IDS005 - Surveillance après commercialisation des dispositifs médicaux
DOI mémoire
https://doi.org/10.34746/nq28-4w94Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

GROELL Agathe 
ZKEIK Hajar 
BENACEUR Kheira
Contacts
Citation
A rappeler pour tout usage : GROELL Agathe, ZKEIK Hajar, BENACEUR Kheira « Surveillance après commercialisation des dispositifs médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2019, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids005 ; https://doi.org/10.34746/nq28-4w94
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2020_04_idsap
Résumé
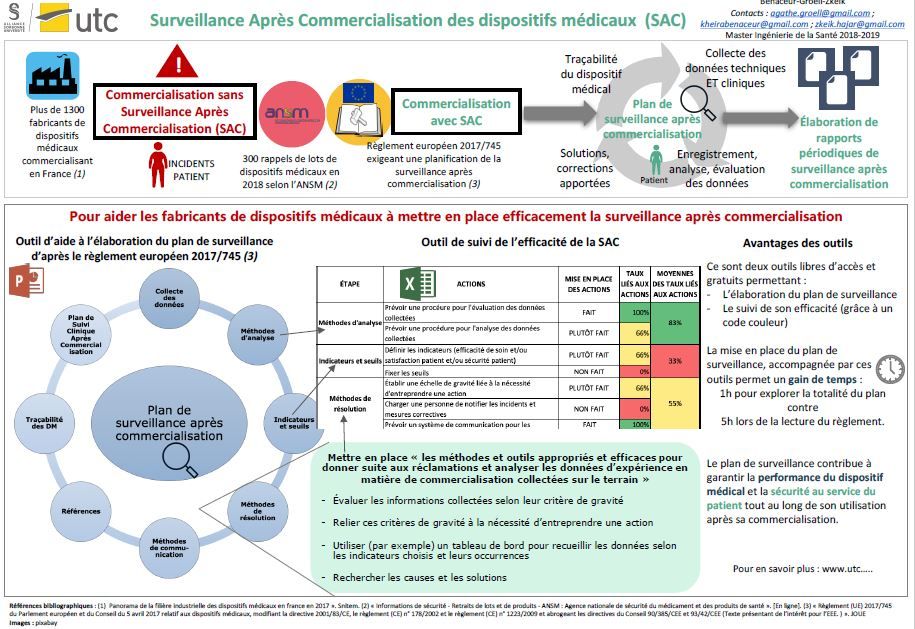
La Directive Européenne 2007/45/CE exige la réalisation d’une surveillance après commercialisation par les fabricants de dispositifs médicaux. Mais en 2018, l’ANSM a effectué 300 rappels de lot suite à des incidents. Il semble donc évident que la surveillance actuelle sur les dispositifs médicaux n’est pas suffisante pour garantir la sécurité et la performance des dispositifs médicaux. C’est pour cela que le nouveau Règlement Européen 2017/745 vient renforcer cette exigence. Cette évolution nécessite que les fabricants puissent s’approprier facilement les nombreux processus liés à la surveillance après commercialisation, deux outils ont été créés dans ce but.
Abstract
The European directive 2007/45/CE requires that the medical device’s manufacturer must carry on a post-market surveillance for his product. Yet, in 2018 the ANSM has recalled over 300 medical devices that let to different incidents. Thus, the post-market surveillance as described in the directive was not so effective. As a result, the European Council launched the regulation 2017/745 regarding the medical devices, that strengthens the post-market surveillance, in order to guarantee the patient and the medical staff security during the use of the device. This project highlights the post-market surveillance as mentioned in the new regulation 2017/745, and offers to the medical device’s manufacturers two tools that may be useful in order to have a good establishment of the post-market surveillance program.
Téléchargements
Poster : la Surveillance Après Commercialisation (SAC) des dispositifs médicaux

Mémoire d’Intelligence Méthodologique : Surveillance Après Commercialisation des Dispositifs Médicaux
Outil d'appropriation de la surveillance après commercialisation
Outil de vérification rapide du degré d'avancement de la mise en place de la surveillance après commercialisation
Mémoire complet :
Surveillance après commercialisation
des dispositifs médicaux
Remerciements
M Jean-Matthieu PROT pour nous avoir accompagné tout au long de ce projet et pour ses réponses à nos questions.
M Gilbert FARGES pour nous avoir orienté durant ce projet et tout particulièrement pour son aide pour la réalisation poster.
Mme Isabelle CLAUDE pour ses remarques pertinentes au cours de nos présentations.
M Marc MAINARDIS pour les éléments de réponses qu’il nous a fourni suite à nos questions.
Nos camarades de promotion pour leur soutien et les échanges d’informations.
Introduction
Ces dernières années, la population a augmenté rapidement, cette augmentation a déclenchée de nouveaux besoins en matière de prise en charge et de soins.
Le dispositif médical faisant partie intégrante du parcours de soin, son utilisation est étroitement surveillée pour assurer la sécurité du patient.
Or, le milieu du dispositif médical compte environ 1 300 entreprises uniquement en France [1], ces entreprises fabriquent des dispositifs médicaux de plus en plus complexes nécessitant un encadrement strict et unifié. C’est pour cela que le marquage CE a été créé pour la mise sur le marché des dispositifs médicaux en Europe.
Aujourd’hui, de plus en plus de dispositifs médicaux sont retirés du marché, au 13 décembre 2018 on compte 290 retraits de lots de dispositifs médicaux (hors dispositifs médicaux de diagnostic in vitro) par l’ANSM seulement pour l’année 2018 [2]. Il semble donc évident que les contrôles actuels portant sur la phase de pré-commercialisation ne sont plus suffisants pour évaluer le potentiel risque du dispositif.
C’est ainsi que le nouveau règlement européen 2017/745 du 5 avril 2017 [3] exige que le fabricant établisse un système de surveillance après commercialisation pour chaque dispositif médical, en fonction de sa classe de risque.
Le système de surveillance après commercialisation permettra de collecter, d'enregistrer et d'analyser, les données sur la qualité, les performances et la sécurité du dispositif médical pendant tout son cycle de vie, afin de bien suivre le dispositif et de définir les mesures préventives et correctives liées au dispositif [4].
La surveillance après commercialisation exige que le
fabricant, d’une part se base sur un plan de surveillance après
commercialisation, et d’autre part établisse un rapport de surveillance après
commercialisation périodique en fonction de la classe du dispositif médical.
I – État des lieux et évolution de la surveillance des dispositifs médicaux après commercialisation
Aujourd’hui, dans le monde ainsi qu’en France la population meurt principalement de maladies cardiovasculaires tels que les cardiopathies ischémiques, comme les insuffisances coronariennes, et les accidents vasculaires cérébraux (AVC). Selon l’Organisation mondiale de la santé (OMS) il a été relevé en 2016, 56,9 millions de décès apparus dans le monde sur ces décès plus de la moitié soit 54% sont dus aux cardiopathies ischémiques et les AVC [5].

Les cardiopathies ischémiques sont diagnostiquées par exemple avec une gamma caméra (classe IIa) [6]. Quant aux traitements, ils peuvent varier mais généralement un traitement médicamenteux est possible, ainsi qu’une stimulation cardiaque par pose de pacemaker (classe III) [6] mais aussi des interventions chirurgicale dans le cas d’un anévrisme, un stent ou coil (classe III) [6] est posé afin de combler la poche de l’anévrisme pour éviter que celle-ci ne se perce.
Dans les pays développés, comme la France, les cancers sont aussi une cause de mortalité très importante. Pour rappel, afin de traiter un cancer il y a une multitude de moyens selon le type de cancer.
Il est par exemple possible d’effectuer une chimiothérapie par voie orale ou veineuse (nécessité de dispositifs médicaux de classe I, voir ci-dessous).
Une autre option pour traiter le cancer est la radiothérapie par émission de rayonnements ionisants à travers un accélérateur de particules (classe IIb) [6].
Ainsi pour ces deux causes de mortalités, le développement des dispositifs médicaux est nécessaire. C’est pour cela qu’au cours de ce rapport toutes les classes de dispositifs médicaux seront traitées.
1- Définitions
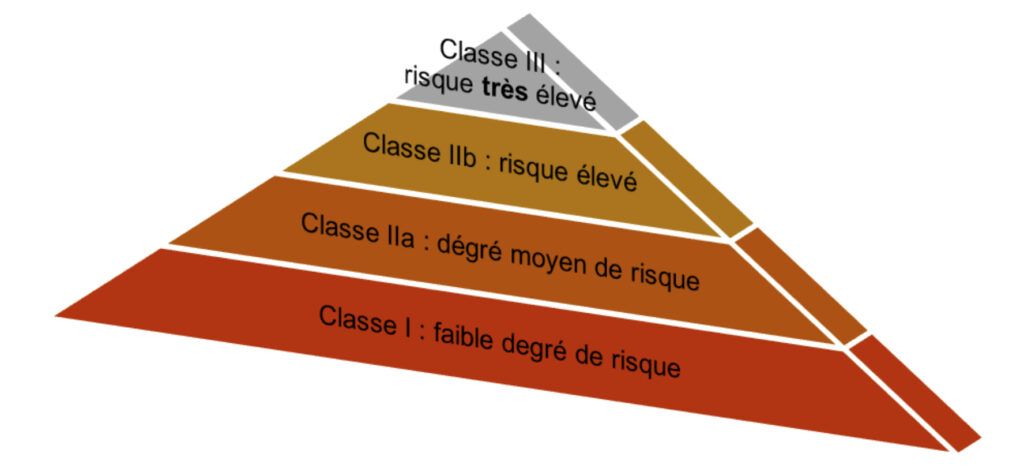
Dispositifs médicaux et classes :
Les dispositifs médicaux sont répartis en quatre classes différentes selon leur niveau de risque associé.
Cette donnée de classe est importante dans la surveillance clinique après commercialisation car des différences significatives de risque entre les différentes classes ont été détectées en Allemagne, où la surveillance clinique après commercialisation est déjà obligatoire [7].
Évaluation clinique [9]
L’évaluation clinique d’un DM se fait avant l’obtention du marquage CE (le marquage CE est obligatoire en Europe pour la mise sur le marché d’un dispositif médical) et tout au long du cycle de vie. Cette évaluation clinique peut se faire selon deux modalités différentes. Il s’agira soit d’une démonstration d’équivalence par rapport à un DM pré-existant, soit d’une surveillance clinique d’investigation basée sur l’expérimentation et non sur un exercice théorique.
Les objectifs de cette surveillance clinique sont :
- De vérifier que le dispositif fonctionne comme escompté
- D’évaluer les effets secondaires éventuels
- Gérer la balance bénéfice/risque pour éventuellement mettre en place des mesures correctives
Matériovigilance [10]
La matériovigilance doit être mise en place sur tous les dispositifs médicaux qui ne nécessite pas de surveillance clinique d’investigation. Les déclarations de matériovigilance doivent être transmises à l’ANSM. Le fabricant et l’ANSM mènent en parallèle l’évaluation de la matériovigilance mais toute personne concernée par le dispositif médical peut faire une déclaration de matériovigilance, que cela soit le premier concerné : le patient ou tout professionnel de santé.
2- État des lieux
Actuellement, seul l’évaluation clinique des DM pré-commercialisation est obligatoire et nécessaire pour la mise sur le marché et donc pour l’obtention du marquage CE.
Quant au suivi clinique des DM après commercialisation, il n’est pas obligatoire, s’il est justifié de ne pas en faire. Cette décision doit être précisément documentée et justifiée.
Ainsi, le fabricant doit fournir un document justifiant de l’absence de surveillance après commercialisation, mais ce suivi est toutefois nécessaire et obligatoire dans les cas suivants :
- Si de nouveaux risques sont identifiés
- Si l’évaluation de la sécurité et des performances à long terme critique
- Si les études pré-marquage CE faites sur un petit échantillon
- S’il est nécessaire d’avoir des études plus longues à cause de la durée de vie du dispositif médical [11]
Les normes et guides en vigueur encadrant la surveillance après commercialisation sont :
- Le guide MEDDEV 2.12.2 « post market clinical »
- La norme ISO 14971 concernant les activités de post-production.
- Directive 92/42/CEE
- Directive 2007/47/CE
- Nouveau règlement européen des dispositifs médicaux 2017/745
Mais face à de plus en plus de dispositifs médicaux retirés du marché, le nouveau règlement européen soumettra les entreprises à effectuer un suivi clinique après commercialisation quel que soit la classe du dispositif médical, afin de protéger au mieux le patient en évaluant la balance/risque tout au long de la vie du dispositif médical.
Or avec l’augmentation des exigences, il est difficile pour une entreprise de mettre en place un tel suivi car l’entreprise doit pouvoir réunir les informations, les traiter, les analyser, organiser au mieux la vigilance, évaluer la balance bénéfice/risques au fur et à mesure des retours pour organiser si nécessaire des actions correctives, et pour finir, organiser les rapports et la communication autour de ce suivi clinique après commercialisation. C’est pour cela qu’un plan de surveillance après commercialisation (PSAC) est nécessaire et rendu obligatoire par le nouveau règlement.
3- Évolution des obligations
Les principales évolutions apportées par le nouveau règlement 2017/745 est l’obligation de constituer un PSUR (rapport périodique actualisé de sécurité), pour les classes IIa, IIb et III ou un rapport de surveillance non périodique pour les dispositifs de classe I, et de prévoir un plan de surveillance clinique en amont du développement du DM.
Ces rapports doivent être établis selon une périodicité bien précise :
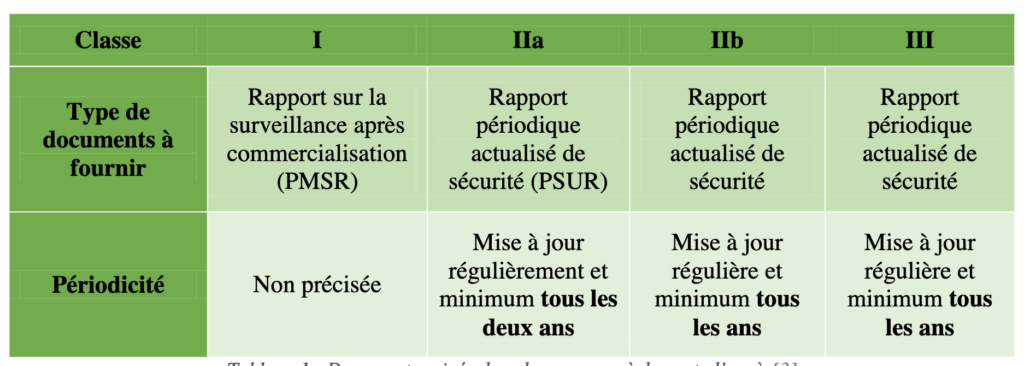
Les PSUR doivent être fournis à l’autorité compétente sur demande et transmis à l’organisme notifié concerné.
Dans le cas des DM de classe III, ils doivent aussi être directement enregistré dans un logiciel informatique destiné à regrouper les informations de vigilance et de suivi clinique après commercialisation, des fabricants, des organismes notifiés et des autorités compétentes de chaque État membre. Ce système permettra de centraliser les données et de favoriser la communication des informations. Un accès approprié sera prévu pour les patients et professionnels de santé.
(Ce logiciel permettra aussi de regrouper les informations sur les incidents graves, les rapports de synthèse périodiques, les rapports de tendance et les avis de sécurité) [3]
4- Enjeux
Ces changements présents dans le nouveau règlement entrainent des enjeux majeurs. En effet, il permet de renforcer la sécurité et la performance par une évaluation en continu du rapport bénéfice/risques, par l’identification des effets secondaires, des contre-indications, des nouveaux risques, ainsi que les erreurs dues à l’utilisation du DM (actuellement, la norme NF EN ISO 62366-1 peut être appliqué pour ce type de risque).
Comme évoqué précédemment l’établissement d’un plan surveillance après commercialisation est nécessaire pour obtenir le marquage CE, ce qui rallonge le temps de constitution du dossier technique et donc la mise sur le marché du DM.
La surveillance après commercialisation implique des ressources financières de la part du fabricant, donc un enjeu économique important, en effet comme vu précédemment, les démarches sont plus lourdes à mettre en place pour un fabricant de dispositifs médicaux de classe III et IIb que pour un fabricant de dispositifs médicaux de classe I.
Pour chaque dispositif médical, les fabricants mettent en place et maintiennent un système de surveillance après commercialisation en fonction de la classe du DM.
Dans le but de réaliser la surveillance après commercialisation, le fabricant va devoir établir un dossier technique de surveillance après commercialisation comprenant aussi une partie de suivi clinique après commercialisation. En effet le fabricant devra à la fois planifier la surveillance concernant des données techniques du dispositif mais aussi des données cliniques.
Un QQOQCP (voir annexe 1) a été réalisé permettant de poser la problématique suivante : comment mettre en place efficacement la surveillance ainsi que le suivi clinique des DM après commercialisation pour le fabricant ?
II – Solutions proposées pour s’approprier le système de surveillance après commercialisation
La surveillance après commercialisation exige que le fabricant se base sur un plan de surveillance après commercialisation pour démontrer qu’il satisfait à l’obligation visée à l’article 83 du règlement.
Ce plan doit faire partie de la documentation technique de l’appareil, il comprend des méthodes et des processus appropriés pour l’évaluation des données collectées, ainsi que les outils appropriés pour donner suite aux réclamations et pour analyser les données d’expérience en matière de commercialisation collectées sur le terrain.
Après la mise en œuvre d’un plan de surveillance après commercialisation conformément à l’annexe III, le fabricant doit établir un rapport qui présentera une synthèse des résultats de l’analyse des données collectées, ainsi que des conclusions exposant la justification et la description de toute mesure préventive et corrective prise.
1- Le plan de surveillance après commercialisation
a. Collecte des données
La première étape d’un plan de surveillance après commercialisation est la collecte des données.
Ces données doivent être collectées tout au long du cycle de vie de dispositif médical.
D’après l’annexe III du règlement ces données peuvent être collectées depuis différentes sources : la vigilance, les retours d’expériences et la littérature.
Les données issues de la vigilance sont collectées grâce aux : rapports d’incidents graves individuels (article 87), rapports de synthèse (article 87), rapports de tendance (article 88) et rapports de surveillance PSUR et PMSR.
Les données issues du retour d’expérience des utilisateurs, distributeurs et importateurs ainsi que les réclamations sont collectées en utilisant des méthodes efficaces pour la communication : rédaction de fiches pratiques, questionnaires et enquêtes.
Ces documents sont à fournir à des intermédiaires (professionnels de santé, ingénieurs biomédicaux…) sélectionnés par les commerciaux faisant régulièrement des réunions pour regrouper les informations.
Les données issues de la littérature sont collectées par une veille réglementaire portant sur des publications, bases de données, registres techniques et dossiers techniques de dispositifs médicaux similaires.
b. Méthodes d’analyse
Après cette collecte de données, le fabricant doit mettre en place des procédures et méthodes appropriés et efficaces pour l'évaluation des données collectées. Ainsi au sein de ces locaux le fabricant pourrait mettre en place des progiciels tels qu’un ERP (Enterprise Ressource Planning) ou le PGI (Progiciel de gestion intégré). Ces progiciels permettent de traiter les données récoltées, comme l’enregistrement des réclamations, ou des incidents liés au dispositifs par exemple. Ils permettent donc de gérer des données relatives aux stocks, gestion commerciale, etc... D’autre outils comme Excel peuvent être utilisés pour l’analyse des données.
L’article 89 « Analyse des incidents graves et des mesures correctives de sécurité » du nouveau règlement européen 2017/745, stipule que le fabricant après une déclaration d’un incident grave doit :
- Mener des investigations comprenant une évaluation des risques liée à l’incident, et aussi les différents éléments ci-dessous :
- La causalité
- Détectabilité
- Probabilité de récurrence du problème
- Fréquence d’utilisation du DM
- Probabilité de survenu du dommage direct ou indirect
- La sévérité
- Le bénéfice clinique du DM
- Les utilisateurs du DM
- Mettre en place des mesures correctives et préventives de sécurité en collaboration avec les autorités compétentes
- Le fabricant doit via le système électronique mentionné (à l’article 92) joindre aux autorités compétentes un rapport de conclusion sur ces incidents
c. Indicateurs et seuils
Afin de pouvoir analyser efficacement les données collectés le fabricant doit fixer suivant des indicateurs, des seuils à ne pas dépasser. Par définition un indicateur est un « ordre de grandeur spécifique observable et mesurable qui peut servir à montrer les changements obtenus ou les progrès accomplis par un dispositif médical dans ce cas » [12]. Ces indicateurs pourront mesurer la qualité de soin, la satisfaction du patient, ou encore la sécurité du patient [13]. Ainsi, il est proposé aux fabricants d’établir un tableau de bord permettant de classer :
- Les différents indicateurs
- De préciser l’intérêt et l’objectif de chaque indicateur
- Les valeurs de seuil seront fixées et visualisées grâce à un code couleur par exemple
- En cas d’éloignement de la valeur du seuil établi le dispositif devra alors être réévalué
- Ne pas négliger que les indicateurs ont un coût non négligeable (collecte, analyse, présentation)
d. Méthodes de résolution
D’après l’annexe III du règlement européen, le fabricant doit mettre en place les méthodes, outils et procédures permettant d’engager des mesures préventives ou correctives cohérentes avec les données collectées concernant les d’incidents graves.
Il s’agira d’évaluer les données recueillies selon le degré de gravité, rechercher les causes et les solutions des incidents détectés.
Comme prévu à l’article 87, le fabricant devra documenter toute incidents grave présents sur le marché ou le dispositif est commercialisé et prévenir les autorités compétentes en France qui est l'ANSM. Les incidents sont notifiés selon leur degré de gravité dans un délai temporel défini :
- Le fabricant possède 15 jours après l’incident grave pour faire la notification.
- Si c’est un incident menaçant la santé publique la notification est faite immédiatement ou maximum deux jour après l’incident
- En cas de décès ou de détérioration grave faire le fabricant doit la faire immédiatement jusqu’à 10 jour maximum.
Si le fabricant n’a pas eu le temps, il transmet un rapport incomplet et transmettra la version définitive après, si le fabricant a un doute sur l’incident il doit faire la notification dans les délais. En cas d'urgence où il doit prendre immédiatement une mesure corrective de sécurité.
Les mesures correctives entreprises devront être notifiées immédiatement et une communication efficace sur toutes ces données devra être mise en place entre organismes notifiés, professionnels de santé et patients, ainsi que l’autorité compétente.
Pour finir l’article 88 précise que tout incident grave nécessite une investigation comportant une évaluation des risques et des mesures correctives.
e. Méthodes de communication
Pour communiquer avec les autorités compétentes, les organismes notifiés et les états membres, le fabricant bénéficiera d’un système électronique auquel il transmettra :
- Les rapports sur les incidents graves et les mesures correctives de sécurité (d’après l’article 89 : ce rapport permettra à l’autorité compétente de prévenir l’utilisateur)
- Les rapports de synthèse périodique
- Les rapports de tendance
- Les PSUR
- Les avis de sécurité
Le fabricant devra aussi prévoir un système pour recevoir les réclamations de tout utilisateur de son dispositif médical.
f. Références
Le fabricant doit mettre en place des procédures permettant de mentionner les normes harmonisées auxquelles il est conforme, et si celles-ci lui ont permis d’effectuer certaines étapes de la surveillance. La norme NF EN ISO 14971 relative à la gestion des risques des dispositifs médicaux en fait partie par exemple.
g. Traçabilité des dispositifs médicaux
Le règlement européen 2017/745 prévoit une mesure particulière pour la traçabilité à l’article 31 : l’IUD (numéro d’enregistrement unique du dispositif médical comparable au code CIP pour les produits pharmaceutiques).
Pour identifier les dispositifs médicaux, le fabricant pourra aussi respecter les recommandations de l’ANSM [14]. Le fabricant devra rendre visible sur le dispositif médical : la dénomination ou référence du produit, le nom du fabricant, un numéro de lot ou de série, un code-barres regroupant ces informations et l’IUD.
Pour assurer la traçabilité, les dispositifs médicaux vendus devront être répertoriés en regroupant le code-barres et les coordonnées de l’acheteur.
h. Plan de suivi clinique après commercialisation
Contrairement aux étapes vues précédemment, cherchant uniquement à collecter et analyser des données techniques (ou précliniques), le plan de suivi clinique après commercialisation (SCAC) est spécifique aux données cliniques.
Pour sa réalisation, les solutions proposées précédemment pour la collecte ou encore l’analyse des données peuvent être appliquées. Cependant la périodicité des actions entreprises devra être précisée par le fabricant.
Ce plan rassemble :
- Une évaluation des données cliniques relatives à des dispositifs équivalents ou similaires
- Un calendrier détaillé des activités à réaliser pour le suivi clinique après commercialisation
- Confirmer la sécurité et les performances du dispositif pendant toute sa durée de vie prévue
- Identifier les effets secondaires inconnus et surveiller ces effets et les contre-indications
- Garantir le caractère constamment acceptable du rapport bénéfice/risque
- Identifier toute mauvaise utilisation ou toute utilisation hors destination éventuelle du dispositif. La norme NF EN 62366 "Aptitude à l'utilisation" peut être appliquée pour éviter les erreurs d’utilisation.
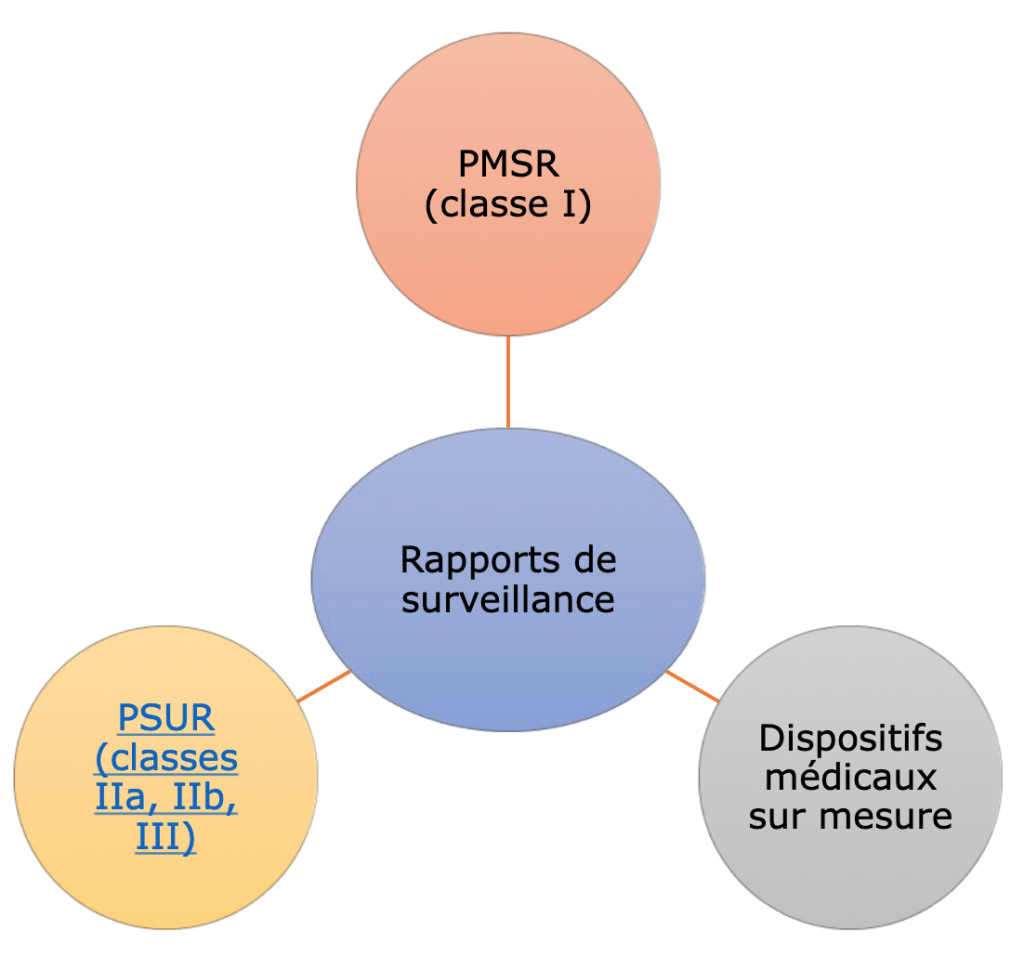
2- Planification des rapports de surveillance après commercialisation
Après la réalisation des plans de surveillance après commercialisation ainsi que le plan de suivi clinique après commercialisation. Le fabricant devra effectuer des rapports de surveillance :
- Le PSUR périodique pour les classes IIa, IIb, et III, contenant des éléments cliniques et techniques liés au dispositif médical
- Le PMSR (rapport non périodique) pour les dispositifs de classe I contenant uniquement des éléments techniques liés au DM
- Un plan spécifique pour les dispositifs sur mesures qui comportera seulement des éléments cliniques. Car pour ces dispositifs un plan de surveillance après commercialisation n’est pas exigé mais un rapport axé sur des éléments cliniques est nécessaire.
- Ces rapports comporteront une synthèse des résultats et des conclusions de l'analyse des données de surveillance après commercialisation qui ont été collectées.
a. PSUR
D’après l’article 86, il doit comporter une synthèse de l’évaluation et l’analyse des données collectées dont différents éléments relatifs aux dispositifs (voir annexe 2) tels que :
- Données administratives
- Date de commercialisation du DM sur le marché, et nombre d’unités vendues
- Taux de plaintes
- Nombre d’événements identifiables
- Nombre de rapports notifiés à l’autorité compétente
- Risques imprévus
- Nombre de rappels de produits
Ces différents éléments doivent être mis en relation avec les procédures utilisées en préclinique telles que la gestion des risque et l’évaluation clinique.
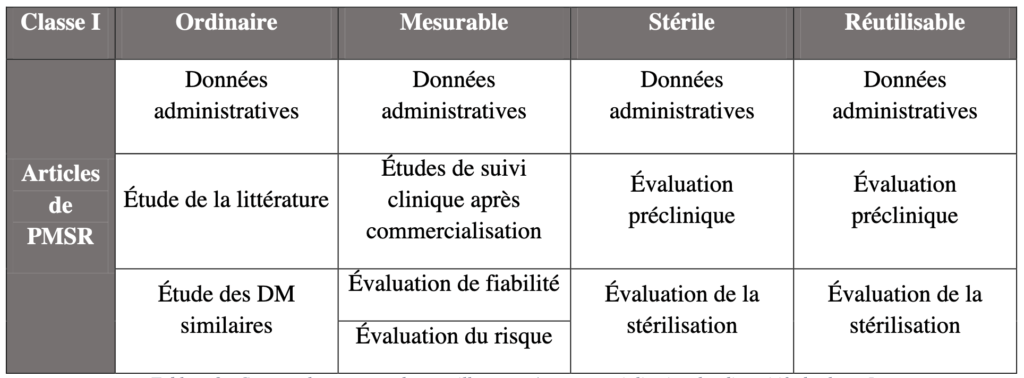
b. PMSR
D’après l’article 85, il doit comporter une synthèse de l’évaluation et l’analyse des données collectées dont différents éléments relatifs aux dispositifs. La ré-évaluation des données précliniques diffère selon le type de dispositif de classe I comme illustré dans le tableau suivant :
III - Outils permettant la prise en main des exigences du règlement 2017-745
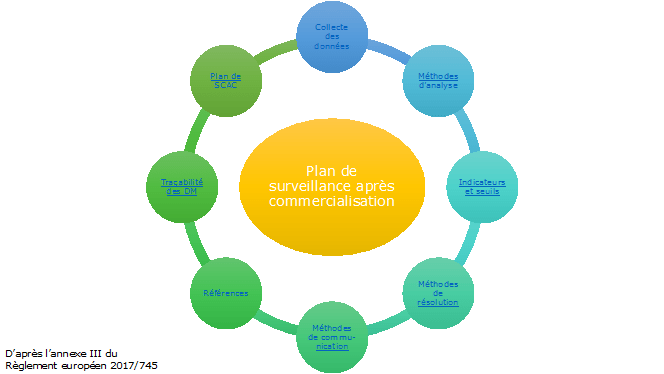
1- Outil d’appropriation de la surveillance après commercialisation (d’après l’annexe III)
Pour faciliter la mise en place de la surveillance après commercialisation du dispositif médical, nous avons voulu mettre au point un outil facile d’utilisation, sur support PowerPoint, se présentant comme une cartographie des processus mis en place.
Il se découpe en deux parties : l’élaboration du plan de surveillance après commercialisation et l’élaboration des rapports de surveillance.
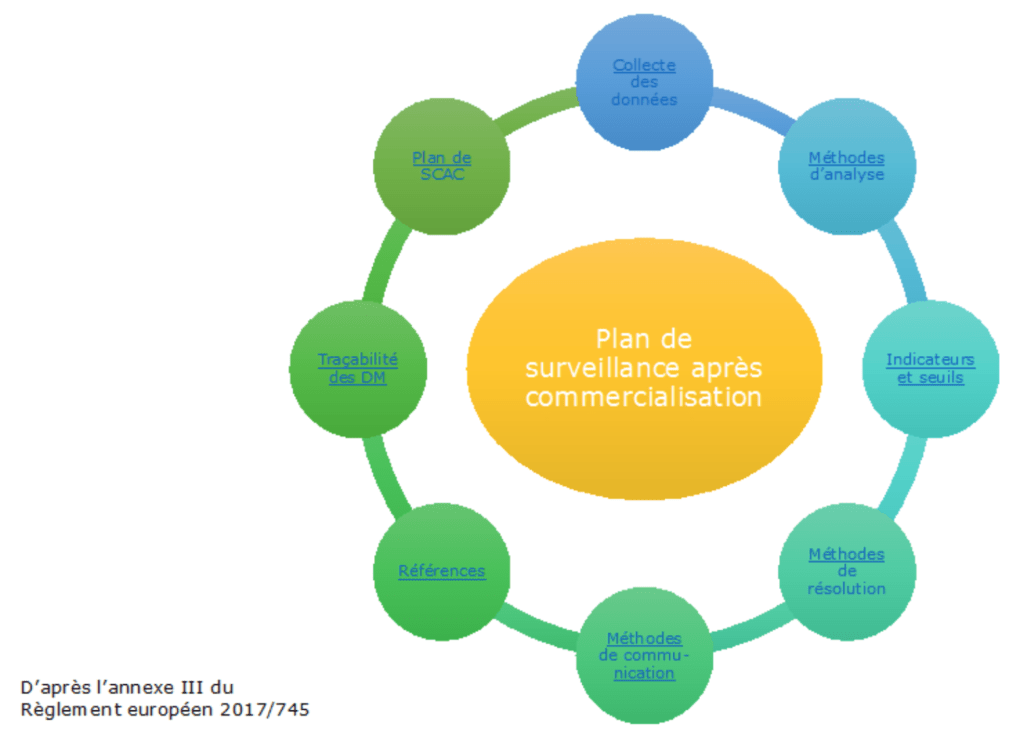
Figure 3 : Plan de surveillance après commercialisation, menu principal
source : auteurs, d’après [3][15]
Chaque partie du plan de surveillance après commercialisation prévue à l’annexe III du règlement est approfondie si l’utilisateur clique sur une des en-têtes.
Les propositions pour établir le plan sont à la fois basées sur les obligations du règlement mais aussi sur les idées du groupe projet.

source : auteurs, d’après [3]
Une flèche en bas à droite de la diapositive permet de revenir au sommaire facilement. Les articles ou les autres éléments cités à d’autres endroits du plan de surveillance sont soulignés en bleu, par un simple clic l’utilisateur de l’outil pourra voir les éléments concernés.
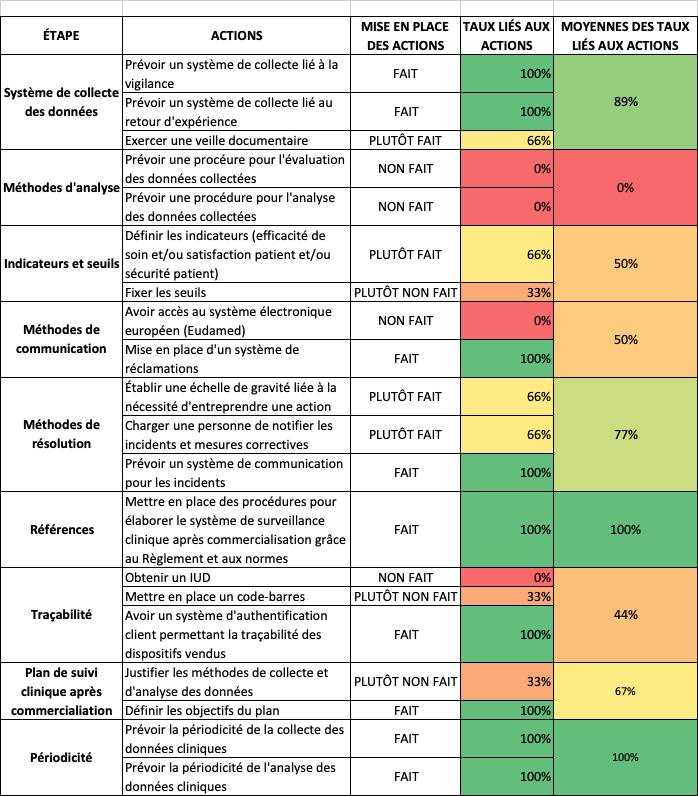
2- Outil de degré d’avancement pour la mise en place de la surveillance
Pour aider le fabricant à mettre en œuvre son plan de surveillance après commercialisation, les principaux items issus du règlement ont été rassemblés dans un tableur Excel, qui est téléchargeable.
Les actions à entreprendre sont reliées à un critère de véracité en 4 modalités : FAIT, PLUTÔT FAIT, PLUTÔT NON FAIT, NON FAIT. Ceux-ci sont liés à des taux de maturité qui permettront à l’utilisateur de visualiser, grâce à une échelle de couleur, les actions qui lui reste à entreprendre.
Conclusion
Ces deux outils permettent à l’utilisateur une prise en main efficace de la surveillance après commercialisation des dispositifs médicaux. En effet, l’outil d’aide à l’élaboration du plan permet un gain de temps de 4h (1h pour explorer l’outil en totalité, contre 5h lors de la lecture du règlement). La réalisation de ce plan de surveillance et la rédaction des différents rapports exigés par le Règlement Européen contribuera à garantir la performance du dispositif médical et la sécurité du patient. Cela permettra d’améliorer l’image des fabricants de dispositifs médicaux à tous les niveaux de la société et de rentrer dans une ère plus sécurisée où les incidents et les retraits de lots se feront de moins en moins nombreux.
Références bibliographiques
[1] « Panorama de la filière industrielle des dispositifs médicaux en France en 2017 ». Ed. Snitem.
[2] « Informations de sécurité - Retraits de lots et de produits, ANSM : Agence nationale de sécurité du médicament et des produits de santé », 2018.
[3] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux ». JOUE, 05-mai-2017.
[4] GMED, « Réglementation Européenne : Réflexions d’un organisme notifié sur la phase dite “post-marché” », 12-avr-2012.
[5] Organisation mondiale de la santé, « Les 10 principales causes de mortalité dans le monde », Organisation Mondiale de la Santé, 24-mai-2018.
[6] V. Franch, « Guide indicatif des dispositifs médicaux soumis à l’obligation de maintenance avec leur classe selon la nomenclature CNEH version 2000 ».
[7] C. Zippel et S. Bohnet-Joschko, « Post market surveillance in the german medical device sector – current state and future perspectives », Health Policy, vol. 121, no 8, p. 880‑886, août 2017.
[8] Abale Iiza Honoré, Nsabimana Diogène, Dago Dogo Maurice, et Bugingo Mbishibishi, « SECURITE ET MAINTENANCE DES DISPOSITIFS MEDICAUX : MISE EN OEUVRE ET SUIVI DE LA NORME NF EN 62353 ».
[9] Haute Autorité de Santé, « Parcours du dispositif médical en France- Guide pratique », nov-2017.
[10] « Qu’est-ce que la matériovigilance ? - ANSM : Agence nationale de sécurité du médicament et des produits de santé ».
[11] Université de Lille - Faculté de Pharmacie, « Le Marquage CE des dispositifs médicaux - Mise en place d’un plan de surveillance du dispositif après commercialisation ». .
[12] Boyer Pauline, « Pilotage des activités pharmaceutiques d’une centrale d’approvisionnement de matériel stérile par la construction d’un tableau de bord », de Lorraine, 2009.
[13] Haute Autorité de Santé, « Qu’est-ce qu’un indicateur de qualité et de sécurité des soins ? »
[14] « Traçabilité des dispositifs médicaux - ANSM : Agence nationale de sécurité du médicament et des produits de santé ».
[15] eCFR — Electronic Code of Federal Regulations, « Food and Drugs - Postmarket surveillance », 20-déc-2018.
Annexe