
IDS194 – Maîtrise des risques des dispositifs médicaux numériques embarquant l'intelligence artificielle
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteure

Contacts
- Meghan MCFADDEN : meghanmcfadden15@gmail.com
Citation
A rappeler pour tout usage : Meghan MCFADDEN. « Maîtrise des risques des dispositifs médicaux numériques embarquant l'intelligence artificielle », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs médicaux et affaires réglementaires (DMAR), Mémoire de Stage, https://travaux.master.utc.fr/, réf n° IDS194, juillet 2023, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids194/
Résumé
L'intelligence artificielle (IA) est une technologie dont le potentiel de transformation du secteur de la santé est considérable, particulièrement par l'amélioration de la précision du diagnostic et la personnalisation du traitement. Cependant, la complexité et la nouveauté de ces technologies créent de risques inédits qui doivent être gérés efficacement pour assurer la sécurité des utilisateurs, engager la confiance du public et protéger la valeur des technologies ainsi que la pérennité des fabricants.
Pour répondre à ces enjeux, une approche systématique basée sur les risques devrait être appliquée à l'ensemble de l'organisation, y compris la direction et les développeurs travaillant sur ces technologies. L’objectif de ce projet de stage étant la mise en place d’un tel système, plusieurs outils sont proposés, y compris une cartographie des risques, une proposition de processus pour les maîtriser et un support de formation destiné aux équipes internes. Enfin, ce rapport se conclut par un cas d’usage dans le contexte d’une entreprise commercialisant un dispositif médical numérique utilisant l’IA pour prédire la concentration du médicament dans le corps du patient en fonction de ses caractéristiques personnelles.
Mots-clefs : dispositifs médicaux numériques, intelligence artificielle, gestion des risques
Abstract
Artificial intelligence (AI) is a technology with tremendous potential to transform the healthcare industry, particularly by improving diagnostic accuracy and personalizing treatment. However, the complexity and novelty of these technologies create unique risks that must be effectively managed to ensure user safety, build public trust, and protect the value of the technology as well as the longevity of the companies developing them.
To address these issues, a systematic risk-based approach should be applied across the entire organization, including upper management and developers working on these technologies. The objective of this internship being the implementation of such a system, several tools are proposed, including a mapping of risks, a proposal for a process to control them and a training presentation intended for the teams working on these systems. Finally, this report concludes with a case study carried out in the context of a company marketing a digital medical device using AI to predict the concentration of drugs in the patient's body according to their personal characteristics.
Keywords : digital medical devices, artificial intelligence, risk management
Téléchargements



Acronymes
AI (anglais) : artificial intelligence
CE : conformité européenne
IA : intelligence artificielle
IT (anglais) : information technology
NIST (anglais) : National Institute for Standards and Technology
OMS : Organisation mondiale de la santé
QCM : questions à choix multiples
RDM : Règlement européen 2017/745 relatif aux dispositifs médicaux
R&D : recherche et développement
RGPD : règlement général sur la protection des données
SAC : surveillance après commercialisation
SCAC : surveillance clinique après commercialisation
SMQ : système de management de la qualité
SOUP (anglais) : software of unknown provenance
UE : Union Européenne
Mémoire Complet : Maîtrise des risques des dispositifs médicaux numériques embarquant l'intelligence artificielle
1. Introduction
L'intelligence artificielle (IA) peut être définie comme la capacité d'un système à effectuer des tâches normalement associées à l'intelligence humaine [1]. Grâce à l'augmentation de la capacité de calcul ainsi que la disponibilité de grandes quantités de données, les algorithmes sont désormais capables d'atteindre ou de dépasser les performances humaines pour certaines tâches [2]. Dans le monde de la santé, l’innovation portée par l’IA est en plein essor dans l’imagerie, la chirurgie robotique, le diagnostic, les essais cliniques, la recherche et développement et la médecine personnalisée [3].
Néanmoins, l’exploitation des technologies d'IA comporte des risques qui peuvent avoir des effets néfastes sur les individus, les organisations, les communautés et la société. Dans l'industrie des dispositifs médicaux, les fabricants doivent gérer ces risques afin de protéger la sécurité des utilisateurs et des patients ainsi que les intérêts de leur organisation et des parties prenantes, y compris des investisseurs, tout en s’adaptant au contexte réglementaire et normatif en pleine évolution.
Ce rapport présente une étude de cas de mise en place d’un système de management des risques au sein de l’entreprise ExactCure, une startup innovante commercialisant un dispositif médical numérique, ExaMed. Cette solution répond à un important problème de santé publique : la mauvaise utilisation des médicaments, qui engendre plus de 10 000 décès par an en France, soit 3 fois plus que les accidents de route [4]. En utilisant des modèles incorporant de l’IA pour simuler et illustrer la posologie des médicaments à partir des données du patient, ExaMed vise à réduire la fréquence des interactions médicamenteuses, des surdoses et des sousdoses [5]. Cette technologie utilise des algorithmes du type système expert, qui se base sur des règles déterministes pour émuler la capacité de prise de décision des experts humains [3].
Le résultat de ce travail est la mise en place d’un système interdisciplinaire de management des risques d’un dispositif médical numérique embarquant l’IA. Afin de mieux comprendre le contexte législatif et normatif de la gestion des risques de ces technologies, un état des lieux des différentes réglementations et des normes clés est réalisé. Ensuite, des outils pour identifier les risques, mettre en place des processus pour les maîtriser et pour former les équipes internes des organisations développant ces systèmes sont proposés. Finalement, un retour d’expérience en tant que stagiaire chez ExactCure est détaillé.
2. Contexte socio-économique des dispositifs médicaux numériques utilisant l’intelligence artificielle
De manière générale, l’expression « intelligence artificielle » implique l’imitation d’un comportement intelligent afin d’effectuer des tâches avec de l’intervention humaine minimale. Les systèmes d'IA peuvent tenter de reproduire avec précision le raisonnement humain pour résoudre un problème ; à l'inverse, ils peuvent ignorer le raisonnement humain et utiliser exclusivement de grands volumes de données pour répondre à la question d'intérêt [3]. Ces systèmes génèrent des sorties telles que des prédictions et des recommandations pour un ensemble d'objectifs définis par l'homme [2].
Il existe de nombreux types et cas d’usage d'IA avec divers degrés d'autonomie et de complexité [Figure 1].
Figure 1. Exemples de méthodes et d’applications d'intelligence artificielle (source : auteure).
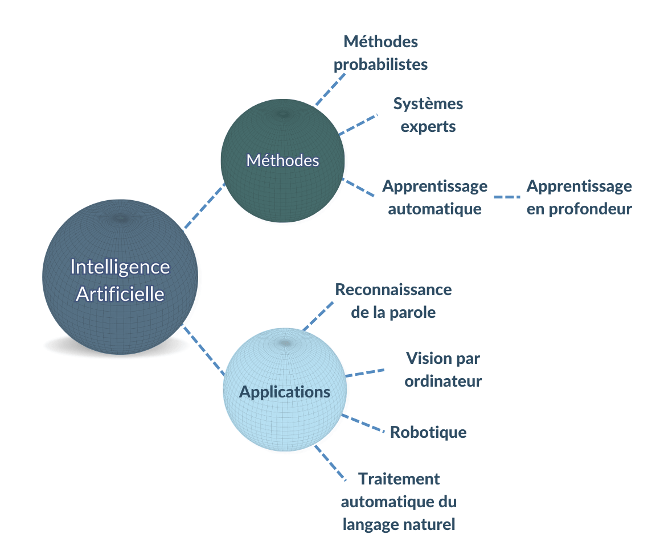
Des exemples importants de méthodes d’intelligence artificielle incluent :
- L'apprentissage automatique : ou machine learning en anglais, regroupe une famille de techniques de modélisation statistique et mathématique qui utilise une variété d'approches pour apprendre et améliorer automatiquement la prédiction d'un état cible, sans programmation explicite [3].
- L’apprentissage profond : ou deep learning en anglais, il s’agit d’un sous-type d'apprentissage automatique qui se base sur des algorithmes d'apprentissage de représentation à plusieurs niveaux. Ils sont obtenus en composant des modules simples mais non linéaires qui transforment chacun la représentation de l’entrée brute en une représentation à un niveau supérieur [6]. Les modèles d'apprentissage profond démontrent une efficacité exceptionnelle dans les tâches de vision par ordinateur, de reconnaissance vocale et de traitement automatique du langage naturel.
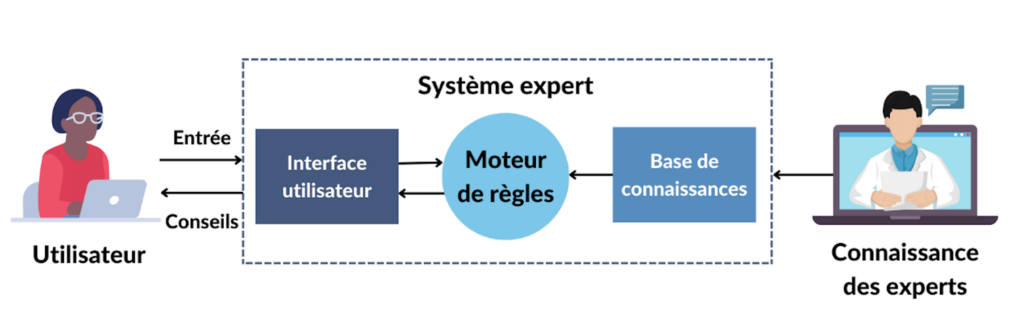
- Les systèmes experts : Les systèmes experts sont un ensemble d'algorithmes informatiques qui s'appuient en grande partie sur des règles booléennes et déterministes pour émuler la capacité de prise de décision des experts humains. Un système expert est divisé en une base de connaissances, qui code la logique du domaine, et un moteur d'inférence, qui applique la base de connaissances aux données présentées au système pour fournir des recommandations ou déduire de nouveaux faits [Figure 2] [3]. Les utilisateurs de ces systèmes peuvent ensuite utiliser les informations obtenues pour aviser des décisions, telles que la bonne dose de médicament à prendre lors de l’automédication.
Figure 2. Fonctionnement d'un système expert (source : auteure).
2.1 Un secteur en croissance
Bien que le concept de l'utilisation d'ordinateurs pour simuler un comportement intelligent ou la pensée critique a été décrite pour la première fois par Alan Turing en 1950, des avancées récentes ont accéléré ce domaine à la pointe de la technologie de l'information [7]. En particulier, les algorithmes d'IA sont souvent formés sur des données représentatives de la population pour fonctionner à grande échelle. Les avancées matérielles récentes permettant une plus grande puissance de calcul ont permis de traiter et d'analyser de grandes quantités de données en temps réel. Les algorithmes d'IA deviennent donc de plus en plus complexes et sophistiqués, désormais capables d'effectuer certaines tâches aussi bien ou même mieux que les humains, telle que la classification d'images [8].
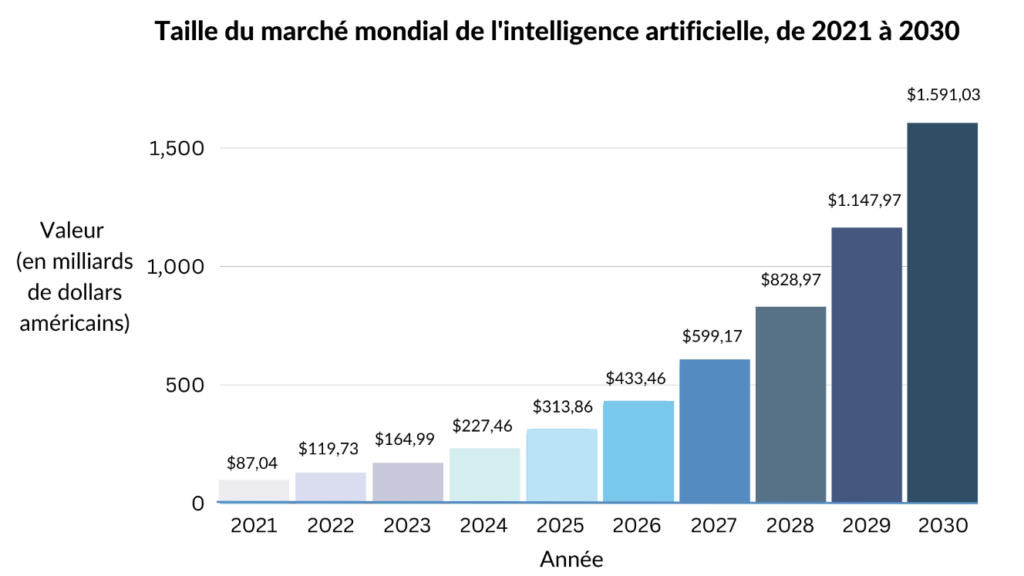
Les entreprises de divers secteurs se tournent vers l'IA pour optimiser l’efficience de leurs opérations, réduire leurs coûts et acquérir un avantage concurrentiel sur le marché. La demande croissante d'automatisation entraine une augmentation fulgurante des investissements dans la recherche et le développement de l'IA, en grande partie par de géants mondiaux de la technologie tels que Google, Microsoft, IBM, Amazon et Apple. Par conséquent, la taille du marché mondial de l'IA était estimée à 120 milliards de dollars américains en 2022 et devrait atteindre près de 1 600 milliards de dollars américains d'ici 2030 [Figure 3] [8].
Figure 3. Graphique projetant la croissance du marché de l'intelligence artificielle dans le monde (adaptée de [8]).
Dans le domaine de la santé, le vieillissement de la population mondiale et la prévalence croissante des maladies chroniques ont contribué à l'augmentation de la demande de diagnostics et à une meilleure compréhension des maladies à leurs stades initiaux. Les algorithmes d'IA sont de plus en plus intégrés dans les systèmes de santé pour prédire avec précision les maladies à leur stade précoce sur la base d'ensembles de données historiques [9].
En 2021, les technologies d'IA dans le domaine de la santé avaient une valeur estimée à environ 11 milliards de dollars américains dans le monde. Les prévisions indiquent que le marché dans ce secteur représentera près de 188 milliards de dollars américains d'ici 2030, augmentant à un taux de croissance annuel composé de 37 % [10].
La part de marché grandissante et la médiatisation de l'IA s'accompagnent de nouvelles préoccupations du public. Selon une enquête menée aux États-Unis, la principale inquiétude concernant l'utilisation accrue de l'IA dans la santé était les menaces liées à la sécurité et à la vie privée. En Europe, une enquête a révélé que les patients seraient plus confiants en utilisation de l'IA en combinaison avec des jugements d'experts plutôt que des décisions prises uniquement par l'IA [10].
2.2 ExactCure : contexte d’une start-up commercialisant un dispositif médical numérique basé sur l’intelligence artificielle
Cette étude a été réalisée dans le cadre d'un stage en tant que chargée affaires réglementaires pour ExactCure, une startup niçoise fondée en 2018 par Fréderic Dayan, président-directeur général, Fabien Astic, directeur scientifique, et Sylvain Benito, directeur de la recherche et développement (R&D).
Organisation de la société
Avec ses 22 employés et deux stagiaires, ExactCure est actuellement classée comme une très petite entreprise. Trois pôles d’activité sont présents au sein de la société :
- Le service R&D : ce service est responsable du développement des ressources et des données utilisées, ainsi que d’autres projets annexes. Ils développent les modèles utilisés dans le cœur de calcul du produit, dont le volet IA.
- Le service IT (information technology en anglais) : ce service est responsable de la conception, du développement et du déploiement des applications qui constituent les interfaces utilisateur du produit (web et mobile). Ils sont également chargés de recevoir les commentaires des clients et d'assurer le fonctionnement du produit.
- Le service qualité et affaires réglementaires : ce service est chargé d'assurer la conformité du produit à toutes les exigences nécessaires pour obtenir et maintenir l'accès au marché. Il est mené par une Responsable Qualité et Affaires Règlementaires, que j’ai accompagnée en tant que stagiaire chargée affaires réglementaires.
Présentation du produit ExaMed
Commercialisée en France depuis 2020, ExaMed, la solution logicielle d’ExactCure, a pour but de répondre à une importante problématique de santé publique : la mauvaise utilisation des médicaments. Qu'elle soit causée par un mauvais dosage, la non-observance du traitement prescrit, ou une interaction inattendue entre plusieurs molécules, le mésusage des médicaments est responsable d'environ 10 000 décès par an en France, soit 3 fois plus que les accidents de la route [4].
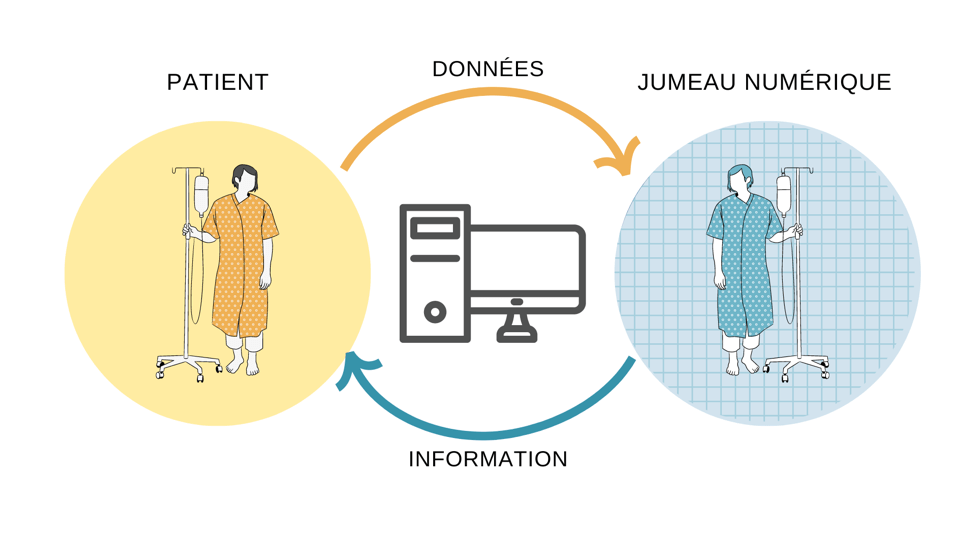
ExaMed fournit des simulations personnalisées de la concentration des médicaments dans le corps de chaque patient en tenant compte des facteurs tels que son âge, son sexe, son état hépatique et son génotype. Ce logiciel est donc un type de « jumeau numérique » : à partir des données dérivées de caractéristiques clairement définies, il permet de modéliser une entité réelle et souvent complexe, telle qu’un patient spécifique [Figure 4] [11].
Figure 4. Schéma du fonctionnement des jumeaux numériques (source : auteure).
ExaMed possède plusieurs finalités médicales qui le qualifient de dispositif médical, notamment [12] :
- Améliorer la compréhension du patient des médicaments ;
- Aider les professionnels de santé à conseiller, prescrire et suivre les traitements médicamenteux ;
- Réduire les risques de mésusage de médicaments ;
- Améliorer l’observance thérapeutique.
ExactCure propose deux modalités d’utilisation d’ExaMed : une application mobile destinée aux patients [Figure 5] et une interface web dédiée aux professionnels de santé [Figure 6].
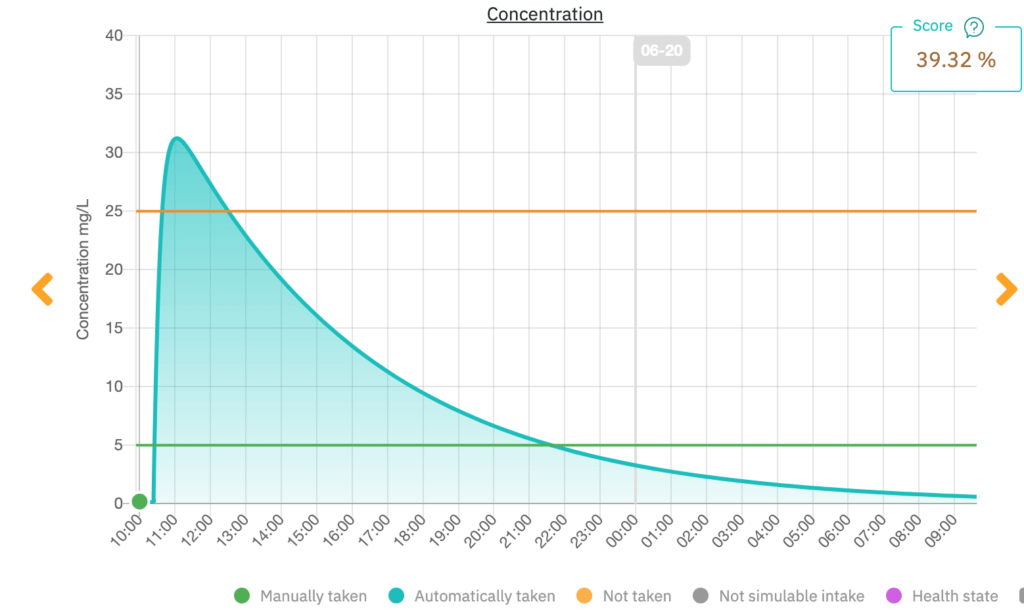
Figure 5. Capture d'écran de l'application mobile ExaMed (source : ExactCure).

Figure 5 montre un exemple du graphique affiché pour un patient qui envisage de prendre une dose excessive de paracétamol. L'horloge indique l'heure à laquelle le médicament prendra effet ainsi que le moment où les effets se dissiperont. Le patient est donc en mesure de comprendre la relation entre la concentration du médicament dans son corps et l'efficacité thérapeutique. Il a également la possibilité de consigner son état de santé dans la rubrique « Comment je me sens maintenant ? ». Cette information est ensuite transmise au professionnel de santé associé à son compte.
Figure 6. Capture d'écran de l'interface du professionnel de la santé affichant la même simulation de médication qu'à la Figure 5 (source : ExactCure).
En montrant la concentration de médicament dans le corps du patient en fonction du temps, cette interface permet aux professionnels de santé de vérifier qu'ils prescrivent une dose sûre et efficace. Ils peuvent également suivre le traitement de leurs patients.
L’application ExaMed fait appel à ExaTwin, un logiciel accessoire de type service Web qui intègre des algorithmes utilisant l'intelligence artificielle du type système expert pour agréger des sources scientifiques et industrielles. Ces algorithmes utilisent les caractéristiques individuelles des patients comme covariables pour créer des simulations pharmacocinétiques personnalisées.
Projets de développement
ExactCure développe de nouvelles fonctionnalités pour élargir les cas d'usage de sa solution. En particulier, un projet appelé Interactwin vise à prédire les interactions polymédicamenteuses en prenant en compte des interactions résultant de prescriptions complexes, souvent composées d’une dizaine de médicaments ou plus. Ce projet a permis à ExactCure de remporter le concours i-Nov 2022 et le prix associé de 4,9 millions d'euros [13].
ExactCure vise également à déployer sa solution dans davantage d'environnements, y compris les hôpitaux. Pour ce faire, il s’est associé avec des acteurs significatifs du logiciel médical. Par exemple, en novembre 2022, ExactCure a noué un partenariat avec Philips à la suite d'un concours organisé au Campus PariSanté, l'emportant sur de nombreuses startups européennes. Ce partenariat leur confère le statut de collaborateur, leur permettant d'intégrer la plateforme de gestion de données Philips HealthSuite et de tirer parti de son écosystème pour fournir une solution de santé complète aux professionnels en milieu hospitalier [13]. Un autre objectif est d’intégrer les produits d’ExactCure dans des logiciels professionnels existants comme le Vidal Sentinel, outil d'analyse du risque des prescriptions hospitalières [14].
2.3 Problématique et enjeux
Dans le cadre d’une entreprise commercialisant un dispositif médical dans l’Union européenne (UE), la mission principale en tant que chargée des affaires réglementaires est de veiller au respect des obligations des réglementations afin d’obtenir le marquage CE (Conformité Européen). Cette certification permet la libre circulation dans le marché européen et atteste la conformité des dispositifs médicaux en termes de sécurité et de performance. Cette tâche est guidée par les normes qui assurent la conformité avec les exigences réglementaires et l’interopérabilité sur les marchés internationaux. La bonne compréhension de ces documents est essentielle pour mener à bien un projet de commercialisation d’un dispositif médical.
Pour ExactCure, le règlement (UE) 2017/745 relatif aux dispositifs médicaux (RDM) est le fil conducteur d’une stratégie de mise sur le marché dans l’UE. Le règlement exige que les DM soient développés et fabriqués conformément à l'état de l'art, qui repose sur les principes de gestion des risques, de sécurité de l'information, de cycle de vie, de vérification et de validation de sa conformité [15].
Mis sur le marché en 2020, ExaMed a été auto-certifié par ExactCure pour obtenir le marquage CE en tant que dispositif médical de classe I au titre de la Directive 93/42/CEE relative aux dispositifs médicaux. Avec la mise en application du RDM, ce produit passera bientôt en classe IIa. Ce changement de classe implique des exigences accrues pour prouver la sécurité et l'efficacité du dispositif, ce qui signifie que ExaMed nécessitera une certification par un organisme notifié pour renouveler son marquage CE [15].
Dans le contexte réglementaire des dispositifs médicaux en constante évolution, les particularités inhérentes à l’IA et l'histoire limitée de l'utilisation de ces technologies présentent des défis uniques pour les développeurs et régulateurs. Les systèmes basés sur l'IA différent des systèmes numériques traditionnels, programmés pour produire des sorties spécifiques en fonction des règles statiques et bien définies, de trois manières [16] :
- Apprentissage : les systèmes peuvent accumuler des données pour évaluer les résultats du traitement et faire des prédictions afin d'améliorer les résultats pour les patients. Vu que le logiciel ExaMed utilise des algorithmes de type système expert sans apprentissage automatique, cette caractéristique n’est pas applicable dans le cadre de ce projet.
- Autonomie : ces systèmes ont le potentiel de fonctionner avec une supervision réduite ou même sans la supervision du clinicien.
- Inexplicabilité : en raison de la complexité des statistiques impliquées et des grands ensembles de données utilisés, la justification des résultats produits par de tels systèmes pourrait ne pas être facilement comprise par les développeurs eux-mêmes ni des professionnels de santé bien formés, et encore moins par le grand public.
Ces différences donnent lieu à un ensemble de risques propres aux systèmes d'IA, notamment des risques « sociotechniques » [17]. Ces risques proviennent de la façon dont les humains interagissent avec ces technologies, y compris la capacité des humains de les juger ou de les évaluer [18]. Des exemples notables incluent :
- La transparence : la capacité de comprendre les résultats donnés et évaluer leur conformité
- L’interprétabilité : la capacité de déterminer si les sorties peuvent être utilisées pour prendre des décisions.
En raison de ces enjeux et de la perception de la fiabilité des dispositifs médicaux embarquant l’IA, l’engagement de la confiance du public est un enjeu clé pour les développeurs de ces technologies. Pour ce faire, ainsi que pour répondre aux exigences réglementaires pour la mise sur le marché des dispositifs médicaux, il est essentiel de comprendre et de gérer efficacement les risques associés.
La gestion des risques est également un élément clé du développement et de l'utilisation responsables des dispositifs médicaux incorporant l'IA. Des pratiques responsables de gestion des risques peuvent aider à aligner les décisions concernant la conception, le développement et les utilisations du système d'IA sur l'objectif et les valeurs visés. La mise en œuvre de la gestion des risques liés à l'IA peut encourager des comportements et des actions responsables au sein des organisations. Les équipes internes impliquées dans la conception, le développement et le déploiement de l'IA peuvent ensuite aborder le processus en tenant compte du contexte et des conséquences potentielles, qu’elles soient positives ou négatives.
La problématique de ce stage est donc la question suivante : comment assurer un système efficace de gestion des risques au sein d’une entreprise commercialisant un dispositif médical numérique basé sur l’intelligence artificielle ?
J’ai dû confronter un certain nombre d’enjeux lors de ce stage :
- Parce que la gestion des risques d’un dispositif médical est intimement liée à son développement, une approche interdisciplinaire avec les équipes des développeurs était nécessaire ;
- Le produit étant déjà sur le marché, un système existant a dû être pris en compte et adapté ;
- En raison de la nature innovante du produit, peu de ressources telles que des études de cas de produits comparables étaient à ma disposition ;
- En tant que fabricant d’un dispositif médical de type logiciel, la société est tenue de se conformer aux règlements et normes détaillés dans le chapitre 3 de ce mémoire, y compris le RDM, le Règlement général sur la protection des données, le Règlement européen 2021/0106 sur l’intelligence artificielle, ISO 13485, ISO 14971, ISO 62304, ISO 62366-1, ISO 23894 et ISO 42001 ;
- Avec une composante d'intelligence artificielle, le système de gestion des risques doit être adapté pour tenir compte des risques uniques de ces systèmes malgré un historique limité de leur utilisation et un manque de normes harmonisées associées.
3. Maîtrise des risques des dispositifs médicaux numériques utilisant l’intelligence artificielle
Comme tous les dispositifs médicaux, les dispositifs médicaux incorporant l'IA peuvent induire des risques importants, tels que des erreurs de diagnostic ou de recommandations de traitement, d'atteintes à la vie privée et d'autres dommages [3]. Les risques liés à l'IA peuvent être classés en fonction de leur durée (à long ou à court terme), de leur probabilité (élevée ou faible), de leur étendue (systémique ou localisée) et impact (élevé ou faible) [4].
La sécurité de dispositifs médicaux intégrant l'IA a récemment été remise en cause dans des solutions déjà disponibles sur le marché aux États-Unis et en Europe. Le manque de validation externe des systèmes (le même ensemble de données étant utilisé pour le développement et la validation) peut conduire à des surestimations de leur précision. L’évaluation des systèmes d’IA par des parties externes est difficile en raison de l'hétérogénéité des paramètres utilisés et du manque de disponibilité des ensembles de données et du codage qui permettraient de reproduire les études effectuées par les fabricants [19].
Ces écueils induisent des risques pour les usagers, patients et professionnels de santé, ainsi que pour les organismes qui commercialisent ces dispositifs. Comme pour les autres risques liés aux dispositifs médicaux, le fournisseur reste responsable de l’identification, contrôle et suivi des risques. La confiance du public dans leur produit dépend donc d'un système de gestion des risques bien planifié et efficace.
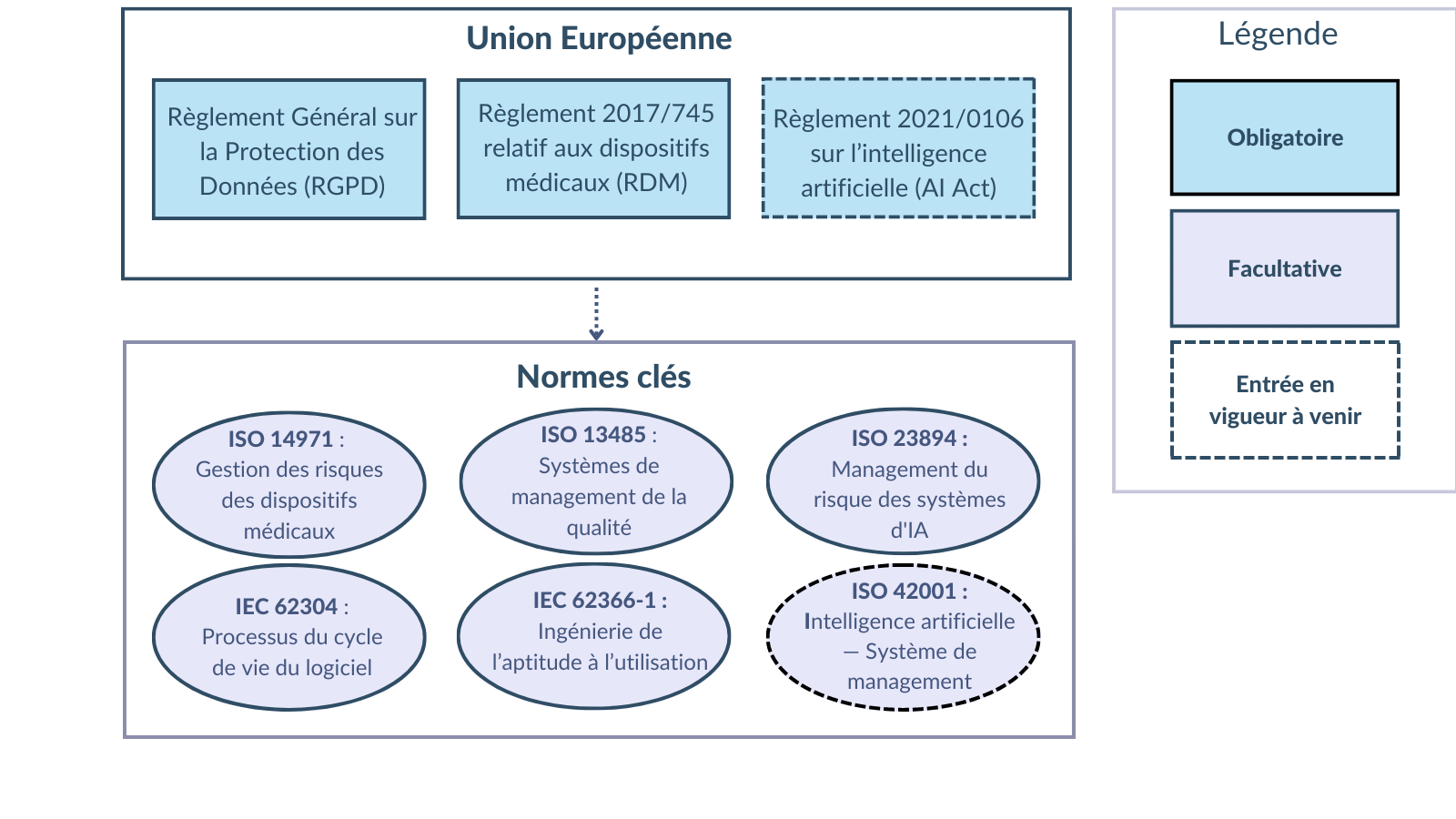
La gestion des risques nécessite une approche systématique, documentée et guidée par la réglementation et les normes pertinentes [Figure 7]. Comme de nombreuses ressources existent pour planifier ce système, cette section ne présente que les principales réglementations et normes et leur pertinence pour les dispositifs médicaux numériques à composante IA.
Figure 7. Schéma du paysage réglementaire et normatif de la gestion des risques des dispositifs médicaux intégrant l'intelligence artificielle (source : auteure).
3.1 Aperçu du paysage réglementaire en Europe
3.1.1 Règlement européen 2017/745 relatif aux dispositifs médicaux
Bien que le RDM ne traite pas spécifiquement les particularités d'IA, la règlementation s'applique à tous les systèmes d'IA qualifiés de dispositifs médicaux. Un grand nombre de DM embarquant l’IA ont déjà reçu le marquage CE en vertu des directives précédentes [20].
De nombreux logiciels qui, selon la Directive 93/42/CEE relative aux dispositifs médicaux, appartenaient à la classe de risque la plus faible passeront désormais à une classe de risque plus élevée sous le RDM. En raison de l’Annexe VIII 6.3, Règle 11 du RDM, la plupart des dispositifs d'IA seront classés dans la classe IIa ou IIb car ils fournissent des informations utilisées pour prendre des décisions à des fins thérapeutiques ou diagnostics. Contrairement aux dispositifs de classe I, ils devront donc subir une évaluation de conformité par un organisme notifié.
L'article 10 du RDM exige de tous les fabricants de dispositifs médicaux qu'ils établissent, documentent, mettent en œuvre et maintiennent un système de gestion des risques. L'Annexe I du règlement fournit plus de détails sur ces exigences, indiquant que ce processus doit être continu, itératif et mené tout au long du cycle de vie d'un dispositif [15].
L'annexe I, chapitre I stipule que les risques doivent être réduits autant que possible sans nuire au rapport bénéfice/risque. Le règlement définit le risque comme « la combinaison de la probabilité d'occurrence d'un dommage et de la gravité de ce dommage ». La détermination du rapport bénéfice/risque est définie comme « l'analyse de toutes les évaluations du bénéfice et du risque pouvant être pertinentes pour l'utilisation du dispositif aux fins prévues, lorsqu'il est utilisé conformément à la destination indiquée par le fabricant ». La gestion des risques doit également prendre en compte les informations sur la production et la surveillance post-commercialisation pour se conformer à ces exigences [15].
Dans l'ensemble, conformément à la réglementation, une documentation approfondie des procédures de gestion des risques est requise. Le dossier technique de chaque dispositif doit inclure les résultats du processus de gestion des risques comprenant l'analyse bénéfice-risque, les mesures de contrôle de risque et le rapport périodique actualisé de sécurité. Toute la documentation sur les risques pour chaque produit doit être tracée, conservée et facilement disponible.
3.1.2 Règlement général sur la protection des données
Entré en application le 25 mai 2018, le Règlement général sur la protection des données (RGPD) vise à protéger la vie privée des citoyens dans l'UE et l'Espace économique européen. Le RGPD est un élément important du droit européen protégeant les droits de l'homme, en particulier l'article 8 de la Charte des droits fondamentaux de l'Union européenne [21].
Ce règlement a trois objectifs principaux [21] :
- renforcer les droits des personnes ;
- responsabiliser les acteurs traitant des données ;
- renforcer la coopération entre les autorités de protection des données.
L'article 5 du RGPD propose les principes du traitement des données et précise que les données doivent être traitées de manière loyale et transparente vis-à-vis de la personne concernée et que les données doivent être exactes et à jour. Dans le domaine de l'IA, les principes d'équité, de transparence et d'exactitude des données sont étroitement liés à ces exigences.
L'article 35 exige également une analyse d'impact sur la protection des données, c'est-à-dire une analyse des risques découlant du traitement de données et une évaluation de la proportionnalité entre les risques existants et les avantages du traitement.
Parmi les critères de mesure du risque, il faut tenir compte des paramètres de protection des données (confidentialité, intégrité et disponibilité) ainsi que de l'impact possible sur les autres droits fondamentaux de la personne.
Dans le cas d'un système d'IA, un exemple de droit fondamental est le principe de non-discrimination, qui doit être évalué dans la phase de test et de programmation de l'algorithme. Dans ces phases, il est nécessaire de vérifier qu'aucune valeur discriminatoire n'a été trouvée intégrée dans le code lui-même.
3.1.3 Règlement 2021/0106 sur l’intelligence artificielle (AI Act)
La Commission européenne juge essentiel d'établir des réglementations au niveau européen pour le développement et déploiement de l’IA, garantissant que les citoyens européens puissent tirer parti des technologies qui adhèrent aux valeurs, droits et principes fondamentaux de l'Union [20].
Le 21 avril 2021, une proposition de règlement européen sur l'IA a été publiée [22]. Au moment de la rédaction de ce rapport, la date d'entrée en vigueur du texte n'est pas précisée. Même si le texte est encore en cours de révision, il est déjà reconnu comme ayant un impact significatif sur le cadre organisationnel des entreprises [19]. En raison de ce que l'on appelle « l’effet Bruxelles », le processus par lequel les réglementations de l'Union européenne influencent l'élaboration des politiques et des réglementations mondiales lorsque les organismes opérant à l'échelle internationale adoptent les normes de l'UE, comme ce fut le cas du RGPD. Ce nouveau règlement façonnera sans aucun doute l'avenir prévisible de la réglementation de l'IA dans l'UE et dans le monde [23].
Comme le règlement sur les dispositifs médicaux, la proposition de règlement sur l’IA utilise une approche basée sur les risques. Les technologies d'IA sont classées en 4 niveaux différents [Figure 8].
Figure 8. Classification de risque selon le règlement européen sur l'IA (source : auteure).
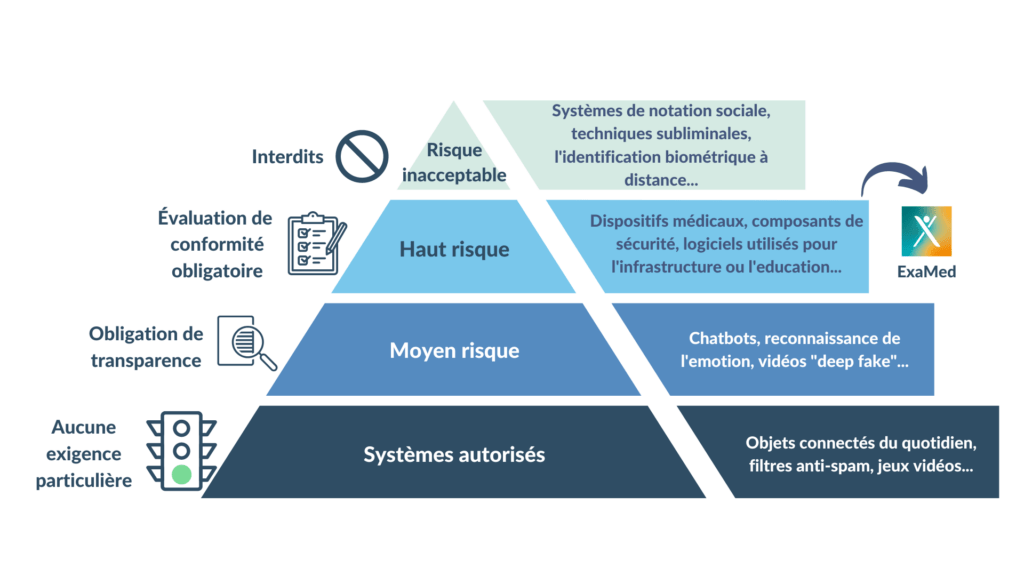
Risque inacceptable : Tous les systèmes d'IA considérés comme une menace pour la sécurité, les moyens de subsistance et les droits fondamentaux des citoyens seront interdits. Des exemples sont la notation sociale par les gouvernements, les bases de données de reconnaissance faciale et les jouets utilisant l'assistance vocale pour encourager les comportements dangereux [24].
Risque élevé : Une technologie est considérée comme étant à haut risque si elle est destinée à être utilisée comme composant de sécurité d’un produit couvert par les actes législatifs d’harmonisation de l’Union énumérés à l’annexe II, ou constitue lui-même un tel produit. Si le produit dont le composant de sécurité est le système d’IA, il est soumis à une évaluation de la conformité par un tiers [25].
Les systèmes d'IA identifiés comme à haut risque comprennent ceux utilisés dans les infrastructures critiques (par exemple, les transports), qui pourrait mettre la sécurité des citoyens en danger ; la formation scolaire ou professionnelle, qui peut déterminer l'accès à l'éducation et le parcours professionnel de la vie d'une personne (par exemple, la notation des examens) ; et les composants de sécurité des produits (par exemple, l'application d’IA dans la chirurgie assistée par robot).
Les systèmes d'IA à haut risque seront soumis à des obligations strictes avant de pouvoir être mis sur le marché, y compris [24]:
- des systèmes adéquats d'évaluation et de contrôle des risques ;
- l'enregistrement des activités pour assurer la traçabilité des résultats ;
- une documentation détaillée fournissant toutes les informations nécessaires sur le système et son objectif pour que les autorités compétentes puissent évaluer sa conformité ;
- des informations claires et adéquates fournies à l'utilisateur ;
- des mesures de surveillance humaine appropriées pour minimiser les risques,
- et un niveau élevé de robustesse, de sécurité et d'exactitude.
Risque limité : Les systèmes d'IA à risque limité, tels que les chatbots, seraient soumis à des obligations de transparence spécifiques. Par exemple, les utilisateurs doivent être conscients qu'ils interagissent avec une machine afin de pouvoir prendre des décisions éclairées concernant ces interactions [24].
Risque minimal : Les systèmes d'IA présentant un risque minimal, tels que les jeux vidéo compatibles avec l'IA ou les filtres anti-spam, pourraient être utilisés sans restriction dans le cadre du règlement. La grande majorité des systèmes d'IA actuellement utilisés dans l'UE entrent dans cette catégorie [24].
Impact sur la gestion des risques des dispositifs médicaux
Dans l'UE, les systèmes d'IA étiquetés « systèmes à haut risque », y compris la plupart des dispositifs médicaux, devront être certifiés sous le nouveau règlement [Figure 9] [26]. Cela signifie que des règles supplémentaires s'appliqueront aux dispositifs médicaux en plus des exigences du RDM. En pratique, l'organisme notifié responsable de l'évaluation de la conformité au RDM procédera également à l'évaluation de la conformité au nouveau règlement sur l’IA [20]. Le RDM est inclus dans la liste de la législation d'harmonisation de l'Union dans l'annexe II du projet de règlement [20].
Figure 9. Processus à réaliser pour les systèmes d'intelligence artificielle à haut risque sous le règlement européen sur l’IA( figure adaptée de [24]).
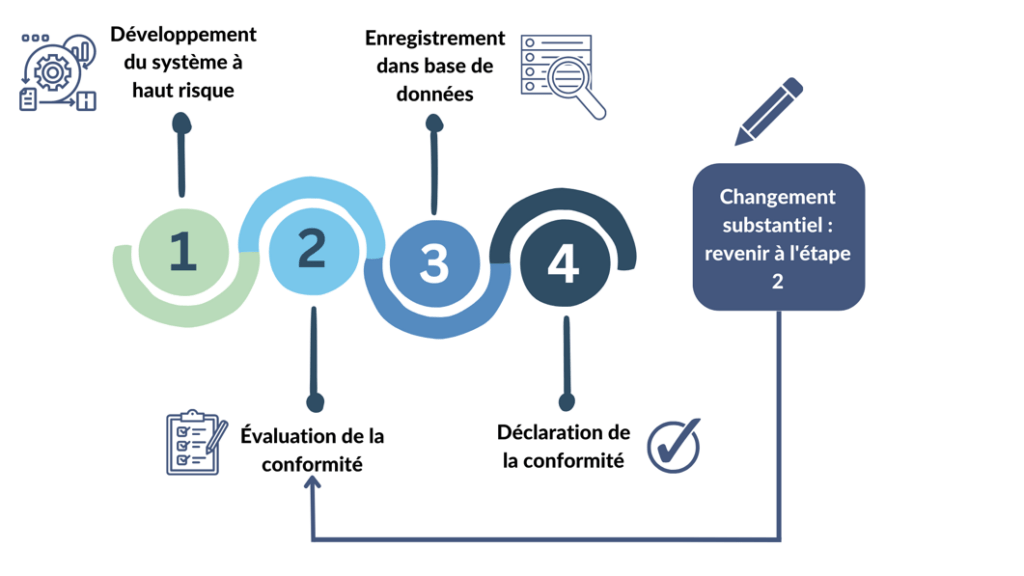
Un changement est considéré comme substantiel lorsqu'il a un effet sur la sécurité et/ou les performances du système, les revendications, l'utilisation prévue, ou la conformité aux réglementations applicables.
Les critères de certification peuvent s'appuyer sur des normes dites « harmonisées » qui définiront en détail les exigences techniques nécessaires [26]. Bien que l’application de ces normes ne soit pas techniquement obligatoire, le respect sera considéré comme une présomption de conformité à la réglementation européenne.
Le processus de gestion des risques à mettre en place pour les technologies à haut risque est défini à l'article 9 du projet de règlement. Conformément aux exigences du RDM, ce processus doit être itératif et continu, se dérouler sur l'ensemble du cycle de vie et impliquer des personnes compétentes vis-à-vis du produit, de sa technologie et de son utilisation. De même, l'acceptabilité des risques résiduels individuels et du risque résiduel global doit être prise en compte.
La démonstration de la maîtrise des risques doit être réalisée au moyen de tests avec des critères de réussite prédéfinis. Les tests ont trois objectifs principaux. Premièrement, ils peuvent aider à identifier les mesures de gestion des risques les plus appropriées en améliorant la compréhension des risques. Deuxièmement, les tests permettent de garantir que les systèmes d'IA fonctionnent de manière cohérente pour l'usage auquel ils sont destinés. Ceci est particulièrement important pour garantir que les données utilisées pour former ces systèmes reflètent avec précision les problèmes rencontrés lors de l'utilisation dans le monde réel. Enfin, les tests garantissent que les systèmes d'IA répondent aux exigences énoncées au chapitre 2 du règlement, notamment la transparence, la précision, la robustesse et la cybersécurité [22].
3.2 Normes clés pour la gestion des risques des dispositifs médicaux numériques
La gestion des risques des dispositifs médicaux est un sujet bien traité par des normes existantes. Comme de nombreuses ressources sont disponibles pour l'interprétation et la mise en œuvre de ces normes, cette section présente seulement un bref résumé de la pertinence de chaque norme pour la gestion des risques des dispositifs médicaux utilisant l’IA. Un processus pour leur application pratique est proposé au chapitre 4.
3.2.1 ISO 13485:2016 « Systèmes de management de la qualité — Exigences à des fins réglementaires »
Considérée comme essentielle et difficilement contournable pour les fabricants de dispositifs médicaux, la norme ISO 13485:2016 « Dispositifs médicaux — Systèmes de management de la qualité — Exigences à des fins réglementaires » met un accent particulier sur la responsabilité d'intégrer les pratiques de gestion des risques [27].
Dans le cadre d'un système de management de la qualité (SMQ) pour assurer un produit de qualité, les principes de gestion des risques doivent être appliqués. Les fabricants doivent déterminer et s'assurer de la compétence des personnes impliquées avant le développement [27].
Selon les exigences relatives aux systèmes informatisés de la norme ISO 13485:2016 4.16, le développement pour la collecte et le traitement des données, pour l'étiquetage, ainsi que pour la formation et le test des modèles doit être validé [27].
3.2.2 ISO 14971:2019 : « Application de la gestion des risques aux dispositifs médicaux »
Obligatoire pour la conformité à l’ISO 13485, la norme ISO 14971:2019 « Dispositifs médicaux – Application de la gestion des risques aux dispositifs médicaux » permet d’évaluer l’ensemble des situations dangereuses associées à l’exploitation d’un dispositif médical.
Le processus de gestion des risques encadré par cette norme consiste à identifier les dangers ainsi que les séquences ou combinaisons d'événements raisonnablement prévisibles et pouvant entraîner une situation dangereuse. Un danger est une source potentielle de dommage, tandis qu'une situation dangereuse est une circonstance dans laquelle des personnes, des biens et/ou l'environnement sont exposés à un ou plusieurs dangers. Pour qu'une situation dangereuse se produise, il doit y avoir une séquence prévisible d'événements qui y conduisent [28].
Au fur et à mesure qu'un dispositif médical embarquant un système d’IA est conçu et développé, des risques apparaissent à chaque étape du cycle de vie. La nécessité de contrôler chaque risque identifié peut être considérée comme une donnée d’entrée de conception. A l'inverse, une spécification d'exigence logicielle résultant d'un besoin utilisateur peut se traduire par un besoin de maîtrise d'un risque correspondant. Chaque entrée de conception est donc un besoin de contrôler un risque, et le processus de conception correspondant est la mesure de contrôle du risque.
Le guide ISO/TR 24971 « Dispositifs médicaux — Recommandations relatives à l'application de l'ISO 14971 » permet de mieux comprendre les recommandations de cette norme [29].
3.2.3 ISO 62304:2006 Logiciels de dispositifs médicaux — Processus du cycle de vie du logiciel
Le processus de gestion des risques des dispositifs médicaux est bien traité par l'ISO 14971, mais certaines exigences supplémentaires en matière de gestion des risques sont nécessaires pour les logiciels. En particulier, il faut identifier des facteurs logiciels liés aux dangers. Pour répondre à ce besoin, l'ISO 62304 utilise ISO 14971 comme référence normative [30]. D’après cette norme, établir la sécurité et l'efficacité d'un dispositif médical contenant un logiciel nécessite de connaître l'objectif du logiciel et de démontrer que son utilisation répond à ces intentions sans entraîner de risques inacceptables [30].
La question de savoir si le logiciel est un facteur contributif à un danger est déterminée au cours de l'activité d'identification des dangers du processus de gestion des risques. Les dangers qui pourraient être indirectement causés par les logiciels (par exemple, en fournissant des informations trompeuses qui pourraient entraîner l'administration d'un traitement inapproprié) doivent être pris en compte pour déterminer si le logiciel est un facteur contributif.
Les fabricants qui utilisent des bibliothèques de logiciels (ce qui est presque toujours le cas pour les logiciels avec l’apprentissage automatique) doivent spécifier et valider ces bibliothèques en tant que SOUP (en anglais : software of unknown provenance).
La décision d'utiliser un logiciel pour contrôler les risques est prise lors de l'activité de contrôle des risques. Le processus de gestion des risques des logiciels requis dans cette norme doit être intégré dans le processus de gestion des risques conformément à la norme ISO 14971 [30].
3.2.4 ISO 62366-1 Dispositifs médicaux — Partie 1 : Application de l'ingénierie de l'aptitude à l'utilisation aux dispositifs médicaux
ISO 62366-1:2015 décrit un processus permettant aux fabricants de DM d'analyser, de spécifier, de développer et d'évaluer l’aptitude à l’utilisation de leurs produits [31]. Ce processus favorise la conception d'une interface utilisateur qui permet aux utilisateurs de comprendre rapidement comment interagir avec un produit de la manière la plus efficace et sans erreur.
La norme se concentre sur les erreurs d'utilisation qui peuvent se produire dans des conditions normales ainsi que sur l'identification et la prise en compte des risques associés lors du processus de conception. Ensuite, des procédures de test fournissent des preuves d'une utilisation sûre. Si la validation montre que les risques liés à l'utilisation sont inacceptables ou que de nouveaux risques d'utilisation ont été introduits, d'autres mesures de contrôle des risques doivent être mises en œuvre. La norme peut être utilisée pour identifier mais n'évalue ni n'atténue les risques associés à une utilisation anormale délibérée par l’utilisateur.
L’ISO 62366-1 exige que les fabricants caractérisent avec précision les utilisateurs prévus, l'environnement d'utilisation prévu et les patients visés, y compris leurs indications et contre-indications. Il faut identifier également la formation, l'expérience dans les domaines médicaux et les connaissances techniques des utilisateurs prévus [31]. Ces informations sont indispensables en tant que données d’entrée pour le processus de conception et l’identification des risques, en particulier pour les systèmes d'IA qui nécessitent des connaissances spécialisées pour comprendre les résultats.
3.3 Contexte normatif de l'intelligence artificielle en évolution
Bien qu'il existe de nombreuses normes et meilleures pratiques pour aider les organisations à atténuer les risques des logiciels traditionnels, les risques posés par les systèmes d'IA sont à bien des égards uniques. Le règlement européen 2021/0106 sur l’intelligence artificielle a prévu un rôle important pour les normes techniques dans la gouvernance de l'IA. Un certain nombre de normes sont en cours d'élaboration ou ont été récemment publiées afin d'aider les fabricants à se conformer aux exigences énoncées. Les deux normes décrites ci-dessous ont été choisies pour la réalisation de ce projet en raison de leur pertinence directe pour la gestion des risques.
3.3.1 Apports de la norme ISO 23894:2023 sur le management des risques
Les normes internationales ont des définitions sensiblement différentes du mot « risque ». Dans la norme ISO 31000 et les normes associées, le « risque » implique un écart par rapport aux objectifs de l’organisation, qu'il soit positif ou négatif [32]. Dans d'autres normes, telles que l'ISO 14971, le « risque » implique uniquement des résultats négatifs potentiels de l’utilisation d’un produit, tels que des problèmes liés à la sécurité. Cette différence d'orientation peut être source de confusion lorsqu'il s'agit de comprendre et de mettre en œuvre un processus de gestion des risques conforme [33]. Cependant, du fait que ces deux approches prennent en compte le management du risque au niveau de l'organisation et la gestion du risque lié à la sécurité des produits, elles sont complémentaires pour les fabricants de dispositifs médicaux.
Publiée en mars 2023 et se basant sur ISO 31000 comme référence normative, l'ISO/IEC 23894 « Technologies de l’information — Intelligence artificielle — Recommandations relatives au management du risque » propose des lignes directrices pour le management des risques liés au développement et à l'utilisation de l'IA. Les directives proposées par cette norme peuvent être adaptées à n'importe quelle organisation et à son contexte commercial. Il fournit également des conseils sur la manière dont les organisations peuvent intégrer le management des risques dans leurs activités et fonctions commerciales axées sur l'IA [33].
Les principaux avantages de l'ISO 23894 pour les fabricants de dispositifs médicaux utilisant l’IA se trouve dans ses annexes, qui incluent des exemples concrets de mise en œuvre et d'intégration efficaces de la gestion des risques tout au long du cycle de vie du développement de l'IA. Ils fournissent également des informations détaillées sur les sources de risques spécifiques à l'IA [33].
Dans l'ensemble, l'utilisation de l'ISO 23894 peut aider les parties prenantes à développer des stratégies efficaces de gestion des risques pour l'IA dans les dispositifs médicaux, en promouvant le déploiement sûr et responsable de ces technologies pour maintenir la confiance dans leur utilisation.
3.3.2 ISO 42001 : Intelligence artificielle — Système de management
Parallèlement à diverses normes abordant les principaux défis de l'IA, le comité ISO pour l'intelligence artificielle élabore actuellement une norme pour l'IA avec la désignation ISO 42001 [34]. Elle sera une norme de « système de gestion », qui fournit un cadre de bonnes pratiques et décrit les processus qu'une organisation peut suivre pour atteindre ses objectifs. L'ISO 42001 met l'accent sur l'intégration d'un système de gestion de l'IA dans les structures existantes de l'organisation afin de favoriser le développement de systèmes d'IA dignes de confiance. De tels systèmes sont capables de répondre aux attentes des parties prenantes de manière fiable et ont des caractéristiques éprouvées telles que la sécurité, la confidentialité, la sûreté et la transparence.
Dans l'annexe B de la norme, qui traite des directives de mise en œuvre des contrôles de l'IA, il est fait mention de mesures de contrôle des risques spécifiques concernant l'IA. En particulier, il est obligatoire que la documentation des données utilisées dans l'organisation inclue le processus de labellisation des données pour la formation et les tests [35].
En termes d'évaluation de l'impact des systèmes d'IA sur les groupes et les individus, la norme mentionne plusieurs domaines tels que l'équité, la responsabilité, la fiabilité, la transparence, l'explicabilité, l'accessibilité et la sécurité. La norme couvre également les effets communs aux systèmes logiciels en général, tels que l'environnement, la désinformation potentielle et les éventuels problèmes de sécurité et de santé [35].
L'annexe C est particulièrement pertinente pour la gestion des risques, car elle traite des objectifs organisationnels et des sources de risques liés à l'IA.
Dans son ensemble, l'ISO 42001 propose aux organisations des conseils sur les normes et les processus nécessaires pour déployer des systèmes d'IA fiables. Cette norme sera sûrement pertinente à mettre en place par tous les développeurs de systèmes d'IA à haut risque, y compris les systèmes intégrés aux dispositifs médicaux.
4. Proposition de méthode de travail pour la maîtrise des risques
Dans le cadre de ce stage, j'ai pu travailler dans un cadre réglementaire et normatif dense et en pleine expansion, mais encore défaillant sur plusieurs fronts. Comme détaillé dans le chapitre précédent, les efforts pour réglementer et harmoniser les bonnes pratiques relatives à l'IA sont en cours. Cependant, il est désormais impératif pour les entreprises développant ces technologies de bien planifier les procédures et les décisions concernant leur conception, leur développement et leurs utilisations prévues afin d'éviter de prendre du retard dans un avenir qui approche à grands pas.
Dans ce chapitre, je propose des stratégies de management et de gestion des risques qui incitent les organisations et leurs équipes qui conçoivent, développent et déploient les dispositifs médicaux intégrant l'IA à réfléchir de manière plus critique aux impacts négatifs et positifs potentiels. Ces démarches contribueront à renforcer la fiabilité et, à son tour, à cultiver la confiance du public dans l’utilisation de ces produits.
4.1. Processus à mettre en œuvre pour la maitrise des risques
Pour engager la confiance des usagers finaux et acheteurs, atteindre les objectifs de croissance, rester compétitif et protéger la valeur des dispositifs médicaux utilisant l’IA, il est crucial de mettre en place des mesures de management et de gestion des risques. Selon un groupe de travail sur la réglementation de l'IA dans la santé mené par l'Organisation mondiale de la santé (OMS), la gestion des risques doit être basée sur une approche qui prend en compte toutes les phases de la vie d'un système d'IA et qui traite les risques associés, tels que les vulnérabilités en matière de cybersécurité, le sous-ajustement (« underfitting » en anglais), et les biais [36].
Pour créer un produit digne de la confiance de ses utilisateurs, il faut que cette approche par les risques intègre tous les niveaux de l'organisation, de sa direction aux équipes développant le produit, ainsi que les parties prenantes externes. Basée sur les normes et règlements décrits ci-dessus, je propose un processus qui s’articule autour de la gestion des risques des systèmes d'IA, y compris les menaces pour les organisations qui les développent [Figure 10]. Cette pratique est encadrée par le processus de pilotage ainsi que les interactions avec les parties prenantes, deux pratiques complémentaires et indispensables pour répondre aux objectifs de responsabilité et de transparence.
Alors que le processus de gestion des risques peut se dérouler séparément du système de management de la qualité et se concentrer uniquement sur les risques de sécurité de leur produit pour les utilisateurs [28], le processus proposé par ce mémoire intègre également les risques pour l'organisation et ses parties prenantes. Cette approche globale est particulièrement pertinente pour les développeurs de dispositifs médicaux utilisant l'IA en raison de l’importance de l’engagement de la confiance du public et des utilisateurs pour la survie de l'entreprise.
Figure 10. Proposition de processus à mettre en œuvre pour la maîtrise des risques (source : auteure).
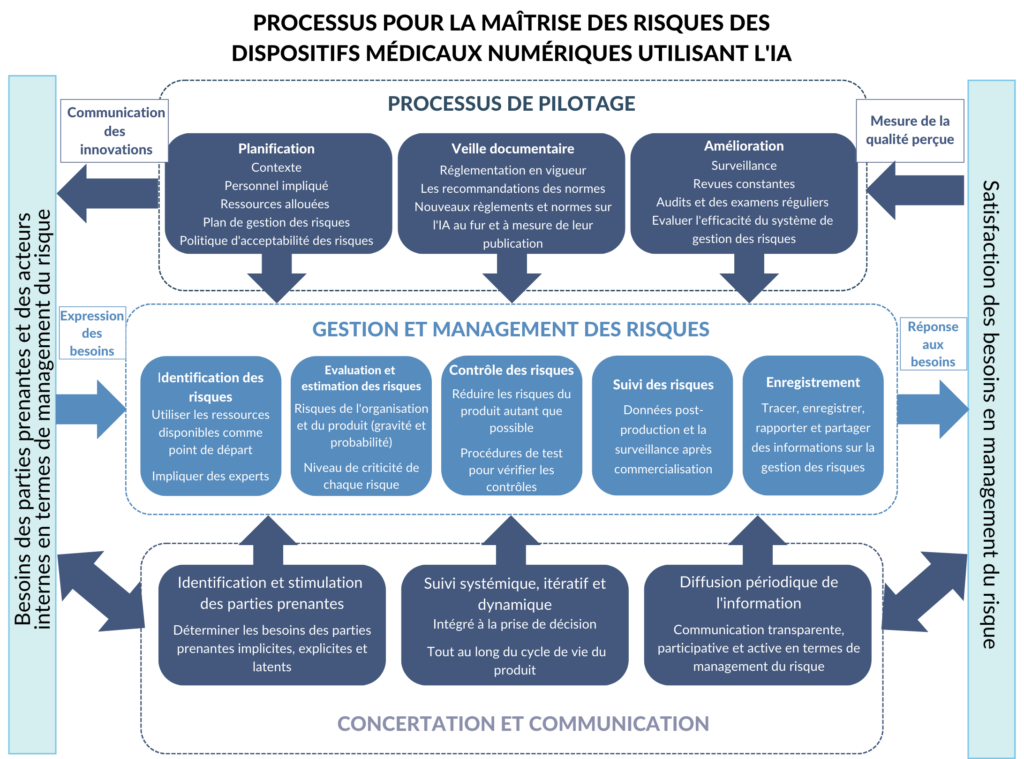
4.1.1 La gestion et le management des risques
Au cœur des efforts de gestion des risques, cette partie du processus repose sur la mise en place de procédures systématiques d'identification, de traitement, d'évaluation et de suivi des risques. Les procédures de test représentent également une partie essentielle. Ces activités doivent être soigneusement documentées dans un format qui facilite la traçabilité et l'examen, y compris lors des audits internes et externes. Un exemple de format est une feuille de calcul qui permet de trier les données par phase de développement, type de risque et/ou composant logiciel.
Pour mettre en place ce système, il est indispensable de collaborer avec l'ensemble du personnel concerné pour s'assurer que les mesures prises sont compatibles avec leur façon de travailler. Ces mesures devraient être intégrées dans les processus existants autant que possible. Par exemple, un système de tickets utilisé par l'équipe de développeurs peut être adapté pour inclure des informations sur la gestion des risques sur chaque ticket.
Identification des risques
Afin de traiter autant de risques prévisibles que possible, un processus d'identification systématique déclenché tôt dans le développement et poursuivi au cours du cycle de vie du produit est essentiel. L’identification précoce des risques permet de définir les besoins de l'utilisateur du produit et les données d’entrée de conception. Cela garantit que les activités de contrôle de conception et de gestion des risques sont synchronisées, favorisant ainsi une conception à sécurité intrinsèque.
Comme pour d’autres dispositifs médicaux, l’identification des risques doit commencer avec l’identification des utilisateurs, des utilisations prévues et des mésusages raisonnablement prévisibles du dispositif [29]. Ensuite, les caractéristiques relatives à la sécurité sont répertoriées [29], tels que la qualité et pertinence des données utilisées pour former le système, l’autonomie du système (est-ce qu’un humain vérifie les résultats ?) et les risques résultants des mesures de sécurité elles-mêmes.
Étant donné que les principes de base de la gestion des risques sont similaires pour l'IA et d'autres technologies, les techniques et méthodes existantes telles que les taxonomies des risques, les bases de données d'incidents et l'analyse de scenarii peuvent être utilisées [37]. En revanche, en raison de la complexité et de la nouveauté de nombreux algorithmes d'IA, cette démarche devrait s’appuyer sur l’expertise les membres de l'équipe impliqués dans le développement du produit.
Selon le National Institute for Standards and Technology (NIST) aux États-Unis, une attention particulière doit être portée à certaines parties sensibles de la gestion des risques pour les systèmes d'IA, y compris [17] :
- Difficulté de compréhension : des risques peuvent survenir de la nature opaque des systèmes d'IA (explicabilité ou interprétabilité limitée), du manque de transparence ou de documentation dans le développement et le déploiement, ou des incertitudes inhérentes aux systèmes d'IA.
- Risques au cours du cycle de vie de l'IA : L'évaluation des risques à différentes étapes du cycle de vie de l'IA peut produire des résultats divers, car certains risques peuvent s'amplifier à mesure que les systèmes d'IA évoluent. De plus, différents participants impliqués dans le cycle de vie de l'IA peuvent avoir des perspectives distinctes sur le risque. Par exemple, un développeur d’un logiciel incorporant l'IA, tels que des modèles préformés, peut voir les risques différemment par rapport à une personne responsable de la mise en œuvre du même modèle dans un scénario spécifique. Ces personnes peuvent ne pas pleinement reconnaître les risques uniques associés à leurs cas d'utilisation particuliers, qui peuvent différer de ceux identifiés par le développeur d'origine.
- Risque dans des contextes réels : bien que la mesure des risques liés à l'IA dans un environnement contrôlé puisse fournir des informations importantes avant le déploiement, ces mesures peuvent différer des risques qui émergent dans des contextes opérationnels réels.
Afin de faciliter le processus d'identification des risques, des listes de sources de risques proposées par les normes pertinentes, telles que l’ISO 24971 et l’ISO 23894 [29], [33], peuvent être utiles. Bien que ces listes ne puissent pas être considérées comme exhaustives, elles peuvent constituer un point de départ pour des discussions avec des personnes spécialisées dans une technologie spécifique. Pour fournir une référence supplémentaire, une cartographie des sources de risque basée sur des normes pertinentes est proposée à la fin de cette section.
Il est important de noter que l’identification des risques doit être mise à jour régulièrement avec les informations résultant de la surveillance post-commercialisation [15], [22].
Estimation et évaluation des risques
Pour les risques liés à la sécurité d'un dispositif médical, l'estimation du risque fait référence à l'estimation de la probabilité d'occurrence d'un dommage et de la gravité de ce dommage [28]. L’évaluation des risques désigne la détermination du caractère acceptable ou non d'un risque [22].
Selon le projet de loi sur l'IA, l'estimation et l'évaluation des risques devraient se concentrer sur les risques susceptibles d'apparaître lorsque le système d'IA à haut risque est utilisé conformément à sa destination et dans des conditions de mauvaise utilisation. Pour préparer cette activité, les fournisseurs doivent utiliser les informations identifiées lors de la première étape, y compris les utilisateurs potentiels, les utilisations prévues et les mésusages raisonnablement prévisibles [22]. Ces derniers peuvent être identifiés grâce à des études d'aptitude à l’utilisation, des retours d'experts ainsi des données historiques du dispositif et des dispositifs similaires.
Traitement des risques
L'annexe I du règlement 2017/745 relatif aux dispositifs médicaux ainsi que la proposition de règlement sur l’IA fournissent l'ordre de priorité que les fabricants doivent prendre en compte lors de la sélection des solutions les plus appropriées [15], [22] :
- Éliminer ou réduire les risques autant que possible grâce à une conception et une fabrication sûre ;
- Mesures de protection adéquates pour les risques qui ne peuvent pas être éliminés (par exemple, alarmes) ;
- Fournir des informations ou une formation des utilisateurs pour la sécurité et divulguer tout risque résiduel.
Le fabricant doit documenter les risques résiduels, en tenant pleinement compte des risques liés au déploiement du produit d'IA et en informant les utilisateurs finaux des impacts négatifs potentiels de l'interaction avec le système [17]. Des stratégies pour informer les utilisateurs des risques résiduels pourraient comprendre des manuels d'utilisation et des instructions qui traitent explicitement des risques résiduels et fournissent des consignes sur la manière d'atténuer ces risques. Un autre exemple est des notifications d’avertissement qui sont facilement visibles et compréhensibles pour les utilisateurs.
Selon le RDM, des mesures doivent être prises pour réduire tous les risques identifiés, pas seulement ceux initialement jugés inacceptables [15]. Il s'agit d'une différence importante avec la loi sur l'IA, qui précise qu'il suffit de documenter les risques acceptables sans prendre de mesures pour les traiter [22].
Procédures de test
Comme énoncée précédemment, la proposition de règlement sur l’IA exige que des procédures de test soient utilisées pour vérifier l’efficacité des mesures de contrôle de risque. Le règlement précise que ces essais doivent avoir lieu le cas échéant, à tout moment du processus de développement et, en tout état de cause, avant la mise sur le marché [22].
Le paragraphe 7 précise que les fournisseurs doivent tester leurs systèmes d'IA à haut risque par rapport à des métriques et des seuils probabilistes préalablement définis qui sont appropriés à l'objectif visé par le système. Les « seuils probabilistes » représentent un type particulier de métrique évaluant une propriété sur une échelle probabiliste avec un ou plusieurs seuils prédéfinis [22].
En raison des applications variées des technologies d’IA et du manque de pratiques établies, aucune ligne directrice universelle ne peut être fournie concernant la sélection des mesures ou des seuils à utiliser. Par conséquent, les fournisseurs doivent naviguer dans l'incertitude et reconnaître que les mesures utilisées précédemment peuvent ne pas être applicables à l'avenir. C'est vraisemblablement la raison pour laquelle la norme parle de « métriques préalablement définies » [37].
Suivi des risques
D’après le RDM, la gestion des risques doit être mis à jour régulièrement en fonction des informations de production et surveillance après commercialisation. Ces dernières devront être utilisées pour identifier la fréquence des dangers et réévaluer ainsi le risque global, le rapport bénéfice/risque et le caractère acceptable du risque [15].
Pour les dispositifs médicaux embarquant l'IA, un système de collecte et d'analyse des données sur les événements indésirables, les plaintes et les commentaires des utilisateurs, des professionnels de la santé et des autres parties prenantes doit être mis en place. Ce système devrait inclure un processus et des canaux de traitement des réclamations pour capturer et évaluer les informations relatives aux performances du dispositif et aux risques. Les performances des algorithmes d'IA dans des contextes cliniques réels doivent être surveillées et évaluées en permanence pour vérifier le bon fonctionnement des algorithmes et identifier tout écart par rapport au comportement attendu. Par exemple, les différences entre les données utilisées dans le développement d'un algorithme et celles appliquées dans le monde réel peuvent entraîner une dérive des données (« data drift » en anglais) [38].
Enregistrement et consultation
L'organisation devrait établir, enregistrer et maintenir un système de collecte et de vérification des informations sur le produit pendant et après le déploiement. Selon la norme ISO 23894, ses informations doivent ensuite être évaluées en rapport à la fiabilité du système d'IA [33].
En particulier, l'organisme doit évaluer s'il existe des risques précédemment non détectés ou si des risques précédemment identifiés ne sont plus acceptables. Ces activités devraient être enregistrées afin d’assurer la traçabilité des facteurs tels que la méthodologie, le personnel impliqué et si les objectifs ont été atteints ou non.
4.1.2 Processus de pilotage
Une gestion efficace des risques est réalisée grâce à un engagement organisationnel aux niveaux supérieurs et peut nécessiter un changement culturel au sein d'une organisation. Les développeurs de dispositifs médicaux intégrant l'IA doivent établir et maintenir les rôles, les responsabilités et la culture appropriés pour que la gestion des risques soit efficace.
Les petites et moyennes entreprises peuvent être confrontées à des défis supplémentaires en fonction de leurs capacités et de leurs ressources [17].
4.1.3 Processus de communication et de concertation
Ce processus comprend l'identification et l'engagement des parties prenantes, la promotion d'une surveillance systémique, itérative et dynamique des risques et la diffusion périodique d'informations. La communication et la consultation servent à aider les parties prenantes à comprendre les risques, les principes de prise de décision ainsi que les raisons pour lesquelles certaines actions sont nécessaires [32].
4.2 Cartographie des risques associés à l’intelligence artificielle
Les entreprises commercialisant des dispositifs médicaux intégrant l’IA doivent identifier et tenir compte des différentes sources de risque au cours du cycle de vie du dispositif. La catégorisation de ces risques à l'aide d'une approche hiérarchique peut aider les organisations à mieux les reconnaître et les gérer.
Selon une étude réalisée par le NIST et basée sur des documents de référence comprenant la proposition du règlement européen sur l’IA, il existe trois catégories principales de sources de risques liées aux systèmes d'IA [17] :
- Caractéristiques techniques de conception : cette catégorie comprend tous les facteurs qui sont sous le contrôle direct des développeurs. Les exemples incluent l'exactitude et les mesures connexes telles que la précision et les taux de faux positifs et de faux négatifs. Un autre exemple est les sources d'erreur statistique qui pourraient être mesurées en appliquant les algorithmes en développement. Enfin, les données générées à partir d'expériences conçues pour évaluer les performances du système entrent également dans cette catégorie et peuvent inclure des tests d'hypothèses causales, des évaluations de la robustesse aux attaques, etc.
- Attributs sociotechniques : Il s'agit de de la perception des représentations de modèles, y compris si le résultat fourni est suffisant pour évaluer la conformité (transparence), si les opérations du modèle peuvent être facilement comprises (explicabilité) et si elles fournissent une sortie qui peut être utilisé pour prendre une décision significative (interprétabilité). En général, tout jugement ou l'évaluation d'un système d'IA, ou de sa sortie, qui est fait par un humain entre dans cette catégorie. Cette catégorie comprend également les biais humains, telles que l’effet expérimentateur et les préjugés sociaux [18]. De plus, les efforts d'identification des risques doivent tenir compte du fait que les humains peuvent supposer que les systèmes d'IA fonctionnent bien ou sont plus objectifs que les humains dans tous les contextes [33].
- Politiques et principes directeurs : Cette catégorie fait référence à des valeurs sociétales plus larges, telles que la vie privée, la responsabilité, l'équité et la justice. Ces facteurs sont difficiles à mesurer de manière cohérente dans tous les domaines en raison de leur dépendance au contexte.
Afin de créer la cartographie des risques, ce cadre a été combiné avec les éléments suivants proposés dans l’annexe B de la norme ISO 23894 [Figure 11] [33] :
- Complexité de l'environnement : La complexité de l'environnement d'un système d'IA détermine la gamme de situations potentielles que le système pourrait rencontrer dans son contexte opérationnel. Au cours du processus de conception et de développement, il est essentiel d'identifier les situations pertinentes que le système est censé gérer afin que les données de formation et de test couvrent ces situations. En cas de compréhension partielle en raison d'une grande complexité, on ne peut pas supposer que toutes les situations pertinentes aient été prises en compte. Ce niveau d'incertitude est une source de risque.
- Niveau d’autonomie : Les systèmes d'IA peuvent fonctionner avec différents niveaux d'automatisation, allant du contrôle total par un opérateur humain à l'automatisation complète. Selon le cas d'utilisation, les décisions automatisées peuvent avoir un effet sur la sûreté, l'équité ou la sécurité.
Figure 11. Catégories de sources de risques liés à l'intelligence artificielle (source : auteure).
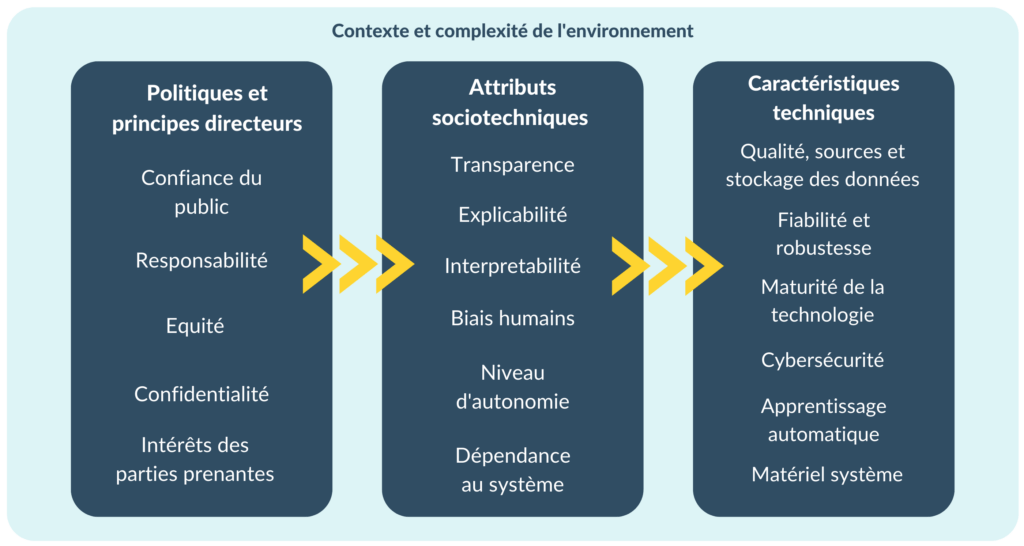
Certains risques liés aux systèmes d'IA sont communs à d'autres types de logiciels, par exemple :
- les problèmes de confidentialité liés à l'utilisation de données pour former des systèmes d'IA ;
- les implications énergétiques et environnementales associées aux demandes informatiques en ressources ;
- les problèmes de sécurité liés à la confidentialité, l'intégrité et la disponibilité du système et de ses données de formation et de sortie ;
- et la sécurité générale du logiciel et du matériel sous-jacents pour les systèmes d'IA [17].
Tous les participants engagés dans le cycle de vie de l'IA portent une responsabilité collective dans la conception, le développement et le déploiement d'un produit fiable qui correspond à son objectif. Compte tenu de la complexité de nombreux systèmes d'IA, il est essentiel d'impliquer les experts qui développent ces technologies dans la gestion des risques, en particulier dans l'identification et l'atténuation des risques. L'une des principales missions de ce stage était donc centrée sur la collaboration interdisciplinaire avec les équipes IT et R&D afin de développer des stratégies efficaces et globales de gestion des risques.
En participant au processus de gestion des risques du DM, les développeurs peuvent contribuer aux décisions concernant les risques liés au logiciel. Ces décisions peuvent inclure, mais sans s'y limiter :
- la fourniture de ressources matérielles adéquates pour prendre en charge le logiciel ;
- le cloisonnement des fonctions entre matériel et logiciel ;
- l'utilisation prévue de l'ensemble du DM et l'utilisation prévue des interfaces utilisateur du logiciel ;
- l'évitement des logiciels inutilement complexes [39].
4.3 Support de formation à la gestion des risques et QCM destinés aux équipes internes
Afin de faciliter l’implication des équipes internes dans la gestion des risques, une formation sur l’introduction à la gestion des risques a été réalisée [Figure 12].
Figure 12. Extrait du support de formation sur la gestion des risques (source : auteure).
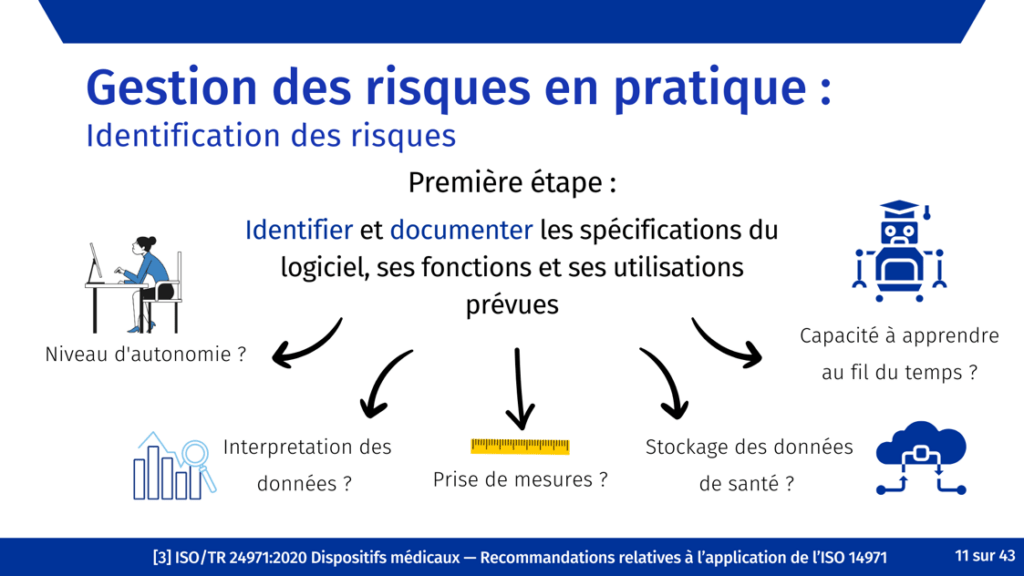
Le but de cette formation était de s'assurer que tous les membres de l'équipe disposaient des informations de base nécessaires pour participer à la gestion des risques, y compris l'identification et l'évaluation des risques.
Le support de cette formation et le formulaire de questions à choix multiples (QCM) qui sert à tester la compréhension de l'équipe interne sont disponibles en téléchargement. Le QCM a été créé en prenant en compte les informations les plus susceptibles d'être source de confusion pour les novices en la matière, comme la nécessité de réduire tous les risques, même ceux qui sont a priori négligeables [Figure 13].
Figure 13. Extrait du QCM accompagnant la formation sur la gestion des risques (source : auteure).
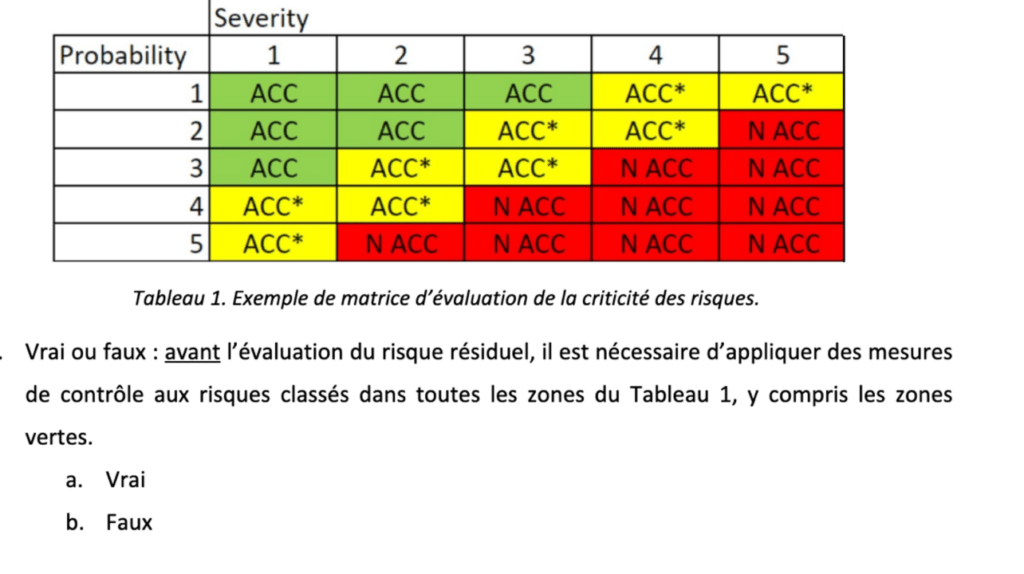
La complétude du QCM peut servir de preuve de la formation continue du personnel lors des audits, par exemple pour la certification ISO 13485 [27].
5. Retour d’expérience en tant que chargée des affaires réglementaires au sein de l’entreprise ExactCure
À la suite d’une première expérience en affaires réglementaires pour une société pharmaceutique américaine, j'étais intriguée par les enjeux liés aux évolutions de la réglementation en Europe, notamment dans le monde des dispositifs médicaux. J'ai choisi d'intégrer la deuxième année du master Ingénierie de la santé, parcours dispositifs médicaux et affaires réglementaires à l'Université de Technologie de Compiègne car ce programme apporterait les connaissances et expériences nécessaires pour devenir une professionnelle dans le domaine. Après un projet explorant les enjeux liés aux thérapies numériques, je me suis rapidement intéressée à la réglementation des dispositifs médicaux numériques et à l'intelligence artificielle en particulier.
Le 20 février 2023, j’intégrais l’entreprise ExactCure en tant que stagiaire Chargée des affaires réglementaires à Nice, France. Avant tout, ce stage était l'occasion idéale d'acquérir une expérience pratique dans la réglementation des dispositifs médicaux numériques embarquant l’intelligence artificielle. Étant donné que le produit ExaMed était déjà commercialisé en tant que produit de classe I en vertu de la Directive 93/42/CEE, j’ai également pu contribuer à la transition vers le RDM et le passage à la classe IIa. Ces deux expériences sont des atouts indéniables pour développer les compétences nécessaires d'un professionnel dans le contexte réglementaire actuel des dispositifs médicaux en Europe.
5.1 Missions et livrables réalisés
Afin d'aborder ces enjeux et de proposer des solutions, j'ai utilisé la roue de Deming pour définir une méthode de travail [Figure 14]. En tant que nouvelle venue dans une structure existante, il convenait de commencer par l’étape d’analyse du système existant et d’identification des écarts avec les objectifs de l’entreprise, notamment le renouvellement du marquage CE (« Check »).
Figure 14. Roue de Deming utilisé pour planifier la démarche de réalisation des missions de ce stage (source : auteure).
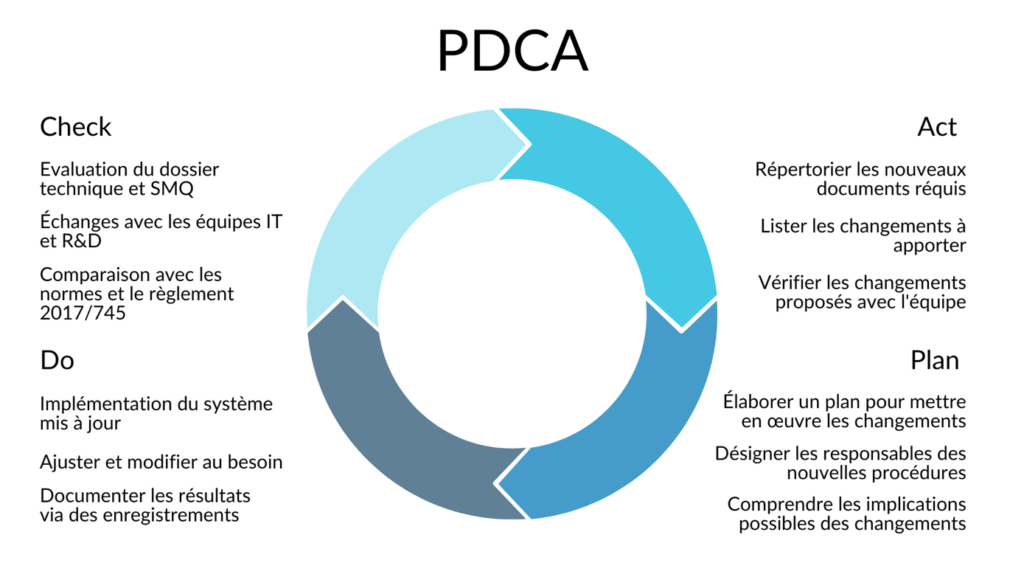
À la suite de l'examen de la documentation existante d'ExactCure et des exigences réglementaires pertinentes pour leur produit, j'ai pu identifier un certain nombre de missions et de documents à créer ou mettre à jour. Ces missions et les livrables associés sont résumés dans Tableau 1.
Tableau 1. Missions et livrables complétés lors de ce stage (source : auteure).
| Mission | Livrable(s) associés |
| Contribuer à l’évolution des dossiers techniques du dispositif médical logiciel d’ExactCure pour une transition vers le nouveau règlement MDR | Processus de gestion des risques Plan de gestion des risques Dossier de gestion des risques Politique d’acceptabilité des risques |
| Participer aux activités d’évaluation clinique | Plan de surveillance clinique après commercialisation |
| Assurer le maintien de la veille réglementaire, la surveillance post commercialisation et la matériovigilance | Plan de surveillance après commercialisation Rapport périodique actualisé de sécurité |
| Réaliser les tests fonctionnels du DM ExactCure selon les scénarios de tests | Rapports de tests |
En travaillant sur l'ensemble de ces livrables, j'ai pu assurer la synchronisation entre les procédures et les plans de gestion des risques, de surveillance après commercialisation et de surveillance clinique après commercialisation. Chaque rapport résultant tient compte des données et des conclusions des autres processus.
Ces mises à jour du dossier technique aideront ExactCure à atteindre les performances prévues en concevant et en développant son produit selon ces procédures. Cela favorisera la sécurité d’ExaMed et garantira que les avantages cliniques l'emportent sur les risques individuels et cumulés.
5.2 Bilan personnel et professionnel du stage
Au cours de ce stage, j'ai acquis un éventail de compétences précieuses. Tout d'abord, j'ai développé une compréhension approfondie du cadre réglementaire régissant les dispositifs médicaux embarquant l'IA, en particulier les exigences en matière de gestion des risques. Cela impliquait d'étudier les textes pertinents, telles que les réglementations et les normes détaillées au chapitre 2. Je suis devenue compétente dans la conduite d'évaluations et l'analyse des risques associés aux technologies d'IA, en tenant compte de facteurs tels que l'utilisation prévue. De plus, j'ai acquis une expertise dans l'élaboration d'un plan de gestion des risques complet pour assurer un processus de gestion des risques continu et itératif tout au long du cycle de vie de l'appareil. En consacrant du temps aux activités connexes à la gestion des risques, j'ai également pu acquérir de l'expérience dans la création de documents importants pour ce processus, notamment le plan de SAC, de SCAC ainsi que le rapport périodique actualisé de sécurité.
Grâce à une collaboration interdisciplinaire avec l'équipe d'ExactCure, comprenant des ingénieurs, des développeurs et la direction de l'entreprise, j'ai développé des compétences en communication efficace sur les questions réglementaires et de qualité. En utilisant ma créativité et mon empathie pour les membres de mon équipe, j'ai pu créer un outil de formation qui facilite la compréhension d'un sujet complexe. En donnant cette formation à l'ensemble de l'entreprise de manière interactive et ouverte, j'ai pu mettre en pratique mes compétences de présentation et mieux comprendre les perspectives de mes collègues.
Afin d'améliorer encore mes compétences dans les affaires réglementaires, il y a des domaines dans lesquels je souhaite acquérir des connaissances supplémentaires. Notamment, je compte approfondir ma compréhension des réglementations internationales sur l’IA et des efforts d'harmonisation. J’aimerais acquérir davantage d’expérience pratique dans la conduite de la surveillance post-commercialisation et l'évaluation des données de performance réelles des dispositifs médicaux embarquant l’IA. De plus, je cherche à renforcer ma compréhension des considérations éthiques de l'IA, telles que les biais algorithmiques et les problèmes de confidentialité, afin d'assurer une mise en œuvre responsable et transparente de l'IA dans le domaine médical. Enfin, je m’engage à me tenir au courant des avancées de la technologie de l'IA et de ses implications pour les affaires réglementaires.
Dans l'ensemble, la combinaison de ce stage et de ma formation théorique m'a permis d'acquérir les compétences et les connaissances nécessaires pour la prochaine étape de ma carrière. Le diplôme résultant de ces efforts atteste de ma volonté de relever les défis uniques des affaires réglementaires, en particulier dans le domaine des dispositifs médicaux numériques embarquant l’IA.
6. Conclusion
L'IA offre des opportunités d'innovation sans précédent dans le domaine des dispositifs médicaux. Néanmoins, l’utilisation de ces technologies introduit également de nouveaux risques, tels que les erreurs potentielles, les risques de biais et inégalités accrues en matière de santé, la manque de transparence et confiance, et la vulnérabilité au piratage et à la confidentialité [40].
Sans contrôles des risques appropriées, les systèmes d'IA peuvent amplifier des résultats inéquitables ou indésirables pour les individus, les organisations, les communautés et la société dans son ensemble. Un manque de confiance résultant comporte des conséquences significatives pour les organisations qui développement ces technologies ainsi que pour leurs parties prenantes. En revanche, avec des contrôles appropriés, les systèmes d'IA peuvent apporter des bénéfices significatifs, créer de la valeur et améliorer la qualité des soins.
Dans le cadre de ce stage, une revue de la réglementation et des normes clés a été entreprise afin d'inventorier les exigences en matière de gestion des risques de l'intelligence artificielle pour les dispositifs médicaux. Grâce à une expérience pratique chez ExactCure, j'ai pu développer une approche systématique pour mettre en œuvre un système de gestion des risques qui répond à ces exigences. Une représentation visuelle de ce processus, une cartographie des risques et une formation destinée aux équipes internes ont été élaborés pour aider d'autres entreprises à mettre en œuvre ces stratégies.
À mesure que les technologies d'IA progressent et deviennent de plus en plus répandues dans l'industrie des dispositifs médicaux, il est essentiel pour les fournisseurs de ces technologies de porter une attention particulière aux risques connus et émergents. Une approche systématique basée sur les risques devrait être appliquée à l'ensemble de l'organisation, y compris la direction et les développeurs travaillant sur ces technologies. En intégrant la gestion des risques à chaque niveau de l'organisation et à chaque étape de conception et de développement du produit, les entreprises commercialisant les dispositifs médicaux utilisant l’IA peuvent assurer la pérennité de leur technologie et protéger le succès de l’entreprise ainsi que la sécurité des patients.