, l’alimentation du dossier technique, mais également la mise en place de la surveillance après-commercialisation et la réalisation de l’évaluation clinique, élément fondamental de la nouvelle réglementation des DM.
Relation entre ma formation théorique et mon expérience professionnelle
La formation à l’UTC est très pertinente dans le domaine de la qualité et des affaires réglementaires. En effet, cette dernière nous offre une vision globale et solide sur les métiers du domaine au travers des divers cours réalisés. La place importante de la qualité dans la formation est très pertinente, car en travaillant dans le domaine, on se rend très vite compte que la qualité est centrale dans l’ensemble des activités effectuées.
Aussi, les nombreux enseignements dispensés par des intervenants du monde professionnel ont été essentiels dans notre appropriation rapide du métier et de ces attendus.
Enfin, il serait intéressant que la formation puisse nous offrir plus d’aspects pratiques sur certains éléments fondamentaux et récurrents du domaine, tels que la rédaction des procédures, processus, mais également de la documentation technique.
Mon projet professionnel à l'issu de mon alternance
L’alternance que j’ai pu réaliser a profondément conforté mon intérêt pour le domaine du DM, et m’a conféré un intérêt particulier pour l’aspect qualité.
En effet, lors du démarrage de mon alternance, j’étais plus axé sur l’aspect règlementaire et je souhaitais donc travailler en tant que Chargé AR. Cependant, mes missions principales concernaient majoritairement la mise en place d’un SMQ, missions que j’ai apprécié accomplir et qui ont grandement développé mon intérêt pour l’aspect qualité.
Ainsi, ayant découvert mon intérêt pour les deux aspects, je compte axer mes recherches d’emploi sur des postes alliant les deux aspects, et chercher des entreprises auprès desquelles je pourrais continuer à travailler sur des missions diversifiées et ainsi parcourir la majorité des activités d’un Chargé affaires réglementaires et qualité des dispositifs médicaux.
Conclusion
Le passage de la classe de risque I à la classe IIa du dispositif médical logiciel I-GEIA à engendrer une quantité importante de démarche à mettre en œuvre pour se conformer à la règlementation applicable et maintenir le marquage CE du DM.
Aussi, à l’issue de mon alternance, la mise en place complète d’un SMQ au sein de ZOS IS n’a malheureusement pas pu être finalisée, cependant de nombreuses exigences ont pu être mise en place et la sensibilisation de l’entreprise à la qualité a pu démarrer.
J’ai également pu travailler en autonomie sur un sujet important de la norme : la validation des applications logicielles. J’ai pu ressortir de cette expérience de nombreuses connaissances qui me serviront sans nul doute dans mes futures expériences professionnelles.
Aussi, par la suite, l’entreprise devra continuer à se conformer aux exigences de la norme ISO 13485 : 2016 et au MDR, pour obtenir le marquage CE (classe IIa).
L’une des phases directes qui précédera mon départ de l’entreprise sera la mise en application de la documentation réalisée et notamment des procédures. L’entreprise devra également s’assurer d’avoir des preuves documentées à montrer aux évaluateurs (rapport de revue de direction ect).
Aussi, l’appel à un consultant pour venir en appuie à la mise en conformité de l’entreprise aux exigences qui lui incombe est une opportunité importante, qui devra permette à l’entreprise d’obtenir le marquage avant la fin de la période dérogatoire.
Références bibliographiques
[1] C. d’expert Ecomundo, « Dispositifs médicaux : La nouvelle réglementation européenne, applicable en 2021 », Ecomundo.eu, 29 août 2019. https://www.ecomundo.eu/fr/blog/dispositifs-medicaux-reglement-2020 (consulté le 26 juillet 2022).
[2] M. Navarro, « Qu’est-ce qu’un système de management de la qualité (SMQ) EN ISO 13485 ? », mauricenavarro, 1 janvier 2022. https://www.mauricenavarro.com/articles/qu-est-ce-qu-un-systeme-de-management-de-la-qualite-smq-en-iso-13485/ (consulté le 26 juillet 2022)
[3] M. Navarro, « Glossaire : Définitions de termes techniques en rapport avec la qualité et le génie logiciel pour DM », mauricenavarro. https://www.mauricenavarro.com/ressources/glossaire/ (consulté le 26 juillet 2022).
[4] L. Millet, « E-santé : une accélération sans précédent », InstitutMontaigne, 24 septembre 2020. https://www.institutmontaigne.org/blog/e-sante-une-acceleration-sans-precedent (consulté le 26 juillet 2022).
[5] L. Meunier, « L’impact de la réglementation concernant les Dispositifs Médicaux sur les différents acteurs de la santé », Synapse-Medicine, 9 décembre 2021. https://synapse-medicine.com/fr/impact-reglementation-dispositifs-medicaux (consulté le 5 août 2022).
[6] Snitem, « Logiciel et application en santé : DM ou pas DM ? », calameo, août 2021. https://www.calameo.com/snitem/read/00061054233b7cebc1141 (consulté le 26 juillet 2022)
[7] Droit technologie, « Quand le logiciel devient un “dispositif médical” », JurisPharma, 20 novembre 2019. https://www.jurispharma.fr/categories/droit-de-la-sante-23/articles/quand-le-logiciel-devient-un-dispositif-medical-991.htm (consulté le 14 juillet 2022).
[8] Elia médical, « Bienvenue chez Elia Médical », Eliamedical-education, 2009. http://www.elia-medical.com/ (consulté le 1 août 2022).
[9] Mythologica, « Mythologie grecque : Hygie », Mythologica, 16 novembre 2021. https://mythologica.fr/grec/hygie.htm (consulté le 25 août 2022).
[10] F. Barlet, « ZOS Information System : Agence de developpement Informatique sur mesure ». http://www.zos-informationsystem.com/ (consulté le 1 août 2022).
[11] Inserm, « Apnée du sommeil : une source de fatigue, mais aussi de maladies cardiovasculaires », Inserm, 1 octobre 2015. https://www.inserm.fr/dossier/apnee-sommeil/ (consulté le 8 août 2022).
[12] Centre d’investigation et de recherche sur le sommeil, « L’apnée du sommeil », CHUV.ch, 22 juin 2021. https://www.chuv.ch/fr/sommeil/cirs-home/patients-et-familles/les-troubles-du-sommeil/lapnee-du-sommeil (consulté le 8 août 2022).
[13] Data Med Care, « Adel santé : le télésuivi optimisé », adel-santé. https://www.adel-sante.fr/ (consulté le 3 août 2022).
[14] Must informatique, « MUST-G5 dans l’assistance respiratoire », Must Informatique. https://www.mustinformatique.com/notre-solution-dans-lassistance-respiratoire/ (consulté le 25 août 2022).
[15] ResMed Pty Ltd, « AirView : Logiciel de gestion des données patients », Resmed, août 2020. https://www.resmed.fr/professionnels-de-sante/insuffisance-respiratoire/airview/ (consulté le 8 août 2022).
[16] Philips, « DreamStation : Appareils de traitement de l’apnée du sommeil », Philips, décembre 2021. https://www.philips.fr/c-e/hs/apnee-du-sommeil/appareils-apnee-du-sommeil.html (consulté le 8 août 2022).
[17] L. Granger, « Concevoir une stratégie : l’analyse SWOT », manager-go.com, 8 juin 2022. https://www.manager-go.com/strategie-entreprise/dossiers-methodes/diagnostic-strategique-swot (consulté le 8 août 2022).
[18] Tribune de Genève, « Maladies respiratoires en hausse dans le monde », Tribune de Genève.ch, 17 août 2022. https://www.tdg.ch/maladies-respiratoires-en-hausse-dans-le-monde-486040706927 (consulté le 8 août 2022).
[19] « Norme NF EN ISO 13485 - Systèmes de management de la qualité - Exigences à des fins réglementaires », Ed.Afnor, Paris, www.afnor.org, 30 avril 2016. Consulté le : 9 août 2022. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/nf-en-iso-13485/FA161550?rechercheID=8535472&searchIndex=5&activeTab=all
[20] « NF EN ISO 13485/A11 - Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », Ed.Afnor, Paris, www.afnor.org, 8 septembre 2021. Consulté le : 22 août 2022. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/nf-en-iso-13485-a11/FA197434?rechercheID=8752749&searchIndex=7&activeTab=all
[21] Adhérent Stratégiqual, « Mise à jour du Système de Management de la Qualité : vous saurez tout le 10 décembre ! », AtlanpoleBiotherapies, 16 novembre 2020. https://www.atlanpolebiotherapies.com/actualites/mise-a-jour-du-systeme-de-management-de-la-qualite-suite-a-lentree-en-vigueur-des-reglements-2017-45-et-2017-746/ (consulté le 26 juillet 2022).
[22] C. Lab, « Comprendre la différence entre les processus et les procédures », Optimiso Group, 15 octobre 2020. https://optimiso-group.com/articles/comprendre-enfin-la-difference-entre-processus-et-procedure/ (consulté le 22 août 2022).
[23] G. Prome, « Approche par les risques dans l’ISO 13485 », Qualitiso, 23 août 2017. https://www.qualitiso.com/approche-par-les-risques-iso-13485-2016/ (consulté le 18 août 2022).
[24] Guillaume Promé, « L’ISO 13485 à l’heure du règlement DM », Qualitiso, 27 novembre 2017. https://www.qualitiso.com/iso-13485-vs-ue-2017-745/ (consulté le 26 juillet 2022).
[25] Nouvelle industrie, « ISO 13485 : Système de Management Qualité pour les dispositifs médicaux », Support.nouvelle industrie. https://support.nouvelleindustrie.com/fr/article/iso-13485-syst%C3%A8me-de-management-qualit%C3%A9-pour-les-dispositifs-m%C3%A9dicaux (consulté le 18 août 2022).
[26] M. Navarro, « Exigences de la norme EN ISO 13485 », mauricenavarro. https://www.mauricenavarro.com/ressources/exigences-de-la-norme-en-iso-13485/ (consulté le 17 août 2022).
[27] Gilbert Farges, « NF EN ISO 13485 : 2016 - Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », UTC, 2021.
[28] « ISO/TR 80002-2:2017 - Logiciels de dispositifs médicaux - Partie 2 : Validation des logiciels pour les systèmes de qualité des dispositifs médicaux », Ed.Afnor, Paris, www.afnor.org, 1 juin 2017. Consulté le : 22 août 2022. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/iso-tr-80002-22017/XS130330?rechercheID=8752803&searchIndex=11&activeTab=all
[29] Replay webinaire - Fabricants de DM : Réussir sa validation des logiciels du SMQ, (23 juin 2022). Consulté le : 22 août 2022. [En ligne Vidéo]. Disponible sur : https://www.youtube.com/watch?v=NZVPhsMtIso
[30] 2017 09 Validation ERP, (5 octobre 2017). Consulté le : 21 août 2022. [En ligne Vidéo]. Disponible sur : https://www.youtube.com/watch?v=5rYCPalhzDA
[31] Guillaume Promé, « Validation des logiciels utilisés dans le SMQ », Qualitiso, 14 juin 2018. https://www.qualitiso.com/validation-logiciels-sqm-13485/ (consulté le 27 janvier 2022).
Annexes
Annexe 1 : Extrait du travail réalisé lors de l'étude de la norme ISO 13485v2016
Annexe 2 : Logigramme du processus de validation des applications logicielles