
IDS139 - Legacy device : Evaluation et suivi clinique sous le Règlement 2017/745
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Autrice

Contact
- Catherine DALLA RIVA : catdallariva@gmail.com
Citation
A rappeler pour tout usage : Catherine DALLA RIVA, « Legacy device : Évaluation et suivi clinique sous le Règlement 2017/745 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire d'alternance, https://travaux.master.utc.fr/, réf n° IDS139, url : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids139/, septembre 2022
Résumé
13 à 18 mois, soit presque 2 ans.
Ceci représente le délai moyen de certification par les Organismes Notifiés, en 2022, sous le titre du Règlement relatif au dispositifs médicaux 2017/745 (MDR). Afin de ne pas ajouter de délai supplémentaire à cet examen de la documentation technique, une totale compréhension des nouvelles exigences est essentielle.
Ce règlement a renforcé de façon substantielle les conditions d’obtention du certificat CE. Parmi ces nouvelles exigences renforcées, l’évaluation clinique. L’évaluation clinique sous MDR complique le recours à l’équivalence, principe sur lequel un bon nombre de legacy devices ont pu obtenir le certificat CE sous directive 93/42/CEE, en se basant sur un autre dispositif lui-même marqué CE.
Désormais, les fabricants doivent générer des données cliniques propres à leurs dispositifs dans l’objectif d’élaborer une évaluation clinique conforme au MDR 2017/745. Ce mémoire propose alors la démarche entrepris par un fabricant français de dispositifs médicaux pour se mettre en conformité avec les exigences cliniques de ce mastodonte.
Abstract
13 to 18 months, almost 2 years.
This represents the average time for certification by Notified Bodies under Medical Device Regulation 2017/745 (MDR) in 2022. To avoid adding additional time to this technical documentation review, a full understanding of the new requirements is key.
This regulation has substantially strengthened the requirements for obtaining the CE certificate. One of the new strengthened requirements is the clinical evaluation. Clinical evaluation under the MDR complicates the use of equivalence, a principle on which many legacy devices have been able to obtain CE certification under the directive 93/42/CEE based on another device that was itself CE marked.
As a result, manufacturers are now required to generate clinical data specific to their devices to establish a clinical evaluation in accordance with MDR 2017/745. This report presents the approach taken by a French medical device manufacturer to comply with the clinical requirements of this behemoth.
Téléchargement

Remerciements
Mes plus profonds remerciements s’adressent à l’équipe d’Aesthetic Group et spécialement Jérôme Baptendier, pour m’avoir fait confiance en m’intégrant dans les projets de manière bienveillante et en renouvelant sa confiance dans le cadre de mon embauche au sein de l’entreprise.
Je remercie tout particulièrement ma responsable et tutrice Valérie BOQUET pour m’avoir accompagné, partagé son expertise mais aussi pour m’avoir laissé voler de mes propres ailes durant cette période d’alternance. Ce fut une expérience aussi enrichissante qu’agréable.
« Le meilleur manager est celui qui sait trouver les talents pour faire les choses, et qui sait aussi réfréner son envie de s’en mêler pendant qu’ils les font » T.Roosevelt.
Merci également à Xavier GROBON, Responsable Qualité pour ses lumières, son aide et son humour qui permet d’instaurer une ambiance joyeuse et créative au sein du service qualité/règlementaire.
Je souhaite bien évidemment remercier mes responsables master IDS, Mme Isabelle CLAUDE ainsi que M. Jean-Matthieu PROT, et M. Gilbert Farges pour leur disponibilité, leur conseil et leurs enseignements.
Grace à la qualité de la formation et de l’équipe pédagogique de l’UTC, j’ai pu construire un bagage professionnel me permettant de continuer à évoluer dans des projets passionnants.
Liste des abréviations
| ANSM | Agence Nationale de la sécurité du Médicaments et des Dispositifs médicaux |
| CER | Clinical Evaluation Report (Rapport d’Évaluation clinique) |
| CNIL | Commission Nationale de l’Informatique et des libertés |
| IC | Investigation clinique |
| MDCG | Medical Device Coordination Group |
| MDR | Medical Device Regulation 2017/745 (Règlement des Dispositifs médicaux 2017/745) |
| MEDDEV | MEDical DEVices Documents |
| ON | Organisme Notifié |
| RIPH | Recherche impliquant la personne humaine |
| RNIPH | Recherche n’impliquant pas la personne humaine |
| SCAC | Suivi clinique Après commercialisation |
| SNITEM | Syndicat national de l'industrie des technologies médicales |
| UE | Union Européenne |
Mémoire complet
Legacy device - Evaluation et suivi clinique sous le règlement 2017/745
Introduction
Contexte
La Communauté européenne est de plus en plus soucieuse de préserver la santé physique de ses citoyens en renforçant ses activités dans le domaine de la santé publique. C’est pourquoi afin d’amener une modernisation mais également une cohérence entre la législation européenne et la forte croissance des innovations de plus en plus sophistiquées en médecine, la Commission Européenne a adopté en mai 2017, deux nouveaux règlements. Ces deux règlements sur les dispositifs médicaux (2017/745) et les dispositifs médicaux de diagnostic in vitro (2017/746) ont pour objectif d’assurer une meilleure protection de la santé publique et de la sécurité des patients.
Ainsi ces deux règlements sont venus abroger les directives 93/42/CEE et 98/79/CE relatives aux dispositifs médicaux ainsi qu’aux dispositifs médicaux in vitro. Contrairement aux directives, ces règlements sont applicables sans transpositions au niveau national, néanmoins chaque État Membre doit revoir son droit afin de le mettre en conformité avec ces deux règlements. L’adaptation de la loi française au règlement 2017/745 (MDR) s’est effectuée par l’ordonnance °2022-582 du 20 avril 2022. Néanmoins, depuis l’entrée en application du MDR en mai 2021, les projets de recherche soumis en France pour évaluation ne relèvent plus de la loi Jardé.
Enjeux
Les dispositifs médicaux sont définis comme tout instrument, appareil, logiciel, matière ou autre article destiné par le fabricant à être utilisé chez l’homme pour diagnostiquer, prévenir, contrôler ou traiter une maladie [1] […] par le biais d’une action mécanique. Une très large gamme de produit répond à cette définition : du produit présent dans l’armoire à pharmacie tel que le pansement qui permet de recouvrir une blessure ou encore le thermomètre qui permet de prendre la température rapidement, jusqu’au produits les plus pointus utilisés dans les blocs opératoires tel que les robots chirurgicaux, assistants 2.0 des chirurgiens du 21ème siècle.
Tous ces produits, aussi bien ceux d’ores et déjà commercialisés sur le marché européen dénommés : legacy devices (ou dispositifs hérités) mais aussi ceux souhaitant l’être, doivent être conformes depuis mai 2021 à la nouvelle règlementation européenne 2017/745. Cependant les opérateurs économiques se heurtent à quelques difficultés et sont envahis d’une grande appréhension. Selon une enquête de Medtech Europe, la mise en œuvre de cette règlementation s’avère difficile pour une « majorité écrasante » de professionnels du secteur. Le processus de l’évaluation clinique s’inscrit ainsi dans le champ de consolidation du règlement et présente une difficulté supplémentaire pour les fabricants. Toutefois, cette difficulté peut s’atténuer en ayant une compréhension totale des attentes cliniques dans le cadre de la réglementation des dispositifs médicaux.
C’est dans ce contexte que s’inscrit mon année d’alternance chez Aesthetic Group, au sein du service Affaires Règlementaires.
Ce mémoire s’est articulé autour de 3 parties majeures. La première partie consiste à introduire le fabricant de dispositifs médicaux que représente Aesthetic Group, y compris les dispositifs médicaux concernés. Ensuite, dans une deuxième partie, le cœur du sujet : l’évaluation et le suivi clinique sous le règlement 2017/745 seront analysés puis ramenés à l’échelle de l’entreprise d’accueil afin de développer la stratégie clinique adoptée par celle-ci. Enfin, pour clôturer ce mémoire, une troisième partie, traitant des acquis personnels et professionnels durant le cursus du master sera établie.
I. Présentation de l'organisme et son environnement
1. Historique
Faisant partie des 93% PME de l’industrie du dispositif médical [2], Aesthetic Group voit le jour en 2008, néanmoins son histoire commence dès 1998 avec sa société mère, EMSI, fabricant de dispositifs médicaux, tels que des prothèses mammaires. Après le scandale des prothèses PIP, comme un bon nombre de fabricant, la société tourne la page des implants mammaires. C’est ainsi qu’est fondé Aesthetic Group, riche en expertise règlementaire et assurant plusieurs fonctions d’opérateurs économiques (figure1) au sens du règlement 2017/745. [1]
Comme l’ensemble des 1440 entreprises de l’industrie du DM, qui réalisent 1/3 de CA à l’export [2], la PME française a su s’imposer dans le marché de niche que représente le marché du tissu adipeux à l’échelle internationale et européenne. Leur certificat ISO 13485 obtenu depuis 2008 mais aussi leur marquage CE renouvelé chaque année, leur a permis d’acquérir la confiance des utilisateurs, et ainsi d’étendre leur marché, aux Etats-Unis en mettant en place la certification 510(k), mais aussi au Brésil avec l’ANVISA.
2. Organisation et analyse de la structure
Aesthetic Group regroupe au sein de ses locaux situés dans l’Oise 18 collaborateurs répartis dans différents services.
Dans l’objectif de mieux comprendre et appréhender l’établissement d’accueil, plusieurs outils d’analyses ont été utilisés :
- L’organigramme
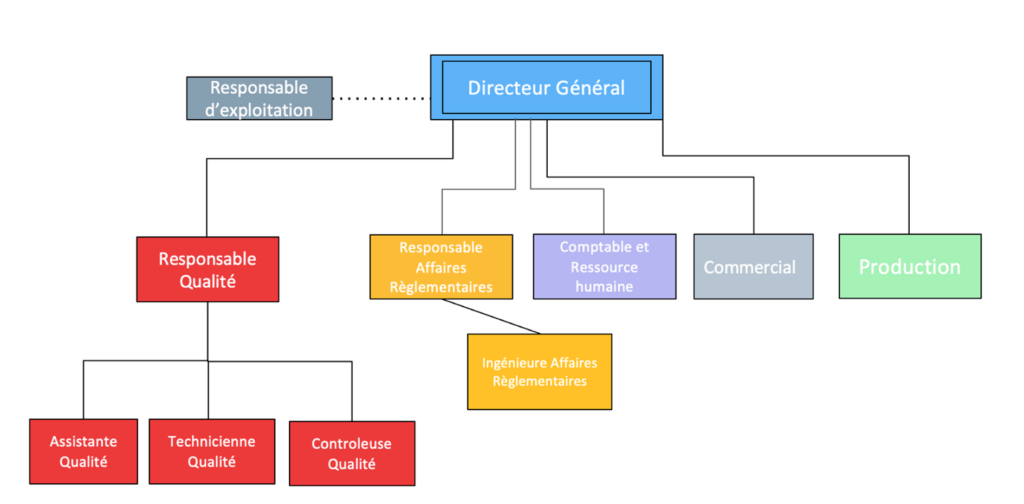
Cet outil permet de faire une représentation schématique (figure2) des liens ainsi que des relations fonctionnelles et hiérarchiques qui existent entre les individus.
- La matrice SWOT
L’acronyme SWOT a pour signification “Strenghts-Weaknesses-Opportunities-Threats” ce qui traduit en français correspond à “Forces-Faiblesses-Opportunités-Menaces”.
Cet outil (figure 3) a pour objectif de mettre en confrontation l’analyse externe de l’environnement de l’entreprise et l’analyse des ressources internes de celle-ci. Cela permet d’identifier les options stratégiques et ainsi définir les objectifs à atteindre.
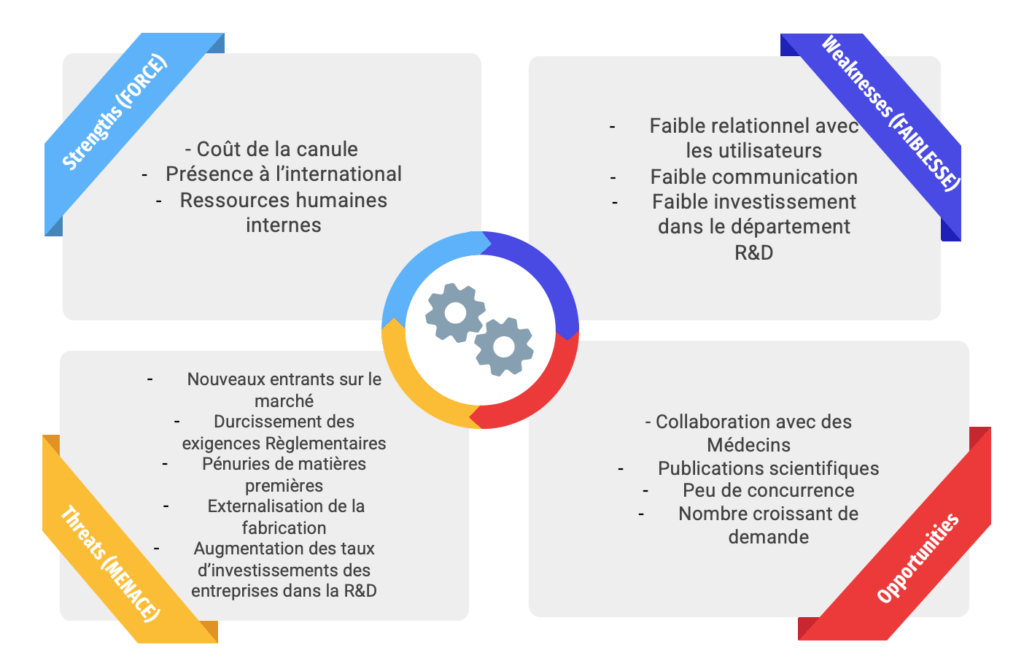
Les principales forces d’Aesthetic Group sont le prix de la canule, qui, par rapport à la concurrence est relativement inférieur. Leur deuxième force est leur présence à l’échelle internationale qui leur octroie un large panel de clients potentiels. Enfin, leur 3ème force est celle des ressources humaines, qui leur permet d’avoir des services structurés et expérimentés.
Quant aux faiblesses, elles concernent notamment le faible relationnel qui existe avec les utilisateurs, de type chirurgien, ne permettant pas de connaître par exemple leurs pratiques habituelles, et/ou leurs besoins.
L’environnement externe de l’entreprise comporte un bon nombre de menaces. Comme toute l’industrie du DM, le renforcement des exigences règlementaires a eu un impact conséquent sur les ressources allouées au service règlementaire. En effet, près de 80% des entreprises du secteur ont engagé des dépenses nouvelles pour se conformer aux nouvelles exigences règlementaires, 79% ont rationnalisé leurs gammes de produits et ainsi plus de 20% d’entre eux voit ce nouveau règlement comme une menace pour leur pérennité [2]. Néanmoins, transformer ce MDR en opportunité est possible, via l’opportunité de créer de nouvelles collaborations.
Le contexte socio-politique de ces dernières années a contribué à l’inflation des prix mais aussi à la pénurie des matières premières, nécessaires à la fabrication des dispositifs. De plus, l’externalisation d’une grande partie de la fabrication est un autre facteur de menace venant de l’extérieur. Une intégration de cette fabrication est une opportunité qui peut être envisagée par Aesthetic Group.
Une augmentation des taux d’investissements du CA des entreprises du DM dans le département R&D [2], tend à penser que les dispositifs seront de plus en plus pointus en termes de qualité, de sécurité et surtout d’innovation. Cette tendance peut amener Aesthetic Group à créer une opportunité en développant elle aussi sa cellule Recherche et Développement.
Toutefois, les opportunités s’offrant à Aesthetic Group ne sont pas en manque : l’environnement règlementaire actuel est propice pour approcher les praticiens en vue de potentielles collaborations, et ainsi inciter les médecins à publier leurs travaux scientifiques relatifs aux dispositifs de l’entreprise. De plus, le nombre de patients voulant bénéficier des opérations chirurgicales effectuées à l’aide des dispositifs proposés par Aesthetic Group est croissant. Ceci est illustré par l’augmentation annuel de 30% du CA de l’’entreprise depuis 2016.
- Le mix Marketing / 4P
Le mix marketing, aussi appelé la politique des 4P (figure 4), permet de représenter l’ensemble des actions et des stratégies déployées par une entreprise pour se positionner sur le marché. Si un produit répond aux besoins des consommateurs, il est susceptible d’exister puis de perdurer.
En effet, ce dernier n’achète pas le produit pour ce qu’il est, mais pour les fonctionnalités qu’il réalise et les bénéfices qu’il tire de son utilisation.
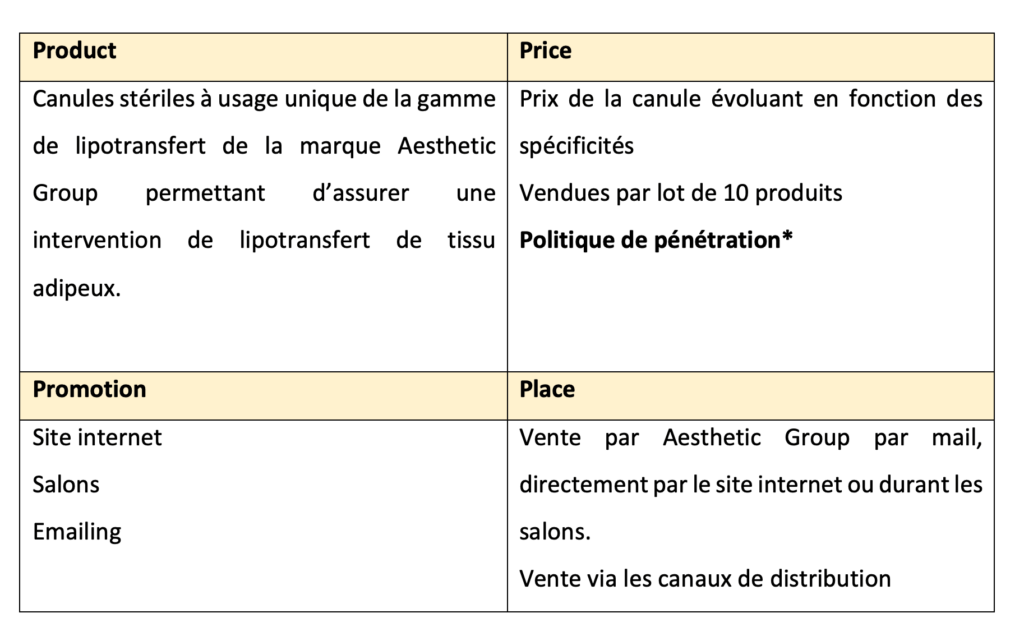
*Politique de pénétration : Aesthetic Group s’est appuyé sur la politique de pénétration, c’est-à-dire que les prix fixés sont plus bas que ceux des concurrents. Cette stratégie permet d’entrer sur le marché.
Place : plusieurs circuits de distribution sont présents pour les dispositifs d’Aesthetic Group. Le canal direct qui permet à l’entreprise de vendre directement au consommateur, le canal long qui fait passer le produit par les distributeurs mais également le canal moderne qui use des nouvelles technologies telles qu’internet ou catalogue. Ce canal est celui qui présente le plus d’avantages car permet au produit d’être disponible 24heures sur 24 et 7jours sur 7.
Aesthetic Group s’est orienté vers la distribution sélective, en d’autres termes la vente du produit n’est accordée qu’aux professionnels de santé.
Après avoir analysé et s’être approprié l’environnement interne mais aussi externe de l’entreprise. Abordons à présent l’essence même de l’entreprise : les canules de lipotransfert.
3. Les canules chirurgicales de lipotransfert
Parmi le portefeuille de produit d’Aesthetic Group, la première gamme à solliciter une certification au titre du MDR est la gamme des canules de lipotransfert. L’explication du terme « lipotransfert » ainsi qu’une brève description du dispositif médical qu’est la canule de lipotransfert est nécessaire.
a) Que savons-nous aujourd’hui du lipotransfert ?
Lipotransfert, lipofilling, transfert de graisse autologue ou encore « fat-grafting » sont les termes interchangeables retrouvés dans la littérature et qui définissent tous la procédure consistant à réutiliser du tissu adipeux préalablement prélevé sur une zone du corps, où elle est généralement en excès, dans le but de la réinjecter dans une autre partie corporelle du patient. Cette greffe présente de nombreux avantages, en effet la réinjection de graisse autologue présente une biocompatibilité accrue car dépourvue d’immunogénicité ce qui lui permet d’être le greffon de référence selon l’état de l’art actuel.
La première « autogreffe de tissu adipeux » remonte au 19ème siècle lorsqu’un chirurgien allemand, Gustav Neuber, afin de corriger des cicatrices faciales dues à une ostéomyélite (une infection des os causée principalement par des bactéries) eu l’idée de prélever de petits greffons de tissu adipeux du bras du patient de manière à le réinjecter au niveau orbital. [3]
Cette technique ne fait sa place qu’un siècle plus tard, dans les années 90, notamment grâce aux publications du Dr. Coleman, permettant de normaliser et d’optimiser la procédure de lipotransfert. En effet, la technique du chirurgien joue un rôle majeur dans le résultat obtenu, les résultats provenant d’études montrent des taux de résorption allants de 30 à 70% dans l’année de la greffe de tissu adipeux. [4]
La technique opératoire optimisée du lipotransfert
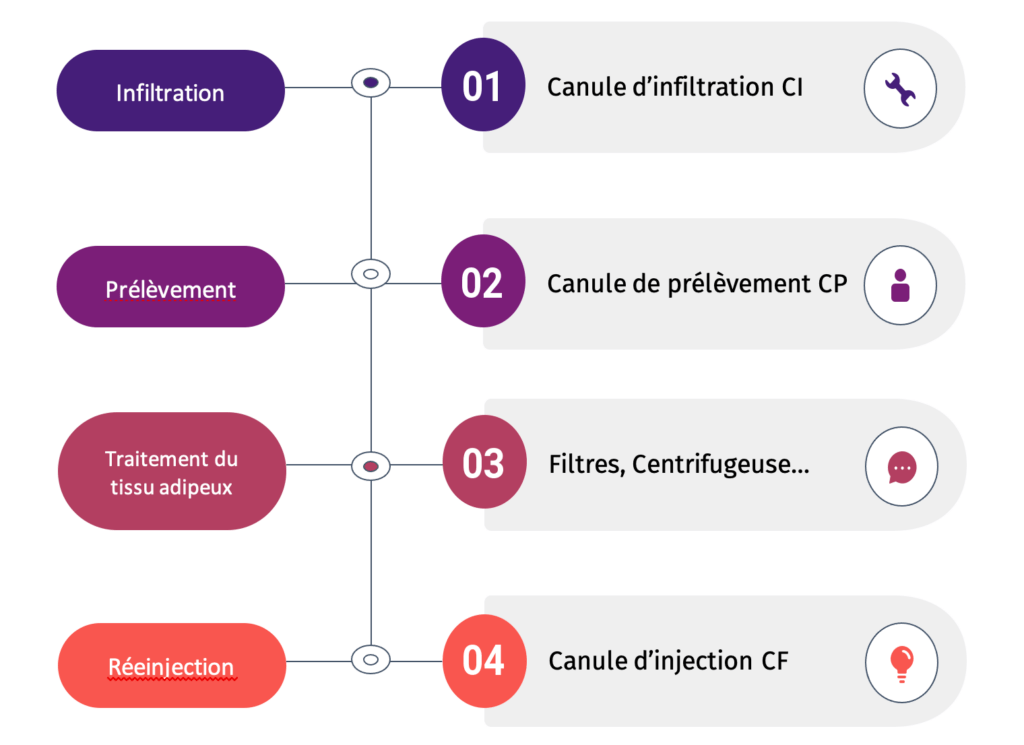
- La technique d’infiltration tumescente
L’infiltration tumescente représente la première étape de la procédure de lipotransfert, en effet cette étape est cruciale et représente en elle-même une technique particulière, c’est pourquoi un dispositif particulier est nécessaire : la canule d’infiltration (CI) (figure 5). Cette technique, mise au point par le Dr. Jeffrey Klein consiste à gonfler ainsi que rendre ferme les cellules adipocytaires par le biais d’une injection locale de solution de tumescence (figure 6) [5] [6]. Cette solution est un mélange généralement composé de lidocaïne, d’adrénaline ainsi que de bicarbonate de sodium ; à noter que les dosages ainsi que l’ajout de médicament(s) supplémentaire(s) sont à l’unique responsabilité et technique du clinicien.

Cette solution de tumescence permet de réduire les saignements, les hématomes et permet d’anesthésier localement le patient. Cette technique a pour avantage de permettre une pratique interventionnelle de lipotransfert en ambulatoire. [5]
Il faut noter que l’infiltration par tumescence est désormais utilisée dans d’autres indications médicales telles que la chirurgie des varices ou encore des tumeurs cutanées [7].
C’est en s’appuyant sur cette pratique courante des médecins qu’Aesthetic Group propose des canules d’infiltration (CI) (figure 7) comprenant de 8 à 24 orifices, offrant ainsi une diffusion tridimensionnelle du produit de tumescence. Plusieurs longueurs ainsi que diamètres appropriés à la zone à traiter sont disponibles : les petites zones auront besoin de canules allant de 100 mm à 150 mm de longueur et 1,25 mm à 2,5 mm de diamètre, contrairement aux zones plus importantes qui nécessiteront des longueurs pouvant aller jusqu’à 200mm.

- Le prélèvement
Suite à l’infiltration du tissu adipeux, le prélèvement, aussi dénommé « liposuccion » (figure 8) [8], peut être effectué manuellement à l’aide de canules (CP) spécifiques à la zone du tissu adipeux à extraire. Plusieurs études nous prouvent que la longueur, le diamètre, la taille mais également le nombre d’orifices de la canule joue un rôle majeur dans la réussite de l’étape de prélèvement des cellules adipeuses [9].
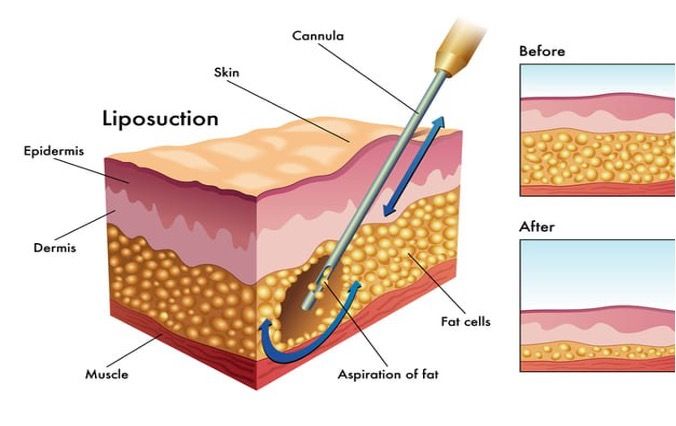
Des gestes brusques ainsi que des techniques électriques telles que les liposuccions laser ou ultrasonores augmentent les risques de destruction des cellules adipeuses. Une viabilité des adipocytes est indispensable afin d’avoir des greffons de meilleure qualité et ainsi un taux de résorption minimale de ceux-ci [4]. Les 40 références de la gamme de canules de prélèvement Aesthetic (figure 8) répondent à ce besoin d’adaptation de la canule à la zone à prélever.

- Le traitement du tissu adipeux
Une fois collecté, le tissu adipeux n’est pas apte à être un greffon viable. Le traitement du prélèvement est nécessaire puisque celui-ci contient non seulement des adipocytes mais aussi des fibres de collagènes, du sang, des débris cellulaires et des résidus de la solution d’infiltration [10]. Injectés dans l’organisme, ces éléments peuvent causer une inflammation du site receveur, ce qui peut être préjudiciable à la greffe postérieure. En plus de cette inflammation, le sang peut accélérer la dégradation du greffon et les débris cellulaires auront pour effet de fausser les volumes injectés, car absorbés après quelques heures après l’intervention par l’organisme [10]. Différentes méthodes de traitements (figure 10) du tissu existent selon l’état de l’art actuel :
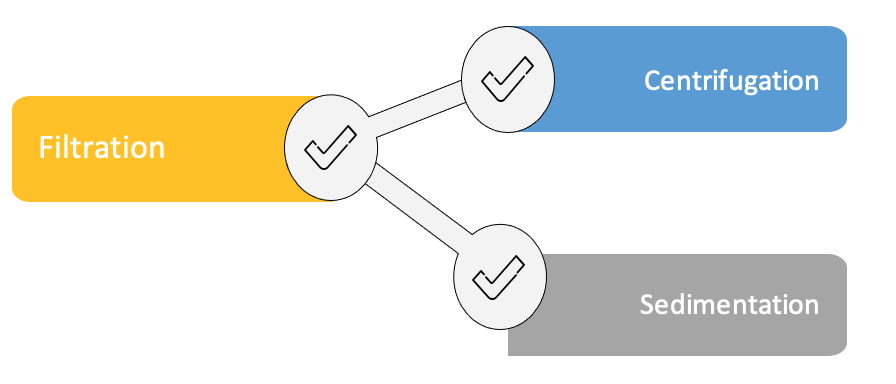
La centrifugation introduite par Dr. Coleman implique la séparation des composants lipoaspirés par centrifugation à grande vitesse. La séparation par sédimentation fait référence au processus consistant à permettre au lipoaspirat de se déposer dans ses trois phases (huiles, tissu adipeux et aqueuse) avec le temps par effet de la gravité. Le chirurgien extrait ensuite uniquement le tissu adipeux avec des seringues. Enfin, le tissu adipeux peut être filtré à l’aide d’un système fermé en passant via des membranes de différentes tailles[10]. D’après une étude comparative même si toutes les méthodes de préparation présentaient une baisse significative des débris cellulaires ainsi que des cellules sanguines, la méthode de filtration (figure 11) dans « un système fermé produit une greffe de graisse avec une viabilité tissulaire plus élevée et une présence plus faible de contaminants par rapport aux greffes préparées par d'autres méthodes » [11].
- La réinjection
A l’aide d’une canule d’injection (CF) (figure 13), le greffon purifié (figure 12) peut être injecté dans le site receveur du patient. Les diamètres, longueurs et orifices des canules d’injection sont, tout comme les canules d’infiltration et de prélèvement, adaptées à la partie de l’anatomie traitée. Toutefois, malgré l’évolution des techniques d’injection, il n’existe pas de consensus scientifique sur la meilleure méthode à utiliser afin d’obtenir une survie accrue des greffons liée à cette étape [12].

La procédure de lipotransfert expliquée, l’état de l’art concernant leurs indications peut être établi.
b) Les différentes utilisations du tissu adipeux
Bien que la vocation primaire de la greffe de tissu adipeux ait été purement esthétique, telle que la restructuration des formes corporelles et faciales, par exemple le comblement des volumes d’un visage dû au vieillissement. Les connaissances concernant le tissu adipeux se sont considérablement enrichies, avec l’avancée de la médecine, les chercheurs trouvent désormais des visées médicales à la réinjection de tissu adipeux.
- Les cellules souches provenant du tissu adipeux
Les cellules souches « adultes » représentent une source infinie de cellules différenciées ayant un intérêt pharmacologique et médical. Ces cellules souches dites multipotentes sont présentes dans les muscles, la moelle osseuse et également dans les vaisseaux sanguins. Néanmoins, celles-ci restent rares et difficiles à utiliser. Le prélèvement de graisse en grosse quantité, sans risque majeur pour le patient, constitue une « source possible et prometteuse de cellules souches multipotentes » [13]
Ces cellules appelées « human multipotent adipose derived stem » (hMADS) présentent les mêmes spécifiés que les cellules souches, c’est-à-dire un fort pouvoir d’auto-renouvellement mais aussi de différenciation cellulaire. [14]
D’après des travaux menés sur ces hMADS, celles-ci sont fonctionnelles et révèlent un potentiel thérapeutique dans les dystrophies musculaires, mais aussi dans la régénération de tout type de tissus, tel que cardiaque, neurologique ou cutané. [15]
D’autres indications thérapeutiques de l’injection de tissu adipeux sont d’ores et déjà établies, comme le démontre la publication « Adipose-derived stem cells for treatment of chronic ulcers : current status » [16] ou bien encore l’article « Adipose tissue-derived stem cells : a comparative review on isolation, culture, and differentiation methods » [15], qui permettent de conclure sur les bénéfices thérapeutiques qu’ont engendré le traitement des ulcères gastriques par le biais des cellules souches dérivées du tissu adipeux. [17]
- Reconstruction mammaire suite à une mastectomie
L’HAS en 2018 comptabilisait près de 100 000 actes de mastectomies dont de 22 000 mastectomie totale. Cette opération chirurgicale est la plus recommandée afin d’éliminer la tumeur lorsqu’un cancer du sein est détecté, en ôtant tout le tissu atteint [18].
« La Santé est un état de complet bien-être physique, mental et social et ne consiste pas seulement en une absence de maladie ou d’infirmité ». (“Constitution - World Health Organization”). C’est sur ce fondement que s’appuie la visée médicale de la reconstruction mammaire. La perte d’un sein modifie profondément l’apparence physique et l’image de soi, c’est pourquoi sa reconstruction est prise en charge à 100% par l’assurance maladie et représente donc un acte médical [19]. Plusieurs chirurgies réparatrices sont disponibles, tel que la pose de prothèses mammaires et l’autogreffe de tissu adipeux. L’avantage que procure cette autogreffe est l’aspect naturel et sans cicatrice. Toutefois, plusieurs séances sont nécessaires [18] [20] . Des contre-indications pour cette autogreffe ont été retenues par l’HAS : « l’absence de rémission locale, une maladie métastatique non contrôlée et un délai de moins de 2 ans après complétion des traitements locaux lorsqu’il existe un fort risque de récidive locale du cancer du sein ». [20]
- Traitement des plaies
Le tissu adipeux est depuis près d’une décennie utilisé pour traiter les plaies profondes. Entre 2013 et 2015, une étude regroupant 10 patients d’une moyenne d’âge de 70 ans présentant une plaie hypersensible et profonde dans la jambe ont bénéficié des effets curatifs de la greffe de tissu adipeux. [21]
D’autres études ont été menées pour confirmer ce pouvoir thérapeutique de la réinjection de graisse dans les plaies, et celles-ci ont pu démontrer une diminution de la douleur associée tout en réduisant le diamètre et la profondeur de la plaie. Près de 60% des cas ont présenté une guérison complète de la plaie.
II. L'évaluation et le suivi clinique sous le règlement 2017/745
1. Contexte de l'évaluation clinique et son suivi
Afin d’obtenir le graal que représente le certificat CE au titre du règlement 2017/745 (MDR), les fabricants doivent passer par un bon nombre d’étapes, plus ou moins critiques, afin de fournir une documentation technique conforme au dit règlement à l’organisme notifié (ON). Ce même organisme a pour objectif d’évaluer la sécurité ainsi que les performances du dispositif dont il est question. Comme évoqué précédemment, la Commission Européenne souhaite par le biais de ce nouveau règlement renforcer les exigences afin d’offrir une sécurité optimale aux patients. Ce désir émane des nombreux scandales de ces dernières décennies liés aux dispositifs médicaux. À titre d’exemple, l’affaire PIP en 2010 qui fit trembler toute l’industrie du DM avec 30 000 femmes, victimes de prothèses mammaires remplies de gel de silicone industriel [22]. Ces dispositifs médicaux, qui présentaient « une fragilité à l’allongement et à la rupture », étaient bien loin du gel revendiqué dans les dossiers de conception. 10 ans après la liquidation du fabricant, plus de 5 000 dysfonctionnements ont été signalés à l’ANSM. [23]
Parmi les critères d’évaluations renforcés, le clinique est un point majeur. Et pour cause, la « confirmation de la conformité aux exigences générales en matière de sécurité et de performances ainsi que l'évaluation des effets secondaires indésirables et du caractère acceptable de la balance bénéfice/risque sont fondées sur des données cliniques apportant une preuve clinique suffisante » [24]
Afin d’apporter ce niveau de preuve suffisant, le fabricant doit alors planifier, réaliser ainsi que documenterune évaluation clinique relative à son dispositif. [1]
2. L'évaluation clinique et son évolution depuis la directive
L’évaluation clinique n’est pas un concept nouveau. Il en était déjà question dans la directive relative aux dispositifs médicaux 93/42/CEE (MDD), mais également dans la norme ISO 13485. Il apparait alors en toute logique dans le nouveau règlement relatif au dispositif médicaux, et est de rigueur pour toutes classes et types de dispositifs, y compris pour les dispositifs n’ayant pas de destination médicale (Annexe XVI).
Ainsi, une évaluation clinique est le terme employé pour décrire un processus dans lequel toutes les données pertinentes concernant un dispositif médical sont collectées, analysées puis évaluées. L’examen de cette évaluation clinique permet de déterminer s’il y a suffisamment de preuves pour démontrer la conformité aux exigences règlementaires.
Ce nouveau texte règlementaire a permis d’éclairer les fabricants sur les exigences en matière d’évaluation clinique. Le MEDDEV 2.7/1 datant de juin 2016 traitant de l’Évaluation Clinique à l’intention des fabricants et des ON introduisait le concept de Plan d’Évaluation Clinique (CEP) mais ne développait pas suffisamment le sujet. Le MDR dans l’annexe XIV partie A énonce clairement les exigences requises dans le CEP :
- La destination du dispositif ;
- Le groupe cible y compris les indications et contre-indications ;
- Les bénéfices cliniques ;
- Les méthodes et paramètres utilisés pour déterminer l’acceptabilité de la balance bénéfice/risque ;
- Les risques résiduels et effets secondaires.
Le MDR exige également qu'un plan de développement clinique soit inclus dans le plan d'évaluation clinique, comprenant une vision à long terme des enquêtes cliniques prévues liées au produit, allant des études de faisabilité aux études de suivi clinique après la mise sur le marché.
Les résultats du processus du plan d’évaluation clinique (CEP) seront centralisés dans le rapport d’évaluation clinique (CER), ce qui conduit alors à leur analyse et aux conclusions qui seront ensuite consignées et documentées dans le rapport (CER).
Tandis que la partie A de l’annexe XIV traite de la partie « pré-commercialisation » du dispositif, la deuxième partie, la partie B concerne une grande nouveauté du règlement : le suivi clinique post-commercialisation(SCAC) ou Post Market Clinical Follow-Up (PMCF). Le SCAC, rendu obligatoire aux legacy device dès l’entrée en application du MDR en mai 2021, est défini comme étant le processus continu de mise à jour de l’évaluation clinique s’inscrivant dans le plan de SCAC établi par le fabricant [1]. Cette surveillance clinique vise à mettre à jour durant toute la durée de vie du dispositif l’évaluation clinique de manière proactive, et doit être inclue dans le plan de surveillance après commercialisation. Ce SCAC, permet entre autres d’avoir une balance bénéfice/risque actualisée, une performance clinique à jour mais permet aussi d’améliorer l’utilisation, la performance et la sécurité du dispositif.
Comment collecter des données cliniques ?
Le règlement énonce 3 différentes façons d’obtenir ces données cliniques nécessaires à l’évaluation clinique :
- La voie de la littérature scientifique
La voie de la littérature scientifique doit être pertinente et relative à la conception, à la destination prévue, à la sécurité ainsi qu’à la performance : Cette voie est la première chronologiquement parlant mais aussi celle privilégiée par les fabricants, car c’est économiquement la moins coûteuse. Elle a pour objectif de collecter les données existantes dans l’état de l’art. Cette première phase de collecte est cruciale et permettra de déterminer la suite de l’évaluation clinique. Ainsi, chaque document consulté, aussi bien ceux retenus que ceux exclus, seront consignés dans le CER.
Une fois cet état de l’art établi, le fabricant pourra, selon les données recueillies, conclure sur l’innocuité ainsi que la performance de son dispositif au sein de son CER. Toutefois, si celles-ci ne sont pas suffisantes, 2 orientations sont envisageables pour le fabricant :
- L’équivalence ;
- Les données propres à son dispositif.
- La voie de la l'équivalence
Dans le cas où un dispositif ayant les mêmes caractéristiques cliniques, techniques et biologiques existe sur le marché, une équivalence peut être revendiquée. Bien que cette voie ait été celle privilégiée par les fabricants sous la directive 93/42/CEE, car elle permettait d’obtenir le marquage en se basant sur les données cliniques d’un autre dispositif médical marqué CE. Dans l’ère du règlement l’équivalence est devenue un des aspects le plus complexes à mettre en place. Toujours dans l’idée de garantir la sécurité du patient, le nouveau règlement réduit drastiquement la possibilité de s’appuyer sur cette voie de récolte de donnée clinique.
Les caractéristiques cliniques, techniques ainsi que biologiques ne doivent pas présenter de « différence cliniquement significative ». L’objectif étant de démontrer que le dispositif évalué est substantiellement équivalent en termes de sécurité et de performance clinique du dispositif. De ce fait, le fabricant doit prouver l’accès total à la documentation technique du dispositif équivalent via un contrat : « les deux fabricants ont conclu un contrat qui accorde explicitement au fabricant du second dispositif un accès total et permanent à la documentation technique ». Ce qui parait pratiquement impossible dans un contexte concurrentiel.
Ainsi le nouveau règlement, tend à pousser les fabricants à générer eux-mêmes les données propres à leur produit.
- Les données propres au dispositif
Les méthodes pour générer des données cliniques sur un dispositif ne sont pas les mêmes en fonction de la présence sur le marché du dispositif ou non mais également en fonction de la classe de risque.
- Investigation clinique
En règle générale, comme précisé dans l’annexe XIII, tous les dispositifs implantables et de classe III doivent avoir recours à une investigation clinique afin de générer des données propres, à ne pas confondre avec l’évaluation clinique, qui elle concerne toutes les classes. L’investigation clinique concerne tout projet de recherche clinique impliquant la personne humaine (RIPH) portant sur les produits mentionnés à l’article 1 du MDR (DM, accessoires, produits sans finalité médicale) [1].
Quelques exceptions exemptent les dispositifs de l’investigation clinique, telle que faire partie de la liste des exceptions définies dans le règlement. Malgré les exigences strictes en matière de preuves cliniques suffisantes, le MDR n'indique pas explicitement quand des investigations cliniques sont requises pour les dispositifs des classes I, IIa et IIb. En 2020, la norme d’investigation clinique sur les DM pour sujets humains – Bonnes pratiques cliniques ISO 14155 : 2020 est devenue une norme harmonisée, elle permet entres autres de définir les bonnes pratiques cliniques [25],mais ne donne pas plus d’indication quant au recours de l’investigation clinique pour ces différentes classes.
Mener une investigation clinique n’est pas la voie la plus facile car implique de prendre en compte un délai assez important, afin de recevoir pour les classes les plus à risque, l’approbation de l’État Membre dans lequel l’investigation sera menée, cela sera en occurrence auprès de l’ANSM en France. L’écriture du protocole d’investigation clinique, la sélection mais aussi l’accord des centres dans lesquels vont avoir lieu l’étude vont repousser d’autant plus le début de l’étude. S’ajoute à ce délai, le cout du processus d’investigation clinique, en effet celui-ci s’élève en moyenne à une centaine de milliers d’euros en fonction de la durée, de la rémunération des participants mais aussi en fonction du caractère multicentrique ou non. Une étude multicentrique, c’est-à-dire qui a lieu simultanément dans plusieurs centres, augmente les couts, cependant c’est celle à privilégier car elle fait appel à plusieurs investigateurs, avec des patients d’origines géographique, sociale et ethnique différentes et permet donc d’augmenter la qualité et la vitesse de l’IC [26].
Pour éviter de passer par le processus long et fastidieux que représente une investigation clinique, d’autres processus de collecte de données « plus légères » sont possibles, comme la recherche n’impliquant pas la personne humaine (RNIPH).
- RNIPH
Les RNIPH représentent toutes les recherches ou encore études, menées exclusivement à partir de données et n’impliquant aucune intervention sur une personne humaine. Ce type d’étude est moins lourde administrativement parlant que l’IC, puisqu’il n’est pas soumis à l’examen de l’ANSM ni du CPP. La loi de modernisation du Système de santé français du 26 janvier 2016, prévoit un cadre législatif et règlementaire pour faciliter l’accès aux données de santé via l’introduction du Système National des données de santé (SNDS). Dans l’objectif d’évaluer les projets, la plateforme en ligne Health Data Hub [27] permet aux fabricants de déposer leur dossier, qui est ensuite examiné par le Comité éthique et scientifique pour les recherches, les études et les évaluations dans le domaine de la santé (CESREES). S’ajoute à cette loi, l’étude RNIPH qui devra également prendre en compte la règlementation relative à la protection des données personnelles (RGPD) dont le respect est contrôlé par la CNIL.
L’accès à ces données personnelles est soumis à plusieurs conditions :
- L’accès à ces données doit être strictement nécessaire afin de répondre aux finalités de la recherche, de l’étude ou de l’évaluation
- L’utilisation des données doit répondre à un motif ou à une finalité́ d’intérêt public. La notion d’intérêt public n’est pas définie par la règlementation mais il ressort des échanges lors de sa mise en place que les recherches, études ou évaluations menées en vue de mettre ou de maintenir sur le marché́ un dispositif médical et d’en obtenir le remboursement, ainsi que les études visant à̀ évaluer l’amélioration de la qualité́ des soins, notamment, répondent à̀ cet intérêt public. [28]
Ce type d’étude est uniquement possible pour les legacy device, au vu de son caractère rétrospectif.
Qui doit effectuer l’évaluation clinique ?
Le MDR dans l’article 61, détaille les compétences et l’expérience que doivent posséder les personnes chargées d’établir le CER. Dans les grandes lignes, les évaluateurs doivent être capable d’effectuer des recherches méthodologiques et de maitriser la technologie du dispositif médical [1].
Ces évaluateurs doivent avoir des connaissances précises concernant le dispositif en question et son application, être dans la capacité de diagnostiquer, traiter l’état clinique dans lequel s’inscrit l’utilisation du dispositif médical mais aussi être au fait des méthodes de traitement courant de celui-ci (état clinique).
3. La stratégie clinique au sein d'Aesthetic Group
a) Une double visée
D’après la partie I. b), il peut être noté qu’il existe deux visées aux canules de lipotransfert : la visée esthétique et médicale. Le règlement 2017/745 ayant introduit les dispositifs n’ayant pas de finalité médicale (annexe XVI), exige dans ce cas uniquement une démonstration de la performance de celui-ci et non pas de démonstration de bénéfice clinique.
Certifier son dispositif selon cette annexe XVI, représente donc un gain de ressources tant humain que pécunier concernant la partie clinique pour les fabricants commercialisant des dispositifs à visée non médicale. Toutefois, le retard de publication des spécifications communes relatives à ce groupe de dispositif, permettant aux organismes notifiés d’évaluer la conformité au MDR des dispositifs sans finalité médicale, a contraint Aesthetic Group à s’orienter vers la stratégie de mise en conformité des dispositifs médicaux. Ainsi, l’évaluation clinique s’oriente vers les revendications médicales.
b) Le cas des legacy devices
Les dispositifs d’Aesthetic Group étant estampillés CE au titre de la directive, ils sont considérés comme des legacy devices. Pour ce type de dispositif, le guide MDCG 2020-6 - Preuves cliniques nécessaires pour les dispositifs médicaux précédemment marqués CE en vertu des directives 93/42/CEE ou 90/385/CEE [29] fournit aux fabricants les orientations sur l’évaluation clinique, afin que celle-ci soit en conformité avec le règlement. Par ailleurs, ce guide permet de préciser la notion de preuve clinique suffisante, nécessaire afin de démontrer la conformité aux exigences générales de sécurité et de performances (EGSP) mais également la notion de donnée clinique.
Durant ce long périple que représente la mise en conformité selon le règlement 2017/745, le service règlementaire s’est vu challenger plusieurs fois.
Tout d’abord par l’évaluation clinique, qui au titre du MDR, exige des données de performances beaucoup plus poussées que sous la directive. Ainsi, les canules de lipotransfert n’ayant pas de normes spécifiques relatives à leur performance, il a fallu rechercher dans la littérature mais aussi être créatif afin de mettre en place des protocoles d’essai afin d’effectuer les tests de performance adéquats aux dispositifs. Ces essais de performances se sont avérés utiles pour alimenter aussi bien l’évaluation clinique que le dossier de conception afin de se coller à la réalité de l’utilisation des dispositifs.
Anticiper est le mot d’ordre de ce nouveau règlement, et pour cause la mise en place des processus et l’obtention des résultats escomptés prennent du temps. La conclusion du CER doit être anticipée puisque celle-ci nous orienterait ou non vers des études cliniques post marché, et nécessiterait alors de collecter les données manquantes. Dans notre cas, la rédaction du CER a pris environ 18 mois. L’anticipation a été faite par le déploiement de plans d’actions (figure 14) pour chaque gamme de dispositifs médicaux, cela a permis de définir l’action à effectuer, la date à laquelle celle-ci doit être finie, la personne chargée de l’action, son état d’avancement et les éventuels commentaires. Un code couleur a été défini afin de connaitre les documents restants en un coup d’œil.
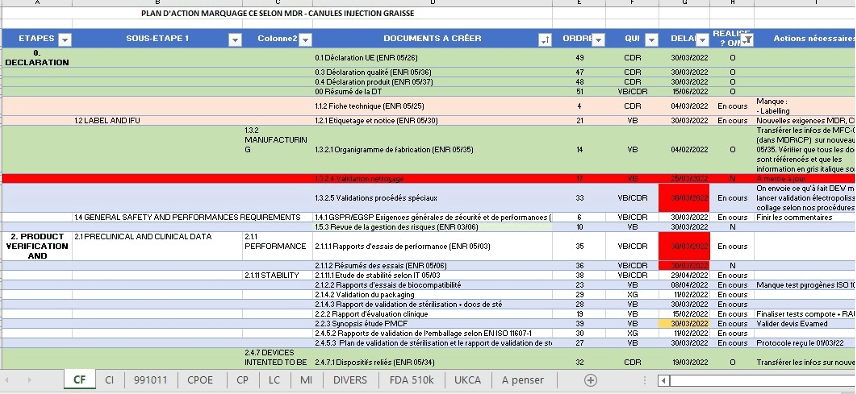
Malgré le fait que les canules soient sur le marché depuis près d’une dizaine d’année, et qu’un système de surveillance post marché via des questionnaires utilisateurs ait été déployé, le manque de données cliniques relatives aux dispositifs a été établi par le CER. Les questionnaires n’ont reçu que très peu de réponse, ce qui ne permet pas d’apporter des preuves cliniques suffisantes. Ce faible taux de réponse est un axe à améliorer via la stratégie de collaboration avec les utilisateurs (voir Figure 3 : Matrice SWOT (source : autrice)
Face à ce besoin de générer des données, choisir la méthode a été fastidieux. Comme expliqué précédemment, les canules étant des dispositifs de faible classe (IIa), ils ne sont pas soumis à l’obligation de mener une investigation clinique. C’est pour cette raison qu’une prospection des différents experts disponibles sur le marché en matière de collecte de donnée clinique s’est effectuée.
Après quelques entretiens avec différents prestataires, de stratégies différentes et de prix allants de 80 000€ à plus de 220 000€, la date de revue des dossiers se rapprochant, le choix de l’entreprise s’est porté sur l’Investigation Clinique.
Pourquoi choisir de mener une Investigation Clinique (IC) ?
Même si, l’investigation clinique représente un coût important en termes de ressources humaines et financières ; appréhender le niveau d’exigence des organismes notifiés sur ce nouveau règlement reste compliqué et difficilement prévisible.
Avec un nombre de seulement 8% de certificats au titre du MDR délivré en un an, mais également un délai de certification de 13 à 18 mois [30], la manœuvre pour une prise de risque est très étroite.
Mener une investigation clinique permet de garantir l’obtention de résultats qualitatifs concernant les critères définis dans le protocole d’investigation clinique [29]. A contrario, les études n’impliquant pas la personne humaine, type étude rétrospective, permettent d’obtenir uniquement les données déjà existantes dans les dossiers patients, et de qualité moindre. Dans le cas où, ces données cliniques récoltées ne sont pas suffisantes pour démontrer la sécurité et la performance du dispositif, le fabricant sera contraint de recommencer le processus de récolte de donnée, et sans doute se résoudre à s’orienter vers une IC, avec pour conséquence, une perte de temps et de budget pour la certification. Néanmoins, pour une PME, à l’image d’Aesthetic Group, une IC constitue un investissement important, près de 5% du CA.
Toutefois, plusieurs leviers existent pour encourager cet investissement, dont un des plus importants :
- Une investigation clinique est une opportunité commerciale [31]
Ce premier levier se base sur la forte collaboration qui va s’établir entre le fabricant et tous les cliniciens impliqués dans l’étude clinique. En effet, cette IC vient compenser le manque de ressource humaine présent sur le terrain aux côtés des utilisateurs (type ingénieur technico-commercial, ou ingénieur d’application). Les chirurgiens inclus dans ce processus pourront faire la promotion de ces travaux entrepris avec Aesthetic Group via leurs réseaux tel que les congrès professionnels, les masters class mais aussi et surtout via ses réseaux sociaux tel que LinkedIn, qui regroupe un bon nombre de potentiels clients. C’est pourquoi choisir des leaders d’opinions en tant que médecins coordinateurs peut être une stratégie intéressante. Accompagner et motiver les médecins dans cette démarche de communication est essentiel.
S’ajoute à cette communication, la publication de ces travaux qui sera une force sur laquelle pourra s’appuyer le département Marketing de l’entreprise afin de promouvoir les dispositifs concernés. Un produit apparaissant dans la littérature scientifique apporte une certaine confiance et est souvent gage de qualité pour les futurs utilisateurs.
- Avoir une meilleure connaissance de son dispositif
L’IC permet également d’améliorer l’utilisation en vie réelle des dispositifs, ce qui permet aux fabricants d’alimenter la gestion des risques mais aussi l’aptitude à l’utilisation. En effet, ces données émanant d’utilisateurs seront indispensables à l’analyse des risques d’utilisations des dispositifs.
En somme le fabricant aura établi de solides relations avec ses utilisateurs, des publications sur son dispositif, aura une vision utilisation dans les conditions réelles du dispositif mais surtout aura généré des données qualitatives pour évaluer la sécurité et la performance du produit.
Comment motiver les médecins à participer à une investigation clinique ?
- Trouver un sujet pour lequel ils portent de l’intérêt afin d’établir, comme dans toute relation, un compromis gagnant-gagnant.
- Une rémunération attractive : de l’ordre de 200 à 400€ par patient suivi, en fonction de la durée mais aussi des actes supplémentaires à réaliser.
c) La stratégie d’opter pour des sous-traitants
Face à la difficulté que représente l’obtention du marquage CE au titre du MDR mais également face au renforcement des exigences règlementaires qui requièrent de plus en plus d’expertises, les fabricants ont de plus en plus recours à des compétences externes. Selon l’enquête annuelle de 2021 du SNITEM, un fabricant de dispositif médical compte en moyenne 15 sous-traitants [2]. Les sous-traitants des DM traitent de nombreux cœurs de métiers, tels que la stérilisation, la conception.
Dans le cas de la partie clinique d’Aesthetic Group, celle-ci a été menée en totalité par des experts externes à l’organisation. En effet, comme démontré dans la partie b), l’évaluation clinique est chronophage et exige une certaine compréhension des attendus règlementaires en termes d’aspect clinique. Ainsi, pour la rédaction du rapport d’évaluation clinique (CER), un bureau d’étude spécialisé dans la règlementation des DM a été inclut dans le processus. Ces experts ont alors pris en charge la rédaction du plan d’évaluation clinique ainsi que le rapport d’évaluation clinique. De plus, le MDR requiert des personnes compétentes en matière d’expertise clinique, la pertinence de leur participation à l’évaluation doit être justifiée. [1]
Quant à l’investigation clinique, la collaboration avec une société de recherche clinique (CRO) s’est avérée indispensable. En effet, dans une industrie française où 93 % [2] des entreprises comptent moins de 250 salariés, inclure un CRO est primordial. Les CRO regroupent très souvent toutes les compétences nécessaires pour mener à bien une investigation clinique : Attaché de recherche clinique, Data Analyst, Biostatisticien [32]. Cette organisation a alors pris en charge la globalité de l’étude clinique :
- La rédaction du protocole d’investigation clinique
- Les soumissions règlementaires auprès du CPP, ASNM et CNIL
- Le pilotage et le suivi de l’investigation clinique
- Suivi des patients et saisie des données
- Analyse statistique des données et rédaction du rapport statistique
- Rapport d’investigation clinique
Choisir de manière optimale ses sous-traitants
Le choix des sous-traitants n’est pas à faire la légère, un bon nombre d’entre eux couvre dans le même temps le domaine pharmaceutique et les dispositifs médicaux. Même si la règlementation des DM est inspirée de celle des médicaments, notamment pour la partie clinique, ils n’en sont pas moins différents. A titre d’exemple les recherches cliniques portant sur un dispositif médical ne doivent pas être abordées de la même façon que des recherches cliniques sur un médicament, qui eux sont menés en double aveugle, ce qui est totalement différent des études réalisées sur un dispositif médical [31]. C’est pourquoi Aesthetic Group traite uniquement avec des sous-traitants spécialisés et expérimentés dans le domaine des dispositifs médicaux. S’ajoute à cette première sélection, les disponibilités des deux parties. En effet, avec 500 000 DM devant être en conformité selon le MDR avant 2024, les experts de l’industrie des DM sont surchargés de demandes de la part des fabricants. Ainsi, les demandes dépassant les offres disponibles sur le marché, le coût des services est répercuté par cette demande qui ne cesse de croitre. Ce qui nous amène à un troisième critère de sélection, le prix. A titre d’illustration, nous avons pu constater une hausse de plus de 40% du prix d’une même prestation de service de 2019 à 2022.
Retour d’expérience sur la mise en place de l’investigation clinique
Le cas des dispositifs d’Aesthetic Group est un cas particulier mais qui est tout de même assez répandu dans le monde du dispositif médical. Comme vu dans la partie I. explication des dispositifs médicaux, les canules n’ont pas de bénéfice clinique « direct » sur le patient. Contrairement à un pacemaker qui va envoyer de petites décharges électriques au cœur afin de remédier aux bradycardies. La canule sert uniquement de vecteur à la récolte et l’injection du tissu adipeux, c’est ainsi le tissu adipeux qui conférera aux patients les bénéfices cliniques escomptés.
Un dispositif médical devant présenter un intérêt clinique pour le patient, les bénéfices cliniques issus de la littérature et énoncés dans le CER sont la satisfaction patient et la satisfaction utilisateur. La satisfaction patient après une procédure de lipotransfert ayant un impact sur sa qualité de vie, celle-ci représente bel et bien un bénéfice clinique [33]. C’est pourquoi les critères principaux de l’investigation clinique présentés dans le plan d’investigation clinique sont la satisfaction patient et utilisateur.
C’est à ce moment-là, que les premiers doutes quant à la pertinence d’investir du temps mais surtout une somme colossale dans cette IC apparaissent. De plus, la satisfaction est un critère qui reste tout de même subjectif selon plusieurs facteurs : la préparation du tissu adipeux, la technique opératoire du chirurgien, mais dépend aussi des différentes niveau d’attentes des patients.
Ces doutes se renforceront lorsqu’il a fallu trouver puis convaincre un médecin coordinateur de l’étude.
Comme nous l’avons évoqué, attiser l’intérêt des chirurgiens est nécessaire pour le bon déroulé de l’étude. Proposer une investigation clinique ayant une durée d’un an afin d’enregistrer uniquement la satisfaction des patients ainsi que des utilisateurs n’a pas réellement d’intérêt thérapeutique, diagnostic, ou tout simplement clinique. Ainsi, le but de cette investigation clinique est uniquement de générer des données pour le marquage CE.
En expliquant au corps médical qu’en cas de non-récolte de ces données de satisfaction, l’accès sur le marché de nos canules ne serait plus possible mais aussi en jouant sur le levier de la rémunération, il nous a été possible d’inclure un chirurgien coordinateur. La rédaction du plan d’IC et l’intégration d’un médecin coordinateur en accord avec l’étude a été réalisé en l’espace de 4 mois. Notre investigation étant multicentrique, un autre défi est à relever : trouver des médecins parmi nos clients intéressés pour participer à l’IC. Réussir à approcher ces médecins reste très compliqué, puisque dans la majorité des cas ce sont les Achats ou les Pharmacies Hospitalières qui passent les commandes, de ce fait nous n’avons pas les contacts directs de nos utilisateurs. Mais aussi, susciter leur intérêt requiert de monopoliser leur temps afin de mettre en place des réunions, chose difficile pour des plasticiens pour lesquels le rendement est le mot d’ordre. En parallèle de cette recherche de participant, le CRO effectue les soumissions règlementaires auprès des autorités compétentes afin d’avoir le top départ de l’inclusion des patients, presque un an après la prise de décision de mener une investigation clinique.
Les leçons qu’on peut tirer de cette première étape de l’investigation clinique :
- Anticiper au maximum l’évaluation clinique et les études qui en découlent si nécessaire
- Être en contact avec les utilisateurs afin d’avoir un accès plus facile et rapide
Pas totalement convaincu par le réel intérêt d’une investigation clinique pour les canules de lipotransfert, cette voie de collecte de donnée s’est rapidement écartée de la stratégie clinique des autres canules d’Aesthetic Group.
Stratégie clinique des autres dispositifs de l’entreprise
Pour en faire une brève description, les 2 autres dispositifs médicaux de l’entreprises, sont des canules de liposuccion et des micro-canules pour comblement par filler synthétique (type acide hyaluronique, acide polyactique) ou autologue (tissu adipeux). Ils présentent le même type d’indication que les canules de lipotransfert mais surtout les mêmes bénéfices cliniques, la satisfaction patient et utilisateur. C’est pourquoi, pour éviter de repasser par la case investigation clinique, la stratégie d’étude RNIPH est en cours d’implémentation. Cette mise en place suit le même plan d’action que celle de l’investigation clinique, c’est-à-dire l’entretien et le demande de devis auprès des différents prestataires de ce type de service. Cette stratégie requiert encore une fois une collaboration avec les utilisateurs des canules d’Aesthetic Group. Mais cette fois ci, cette collaboration a lieu en amont de l’étude, afin d’analyser la faisabilité du projet via des questionnaires auprès de tous les utilisateurs dans l’objectif de connaitre : leur potentielle participation à l’étude, la nature des données collectées dans les dossiers patients mais aussi le nombre d’interventions effectuées dans un délai imparti. Cette première étape menée en interne permettra de savoir si la stratégie rétrospective est viable, ou si le recours à l’investigation clinique est indispensable afin d’obtenir les données cliniques manquantes.
Que ce soit dans le cadre d’une étude prospective telle que l’investigation clinique ou l’étude rétrospective, établir une collaboration avec les utilisateurs est nécessaire afin de collecter les données cliniques nécessaires à la conformité aux exigences essentielles du règlement.
III. Bilan personnel et professionnel de l'expérience professionnelle
1. Compétences et comportements
a) Acquis
Cette première expérience dans le domaine des affaires règlementaires a été très instructif car intégrée chez un fabricant de DM de type PME j’ai pu exploiter tous les aspects règlementaires. De la conception au suivi post marché, cette année d’alternance a été une opportunité au vu de la diversité des missions, d’acquérir des connaissances et compétences approfondies (figure 14) sur le métier d’ingénieur affaires règlementaires. L’un des premiers acquis a été l’appropriation du règlement européen et des normes associées, compétence indispensable pour le métier visé. En effet son application sur des dispositifs m’a permis de mieux appréhender les exigences règlementaires et normatives. Ensuite, il m’a fallu maitriser les aspects plus techniques du dossier de conception mais aussi l’aspect clinique des dispositifs. Tous ces savoirs et savoir-faire vont de pair avec les savoirs-êtres qui permettent le team work, nécessaire à l’efficience des projets.
| Savoirs | Savoir-faire | Savoirs être |
| Meilleure maitrise et compréhension du MDR Meilleure maitrise des normes ISO Savoirs technique sur les matières premières Technique chirurgicale de lipotransfert Langue Anglaise | Élaboration et mise à jour des dossiers technique sous règlement 2017/745 Gestion et analyse des risques selon l’ISO 14971 Mise en pratique de l’aptitude à l’utilisation selon l’ISO 62366-1 Conduite de réunion Gestion des priorités | Capacité d’adaptation à l’environnement Créativité Curiosité Esprit d’équipe Polyvalence Prise d’initiative Autonomie Organisation Vision globale Prise de recul |
b) En cours de développement
Durant cette année d’apprentissage mes compétences se sont rapidement développées. Le service règlementaire d’Aesthetic Group étant composé d’une seule personne (excepté moi-même) le challenge qu’a été de fournir les 3 dossiers techniques (canules d’injection, prélèvement et infiltration) en 5 mois a nécessité une réelle proactivité, une autonomie ainsi que d’un fort esprit d’initiative. Ainsi j’ai pu prendre part à la totalité du dossier technique : la gestion des risques, le dossier de conception, ou l’aptitude à l’utilisation à titre d’exemples, toutefois mon intérêt pour l’évaluation clinique m’a poussé à prendre en charge cet aspect. Aesthetic group n’ayant pas de service R&D, les protocoles d’essai, les essais de performances et les rapports d’essais sont à la charge du service règlementaire, j’ai pu de ce fait améliorer mes compétences techniques et analytiques.
Aesthetic Group m’a alors permis de développer mes capacités à communiquer, mobiliser et convaincre en m’octroyant de réelles responsabilités tout en mettant en œuvre ma qualité d’ingénieur affaires règlementaires. Le domaine des affaires règlementaires, est en perpétuelle évolution, c’est pourquoi nos compétences doivent elles-aussi évoluer avec les besoins de l’industrie.
c) Mobilisation des acquis de la formation théorique
Cette expérience professionnelle m’a permis de constater l’importance qu’est d’avoir eu une formation multi disciplinaire. La formation de l’UTC donne l’opportunité d’accéder à tout le panorama des métiers du dispositif médical. Le tronc commun s’est articulé autour des grandes technologies biomédicales actuelles tout en nous faisant développer notre capacité à manager les projets. La pluridisciplinarité de cette formation s’est avérée utile dans l’appropriation des missions mais aussi dans l’intégration du monde professionnel. Cette pluridisciplinarité s’illustre dans les unités d’enseignement (UE) tel que la physiopathologie humaine, ou encore les traitements et soins au bloc opératoire, d’aspect plutôt scientifique, contre balancé par des enseignements plus stratégiques comme le management de la qualité et la gestion de projet. Desenseignements uniques dans le cadre d’une formation d’ingénieur biomédical tels que l’UE Geste, Parole et Savoir être et également Intelligence Collective et Organisationnelle donnent sens aux exigences techniques, mais surtout humaines des métiers visés, que l’on a tendance à oublier.
Le choix des autres enseignements n’étant pas imposé, chaque étudiant peut se construire ses propres connaissances en fonction de son projet professionnel mais aussi selon ses attraits personnels. Pour ma part, les connaissances apportées par l’enseignement Marketing de l’Innovation m’a permis d’avoir des outils pour structurer ma vision des projets mis en place en entreprise. Couplé aux enseignements de Droit qui m’ont permis de comprendre rapidement le contexte et les enjeux de la règlementation européenne relatifs aux DM.
Ces acquis théoriques ont été une plus-value sein de l’entreprise d’accueil car ils ont permis d’apporter un regard neuf sur les projets en cours mais aussi ceux à venir, de cette manière j’ai pu prendre part à la définition des stratégies opérationnelles. Aussi, la flexibilité des PME tel qu’Aesthetic Group m’a donné l’occasion de sortir de la zone règlementaire et de proposer des projets « terrains » tel qu’un ingénieur d’application pourrai effectuer.
2. La place de l'ingénieur affaires règlementaires chez un fabricant de DM
Le milieu de l’ingénierie biomédicale, et plus précisément celui des affaires règlementaires requiert d’avoir des connaissances et des aptitudes relatives à l’entièreté du cycle de vie du dispositif médical. Le métier d’ingénieur Affaires Règlementaires occupe une place centrale dans chez Aesthetic Group, même s’il a été longtemps considéré comme un métier qui joue un rôle de soutien et même considéré comme étant une lourdeur administrative. Le règlement via l’obligation d’avoir une personne chargée de veiller au respect de la règlementation (PCVRR), place ce métier au cœur de l’entreprise et des préoccupations de la direction. Et pour cause, sans la certification assurée par la PCVRR, qui permet la commercialisation du dispositif médical, le travail fournit par le département R&D est en vain, de même pour les services commerciaux et marketing, mais aussi pour la production. Cependant, ¾ des entreprises de l’industrie du DM rencontrent des difficultés à recruter des cadres règlementaires.[2]
Être ingénieur règlementaire nécessite d’avoir une combinaison de savoirs et de savoir-faire technique mais aussi des savoirs-êtres humains. Une pression sur les affaires règlementaires peut être instaurée par la direction dans l’objectif d’obtenir les certifications le plus rapidement possible, en réduisant au maximal les coûts et tout en se conformant aux critères exigeants du MDR mais également des ON. Il faut alors savoir être stratégique dans ces prises de positions et ainsi mobiliser les équipes pluridisciplinaires afin de déployer avec efficacité la stratégie règlementaire.
Afin de ne pas interpréter individuellement les textes règlementaires mais aussi faire face aux exigences, plusieurs associations et syndicats existent tel que le SNITEM, Team-PRRC, DM expert à l’échelle français, mais également au niveau Européen tel que Medtech Europe. Aussi, afin de se tenir au courant des technologies, des congrès mais aussi des conférences relatives aux sujets traités par les différents DM existent. Ces événements ne doivent pas être mis sur le côté du service règlementaire puisque ceux-ci permettent une veille concurrentielle, nécessaire dans le cadre d’un bon nombre de document du dossier technique tel que le suivi après commercialisation mais aussi l’évaluation clinique.
Conclusion
En vertu du nouveau règlement relatif aux dispositifs médicaux (DM) (MDR), les fabricants sont tenus de planifier de manière proactive la collecte de données cliniques relatives à leurs dispositifs afin de les enregistrer et analyser tout au long de leur cycle de vie. Ce suivi clinique après commercialisation (SCAC) a pour objectif de confirmer les performances cliniques et la sécurité du dispositif et ainsi assurer l’acceptabilité de la balance bénéfice/risque.
Pour se faire, la mise à jour des données cliniques par le biais d’étude post-commercialisation est devenu la norme, même si dans certains cas celles-ci ne sont pas nécessaires (voir Annexe 1). Les legacy devices qui sont sur le marché depuis un certain temps mais ayant des données cliniques limitées, ou ne pouvant plus tirer avantage des données de l’équivalence en raison des exigences règlementaires accrues de l’ère du règlement, doivent effectuer une revue de toutes les preuves cliniques disponibles dans le rapport d’évaluation clinique (CER) afin de décider si les études SCAC sont justifiées ou non. Ces études peuvent prendre plusieurs formes, plus ou moins longues et coûteuses, telles que l’investigation clinique ou les études rétrospectives. Toutefois même si le guide MDCG 2020-7 propose un modèle précis du plan de Suivi Clinique Après Commercialisation [34] et que le MDCG 2020-8 [35] offre un guide permettant d’établir le rapport SCAC, celui-ci reste difficile à mettre en place sans faire appel à des experts dans le domaine des études cliniques.
Même si de nouvelles pratiques d’études cliniques sur les DM prennent place ces dernières années, comme la digitalisation de la collecte de données (via les eCRF) et la future procédure de soumission des études cliniques via la base EUDAMED, qui permettrait de raccourcir les délais de réponses des autorités compétentes, les besoins en termes de données cliniques doivent être anticipés et identifiés le plus rapidement. Cette anticipation doit être fait afin d’éviter la perte du certificat CE étant donné le délai de certification actuel, dû au nombre insuffisant d’ON et au nombre croissant de dossiers à certifier mais aussi au vu du délai nécessaire à l’inclusion des investigateurs et des patients dans les études cliniques.
Néanmoins, les études cliniques restent un important investissement pour les PME de l’ordre d’une centaine de milliers d’euros, s’ajoute à ça le coûts des prestataires externes et celui de la certification CE. Nous pouvons alors nous demander si les PME qui composent à elles seules 93 % de la filière du DM, ont les ressources nécessaires pour faire face à ce nouveau règlement, où tomberont nous dans la pénurie de DM, comme l’alertait en mai dernier les Académies nationales de médecine, chirurgie et pharmacie. Cette pénurie aura pour conséquence de « ne plus garantir la prise en charge interventionnelle et chirurgicale des patients. »[36]
References bibliographiques
[2] SNITEM, Snitem Panorama et analyse qualitative de la filière industrielle des dispositifs médicaux en France en 2021 [3]
[22] W. Mokni, Le cas des prothèses mammaires (PIP), p. 2.
[25] AFNOR, « NF EN ISO 14155 », Cobaz, 19 août 2020. https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/nf-en-iso-14155/FA190411?rechercheID=3945069&searchIndex=4&activeTab=all (consulté le 23 novembre 2021).
[28] Snitem-Guide-Pratique-RNIPH-version octobre 2020.pdf
[31] [Livre Blanc] Comment Transformer une Étude PMCF en une Opportunité Commerciale - EasyMedStat.pdf
[33] NF EN ISO 14971 .pdf