IDS251 - Mise sur le marché des dispositifs médicaux en Europe et aux Etats-Unis : Guide d’accompagnement pour les dispositifs médicaux logiciels de Classe II
DOI mémoire
https://doi.org/10.34746/ids251Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- BOURMDANE Manal : manalbouramdane7@gmail.com
- DZEGANG Kevine : kevinedzegang@gmail.com
- KADIRI Haitam : haitam.kadiri07@gmail.com
- SLIKA Hoda : slikahoda2001@gmail.com
- YANZE Charles : charlesyanze01@gmail.com
Citation
BOURAMDANE Manal, SLIKA Hoda, DZEGANG Kevine, KADIRI Haitam et YANZE Charles, "Mise sur le marché des dispositifs médicaux en Europe et aux États-Unis : Guide d’accompagnement pour les dispositifs médicaux logiciels de Classe II", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, parcours Dispositif Médical et Affaires Règlementaires, Mémoire de Projet, Janvier 2025, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids251, DOI : https://doi.org/10.34746/ids251.
Résumé
Les dispositifs médicaux (DM) logiciels de classe II jouent un rôle clé dans l’innovation médicale, mais leur mise en conformité reste un défi en raison de leur niveau de risque modéré nécessitant des contrôles stricts. Une étude a été menée pour comparer les exigences réglementaires en Europe (Règlement (UE) 2017/745) et aux États-Unis (Titre 21 du Code de Règlementation Fédérale ou CFR), en analysant les disparités procédurales, les délais et les coûts liés à leur mise sur le marché.
Des recherches documentaires et des interviews avec des experts en qualité et affaires réglementaires ont permis d’identifier les principaux défis : documentation technique, évaluation clinique et traçabilité. En réponse, un guide interactif a été conçu, détaillant les étapes clés et les normes actualisées pour simplifier l’accès aux marchés européen et américain.
Mots-clés : Dispositifs médicaux logiciels, Classe II, Règlement (UE) 2017/745, Code of Federal Regulations - Title 21, FDA, Guide interactif.
Abstract
Class II medical software devices are pivotal in medical innovation but present compliance challenges due to their moderate risk requiring stringent controls. A study was conducted to compare regulatory requirements in Europe (Regulation (EU) 2017/745) and the United States (Code of Federal Regulation or CFR, Title 21), focusing on procedural disparities, timelines, and costs for market entry.
Documentary research and interviews with Quality and Regulatory Affairs (QARA) experts identified key challenges : technical documentation, clinical evaluation, and traceability. In response, an interactive guide was developed, outlining key steps and updated standards to facilitate access to European and US markets.
Keywords : Medical software devices, Class II, Regulation (EU) 2017/745, Code of Federal Regulations - Title 21, FDA, Interactive guide.
Téléchargements


Remerciements
Avant tout, nous souhaitons exprimer notre profonde gratitude à toutes les personnes qui ont, de près ou de loin, contribué à la réalisation de ce projet.
Nous adressons nos remerciements les plus sincères aux responsables du Master Ingénierie de la Santé de l’Université de Technologie de Compiègne, Madame Isabelle CLAUDE et Monsieur Jean-Matthieu PROT, pour leur engagement, leur accompagnement bienveillant et leurs précieux conseils tout au long de ce semestre. Leur disponibilité et leur soutien constant ont été des sources de motivation et ont joué un rôle clé dans l’avancement de ce travail.
Nous tenons également à exprimer toute notre reconnaissance à Madame Julie FOLLET, notre encadrante de projet, pour son regard critique, ses conseils éclairés et son suivi attentif. Son approche méthodique et son accompagnement patient nous ont guidés à chaque étape et ont fortement contribué à structurer et à enrichir notre travail.
Nous remercions chaleureusement Madame Béatrice KONIG pour sa relecture minutieuse de notre bibliographie et ses remarques constructives, qui ont été d’une grande aide pour améliorer la qualité de notre rapport.
Enfin, nous exprimons notre sincère reconnaissance aux intervenants qui ont participé à nos entretiens et partagé leurs retours sur notre guide. Leur expérience et leurs connaissances ont été d’une grande valeur pour approfondir notre analyse, et nous leur sommes profondément reconnaissants pour le temps qu’ils ont pris pour nous accompagner dans ce projet.
Liste des abréviations
CDRH Portal : Send Medical Device Premarket Submissions and Small Business Determination Requests Online/ Envoi en ligne des demandes d'autorisation de mise sur le marché pour les dispositifs médicaux et des demandes de détermination pour les petites entreprises
CFR : Code of Federal Regulations / Code de Règlementation Fédérale DM : Dispositif médical / Medical devices
eSTAR : Electronic Submission Template for Medical Device 510(k) Submissions / Modèle de soumission électronique pour les soumissions de dispositifs médicaux 510(k)
EUDAMED : European Database on Medical Devices / Base de données européenne sur les dispositifs médicaux
FDA : Food and Drug Administration / Administration des aliments et des médicaments
GUDID : Global Unique Device Identification Database / Base de données mondiale d'identification unique des dispositfs
GS1 : Global Standards 1 / Normes Mondiales 1
HDE : Humanitarian Device Exemption / Exemption relative aux dispositifs humanitaires
HIBCC : Health Industry Business Communications Council / Conseil de Communication pour les Affaires de l'Industrie de la Santé
ICCBBA : International Council for Commonality in Blood Banking Automation / Conseil International pour l’Harmonisation de l’Automatisation des Banques d’échantillons sanguins
IUD : Identifiant Unique des Dispositifs / Unique Device Identifier
MAUDE : Manufacturer and User Facility Device Experience / Expérience du fabricant et de l'installation de l'utilisateur en matière de dispositifs
ON : Organismes Notifiés / Notified bodies
PMA : Premarket approval / Approbation préalable à la mise sur le marché
PME : Petites et Moyennes Entreprises
QMS : Quality Management System / Système de Management de la Qualité
SNITEM : Syndicat National de l'Industrie des Technologies Médicales / National Union of the Medical Technology Industry
Liste des Figures
Figure 1 : Histogramme de la répartition des parts de marché des dispositifs médicaux par aire géographique (en %) [4]
Figure 2 : Impact du Règlement (UE) 2017/745 vu par les entreprises du dispositif médical en Europe selon le rapport du Syndicat National de l'Industrie des Technologies Médicales de décembre 2023 [1]
Figure 3 : Processus de classification des dispositifs médicaux logiciels selon le Règlement (UE) 2017/745 et le Titre 21 du Code de Règlementation Fédérale aux États-Unis
Figure 5 : Synthèse des exigences applicables aux dispositifs médicaux logiciels selon le Règlement (UE) 2017/745
Figure 6 : Documentation technique des dispositifs médicaux logiciels requise par le Règlement (UE) 2017/745
Figure 7 : Modules disponibles dans la base de données européenne sur les dispositifs médicaux EUDAMED
Figure 8 : Différentes procédures d’évaluation des DM (Dispositifs Médicaux) logiciel de classe II
Figure 9 : Exemple d’étiquette avec code IUD [2]
Figure 10 : Logigramme des différentes étapes du projet
Figure 11 : Diagramme de décision : risques identifiés et solutions retenues
Figure 12 : Captures écran des pages d’accueil du guide d’accompagnement pour la mise sur le marché de dispositifs médicaux sous le règlement MDR 2017/745 [IDS 203]
Figure 13 : Aperçu page d’accueil du guide
Figure 14 : Aperçu des étapes de mise sur le marché proposé dans le guide
Figure 15 : Liste des icônes utilisées pour naviguer dans le guide interactif d’accompagnement
Liste des tableaux
Tableau 1 : Délai de notification des incidents graves liés à l’utilisation de dispositifs médicaux commercialisés selon le type d’incident, comme exigé par le règlement (UE) 2017/745
Tableau 2 : Tableau comparatif des exigences réglementaires entre l’Europe et les Etats-Unis
Tableau 3 : Questions et réponses variées recueillies lors des entretiens avec les professionnels.
Tableau 4 : Cahier des charges du guide d’accompagnement pour la mise sur le marché de dispositifs médicaux logiciels en Europe et aux Etats-Unis
Mise sur le marché des dispositifs médicaux en Europe et aux Etats Unis : Guide d’accompagnement pour les dispositifs médicaux logiciels de Classe II
Introduction
En Europe, l’entrée en vigueur du Règlement (UE) 2017/745 en mai 2021 a marqué un tournant dans la mise sur le marché des dispositifs médicaux (DM), avec des exigences strictes pour garantir leur sécurité et leur efficacité. Ce cadre réglementaire impose des normes rigoureuses, notamment en matière d’évaluation clinique, de documentation technique et de surveillance post-commercialisation, et s’appuie sur un système décentralisé d’organismes notifiés responsables de l’évaluation de la conformité.
Aux États-Unis, la Food and Drug Administration (FDA) adopte une approche centralisée, avec des processus d’approbation basés sur une évaluation des risques et une démonstration de l’efficacité des dispositifs avant leur mise sur le marché. Le mécanisme de soumission 510(k), qui permet de prouver l’équivalence d’un nouveau produit avec un dispositif déjà commercialisé, est une voie fréquemment empruntée, souvent moins contraignante que la procédure de mise sur le marché européennes. Cependant, cette flexibilité pose des questions sur l’équilibre entre rapidité d’accès au marché des innovations et sécurité des dispositifs.
Ces écarts entre les réglementations européenne et américaine soulignent des priorités différentes : d'une part, la protection accrue des patients, d'autre part, la volonté de faciliter l'accès aux innovations impactant la santé des patients et les conditions d’exercice des professionnels de santé dans les zones géographiques les plus consommatrices de DM.
Bien que les deux systèmes classifient les DM selon leur niveau de risque, les différents niveaux d’exigences pour leur mise sur le marché entraînent des délais d'approbation variables, des coûts de mise en conformité contrastés, et des stratégies de surveillance post-commercialisation distinctes. La question centrale à laquelle nous répondrons est la suivante : "Comment soutenir les petites entreprises dans la mise sur le marché des dispositifs médicaux logiciels de classe II en Europe et aux Etats-Unis ?"
Pour ce faire, un guide pratique et interactif a été conçu pour simplifier l’appropriation des deux systèmes réglementaires par les fabricants.
Chapitre 1 : Contexte, enjeux et cadre réglementaire des dispositifs médicaux logiciels
1.1 Situation du marché des dispositifs médicaux
Le marché mondial des DM connaît une croissance continue, avec une augmentation moyenne d’environ 5 % par an entre 2016 et 2023, et des projections similaires jusqu’en 2028. Cette progression touche une large gamme de dispositifs, allant des appareils d’imagerie médicale aux équipements de cardiologie et de chirurgie, en passant par d’autres technologies médicales innovantes.
Cette expansion du marché s’explique par plusieurs facteurs. D’une part, les besoins en matière de santé évoluent en raison de l’apparition de nouvelles pathologies, de l’augmentation des maladies chroniques, et de la demande accrue pour des soins personnalisés et accessibles. D’autre part, le vieillissement de la population mondiale entraîne une hausse des besoins en soins de santé spécialisés. Enfin, les avancées technologiques rapides dans le domaine médical, telles que l’intelligence artificielle et les dispositifs connectés, jouent un rôle clé dans le développement de solutions innovantes répondant à ces nouveaux enjeux [1].
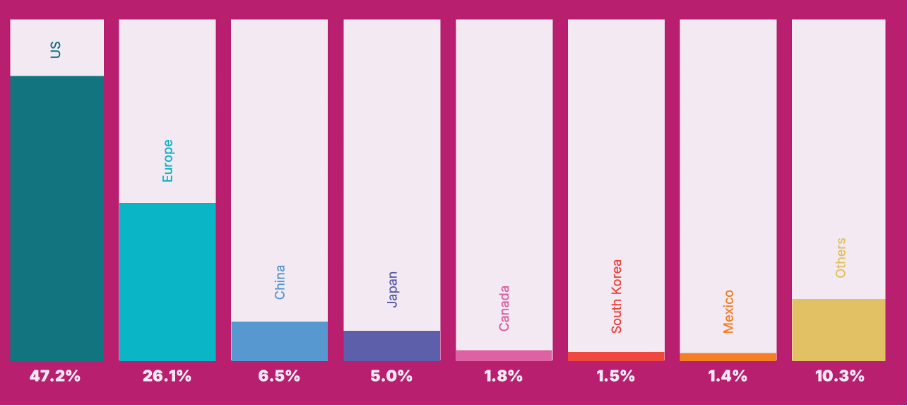
Les États-Unis et l'Europe rassemblent près de 70 % du marché mondial des DM (voir figure 1). En 2022, les États-Unis dominent ce secteur avec un chiffre d'affaires de 160,6 milliards de dollars, et une projection atteignant 225,37 milliards de dollars d'ici 2028. Cette position de leader s'explique par une infrastructure de santé avancée, des investissements conséquents en recherche et développement, ainsi qu'un cadre réglementaire favorable qui stimule l'innovation et la croissance [4].
Avec un chiffre d’affaires de 132,80 milliards de dollars en 2022 et une projection atteignant 170,88 milliards de dollars en 2028, l’Europe se positionne comme le deuxième plus grand marché des DM. Les principaux contributeurs européens à ce marché sont l’Allemagne, la France, le Royaume-Uni, l’Italie et l’Espagne, l’Allemagne se distinguant grâce à sa capacité de production et à ses infrastructures de santé avancées. L'Europe et l'Amérique du Nord appliquent des réglementations strictes pour la mise sur le marché des DM, exigeant une conformité rigoureuse de la part des fabricants afin de garantir la sécurité des patients et l'efficacité des DM, et de renforcer la confiance des des utilisateurs finaux dont les professionnels de santé [4].
Figure 1 : Histogramme de la répartition des parts de marché des dispositifs médicaux par aire géographique (en %) [4]
Avec l'évolution rapide des technologies numériques, les dispositifs médicaux logiciels jouent un rôle clé dans la transformation des soins de santé. La pandémie de COVID-19 a accéléré l’adoption des technologies de santé numérique, transformant le secteur des DM. Les systèmes de santé ont massivement recours à la télémédecine et à la télésurveillance des patients, les consultations en ligne étant passées de moins de 1 % avant 2020 à plus de 40 % au plus fort de la pandémie aux États-Unis, avec une adoption également rapide en Europe (France, Allemagne, Royaume-Uni). Le secteur de la télémédecine et des technologies de télésurveillance des patients a connu une croissance de 20 % en 2020 et devrait poursuivre à un rythme annuel de 12 % jusqu'en 2027. Ces technologies permettent la gestion à distance des maladies chroniques via des dispositifs mesurant notamment la fréquence cardiaque, la saturation en oxygène et la glycémie. Les fabricants investissent donc davantage dans les logiciels, devenus essentiels [5,6]. La mise sur le marché des DM logiciels requiert une stricte conformité aux cadres règlementaires en vigueur, notamment le Règlement (UE) 2017/745 pour l'Europe et les exigences du 21 CFR pour les États-Unis.
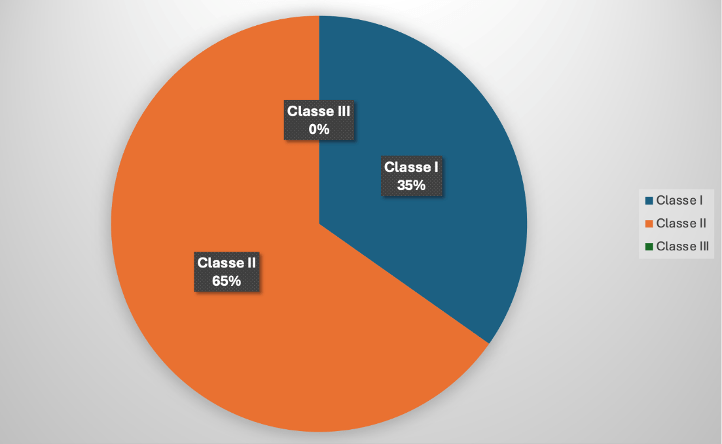
À la suite d’une analyse réalisée le 18 novembre 2024 de la base de données EUDAMED, qui recense les DM commercialisés en Europe, il apparaît que parmi l’ensemble des dispositifs enregistrés, 65 % des logiciels médicaux appartiennent à la classe II (1 088 logiciels enregistrés), 35 % à la classe I (551 logiciels enregistrés), mais aucun à la classe III (voir figure 2). La prédominance des dispositifs de classe II conjuguée à l’absence des DM de classe III peut s’expliquer par l’utilisation des DM de classe II généralement circonscrite à l’aide au diagnostic ou à la télésurveillance sans impliquer de décisions critiques immédiates pour la survie du patient, ces décisions restant sous la responsabilité du médecin, contrairement à celle des DM de classe III associés à des risques très élevés pouvant entraîner la mort directe du patient. La mise sur le marché de ces derniers est conditionnée à la conformité à des exigences réglementaires strictes, ce qui élève les coûts de développement, ce qui peut constituer un frein pour certains fabricants, notamment les petites et moyennes entreprises (PME).
Figure 2 : Répartition des DM logiciels commercialisés en Europe et recensés dans la base de données européenne EUDAMED par classe de risque
Plusieurs facteurs ont été identifiés lors des discussions avec des fabricants, des spécialistes de la réglementation et des acteurs impliqués dans la commercialisation des DM en Europe et aux États-Unis :
- Complexité croissante des dispositifs : Les DM deviennent de plus en plus sophistiqués, intégrant des technologies avancées telles que l'intelligence artificielle, l'Internet des objets (IoT) et la nanotechnologie.
- Adaptation aux nouvelles réglementations : Les règlementations, comme le Règlement (UE) 2017/745 et les directives de la FDA, évoluent pour s'adapter à ces évolutions technologiques.
- Les guides méthodologiques doivent refléter ces changements réglementaires pour aider les fabricants à se conformer aux nouvelles exigences d'évaluation de la sécurité et l'efficacité de ces nouveaux dispositifs.
Un rapport du Syndicat National de l'Industrie des Technologies Médicales (SNITEM) présente les impacts de la réglementation européenne sur les fabricants de dispositifs médicaux (voir figure 3).
Figure 3 : Impact du Règlement (UE) 2017/745 vu par les entreprises du dispositif médical en Europe selon le rapport du Syndicat National de l'Industrie des Technologies Médicales de décembre 2023 de décembre 2023 [1]

Compte tenu de la situation actuelle du marché des logiciels et des évolutions réglementaires, nous nous sommes interrogés sur la question suivante : Comment soutenir les petites entreprises dans la mise sur le marché des dispositifs médicaux logiciels de classe II en Europe et aux Etats-Unis (voir annexe 1) ?
1.2 Définition et classification des dispositif médicaux logiciels
1.2.1 Définition d’un dispositif médical logiciel
Selon le Règlement (UE) 2017/745
Un DM désigne n'importe quel produit (instrument, logiciel, implant, etc) conçu pour être utilisé dans le domaine médical chez l'être humain, dans les buts suivants :
Objectifs médicaux principaux :
- Diagnostic, prévention, ou traitement d'une maladie.
- Contrôle ou compensation d'une blessure ou d'un handicap.
Modifications physiologiques ou anatomiques :
- Investigation, remplacement, ou modification d'une fonction ou structure anatomique ou physiologique.
Examens in vitro :
- Analyse d'échantillons corporels pour fournir des informations médicales.
Produits annexes considérés comme DM :
- Dispositifs de maîtrise ou assistance à la conception.
- Produits destinés au nettoyage, désinfection, ou stérilisation de DM [7].
Selon la section 860.38 du Titre 21 du CFR (Dispositif Médical de Classe II)
Un DM appartient à la classe II si les contrôles généraux ne suffisent pas à fournir une assurance raisonnable de sa sécurité et de son efficacité et s'il existe suffisamment d'informations pour établir des contrôles spéciaux, y compris la promulgation de normes de performance, la surveillance post-commercialisation, des registres de patients, l'élaboration et la diffusion de lignes directrices (y compris des lignes directrices pour la soumission de données cliniques dans les demandes de notification préalable à la mise sur le marché conformément à la section 510(k) du Federal Food, Drug, and Cosmetic Act), des recommandations et d'autres actions appropriées que le commissaire juge nécessaires pour fournir une telle assurance. Pour un dispositif qui est censé être utilisé pour soutenir ou maintenir la vie humaine, le commissaire examine et identifie les contrôles spéciaux, le cas échéant, qui sont nécessaires pour fournir une assurance adéquate de sécurité et d'efficacité, et décrit comment ces contrôles fournissent une telle assurance [8].
1.2.2 Processus de classification d’un dispositif médical
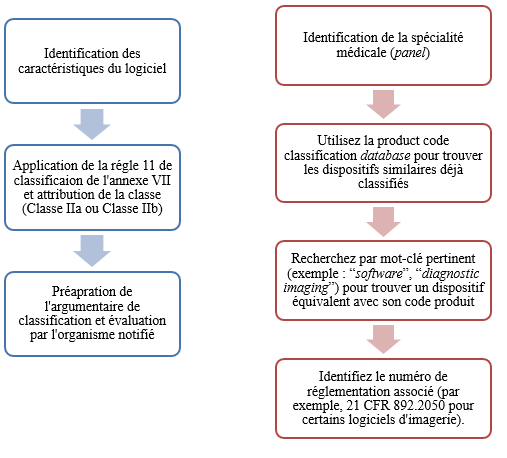
En Europe, les logiciels de Classe II sont classés selon le Règlement (UE) 2017/745, qui évalue les dangers associés à leur utilisation, en particulier selon la règle 11. Aux États-Unis, la Food and Drug Administration (FDA) gère cette classification en se basant sur une approche par les risques liés à l’utilisation et l'usage prévu. La Figure 4 ci-dessous compare ces deux systèmes afin de mettre en évidence les particularités qui s'appliquent aux logiciels de Classe II [7, 8].
Figure 4 : Processus de classification des dispositifs médicaux logiciels selon le Règlement (UE) 2017/745 et le Titre 21 du Code de Règlementation Fédérale aux États-Unis
1.3 Procédures de mise sur le marché des dispositifs médicaux de classe II logiciels en Europe
1.3.1 Les exigences applicables aux dispositifs médicaux logiciels
- Exigences Qualité
Le Règlement (UE) 2017/745 impose à tous les fabricants de DM d'établir, documenter, mettre en œuvre, maintenir, mettre à jour et améliorer en permanence un système de gestion de la qualité avant la mise sur le marché de leur dispositif. La norme ISO 13485 relative au système de management de la qualité est une norme harmonisée dont la conformité présume la conformité aux différentes exigences du Règlement (UE) 2017/745. Cette norme comprend de nombreux concepts clés comme la mise en place et la documentation des procédures pour les différents processus et l’approche par risque du design de la conception à la fabrication. En France, la certification ISO 13485 :2016 est délivrée par un organisme certificateur idéalement accrédité dès lors que le système de gestion de la qualité mis en place par le fabricant répond à toutes les exigences de la norme. La certification ISO 13485 joue donc un rôle crucial dans le cadre de l'audit de conformité au règlement (UE) 2017/745. En plus de garantir la conformité réglementaire, la certification renforce la confiance des utilisateurs, des clients et des partenaires à l'échelle de l'Union Européenne [7, 9].
- Exigences générales en matière de sécurité, conception et informations fournies
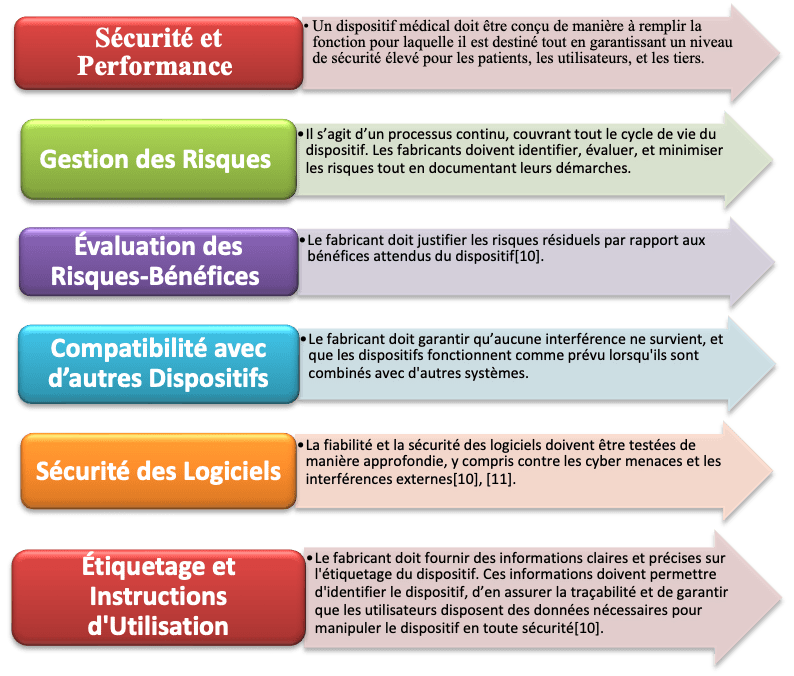
Les dispositifs médicaux doivent respecter un ensemble d'exigences strictes pour garantir leur sécurité, leur efficacité et leur conformité au Règlement (UE) 2017/745. Ces exigences couvrent plusieurs aspects essentiels, notamment la gestion des risques, l'évaluation des bénéfices-risques, la compatibilité avec d'autres dispositifs, et la sécurité des logiciels. Elles imposent également des normes précises concernant l'étiquetage et les instructions d'utilisation. Chaque élément contribue à assurer que les dispositifs médicaux répondent aux attentes des utilisateurs tout en protégeant la santé et la sécurité des patients (voir Figure 5).
Figure 5 : Synthèse des exigences applicables aux dispositifs médicaux logiciels selon le Règlement (UE) 2017/745.
1.3.2 Documentation technique
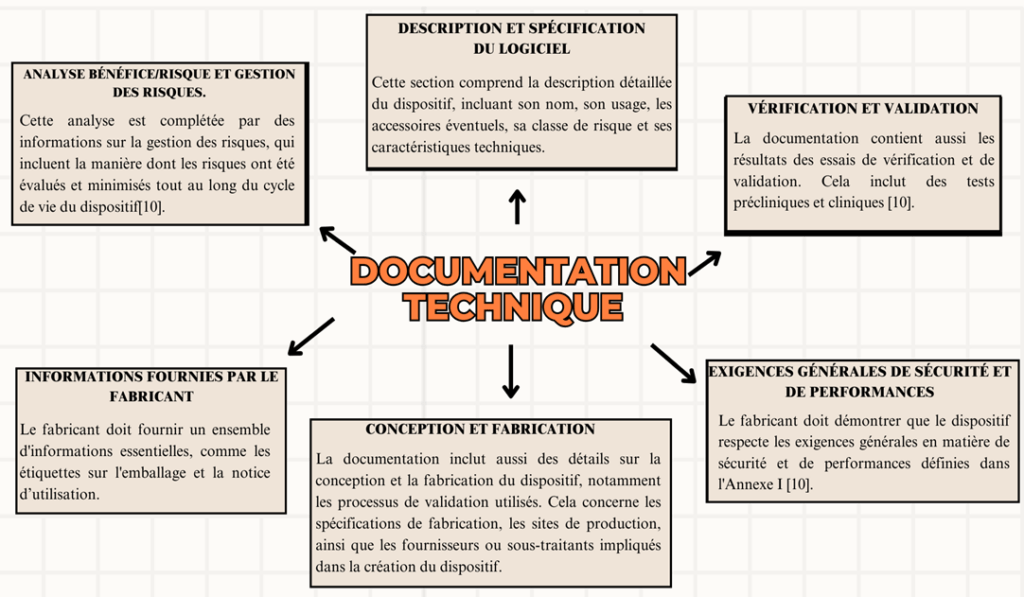
La documentation technique constitue une partie essentielle du processus de conformité des DM, en particulier pour les logiciels médicaux. La figure 6 ci-dessous illustre les différentes composantes clés de la documentation technique, notamment les exigences de sécurité, la description du logiciel, l'analyse des risques, les informations fournies par le fabricant, ainsi que les résultats des vérifications et validations. Chaque élément joue un rôle crucial dans l'assurance de la conformité et de la fiabilité du dispositif tout au long de son cycle de vie.
Figure 6 : Documentation technique des dispositifs médicaux logiciels requise par le Règlement (UE) 2017/745
1.3.3 Identification Unique des Dispositifs
1.3.3.1 Procédure d'obtention
Il s’agit de la dernière étape avant la mise sur le marché, obligatoire au sein des deux systèmes. L’identifiant unique des dispositifs (IUD) est un code numérique ou alphanumérique unique lié à un dispositif médical qui permet l'identification claire et formelle de dispositifs donnés sur le marché. Cet IUD est constitué de :
- UID-ID : c’est un code numérique unique permettant d’accéder aux informations du DM (classe de risque, type de dispositif, etc), il figure sur les certificats et la déclaration de conformité UE.
- UID-IP : c’est un code numérique ou alphanumérique unique identifiant l'unité de production d'un dispositif par le numéro de série, le numéro de lot, l'identifiant de logiciel et la date de fabrication ou d'expiration ou ces deux types de date.
Pour obtenir un IUD, les fabricants doivent s’enregistrer auprès d'une agence d’émission agréée par la Commission européenne et la FDA. Parmi ces agences reconnues, on retrouve notamment GS1 (Global Standards 1), HIBCC (Health Industry Business Communications Council), ICCBBA (International Council for Commonality in Blood Banking Automation) et IFA (International Federation of Associations). Une fois l’agence sélectionnée par le fabricant et l’enregistrement validé, celle-ci fournit au fabricant un code IUD unique, adapté aux spécificités du dispositif en question. Ce code unique doit ensuite être apposé, sous forme de code-barres, sur l’étiquette du dispositif ou sur son emballage [2].
1.3.3.2 L’IUD en Europe
Avec l'entrée en vigueur du Règlement (UE) 2017/745, de nouvelles obligations s'imposent aux fabricants :
- Les fabricants, importateurs, et mandataires (autres que dans le cas de dispositifs sur mesure), doivent enregistrer leurs activités avant la mise sur le marché des dispositifs conformément à l’article 31.
- Les fabricants doivent s’enregistrer ainsi que leurs dispositifs sur la base de données Eudamed (autre que les dispositifs sur mesure), avant leur mise sur le marché. Cette obligation n’est relégable à aucun autre opérateur conformément à l’article 10 et 29 du règlement.
- Le fabricant veille à ce que les informations concernant le dispositif en question visées à l'annexe VI partie B soient correctement soumises dans la base de données européenne EUDAMED (European Database on Medical Devices). Cette base de données est composée de six modules (acteurs, dispositifs, certificats, évaluations cliniques, vigilance et surveillance du marché) interconnectés dont trois sont déjà disponibles (voir figure 7).
Figure 7 : Modules disponibles dans la base de données européenne sur les dispositifs médicaux EUDAMED

La Commission européenne prévoit un déploiement progressif des différents modules d'EUDAMED, avec une mise en œuvre complète attendue au deuxième trimestre 2027. Jusque-là, l'enregistrement sur EUDAMED se fait sur base volontaire des fabricants, tant que la base de données n'est pas totalement opérationnelle. Il sera utilisé lors d’une demande d’évaluation de la conformité auprès d’un Organisme Notifié (ON) [10].
1.3.4 Évaluation ou investigations clinique
1.3.4.1 Les évaluations cliniques
L’évaluation clinique représente une exigence fondamentale imposée par le règlement (UE) 2017/745, s’appliquant à l’ensemble des dispositifs médicaux, quelle que soit leur classe ou leur type. Elle englobe non seulement les dispositifs pour lesquels la démonstration de conformité aux exigences générales de sécurité et de performance, fondée sur des données cliniques, ne serait pas jugée appropriée, mais également ceux énumérés à l'Annexe XVI du même règlement, même si leur destination n’est pas médicale. En d’autres termes, cette évaluation vise à garantir que tous les dispositifs, qu’ils soient à usage médical ou non, respectent les exigences de sécurité et de performance. L’évaluation clinique est un processus continu et dynamique, qui doit être réalisé tout au long du cycle de vie du dispositif. Elle n’est pas ponctuelle, mais évolutive, s’adaptant aux différentes phases de développement et d’utilisation du produit. Elle doit être menée en fonction des caractéristiques propres de chaque dispositif, de ses propriétés spécifiques et de sa destination prévue. Ainsi, il est essentiel que cette évaluation soit adaptée de manière à refléter les risques et les bénéfices pour les utilisateurs, afin de garantir la sécurité et la performance continues du dispositif. Cette approche proactive et flexible assure une surveillance permanente, contribuant ainsi à l'amélioration de la sécurité des patients et de l’efficacité des dispositifs médicaux sur le marché européen [12].
1.3.4.2 Les investigations cliniques
Les investigations cliniques en Europe sont réalisées dans les cas suivants :
- Pour les DM de classe III et les dispositifs implantables, sauf exceptions spécifiques.
- Lorsque l'approche par équivalence n'est pas applicable pour les dispositifs innovants.
- Lorsque les données cliniques existantes sont insuffisantes pour démontrer la conformité aux exigences réglementaires.
Ces investigations doivent être conduites dans des conditions normales d’utilisation du dispositif, conformément à un protocole d’investigation qui rappelle au fabricant les impératifs relatifs à la sécurité, à la performance et à l’évaluation du rapport bénéfice/risque, tels que définis à l’article 62 du Règlement (UE) 2017/745. Ce protocole doit également garantir la protection des droits, de la sécurité, de la dignité et du bien-être des participants. Le promoteur de l’investigation clinique est tenu de soumettre une demande auprès des autorités compétentes du ou des États membres concernés. Cette demande doit être accompagnée d’une documentation détaillée, conformément aux exigences énoncées dans l’annexe XV, chapitre II, du règlement [13].
1.3.5 Évaluation de la conformité et marquage CE
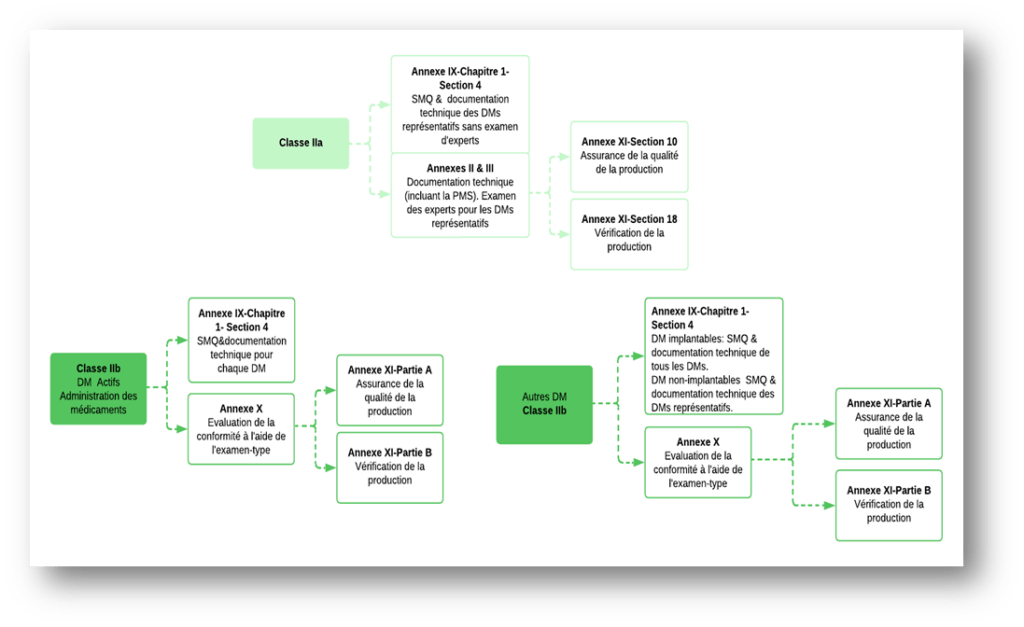
Les fabricants de DM déterminent la procédure d’évaluation de la conformité en Europe en fonction de la classe de risque de leur dispositif, des exigences réglementaires détaillées dans le Règlement (UE) 2017/745 et des ressources nécessaires pour satisfaire ces exigences. Ce choix est guidé par des critères tels que la complexité du dispositif, son usage prévu, et les exigences associées aux annexes réglementaires applicables. Une fois la procédure choisie, ils contactent un Organisme Notifié (ON) pour entamer l’évaluation de conformité. Les annexes et les exigences spécifiques pour chaque classe de DM sont détaillées dans la figure 8, garantissant la sécurité et la performance des dispositifs avant leur mise sur le marché [7].
Figure 8 : Différentes procédures d’évaluation des DM (Dispositifs Médicaux) logiciel de classe II (source auteurs).
1.3.6 Surveillance post-commercialisation et vigilance
1.3.6.1 Surveillance après commercialisation
Le système de surveillance après commercialisation (PMS) consiste à collecter, enregistrer et analyser de manière active et systématique les données sur la qualité, les performances et la sécurité des DM tout au long de leur cycle de vie.
Les informations recueillies proviennent notamment des réclamations, des retours d’expérience des utilisateurs, des données de production et des incidents signalés. Elles servent à réévaluer le rapport bénéfice/risque, à actualiser les informations techniques et cliniques du dispositif, ainsi qu’à identifier des opportunités d’amélioration en matière de sécurité, de performance et de facilité d’utilisation.
Les fabricants documentent et mettent à jour ce système dans un rapport périodique actualisé de sécurité (PSUR) en fonction de la classe de risque du dispositif et le soumettent aux autorités compétentes pour assurer un suivi continu et rigoureux. Pour les dispositifs de classe IIa, le PSUR doit être soumis au moins tous les deux ans, tandis que pour les dispositifs de classe IIb, la soumission est requise au moins tous les ans. Ces rapports permettent aux autorités de s'assurer que la surveillance post-commercialisation est adéquate et que les dispositifs restent conformes aux exigences de sécurité et de performance [7].
1.3.6.2 Vigilance
Les fabricants doivent notifier aux autorités compétentes tout incident grave survenu avec leurs dispositifs sur le marché de l'Union, sauf si ces effets secondaires sont attendus, clairement documentés et déjà inclus dans un rapport de tendances. Ils doivent également signaler toute mesure corrective de sécurité prise, que ce soit sur le marché de l'Union ou dans un pays tiers, si elle concerne un dispositif également disponible dans l'Union (voir tableau 1). Le délai de notification varie en fonction de la gravité de l'incident [7].
Tableau 1 : Délai de notification des incidents graves liés à l’utilisation de dispositifs médicaux commercialisés selon le type d’incident, comme exigé par le règlement (UE) 2017/745
| Fréquence | Deux jours | Dix jours | Quinze jours |
| Type d’incident survenu | Menace grave pour la santé publique | Décès ou de détérioration grave inattendue de l'état de santé d'une personne | Tout incident grave |
1.4 Procédures de mise sur le marché des dispositifs médicaux logiciels de classe II aux Etats-Unis
1.4.1 Exigences applicables aux dispositifs médicaux aux USA
La FDA régule la mise sur le marché des DM aux États-Unis dans le cadre du 21 CFR Part 820 (Quality System Regulation), qui impose des exigences relatives à la conception, la fabrication et la distribution des dispositifs médicaux.Cette réglementation impose les mêmes exigences que celles appliquées à l'international (En Europe) en conformité avec les normes de sécurité, d'efficacité et de qualité. Les DM doivent donc répondre aux mêmes standards rigoureux, garantissant leur fiabilité pour les utilisateurs et les patients[8]. Les fabricants de DM basés aux États-Unis doivent établir et maintenir un système de gestion de la qualité conformément au règlement QSR (Quality System Regulation) pour s'assurer que leurs dispositifs respectent en permanence les exigences et spécifications applicables, conformément à la partie 820 du titre 21 du CFR.Dans le cadre de ses efforts pour harmoniser les exigences du QSR avec la norme internationale ISO 13485 : 2016 adoptée dans de nombreux pays, la FDA a publié le 2 février 2024 une version révisée du règlement dédié au système de management de la qualité (QSRM pour Quality Management System Regulation) des DM qui entrera en vigueur le 2 février 2026. Les fabricants disposent ainsi de deux ans pour adapter leurs systèmes de gestion de la qualité à ces nouvelles dispositions à savoir : harmoniser les processus et procédures internes, apporter les changements appropriés au sein de leur organisation, et mettre à jour la documentation conformément aux exigences du QMSR. Ce nouveau Règlement intègre également les termes et définitions nécessaires à l'application de la norme ISO 13485 propres aux contrôles des dossiers, ainsi que de nouvelles exigences en matière d'étiquetage et emballage des dispositifs médicaux [14].
Cependant La FDA confirme que la seule conformité à la norme ISO 13485 ne satisfait pas pleinement au QMSR. Les Etats-Unis ont intégré le Medical Device Single Audit Program incluant l’Australie, le Brésil, le Canada et le Japon et dont la finalité est de mutualiser l’évaluation des systèmes de management de la qualité des fabricants de dispositifs médicaux à l’échelle internationale [15]. Dans ce programme les fabricants sont audités sur la conformité de leur système de management de la qualité conformément aux exigences règlementaires de tous les pays membres et un rapport d’audit sera transmis à chaque autorité des pays membres. L’audit MDSAP peut dans un cas se substitue à l’audit local lorsque les autorités nationales estiment que l’audit global est suffisant pour garantir la conformité aux exigences, mais il peut aussi dans un autre cas être complété par un audit local si des exigences supplémentaires ou des spécificités nationales doivent être vérifiées [16]. Cela permet un équilibre entre la réduction de la duplication des efforts et la garantie du respect des normes locales.
1.4.2 Aperçu des démarches d'approbation réglementaire aux Etats-Unis
Contrairement à l'Europe, où le marquage CE médical est requis, la FDA propose deux principales voies d’approbation pour les logiciels de classe II :
- 510(k) (Équivalence substantielle) : Pour les dispositifs médicaux (DM) de classe I et II, le fabricant doit démontrer que le dispositif est "substantiellement équivalent" à un dispositif de référence, appelé prédicat, déjà commercialisé légalement aux États-Unis. Cette équivalence repose sur des caractéristiques similaires en matière de sécurité et d’efficacité. Lorsque cette équivalence est prouvée, le fabricant peut accéder à une procédure simplifiée, sans nécessiter d’études cliniques approfondies, sauf si des caractéristiques uniques ou des modifications significatives du dispositif rendent ces études nécessaires [18].
- De Novo : Pour les dispositifs innovants de classe II sans "prédicat" existant. Ces dispositifs présentent un "risque modéré", c’est-à-dire que des contrôles spécifiques sont nécessaires pour garantir leur sécurité sans exposer à des risques graves (ex. : logiciel d’aide au diagnostic). L’innovation inclut l’introduction d’une technologie ou fonctionnalité nouvelle non existante sur le marché.
1.4.3 Évaluation clinique
Pour mener une évaluation clinique aux Etats-Unis, le fabricant doit obtenir une autorisation spéciale (Investigational Device Exemption) permettant aux fabricants d’utiliser leur DM expérimentalement, dans le cadre d’un essai clinique, alors qu’il n’est pas encore approuvé pour la commercialisation. Cette étude a pour but de recueillir des données i sur la sécurité d’utilisation du dispositif et son efficacité (fonctionne-t ’il comme prévu pour traiter ou diagnostiquer un problème de santé ?). Les études cliniques doivent se faire selon Bonnes Pratiques de recherche Clinique (BPC) qui se réfèrent aux différentes réglementations et exigences que doivent respecter les fabricants tout au long de l’étude clinique dont la loi 21 CFR 812 qui couvre les procédures de conduite d'études cliniques sur des DM [20]. Les évaluations cliniques sont menées sur :
- Des dispositifs à risque significatif, qui présentent donc un Risque accru d'effets indésirables graves, pouvant entraîner une dégradation grave de l'état de santé du patient(dispositifs de classe III).
- Des dispositifs déjà commercialisés ayant subi certaines modifications affectant la sécurité et la performance du dispositif ou seront destinés à de nouvelles utilisations prévues.
- Certains dispositifs faisant l'objet d’une procédure 510(k) dont le prédicat identifié fait l’objet d’une investigation clinique ou sur la demande de la FDA.
1.4.4 Documentation technique
La structure des documents techniques relatifs aux DM pour le marché américain est globalement similaire à celle exigée en Europe, avec une grande partie du contenu en commun. Les deux marchés partagent des normes de qualité, de sécurité et de performance. Cependant, des différences existent, principalement en ce qui concerne certaines informations spécifiques, qui varient en fonction de la classification du DM et de la méthode d'approbation suivie (510(k), De Novo). Ces variations reflètent les exigences réglementaires propres à chaque région, notamment en termes de classification des risques et des processus d'homologation ou de certification [12,13,17]
Cette documentation technique doit être structurée conformément au modèle de soumission eSTAR, qui définit l'organisation des informations, les formats requis, et les templates à utiliser. Elle doit ensuite être déposée via le portail CDRH pour garantir une soumission conforme aux exigences réglementaires.
1.4.5 L’Enregistrement des produits
La FDA a défini des exigences obligatoires que les fabricants doivent respecter concernant le système d’identification unique des dispositifs (IUD). Chaque fabricant est tenu d’obtenir un numéro IUD, conformément aux étapes détaillées dans la section
Une fois le numéro obtenu, les fabricants doivent apposer le numéro sur le dispositif ou son conditionnement (voir figure 9)et renseigner la base de données mondiale d'identification unique des dispositifs (GUDID) administrée par la FDA avant la mise sur le marché. La base de donnée GUDID comprends un ensemble standard d'éléments d'identification de base pour chaque dispositif qui contient uniquement l'identifiant du dispositif (UID-ID), qui sert de clé pour obtenir des informations sur le dispositif dans la base de données, elle n'inclut pas l'identifiant de production (IP) [19].
Figure 9 : Exemple d’étiquette avec code d'identification unique du dispositif qui doit être apposé sur le dispositif [2].
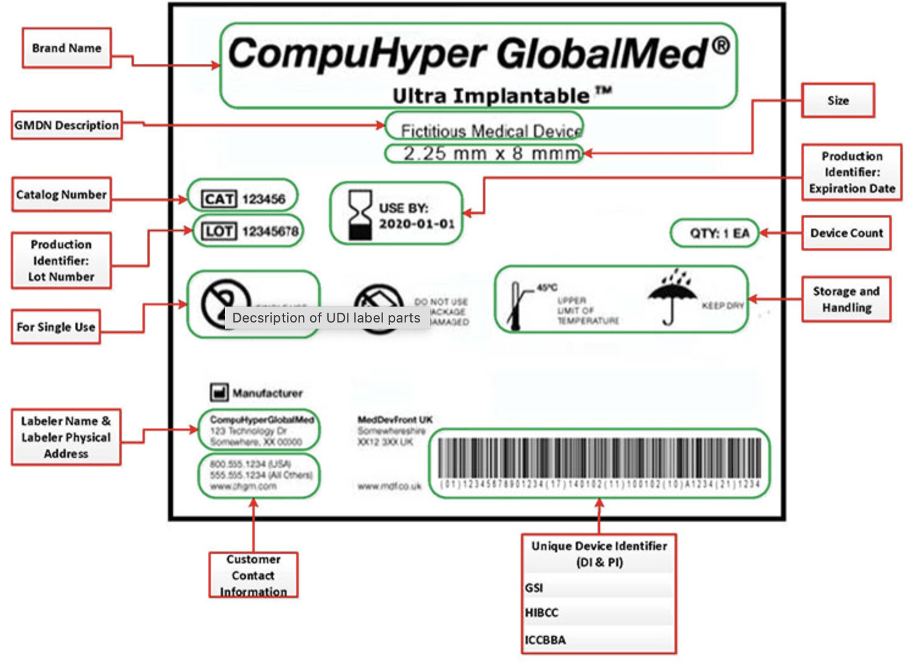
1.4.6 Surveillance post-commercialisation
Cette phase implique obligatoirement la collecte, l’analyse et l’interprétation systématiques des données sur les DM de classes II et III aux Etats-Unis.
Les fabricants reçoivent une ordonnance de la FDA précisant les dispositifs concernés et doivent soumettre un plan de surveillance dans un délai de 30 jours.
Le plan doit inclure les objectifs de la surveillance, les critères d’évaluation, la méthodologie, la taille de l’échantillon, les sources de données, les plans de collecte et d’analyse, ainsi que les délais et rapports prévus. L’examen de ce plan par la FDA peut prendre jusqu’à 60 jours. Selon les conclusions et objectifs, des actions spécifiques peuvent être mises en œuvre pour assurer la sécurité et l’efficacité des dispositifs [21].
Tous les dispositifs médicaux ayant causé un incident doivent être signalés à la FDA, avec des délais de soumission variant selon la gravité de l’incident et le rôle de l’organisme signalant (fabricant, importateur, ou utilisateur). Les incidents graves, tels que les décès ou blessures graves, doivent être rapportés dans un délai de 10 jours par les établissements utilisateurs et de 30 jours par les fabricants et importateurs.
Ces rapports sont collectés et centralisés dans la base de données Manufacturer and User Facility Device Experience (MAUDE), un système qui analyse et détecte les tendances liées aux incidents déclarés. Les rapports sont soumis via le formulaire FDA 3500, permettant une gestion standardisée et un suivi des événements indésirables pour protéger la santé publique [22].
1.5 Comparaison des procédures de soumission entre les États-Unis et l’Europe
Étapes de mise sur le marché
Après une analyse approfondie des différentes étapes des procédures de commercialisation des DM en Europe et aux États-Unis, nous avons identifié un ensemble de similitudes et de divergences entre les cadres règlementaires afférents illustrées dans le tableau 2 ci-dessous.
Tableau 2 : Tableau comparatif des exigences réglementaires entre l’Europe et les Etats-Unis

Les procédures de commercialisation des DM en Europe et aux États-Unis partagent de nombreuses similitudes qui offrent des avantages considérables aux fabricants de dispositifs médicaux. Par exemple, l’adoption d’un numéro UDI harmonisé dans les deux systèmes renforce la traçabilité des dispositifs, ce qui contribue à réduire les risques de rappel de produits tout en renforçant la confiance des patients. Cette harmonisation simplifie également l’intégration des informations dans les bases de données réglementaires des deux régions, optimisant ainsi la gestion des dispositifs.
En outre, les similitudes dans la structure des dossiers techniques permettent aux fabricants de réduire à la fois les coûts et les efforts nécessaires à leur préparation, les mêmes informations pouvant être utilisées pour répondre aux exigences réglementaires de commercialisation sur les deux marchés.
Cependant, les nombreuses différences entre les deux réglementations imposant aux entreprises des efforts et des coûts supplémentaires qui peuvent avoir un impact significatif sur leurs activités. Ces différences incluent, d'une part, des exigences renforcées en matière de système de gestion de la qualité (SMQ) aux États-Unis. Bien que la certification ISO 13485 soit une norme internationale reconnue pour les dispositifs médicaux, elle ne suffit pas à répondre pleinement aux attentes de la FDA (Food and Drug Administration). En effet, les fabricants doivent se conformer au 21 CFR Part 820 (Quality System Règlement), qui inclut des exigences spécifiques telles que des protocoles de contrôle des modifications, une documentation plus détaillée des processus de conception et une gestion des risques plus stricte. D'autre part, des divergences importantes existant dans les contenus requis pour les dossiers techniques soumis aux autorités réglementaires. Par exemple, la FDA exige un « 510(k) » ou une autorisation préalable à la mise sur le marché (PMA) pour prouver l'équivalence substantielle ou démontrer la sécurité et l'efficacité d'un dispositif. Cela diffère considérablement du marquage CE en Europe, où le dossier technique suit les spécifications du règlement (UE) 2017/745 et met davantage l'accent sur les données cliniques et la performance du produit.
Ces écarts obligent les fabricants à adapter leurs processus et leur documentation en fonction des exigences locales, ce qui génère des délais, des coûts de mise en conformité supplémentaires et une complexité accumulée dans la gestion réglementaire.
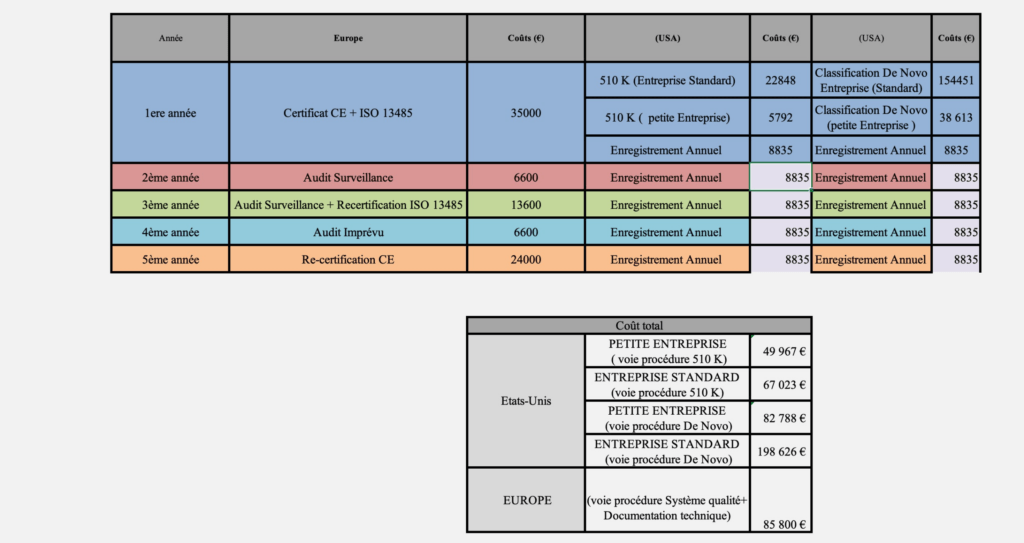
Comparaison des coût entre l’Europe et les États-Unis
- Les coûts administratifs en Europe et aux États-Unis
En Europe, les coûts administratifs pour obtenir un marquage CE s’élèvent autour de 85 800 € sur une période de cinq ans, incluant l'évaluation de la documentation technique, des audits(de surveillance , de renouvellement du marquage CE et du système qualité), et d’autres de frais de gestion liés aux organisme notifiés . Ces coûts sont fixes, quelle que soit la taille de l’entreprise, ce qui garantit une transparence mais peut représenter une charge importante pour les petites entreprises.
Aux États-Unis, les coûts administratifs incluent les frais de la procédure d’autorisation ainsi que les frais d’enregistrement annuel auprès de la FDA, calculés sur une période de cinq ans. Si le dispositif est similaire à un produit déjà commercialisé(prédicat), les entreprises peuvent recourir à la procédure simplifiée 510(k), ces coûts s’élèvent à environ 67 023 € pour une entreprise standard et à 49 967 € pour une petite entreprise. En revanche, la procédure De Novo, applicable en l’absence de prédicat, entraîne des coûts administratifs beaucoup plus élevés, atteignant 82 788 € pour une petite entreprise et jusqu’à 198 626 € pour une grande structure (voir annexe 2 )
- Les coûts des évaluations cliniques : un facteur décisif pour les Petites et moyenne entreprise
Les coûts administratifs ne représentent qu’une partie des dépenses. En Europe, l’évaluation clinique obligatoire est un facteur clé, car elle nécessite des études approfondies pour démontrer la sécurité et la performance du dispositif dans des conditions réelles. Ces études, souvent longues et coûteuses, peuvent représenter un investissement supplémentaire de plusieurs centaines de milliers d’euros selon la complexité du dispositif et les exigences réglementaires.
Aux États-Unis, grâce à la notion de prédicat, l’évaluation clinique peut être évitée pour les dispositifs entrant dans la procédure 510(k). Cela constitue un avantage décisif pour les petites entreprises, car cela réduit non seulement les délais d’accès au marché mais également les dépenses globales. En revanche, pour les dispositifs sans prédicat, l’obligation de mener des études cliniques dans le cadre de la procédure De Novo alourdit considérablement les coûts totaux.
Impact sur les petites entreprises et l’innovation
Pour les PME, le modèle européen impose des coûts fixes élevés, notamment en raison des évaluations cliniques obligatoires, ce qui peut freiner leur capacité à innover ou à accéder rapidement au marché. Aux États-Unis, la flexibilité offerte par la procédure 510(k) représente un avantage compétitif pour les startups disposant de ressources limitées, à condition qu’un prédicat approprié existe.
Comparaison des délais entre l’Europe et les États-Unis
Les délais d’évaluation des dispositifs médicaux logiciels de classe II ont un impact d’autant plus crucial qu’ils viennent s’ajouter au temps nécessaire au développement du logiciel lui-même. Concevoir un logiciel médical de classe II est un processus complexe et coûteux, souvent étalé sur plusieurs années, avec des étapes critiques comme la recherche, les tests cliniques, et la validation technique. Ce travail représente déjà un investissement financier et humain considérable pour l’entreprise.
En Europe, les 12 à 16 mois supplémentaires requis pour l’approbation du certificat de la mise sur le marché allongent encore ce cycle, retardant d’autant la capacité de l’entreprise à générer des revenus. Pendant cette période, l’entreprise continue de mobiliser des ressources financières pour maintenir ses équipes et couvrir les frais opérationnels, sans pouvoir compenser ces dépenses par une entrée d'argent.
Par exemple, un produit innovant lancé avec un retard d’un an peut non seulement perdre l’avantage du "premier entrant", mais aussi permettre à des concurrents d’occuper une position dominante. Ce décalage se traduit par un manque à gagner significatif, qui peut se chiffrer en millions d’euros pour certaines entreprises, surtout dans des secteurs où les marges sont élevées.
En revanche, aux États-Unis, la procédure 510(k), avec un délai moyen de 3 mois, s’intègre mieux dans le cycle de développement global. Elle permet de réduire le temps total nécessaire pour transformer une idée innovante en produit commercialisable. Ce gain de temps permet non seulement de générer des revenus plus rapidement, mais également de réinvestir plus tôt dans le développement de nouveaux produits ou l’amélioration des logiciels existants. Cependant, lorsqu’il n’existe pas de prédicat pour justifier l’équivalence substantielle, la procédure De Novo s’applique. Bien que cette procédure puisse allonger le délai global de 4 mois, elle offre un cadre permettant d'introduire des dispositifs véritablement innovants sur le marché tout en respectant les exigences de sécurité et de performance.
En somme, dans un secteur où le développement est déjà long et exigeant, les délais d’évaluation deviennent un facteur déterminant. Ils peuvent freiner la croissance de l’entreprise en Europe, alors qu’une procédure plus rapide, comme celle des États-Unis, offre des perspectives plus favorables pour maximiser le potentiel commercial des logiciels médicaux.
La comparaison approfondie des systèmes réglementaires européen et américain pour les dispositifs médicaux révèle des différences significatives, notamment en termes de processus d'approbation, de classification des produits et d'exigences post-commercialisation. Ces disparités posent des défis particuliers pour les petites entreprises innovantes qui cherchent à commercialiser leurs produits sur ces deux marchés majeurs.
Face à ce constat, il apparaît nécessaire d'élaborer un guide d'accompagnement spécifique, permettant aux petites entreprises innovantes de naviguer efficacement dans les complexités réglementaires des marchés européen et américain. Ce guide vise à combler une lacune importante en fournissant un outil pratique et accessible, adapté aux contraintes et aux besoins particuliers de ces acteurs. Dans le chapitre suivant, nous détaillerons la méthodologie employée pour développer ce guide d'accompagnement, en nous appuyant sur l'analyse comparative effectuée et en tenant compte des spécificités des petites entreprises innovantes dans le secteur des dispositifs médicaux.
Chapitre 2 : Méthodologie adoptée pour le développement du guide d’accompagnement
2.1 Démarche employée par le groupe
2.1.1 Schéma général
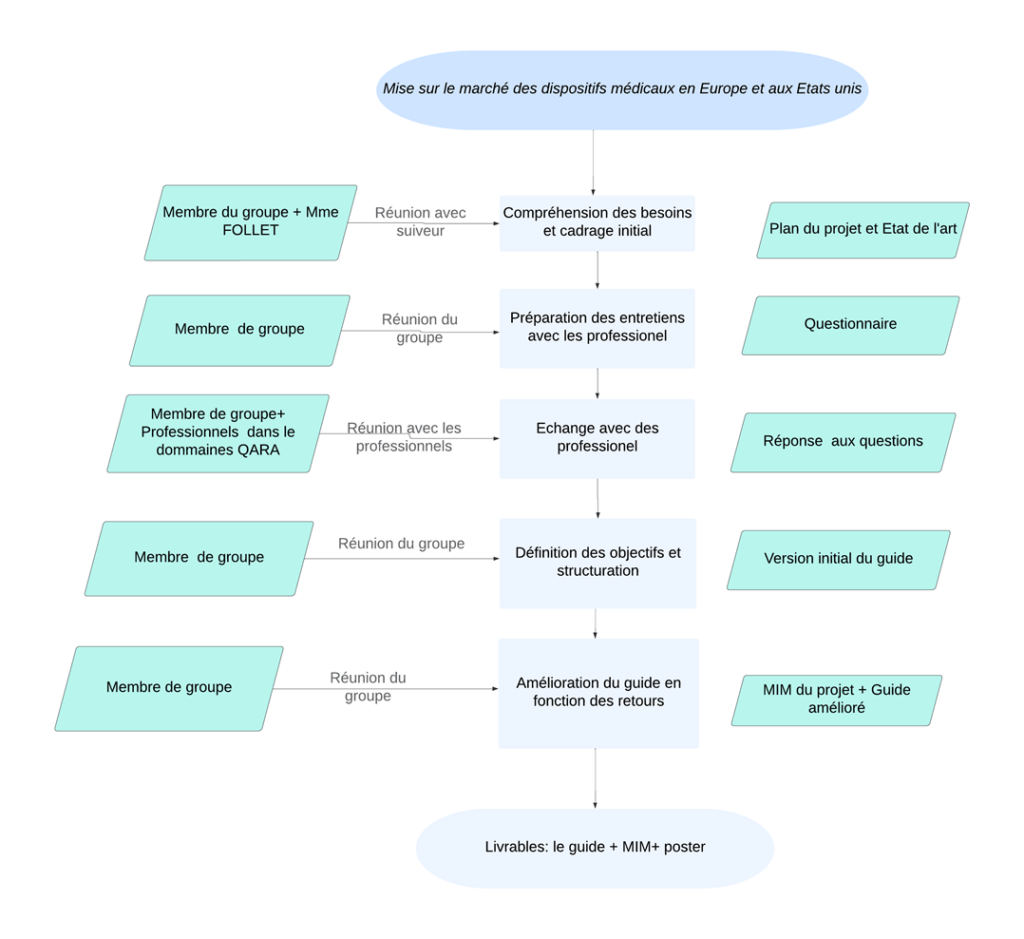
Un logigramme figure 10 a été mis en place afin d’avoir une visualisation sur les différentes étapes du projet.
Figure 10 : Logigramme des différentes étapes du projet
2.1.2 Evaluation des risques
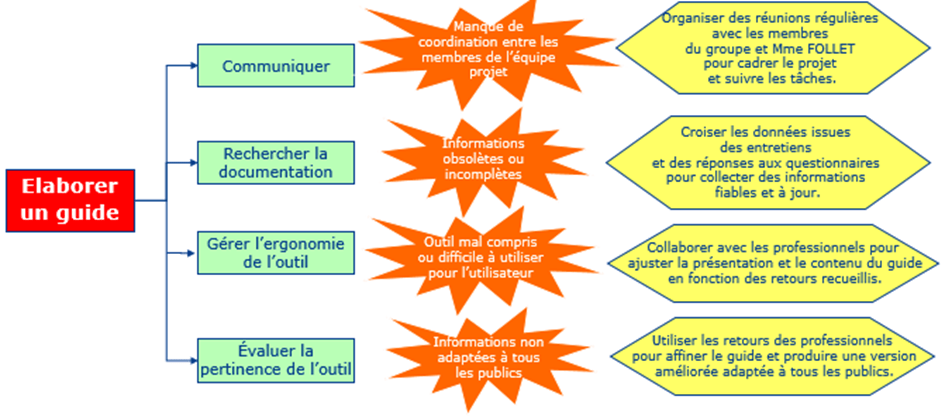
Tout au long de cette démarche, des risques liés l'exécution des différentes phases du projet ont été identifiés. Nous avons donc élaboré au préalable un diagramme de décisions ( voir figure 11). Le diagramme de décisions est une représentation graphique qui permet d’identifier les risques associés à chaque action et de proposer des actions alternatives qui élimineront ou réduiront les risques. Il structure précisément les options disponibles pour faciliter la compréhension des situations et prendre des décisions.
Figure 11 : Diagramme de décision : risques identifiés et solutions retenues
2.2 Etude des outils existants et choix de l’outil
2.2.1 Etat de l’art des différents outils existants
L'objectif de ce projet est de fournir un outil qui présentera une méthode claire et concrète pour aider les fabricants à mettre leurs DM sur les marchés européens et américains en respectant les réglementations et normes en vigueur dans les deux régions.
La stratégie utilisée pour réaliser ce guide était de faire dans un premier temps une étude de tous les outils existants. Lors de notre recherche, nous nous sommes rendus compte que les différents outils existants portaient principalement sur la procédure en Europe. En effet, de nombreuses ressources sont disponibles pour faciliter la mise sur le marché des dispositifs médicaux en Europe. En comparaison, les outils spécifiques pour le marché américain semblent moins nombreux et moins accessibles.
Cette disparité dans la disponibilité des ressources entre les marchés européen et américain justifie la nécessité de développer un guide complet couvrant les deux régions. Notre outil vise ainsi à combler cette lacune en fournissant une approche intégrée pour la mise en conformité des dispositifs médicaux sur les deux marchés. Un outil a principalement attiré notre attention celui du IDS 203- Bilan du nouveau règlement européen sur les dispositifs médicaux 2017/745 : de l’entrée en vigueur à l’application (voir figure 12) .
Figure 12 : Captures écran des pages d’accueil du guide d’accompagnement pour la mise sur le marché de dispositifs médicaux sous le règlement MDR 2017/745 [IDS 203]

Ce guide d’accompagnement, présente la procédure de mise sur le marché d’un dispositif médical en Europe, comment effectuer le choix d’un organisme notifié, un zoom sur les différentes aides financières et le calendrier transitoire des dispositifs médicaux .
Il n’y a donc pas d’outils qui regroupe les procédures de mise sur le marché dans les deux régions. Pour restructurer le travail, des échanges ont étés planifiées avec des professionnels du secteur afin de recueillir les différents besoins pour mieux alimenter notre travail.
2.2.2 Justification du choix de l’outil proposé
Le tableau 3 ci-dessous regroupe les personnes qui ont été interviewées, les questions qui ont été posées et les réponses obtenues :
Tableau 3 : Questions et réponses variées recueillies lors des entretiens avec les professionnels.
| Type d’entreprise | Personnes Interviewées | Questions | Réponses |
| Fabricant de cathéter implantable 15-20 employés | Directeur qualité et affaires réglementaires | Quels sont les principaux défis pour un fabricant pour obtenir le marquage CE en Europe ? | Les principaux défis incluent la complexité des exigences réglementaires (règlement (UE) 2017/745), la nécessité d'une documentation technique détaillée, l'implication d'un organisme notifié pour l'évaluation de la conformité, et la gestion des exigences de surveillance post-commercialisation. Le processus peut être long et coûteux, notamment pour les dispositifs de classe II et au-delà. |
| Cabinet de conseil en Tunisie | Consultante assurance qualité sénior | Comment un fabricant peut-il s’assurer que son dispositif respecte les exigences des normes européennes et américaines ? | Le fabricant doit comprendre les différences entre les deux systèmes de régulation et s'assurer que son produit est conforme aux exigences locales. Par exemple, le marquage CE est nécessaire en Europe, tandis qu'aux États-Unis, la soumission FDA 510(k) est souvent requise. Les normes ISO, telles qu'ISO 13485 pour la gestion de la qualité, sont essentielles dans les deux régions. |
| Fabricant de logiciel de télésurveillance cardiaque 60-80 employés | Responsable qualité et affaires Règlementaires | Quels sont les principaux défis financiers pour un fabricant de dispositifs médicaux dans le processus de mise sur le marché ? Comment une startup peut-elle gérer les coûts associés à la certification et à la mise sur le marché d’un dispositif médical ? | Les coûts de conformité réglementaire sont souvent élevés, en particulier pour les dispositifs de classe II et III. Cela inclut les frais pour l'évaluation par un organisme notifié, la réalisation d'études cliniques, le développement de la documentation technique, et la gestion de la surveillance post-commercialisation. Les coûts peuvent être un obstacle majeur, en particulier pour les startups qui ont un budget limité. Une startup peut rechercher des subventions ou des financements publics pour soutenir les coûts de conformité. Elle peut également faire appel à des consultants spécialisés pour éviter les erreurs coûteuses dans le processus de soumission. Le recours à des partenariats avec d'autres entreprises ou des investisseurs en capital-risque peut également faciliter le financement du processus. |
| Fabricant de stent (classe 3) 80-100 employés | Responsable qualité | Quels défis un fabricant rencontre-t-il pour classer son dispositif médical sous la réglementation UE 2017/745 en Europe ? Quelles étapes sont les plus critiques pour obtenir une autorisation 510(k) aux États-Unis ? | Les défis incluent la démonstration de la "substantiel équivalence" pour les dispositifs de classe II via la soumission 510(k), les coûts associés aux études cliniques, la gestion de la documentation technique et le respect des Good Manufacturing Practices (GMP). De plus, le processus de soumission peut être complexe et nécessite une coordination étroite avec des experts réglementaires. Identifier un dispositif de prédicat approprié, effectuer des tests de conformité aux normes FDA, et répondre rapidement aux demandes d'informations supplémentaires de la FDA. |
| Fabricant logiciel permettant de réaliser une réfraction subjective, simplement et rapidement 20-40 employés | Chargée qualité et affaires réglementaires | Est-ce qu’un guide FDA et MDR est un outil utile pour votre entreprise ? | Absolument. Pour nous, en tant que petite entreprise en plein essor et désireuse de vendre ses produits en Europe et en Amérique, ces guides sont indispensables. Il est essentiel de suivre le guide FDA afin de saisir et mettre en œuvre les normes américaines, tandis que le MDR est essentiel pour répondre aux exigences du marché européen. |
| Cabinet de conseil en Ile de France, accompagne des entreprises dans la mise sur le marché des DM | Responsable affaires règlementaires | Qu’attendez vous spécifiquement d’un guide FDA ou MDR pour soutenir votre petite entreprise ? Quels sont les impacts des délais liés aux organismes notifiés en Europe sur le marché des DM ? | Il est nécessaire d'avoir des guides qui répondent aux besoins des petites entreprises, c'est-à-dire qui soient simples, pratiques et faciles à appréhender. Il serait extrêmement bénéfique d'avoir des exemples concrets ou des checklists pour nous accompagner étape par étape, afin de minimiser les risques d'erreur. Nous désirons également obtenir des données à jour afin de prévoir les modifications réglementaires sans avoir besoin de mobiliser une quantité excessive de ressources. Les retards augmentent les coûts, retardent l'accès au marché et réduisent la compétitivité des entreprises européennes. |
| Fabricant de dispositif pour l’apnée du sommeil avec logiciel embarqué Start up : 8-10 employés | Chargée Qualité | Est-ce qu’un guide pour les logiciels dispositifs médicaux (DM) serait utile pour votre entreprise ? Quelle est l'importance de la documentation technique pour la mise sur le marché des dispositifs médicaux dans les deux régions ? | Effectivement, un tel guide serait très bénéfique. Un guide clair pourrait nous permettre de mieux appréhender les attentes des autorités telles que la FDA ou celles du MDR en Europe en ce qui concerne les logiciels DM. Cela rendrait la mise en conformité plus facile et diminuerait le risque d'erreurs. En Europe : la documentation technique est essentielle pour démontrer la conformité avec le Règlement (UE) 2017/745 et pour le marquage CE. Elle doit être complète, incluant des preuves cliniques et techniques. Aux États-Unis : bien que la FDA exige des soumissions spécifiques (510(k) ou PMA), la documentation technique est également cruciale pour prouver la sécurité et l'efficacité du produit. |
| Fabricant de logiciel de modélisation cardiaque 20-40 employés | Chargée assurance qualité et affaires réglementaires | Quels aspects spécifiques aimeriez vous voir dans ce guide ? Quels sont les critères spécifiques que la FDA et l'UE prennent en compte lors de l'évaluation de la sécurité et de l'efficacité des dispositifs médicaux ? | Nous souhaiterions qu'il englobe divers aspects : Les catégories de logiciels DM établies par la FDA et le MDR, les différentes étapes pour valider et vérifier les logiciels, les normes de sécurité informatique et de gestion des risques. En Europe : le processus évalue la conformité du dispositif aux exigences essentielles, incluant la sécurité et la performance sur la base des études cliniques, des tests précliniques, et de la gestion des risques. Aux États-Unis : la FDA se concentre sur des preuves cliniques ou des données de performance (dossiers 510(k) ou PMA) qui montrent que le produit est aussi sûr et efficace que les produits existants ou qu'il représente une amélioration substantielle. |
Après avoir analysé les ressources existantes et pris en compte les retours reçus, nous augmenterons le guide d’accompagnement pour la mise sur le marché de dispositifs médicaux sous le règlement MDR 2017/745 publié en janvier 2024 [IDS 203]. Dans notre version du guide, nous intégrerons la procédure de mise sur le marché des dispositifs médicaux logiciels de classe II en Europe et aux États-Unis, ainsi que les normes applicables à ces dispositifs (voire le tableau 4).
Tableau 4 : Cahier des charges du guide d’accompagnement pour la mise sur le marché de dispositifs médicaux logiciels en Europe et aux Etats-Unis
| Catégorie | Description |
| Objectif | Fournir un guide clair et structuré pour accompagner les fabricants de logiciels médicaux de classe II dans leur mise sur le marché en Europe et aux USA. |
| Public cible | -Petites entreprises fabricants de dispositifs médicaux -Responsables qualité et affaires réglementaires. |
| Structure et contenu | - Procédure de mise sur le marché en Europe - Choix d’un organisme notifié - Procédure de mise sur le marché aux Etats-Unis - Normes applicables aux logiciels DM de classe II |
| Format et langue | Langue : Français Version numérique : PDF interactif |
| Méthodologie et réalisation | - Recherche et structuration : Analyse de l’existant et échange avec les professionnels - Rédaction et conception : Création d’une Version 1 du guide - Relecture et validation : Contrôle par des professeurs, des étudiants UTC et professionnels du domaine |
| Contraintes et exigences | - Respect des dernières Règlementations (règlement (UE) 2017/745 et FDA |
| Livrables | Guide complet |
Chapitre 3 : Présentation du guide d’accompagnement des fabricants de logiciel de classe II
3.1 Vue d’ensemble du guide d’accompagnement
Ce guide s’adresse spécifiquement aux entreprises développant des dispositifs médicaux logiciels de classe II, qu’elles soient des start-ups, des PME ou des grandes organisations, et couvre les logiciel de classe II en phase de mise sur le marché (voir figure 13). Conçu pour répondre aux besoins concrets des fabricants à chaque étape critique, il offre une méthodologie claire et pratique, adaptée à la réalité des exigences réglementaires en Europe (règlement UE 2017/745) et aux États-Unis (21 CFR Part 820). En mettant l’accent sur des solutions applicables et détaillées, ce guide vise à simplifier les démarches de classification, de documentation technique, de mise en place du système de gestion de la qualité (SMQ), et d’enregistrement dans les bases EUDAMED et GUIDID.
Figure 13 : Aperçu page d’accueil du guide (source auteurs)

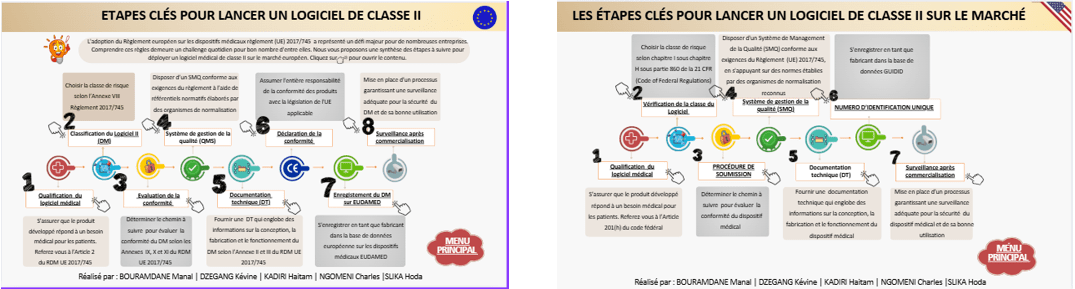
Ce guide est structuré pour permettre une navigation intuitive et suivre une progression logique qui reflète le processus de mise sur le marché des dispositifs médicaux logiciels de classe II. Il accompagne les fabricants à travers des étapes chronologiques (Voir figure 14), de la qualification initiale du produit à sa surveillance post-commercialisation, en garantissant une clarté à chaque phase. L’organisation du guide facilite l’accès rapide aux informations pertinentes selon l’étape où se trouve l’entreprise.
Figure 14 : Aperçu des étapes de mise sur le marché proposé dans le guide (source auteurs)
La première étape aborde la qualification et classification du dispositif médical logiciel, une étape essentielle pour déterminer le niveau de risque du produit en fonction de son usage prévu et de son impact potentiel sur la santé. Cette section explique comment utiliser les critères définis par le règlement UE 2017/745 et les réglementations américaines (21 CFR), tout en illustrant ces concepts avec des exemples concrets.
Le guide enchaîne avec l’élaboration du dossier technique, une étape clé pour démontrer la conformité réglementaire. Cette partie décrit comment structurer et documenter les informations sur la conception, la fabrication, les performances, et la gestion des risques, conformément aux exigences des annexes II et III du MDR ou du 21 CFR Part 807.
Ensuite, le guide couvre la mise en place du système de gestion de la qualité (SMQ). Il explore comment intégrer des normes ISO, telles que l’ISO 13485, pour garantir la qualité des processus internes. Cette section détaille les étapes à suivre pour aligner les systèmes de gestion sur les attentes des régulateurs tout en maintenant une efficacité opérationnelle.
Enfin, la section sur la surveillance post-commercialisation (PMS) clôture le guide, en insistant sur l’importance de collecter et d’analyser les données relatives à la sécurité et aux performances après la mise sur le marché. Cette phase assure un suivi continu du produit pour répondre aux exigences de sécurité et garantir une conformité durable.
L’ordre des sections est conçu pour refléter une progression logique, permettant aux fabricants de se concentrer sur une étape à la fois tout en anticipant les exigences des phases ultérieures. Ce format chronologique garantit que les entreprises peuvent naviguer facilement dans le guide et appliquer ses recommandations de manière efficace, en fonction de leur stade de développement.
Voici les principales fonctionnalités du guide :
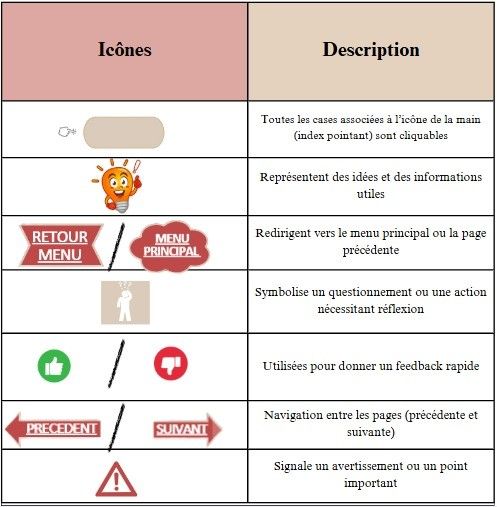
Icônes interactives : Pour faciliter la navigation dans le guide, des icônes cliquables ont été intégrées. Chaque icône mène l’utilisateur à une section spécifique ou met en avant des points critiques à ne pas négliger (voir figure 15) Voici une explication de leur rôle :
Figure 15 : Liste des icônes utilisées pour naviguer dans le guide interactif d’accompagnement (source auteurs)
3.2 Cas d'utilisation d'un logiciel classe II et perspectives
3.2.1 Cas d'utilisation d'un logiciel classe II
Un fabricant de logiciels d’analyse de données médicales, destiné au suivi à distance des patients et à la prédiction des risques de maladies chroniques, souhaite commercialiser son produit sur les marchés européen et américain. En utilisant le guide, il suit une démarche claire et structurée pour garantir la conformité réglementaire et s’assurer que son logiciel respecte les exigences des deux régions.
La première étape consiste à accéder au menu principal du guide, où le fabricant peut explorer différentes sections correspondant aux étapes clés : qualification, classification, documentation technique, soumission et surveillance post-marché. Pour commencer, il sélectionne la section « Qualification » afin de vérifier que son logiciel relève de la définition d’un dispositif médical logiciel. Grâce aux outils interactifs du guide, le fabricant se réfère aux textes européens, tels que l’Article 2 du Règlement (UE) 2017/745, et aux définitions américaines, comme celles du CFR Article 201(h). Il confirme que son produit, influençant directement les décisions médicales, est effectivement un dispositif médical logiciel.
Le fabricant passe ensuite à la classification du produit pour déterminer la classe de risque. Le guide le dirige vers l’Annexe VIII et la Règle 11 du Règlement (UE) 2017/745 pour l’Europe, qui classent les logiciels influençant des décisions médicales en fonction de leur impact sur les patients. Pour le marché américain, il suit les recommandations des FDA Panels, qui permettent de classer le produit en classe II. Ces étapes lui permettent de mieux comprendre les implications réglementaires spécifiques à sa classe de risque.
Dans la section « Documentation Technique », le guide fournit des conseils pour structurer le dossier technique de manière conforme aux annexes II et III du Règlement (UE) 2017/745. Le fabricant compile les informations essentielles, notamment une description détaillée du produit, les résultats des tests de performance démontrant sa fiabilité, un plan de gestion des risques conforme à l’ISO 14971, et des preuves de conformité aux normes IEC 62304 et ISO 13485. Cette préparation méthodique garantit que le dossier répond aux exigences des organismes notifiés en Europe et de la FDA aux États-Unis.
Une fois le dossier technique complété, le fabricant procède à la soumission du produit. En Europe, il enregistre le logiciel dans la base EUDAMED pour répondre aux exigences européennes. Aux États-Unis, il utilise l’outil eSTAR pour soumettre une demande 510(k), démontrant l’équivalence substantielle de son produit avec un dispositif déjà approuvé. Ces démarches lui permettent de déposer son dossier pour évaluation et d’entamer le processus de mise sur le marché.
Après la commercialisation, le fabricant met en œuvre un plan de surveillance post-marché (PMS) conformément aux recommandations du guide. Ce plan inclut la collecte et l’analyse de données en conditions réelles, la gestion des signalements d’incidents via les plateformes réglementaires (EUDAMED et MDR aux États-Unis), et l’évaluation continue des risques pour garantir la sécurité et la performance du logiciel tout au long de son cycle de vie.
En suivant les étapes décrites dans le guide, le fabricant parvient à naviguer efficacement dans les processus réglementaires complexes et à garantir la conformité de son logiciel aux exigences européennes et américaines. Ce scénario illustre la valeur pratique du guide en tant qu’outil d’assistance pour les fabricants de dispositifs médicaux logiciels de classe II.
3.2.2 Limites et perspectives du guide
Le guide présente plusieurs limites qui réduisent son efficacité dans certains contextes spécifiques. Tout d'abord, il manque une contextualisation approfondie pour aborder les disparités locales au sein de l'Union européenne et des États-Unis, ce qui le rend parfois insuffisant pour des entreprises confrontées à des cadres législatifs complexes ou spécifiques. En termes d’ergonomie, bien que la navigation interactive soit une valeur ajoutée, elle peut poser des difficultés aux utilisateurs peu familiers avec les outils numériques ou les novices en affaires réglementaires. De plus, le guide ne propose pas de mécanisme de mise à jour régulier, ce qui expose certaines sections au risque d’obsolescence rapide face à l'évolution des réglementations, comme les amendements récents du Règlement UE 2017/745 ou les nouvelles directives de la FDA. Enfin, bien que conçu pour l’Europe et les États-Unis, il ne couvre pas d’autres marchés importants, limitant ainsi son audience et son utilité pour des fabricants ayant une ambition globale.
Les retours des usagers sont globalement très positifs et mettent en avant plusieurs points forts du guide. La clarté et la structuration des étapes sont largement saluées, notamment la présentation méthodique des exigences de conformité et l’intégration de menus interactifs avec des hyperliens vers les textes réglementaires. Les usagers apprécient particulièrement les exemples pratiques et les études de cas qui rendent les concepts complexes plus accessibles, même pour les novices. La section sur la documentation technique, avec des détails précis sur les annexes II et III du Règlement UE 2017/745, est jugée très utile. En outre, le guide est reconnu pour son application pratique, notamment grâce aux références directes aux articles des règlements et guides, qui permettent un gain de temps significatif dans les recherches. Enfin, la pédagogie et l’accessibilité du langage rendent les réglementations, souvent intimidantes, plus compréhensibles, ce qui améliore l’expérience utilisateur.
Pour renforcer son efficacité et répondre aux attentes des usagers, plusieurs évolutions peuvent être envisagées. Tout d'abord, une personnalisation accrue du guide pourrait être mise en place, par exemple en intégrant un questionnaire interactif ou un arbre décisionnel pour adapter le parcours en fonction de la classe de risque ou du marché visé. Une section dédiée aux startups et aux petits fabricants, avec des ressources et outils spécifiques, permettrait également de mieux répondre à leurs besoins.
La mise en œuvre d’un système de mise à jour régulière, avec des notifications ou des sections dédiées aux évolutions des réglementations (comme les amendements futurs au Règlement UE 2017/745 ou les directives de la FDA), renforcerait la pertinence du guide à long terme. Par ailleurs, une extension géographique pour inclure des marchés comme l’Asie ou l’Amérique latine serait bénéfique, notamment pour les entreprises exportatrices. Cela pourrait être accompagné d’une carte interactive permettant de visualiser les obligations réglementaires par pays ou région. Enfin, le guide pourrait améliorer sa visualisation grâce à des schémas plus détaillés, des diagrammes interactifs ou des infographies pour illustrer les flux de processus. Proposer des modèles téléchargeables pour les documents réglementaires ou la gestion de la qualité ajouterait une valeur pratique supplémentaire. Ces perspectives, combinées aux points forts actuels du guide, contribueraient à en faire un outil encore plus pertinent et utile pour les fabricants de dispositifs médicaux logiciels.
Conclusion
L'analyse détaillée des exigences réglementaires pour la commercialisation des dispositifs médicaux logiciels de classe II, ciblés en raison de leur rôle croissant dans l'innovation médicale, de leur niveau de risque modéré nécessitant des contrôles stricts, et parce que la majorité des dispositifs médicaux logiciels commercialisés, que ce soit en Europe ou aux États-Unis, appartiennent à cette catégorie, a révélé des similarités. Les autorités des deux régions visent à garantir la sûreté et l'efficacité des dispositifs logiciels mis sur le marché, mais également des divergences, notamment procédurales (exigences en matière d’évaluation clinique, délais et coûts associés).
En Europe, l’entrée en application du Règlement (UE) 2017/745 a permis d'introduire des normes plus sévères, notamment grâce à la participation des Organismes Notifiés, ce qui a allongé les délais nécessaires pour se conformer. Cette situation constitue un enjeu significatif pour les producteurs, qui font face à des coûts administratifs et financiers considérables. Des études montrent que ces coûts peuvent représenter une part importante du budget global, en particulier pour les petites et moyennes entreprises, et que les délais de certification varient en fonction de la disponibilité des Organismes Notifiés et de la complexité des dispositifs. Par opposition, la FDA aux États-Unis offre des processus accélérés comme la procédure 510(k), permettant souvent un accès plus rapide au marché pour les dispositifs de classe II. Bien que cette procédure soit perçue comme plus flexible, elle nécessite toujours une démonstration rigoureuse de l’équivalence substantielle avec un dispositif déjà commercialisé.
Dans le cadre de ce projet, un guide interactif a été conçu pour aider les fabricants à se conformer aux exigences réglementaires. Il offre des outils pratiques pour naviguer efficacement entre les réglementations européennes et américaines, en prenant en compte les procédures, les exigences documentaires et les démarches d’évaluation clinique tout en proposant des réponses pratiques.
La comparaison des exigences entre l'Europe et les États-Unis souligne l'importance d'adopter des stratégies adaptées aux spécificités de chaque région. Le guide interactif développé dans ce projet pourrait être encore amélioré en intégrant des mises à jour en temps réel des évolutions réglementaires, en recueillant les retours d'expérience des utilisateurs et en étendant son application à d'autres marchés internationaux. Ces améliorations visent à maximiser l'efficacité de l'outil et à mieux accompagner les fabricants dans leurs démarches de conformité.
Bibliographie
[1] Snitem, « Panorama de la filière DM : les chiffres clés », Snitem,2023. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.snitem.fr/presse/panorama-de-la-filiere-dm-les-chiffres-cles/
[2] C. for D. and R. Health, « UDI Basics », FDA, août 2023, Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/udi-basics
[3] Statista, « Medical Devices : market data & analysis », janv. 2024. Consulté le : 24 septembre 2024. [En ligne]. Disponible sur : https://www.statista.com/study/107264/medical-devices-market-data-and-analysis/
[4] Medtech, « MedTech Europe’s Facts & Figures 2024 ». 2024. Consulté le : 26 septembre 2024. [En ligne]. Disponible sur : https://www.medtecheurope.org/resource-library/medtech-europes-facts-figures-2024/
[5] R. Bartlett, A. Somauroo, et C. Zerbi, « How the medtech industry can capture value from digital health », p. 9, juill. 2021. https://www.mckinsey.com/industries/life-sciences/our-insights/how-the-medtech-industry-can-capture-value-from-digital-health
[6] D. Valdes, L. Alqazlan, R. Procter, et J. Dale, « Global evidence on the rapid adoption of telemedicine in primary care during the first 2 years of the COVID-19 pandemic : a scoping review protocol », Syst. Rev., vol. 11, no 1, p. 124, juin 2022, doi : https:10.1186/s13643-022-01934-3
[7] Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ), vol. 117, Avril 2017. Consulté le : 30 septembre 2024. [En ligne]. Disponible sur:http://data.europa.eu/eli/reg/2017/745/oj/fra
[8] FDA, « CFR - Code of Federal Regulations Title 21 », août 2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm
[9] S. Remy, « De l’enregistrement à la surveillance sur le marché des dispositifs médicaux. modele comparatif entre les Etats unis et l’Europe », Université de Strasbourg, Novembre 2023. Consulté le : 15 octobre 2024. [En ligne]. Disponible sur : https://publication-theses.unistra.fr/public/theses_exercice/PHA/2023/2023_REMY_Sebastien.pdf
[10] « Guidance - MDCG endorsed documents and other guidance - European Commission »,Novembre 2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en
[11] C. for D. and R. Health, « 510(k) Submission Process », FDA, août 2023, Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/premarket-notification-510k/510k-submission-process
[12] FDA, « CFR - Code of Federal Regulations Title 21-814 », août 2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=814&showFR=1
[13] « FDA QSR vs. QMSR : What Medical Device Manufacturers Need to Know – Oriel STAT A MATRIX – ELIQUENT Life Sciences Blog », Oriel STAT A MATRIX - ELIQUENT Life Sciences Blog, Juin 2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.orielstat.com/blog/fda-qsr-vs-qmsr/
[14] A. Lyx, « La réglementation internationale des dispositifs médicaux », UFR des sciences pharmaceutiques et biologiques de Montpellier ,Juin 2023.Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-04131370v1/document
[15] C. for D. and R. Health, « Medical Device Single Audit Program (MDSAP) », FDA, Février.2024 . Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/cdrh-international-affairs/medical-device-single-audit-program-mdsap
[16] C. for D. and R. Health, « De Novo Classification Request », FDA, sept. 2024, Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/de-novo-classification-request
[18] « Unique Device Identification System », Federal Register, sept.2013. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.federalregister.gov/documents/2013/09/24/2013-23059/unique-device-identification-system
[19] FDA, « CFR - Code of Federal Regulations Title 21-812 », août.2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=812&showFR=1
[20] FDA, « CFR - Code of Federal Regulations Title 21-820 »,août.2024. Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820
[21] FDA, « CFR - Code of Federal Regulations Title 21-803 ». Consulté le : 18 novembre 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=803.3
[22] P. Guillaume, « Comparatif : tarifs des Organismes Notifiés 2017/745 », Qualitiso, sept. 2022. Consulté le : 16 décembre 2024. [En ligne]. Disponible sur : https://www.qualitiso.com/comparatif-des-tarifs-des-organismes-notifies-2017-745/
[23] Commission européenne, « Published fees on notified bodies websites for MDR (Table 1.) and IVDR (Table 2.) related services. » 10 août 2024. [En ligne]. Disponible sur : https://health.ec.europa.eu/document/download/ff5716d5-fe77-4f45-b883-fcf3da4acd15_en?filename=md_nbs_fees_en.pdf
Annexes
Annexe 1 : QQOQCP

Annexe 2 : Récapitulatif des coûts administratifs en Europe et aux Etats unis pour la mise sur le marché des dispositifs médicaux