
IDS040 - Dispositifs médicaux — Informations à fournir par le fabricant selon la norme PR NF EN ISO 20417
DOI mémoire
https://doi.org/10.34746/f4eg-rn84Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

CHEDJOU TAKAM Jean Ernest 
CHEN Yihan 
OUEDRAOGO Ismaila
Contacts
Citation
A rappeler pour tout usage : CHEDJOU TAKAM Jean Ernest, CHEN Yihan, OUEDRAOGO Ismaila « Dispositifs médicaux : Informations à fournir par le fabricant selon la norme PR NF EN ISO 20417 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids040/ ; https://doi.org/10.34746/f4eg-rn84
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_01_idsap
Résumé
Les nouveaux Règlements européennes 2017/745 et 2017/746 sont entrées en vigueur en Mai 2020 pour remplacer les anciennes Directives afin d’améliorer la transparence et traçabilité des dispositifs médicaux dans le but d’assurer la sécurité du patient. Dans ce cadre du changement réglementaire, la norme harmonisée en projet NF EN ISO 20417 Dispositifs médicaux — Informations à fournir par le fabricant vient remplacer la norme EN ISO 1041. En effet cette nouvelle norme fournie les informations nécessaires pour la conception d’un dispositif et mise sur le marché du dispositif médical. Pour maitriser les informations le contenu dans cette norme, deux outils ont été développés : une cartographie pour faciliter compréhension de la norme et un outil de diagnostic pour évaluer sa conformité .
Abstract
The new European Regulations 2017/745 and 2017/746 will come into force in May 2020 to replace the old Directives in order to improve the transparency and traceability of medical devices in order to ensure patient safety. In this context of the regulatory change, the harmonized standard in draft NF EN ISO 20417 Medical devices - Information to be provided by the manufacturer replaces the standard EN ISO 1041. Indeed this new standard provides the information necessary for the design of a device and placing the medical device on the market. To master the information contained in this standard, two tools have been developed : a map to facilitate understanding of the standard and a diagnostic tool to assess its compliance.
Téléchargements

Poster:Dispositifs médicaux — Informations à fournir par le fabricant selon la norme PR NF EN ISO 20417

Mémoire d’Intelligence Méthodologique : Dispositifs médicaux —Informations à fournir par le fabricant selon la norme PR NF EN ISO 20417
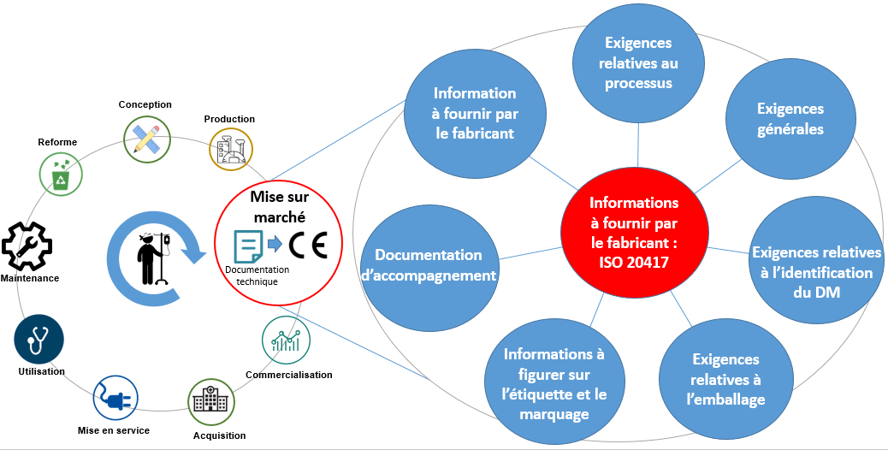
Cartographie pour faciliter la compréhension rapide de la norme
Outil de diagnostic : Connaitre rapidement son niveau de respect des exigences
Mémoire complet :
Dispositifs médicaux — Informations à fournir par le fabricant selon la norme PR NF EN ISO 20417
Remerciements
M. Dan ISTRATE pour nous avoir accompagné pour sa disponibilité et surtout ses judicieux conseils tout au long de ce projet.
M. Gilbert FARGES pour nous avoir orienté durant ce projet et tous ses soutiens multiformes.
Nos camarades de promotion pour leur soutien et les échanges d’informations.
Introduction
Le monde des dispositifs médicaux au sein de l’Union Européenne traverse une période transitoire à la fois difficile et importante de son histoire. Pour mettre un produit sur le marché, les fabricants de DM et DMDIV devront respecter certaines exigences documentaires afin d’obtenir le marquage CE et pouvoir accompagner le produit sur le marché. C’est à ce moment que la norme harmonisée en projet PR NF EN ISO 20417 concernant les informations à fournir par fabricants entre en jeux. Cette norme s’inspire des deux nouveaux règlements européens 2017/745[1] et 2017/746[2] relatifs respectivement aux dispositifs médicaux et dispositifs médicaux de diagnostic in vitro et également des directives 93/42/CEE[3] 90/385/CEE[4] et 98/79/CE[5]. Pour garantir la sécurité des patients pendant tout le cycle de vie du dispositif médical, l’une des modalités importantes c’est la maîtrise de l’information fournie à l’utilisateur.
Pour une prise en main facile, rapide et efficace des informations contenues dans la norme en question, une cartographie interactive vous est proposée. Et pour permettre au lecteur de se positionner et connaître son écart vis-à-vis du respect des exigences normatives et règlementaires liées aux informations à fournir, un outil de diagnostic vous est également proposé. Alors pour réaliser toutes ces taches, la démarche adoptée est la suivante :
- D'abord contextualiser le travail, ressortir les intérêts, enjeux et problématiques liées à notre sujet afin de bien le cerner et le situer dans son environnement.
- Ensuite, une synthèse des règlements 2017/745[1]sur les dispositifs médicaux, 2017/746[2]sur les dispositifs médicaux in vitro et de la norme PR NF EN ISO 20417[6] concernant les informations à fournir par le fabricant, afin de rassembler les éléments de connaissance et d’en dégager une vue générale.
- Ensuite une analyse des points critiques et des alternatives possibles afin d’obtenir une mise en œuvre rapide et efficiente.
- Ensuite une cartographie interactive des processus pour mettre en œuvre les exigences réglementaires / normatives et faciliter aux fabricants la prise en main et l’usage de ces informations.
- Enfin un outil d'autodiagnostic pour permettre aux fabricants de se positionner vis à vis du respect des exigences réglementaires en s'auto-évaluant.
I. Contexte : Dispositif médical et la sécurité du patient
1. Contexte général
En 2017, parmi les industries de santé en France, le secteur du dispositif médical est le secteur le plus dynamique avec un chiffre d'affaire d'environ 28 Md€, soit une augmentation de 4% chaque année. C'est un secteur qui rassemble plus de 1300 entreprises dont 92 % sont des PME pour près de 85 000 emplois créés [7].
Dorénavant tous les fabricants de dispositifs médicaux au sein de l'union européenne ne devront concevoir et commercialiser que des équipements certifiés d'un marquage CE. C'est dans ce contexte que la norme en projet PR NF EN ISO 20417[6] s'applique afin d'aider les fabricants de dispositifs médicaux à constituer la partie informations à fournir, qui est un élément indispensable de la documentation technique pour l'obtention du marquage CE. Après évaluation de cette documentation technique par un organisme notifié, ce dernier délivre le marquage CE et donne l'autorisation pour que ces informations accompagnent le dispositif sur le marché afin de garantir la sécurité du patient. En Europe, la production est une activité qui est exercée par la plupart des entreprises du secteur des dispositifs médicaux (dont 60% s'y consacre exclusivement) et 13% environ exerce la R&D. Selon les chiffres de 2015, il en ressort que les dispositifs médicaux sont très utilisés en France, entre 800 000 et 2 millions de références de DM ont été recensé avec environ 8,7 milliards d'euros remboursés, ce qui témoigne une nette évolution de 2,8 % par rapport à 2014 [8].
2. Contextes associés à la sécurité et l’usage de l'information
a. Impact de l'information dans le cycle de vie du dispositif médical
Dans le cycle de vie d'un dispositif médical, le patient est au centre de tout le processus de la conception à la reforme. Les informations de la norme en question se situent entre la fin de la phase de production et le début de la phase de commercialisation. En effet après la production d'un dispositif médical, pour obtenir le marquage CE qui le permet d'avoir accès au marché, on utilise la norme PR NF EN ISO 20417 pour constituer la partie informations à fournir qui est un élément indispensable de la documentation technique pour obtenir le marquage CE et qui accompagnera le dispositif sur le marché. Notons que le patient est également au centre d'un vaste écosystème constitué : des dispositifs médicaux, des fabricants, des chercheurs, des distributeurs, des exploitants, des réformateurs. Alors pour assurer sa sécurité, les informations contenues dans les règlements et les normes ont vu le jour, afin de réguler et de standardiser ce vaste système et ces informations sont très diverses et variées.
- Conception :
C’est une phase très importante du cycle de vie du dispositif médical qui consiste à déployer des équipes afin de créer des solutions médicales différentes de celles existantes et qui répondent aux besoins collectifs des patients et des utilisateurs. Elle est effectuée par des chercheurs dans des laboratoires et centres de recherches, par des docteurs et professeurs dans des universités…
- Production :
C’est une phase du cycle de vie du dispositif médical qui consiste à réaliser les travaux de conceptions effectués par chercheurs et les universitaires ou encore l'ensemble des opérations de fabrication indispensable à la réalisation d'un produit jusqu'à sa mise sur le marché (développement, marquage CE et documentation d’accompagnement). Elle est réalisé par les fabricants, les industrielles...
- Mise sur le marché :
C'est une phase aussi importante que les autres phases du cycle de vie du dispositif médical, qui consiste à mettre le produit développé à dispositif sur le marché pour les utilisateurs afin qu'il puisse servir pour le traitement des patients selon la destination revendiquée par le fabricant. Elle est effectuée par les fabricants eux-mêmes, les distributeurs, les importateurs…
- Acquisition :
C'est une phase du cycle de vie du dispositif médical qui consiste à mettre en service les dispositifs médicaux et à assurer la formation des utilisateurs. Pour cela les acteurs devront répondre aux appels d'offre, monter le dossier selon les besoins exprimés et soumettre pour étude et validation. Elle est effectuée par les fabricants eux-mêmes, les distributeurs, les importateurs…
- Utilisation :
C'est une phase du cycle de vie du dispositif médical qui consiste, à l'utilisation des dispositifs médicaux (sûr et performants) sur les patients, pour les apporter des soins adéquats, pour leur prise en charge et rétablir leur état de santé. Elle est effectuée par les personnels de santé (médecin, chirurgien, infirmier…
- Maintenance :
C'est une phase du cycle de vie du dispositif médical qui consiste maintenir et rétablir la stabilité d'un dispositif médical afin qu'il puisse conserver ses caractéristiques de sécurité et de performance pendant toute la période de validité. Pour une gestion plus efficacement, on utilise une GMAO qui est un outil indispensable pour améliorer la maintenance et assurer sa traçabilité.
- Réforme des dispositifs médicaux :
C'est une phase du cycle de vie du dispositif médical qui consiste à protéger l'environnement des pollutions ou contaminations (terre ou eau). En effet, la reforme favorise l'élimination des dispositifs médicaux ou le reconfigure pour un nouvel usage. On retrouve ainsi un cycle où le dispositif médical n'est pas seulement un produit mais un écosystème qui tient compte de terre, de personnes, la sécurité du patient.
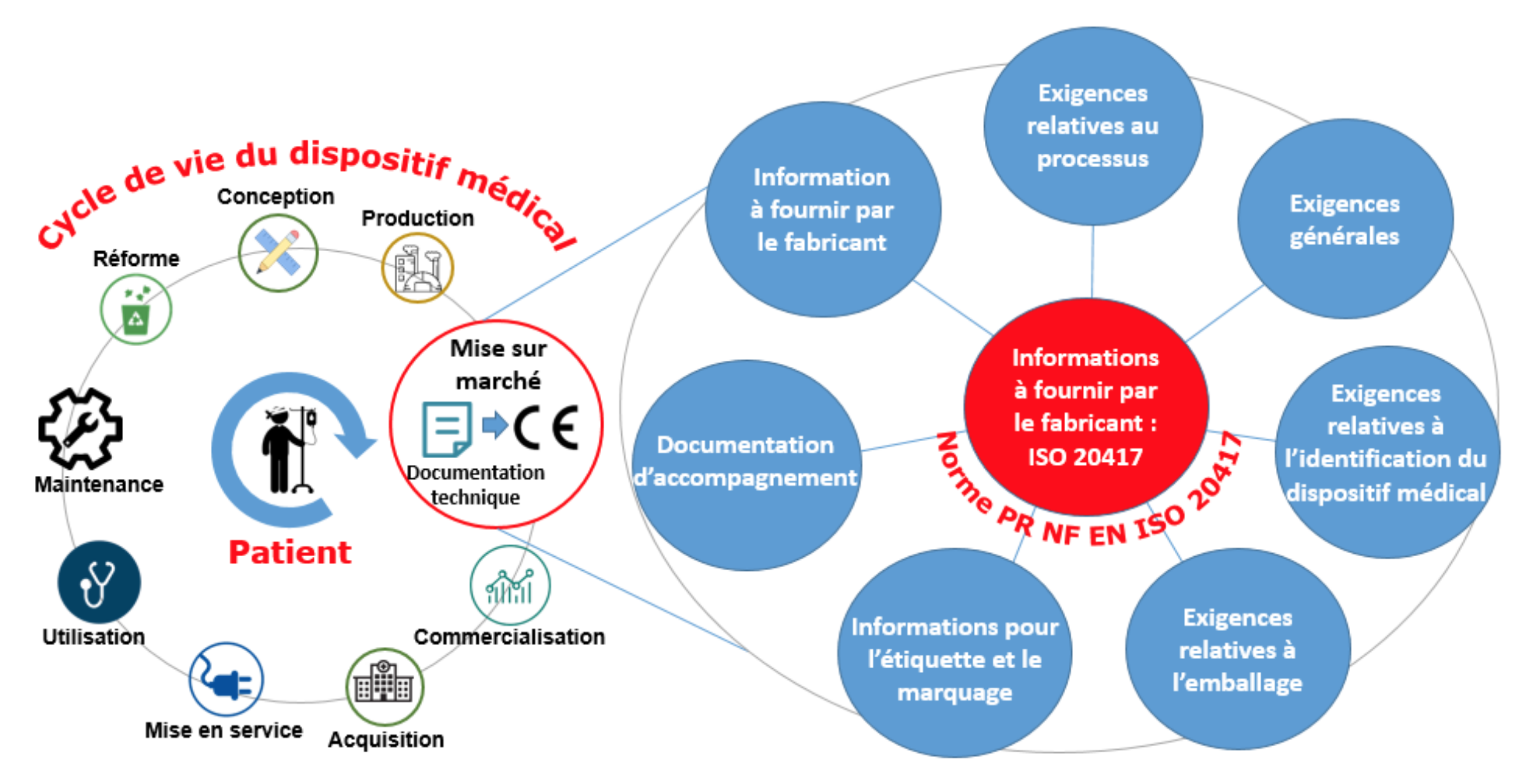
b. Rôle de l'information
Quand l'information est bien transmise, elle est un facteur de cohésion, innovation, implication, réactivité, adaptabilité, motivation, efficacité. Cependant quand elle n'est pas bien transmise, elle est un facteur de division, stress, inquiétudes, doute, divergences, conflits, tensions. Le management de linformation permet d'éviter beaucoup de tension et de gaspillage d'énergie. Une attention particulière doit être accordée à la formalisation et à la circulation de l'information.
3. Contexte lié à la norme PR NF EN ISO 20417
La toute première édition de la norme PR NF EN ISO 20417 (norme harmonisée) viendra remplacer l'ancienne norme EN1041[9]. Cette norme définit les exigences en matière de conception et de mise en œuvre de la documentation d'accompagnement, des étiquettes sur les dispositifs médicaux ou leur emballage et le marquage des dispositifs médicaux.
Elle vise à remplacer ou compléter les exigences d'étiquetage redondantes, qu'on retrouve dans les normes des dispositifs médicaux. Son objectif majeur est qu'il soit une référence pour ces exigences génériques communes, en permettant aux prochaines normes de dispositif médical de se focaliser sur ses exigences applicables. Les exigences des normes propres aux dispositifs médicaux peuvent compléter ou modifier ces exigences générales. Cette norme ne doit donc pas être utilisée seul mais en accord avec ces normes de dispositifs médicaux s'il en existe à moins que la norme en question spécifie le contraire. Les exigences de la norme de produit (dispositif médical) l'emportent sur le présent document. Cette norme a été conçue pour venir en renfort, en matière d'information à fournir par les fabricants de dispositif médical, des principes essentiels de sécurité et performance conforme à l'ISO 16142.
La norme en projet PR NF EN ISO 20417 est un outil exceptionnel de pilotage des informations à fournir par un fabricant et d'amélioration de la sécurité dans tout le cycle de vie du dispositif médical. Cette norme assure aux entreprises une gestion conforme des informations et est composée de 10 chapitres, auxquels viennent s'ajouter 10 annexes. Comme toutes normes, son utilisation n'est pas obligatoire, les fabricants sont libres de choisir un autre référentiel pour démontrer la conformité aux exigences réglementaires applicables. Notons que cette norme fait présomption de respect aux exigences des nouveaux règlements européens 2017/745 et 2017/746.
Ce document regroupe les exigences concernant les informations à fournir par les fabricants de dispositif médical ou d'accessoire et contient les exigences qui sont généralement appliquées pour la documentation d'un dispositif médical ou d'un accessoire, son identification et son marquage. Mais ce dernier n'indique ni la langue ni le moyen de transmission de l'information.
La problématique que soulève ce sujet et qui a été résolue tout au long de ce travail est la suivante : comment faciliter la prise en main des informations à fournir par un fabricant conformément à la norme harmonisée PR NF EN ISO 20417 ?
II. Maitrise de l'information : un facteur important pour la mise d'un dispositif médical sur le marché
L'information quelques soit sa nature, a pour but d'informer, de s'informer ou de renseigner sur quelque chose à quelqu'un. Cela demande de bonnes compétences rédactionnelles, communicationnelles, de synthèse, de clarté, de simplicité, de rigueur, de logique et d'illustration pour être sûr que le message qu'on souhaite véhiculer sera effectivement celui qui sera interpréter par le destinataire. Alors il convient d'être vigilant, car si ce n'est pas le cas, ça peut produire les effets inverses. Les enjeux sont donc majeurs, variés et peuvent être économique, financière, juridique, sociale, environnementale et autres.
La mauvaise gestion des informations contenues dans cette norme peut entraîner de nombreux risques à savoir :
- Mauvaise interprétation de la norme ou du règlement
- Accident lié à la mauvaise utilisation de la documentation (notice…)
- Documentation d’accompagnement du produit erroné
- Mauvaise publicité du dispositif médical
- Dossier de marquage CE incomplet
- Documentent technique non conforme
Pour pallier à ces risques, il vous a été proposé une cartographie interactive de cette norme harmonisée. En plus de la cartographie, il est également possible de s'auto évaluer à l'aide d'un outil autodiagnostic développé.
III. Proposition d'outils pour faciliter la prise en main des informations et documentations à fournir par un fabricant
L'outil utilisé pour synthétiser les deux règlements européens et la norme harmonisée en projet est l'ANO (analyse Normative Opérationnel), qui a permis transformer les exigences complexes de ces différents documents en objectifs clairs et mesurables. Ce sont ces exigences mesurables qui été utilisés pour développer la cartographie interactive et l'outil d'autodiagnostic.
1. Cartographie de la norme PR NF EN ISO 20417
Pour une compréhension claire et globale des informations à fournir par un fabricant, une cartographie a été élaboré dans but de faciliter la compréhension et la prise en main de ces informations aux utilisateurs confirmés et novices. C'est un outil simple à utiliser qui offre une vision globale de la norme et la possibilité de la parcourir article par article.
Pour réaliser cette cartographie, il a fallu premièrement schématiser, organiser, designer, embellir, structurer les différentes interfaces de l'outil sur un fichier Powerpoint. Ce qui a demandé beaucoup d'imagination et d'expérience. Après cette étape, pour pouvoir naviguer librement dans l'outil, il a fallu intégrer la notion de liens hypertextes, pour le rendre interactif. Cette dernière étape terminée, de nombreux tests ont été réalisés afin de s'assurer du bon fonctionnement de l'outil. Alors pour qu'il ne soit pas modifié par l'utilisateur, nous avons transformé ce fichier PowerPoint en document PDF.

La cartographie a été conçu de telle manière à pouvoir être utilisé même par les néophytes, c'est-à-dire son utilisation est intuitive, laissez-vous juste guider. Mais par mesure de prudence, il est conseiller de lire le mode d'emploi qui y est associé afin de l'utiliser efficacement.
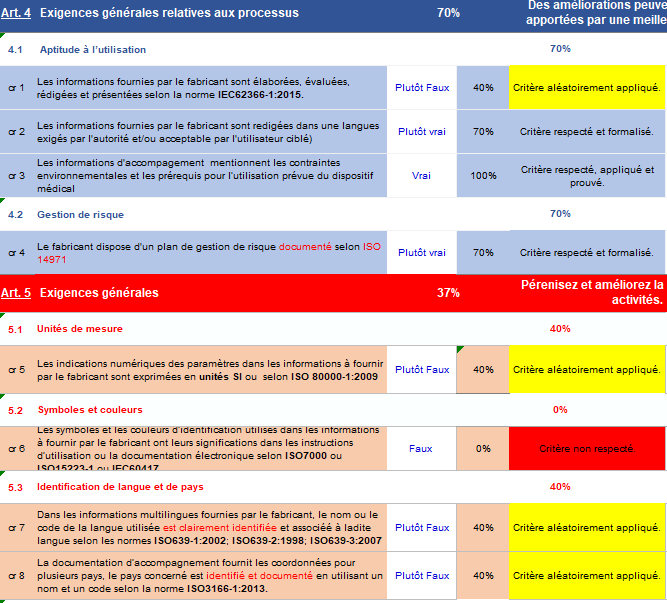
2. Outil d'autodiagnostic
Après la prise en main des informations à fournir par le fabricant, pour permettre à l'utilisateur de se s'autoévaluer (se positionner vis-à-vis du respect des exigences en matière d'information à fournir), un outil de diagnostic a été élaboré dans but de faciliter à l'utilisateur cette évaluation. Comme la cartographie, l'outil d'autodiagnostic est simple à utiliser et offre une évaluation rapide et efficace article par article.
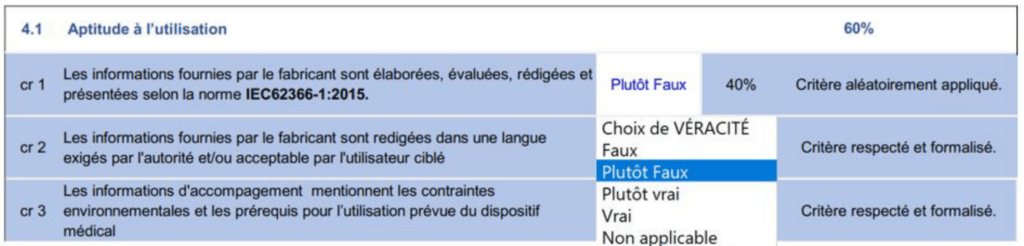
L'outil autodiagnostic a été réalisé à l'aide du tableur Windows nommé EXCEL. Il regroupe l'ensemble des exigences essentielles en matière d'information de manière visuelle et subdivisé en critères précis. Ces critères sont formulés sous forme d'affirmation et subdivisés en plusieurs sous-groupes différents. L'outil est divisé en 4 onglets différents détaillés dans la figure suivante :
En plus des quatre niveaux de véracité, une possibilité de répondre par « non applicable » a été ajoutée afin de permettre à l'utilisateur de répondre par « non applicable » s'il estime ne pas être concerné par le critère. Dans certains critères des mots ont été écrits en rouge afin d'attirer l'attention de l'utilisateur sur l'importance du critère. En fonction du niveau de conformité à une exigence, différents libellés d'évaluation ont été choisie afin d'attirer l'attention de l'utilisateur sur cet aspect.
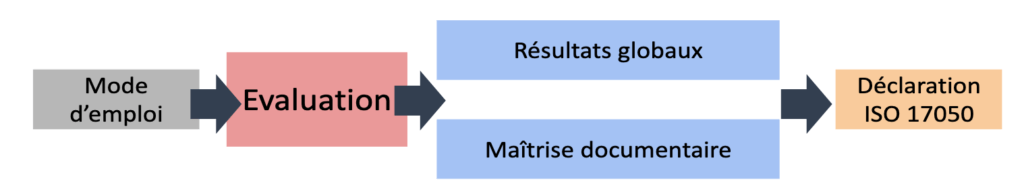
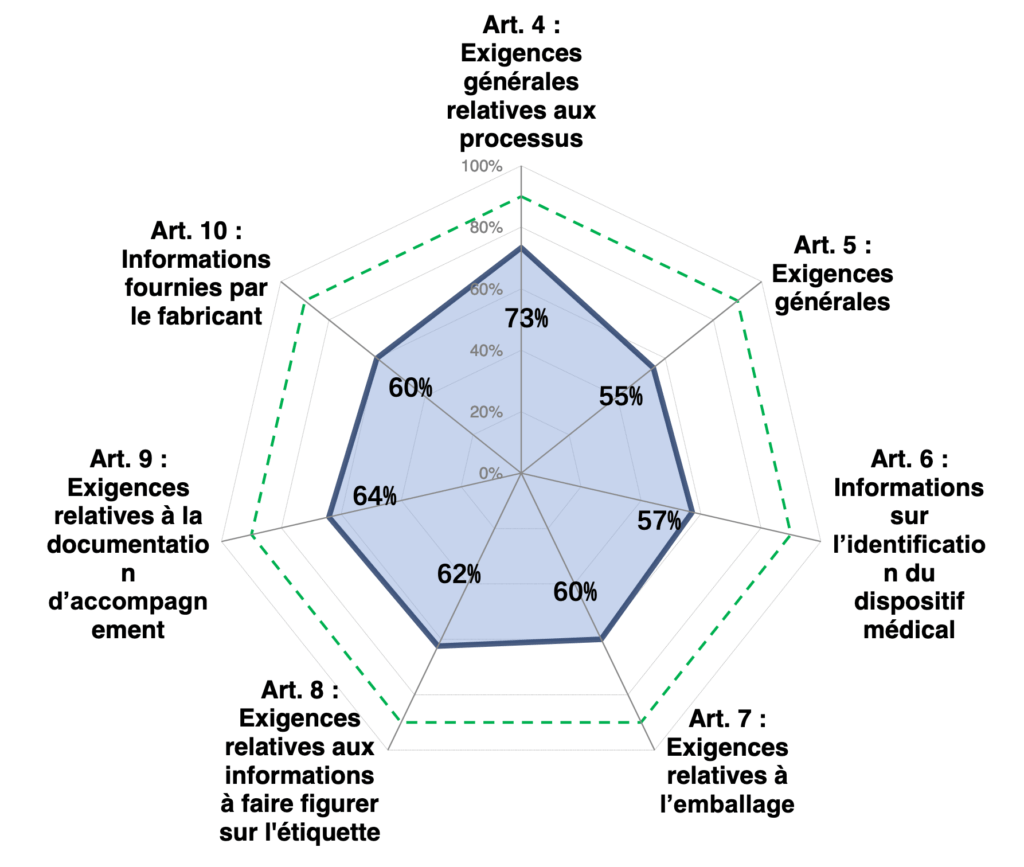
Après évaluation, résultats globaux présente la synthèse globale sous forme de graphe radar permet d'identifier très rapidement les non-conformités et de prioriser les plans d'actions élaborées.
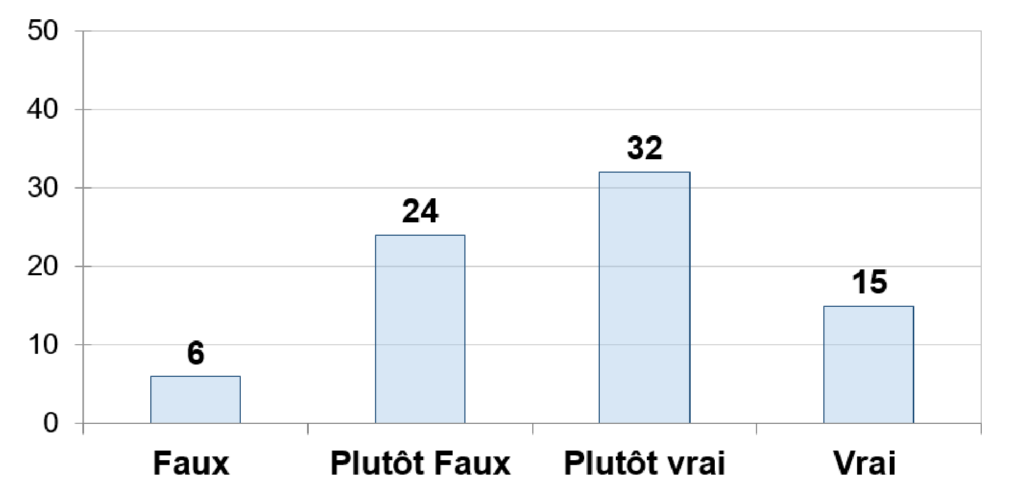
Également, sur le même onglet, nous pouvons visualiser les niveaux de véracité et de conformités aux exigences vis-à-vis de la norme étudiée. Ces résultats permettent d'identifier très rapidement le nombre d'article ayant des résultats satisfaisants ou non . Ce graphe aide l'utilisateur à l'élaboration de son plan d'action pour améliorer les pratiques de son entreprise.
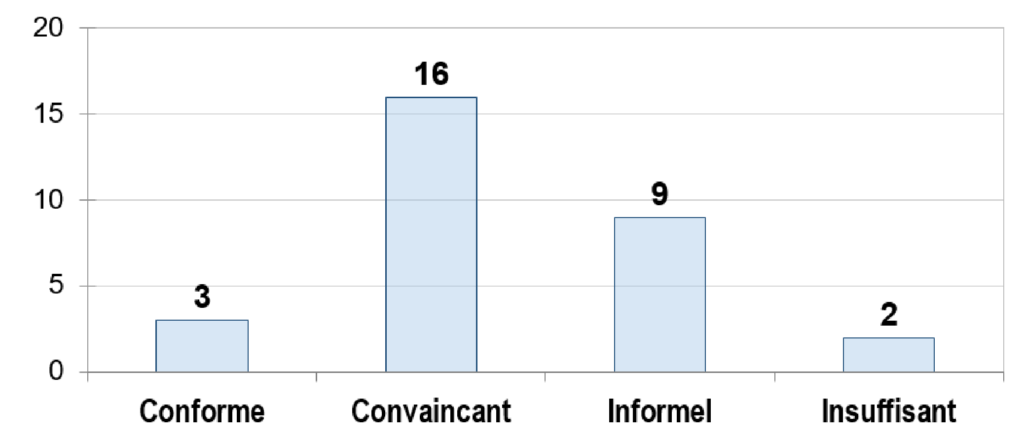
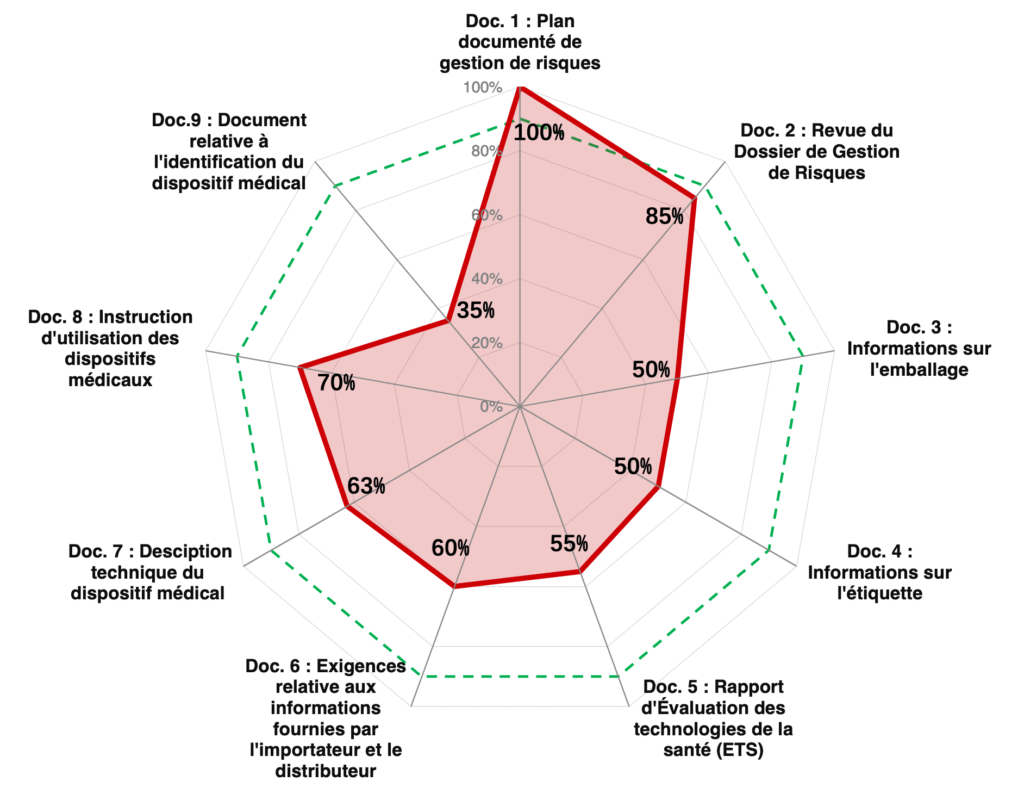
L'onglet concernant la « maitrise documentaire » permet de vérifier très rapidement l'état de conformité des documents du fabricant par rapport aux documents à fournir. (voir figure 8).
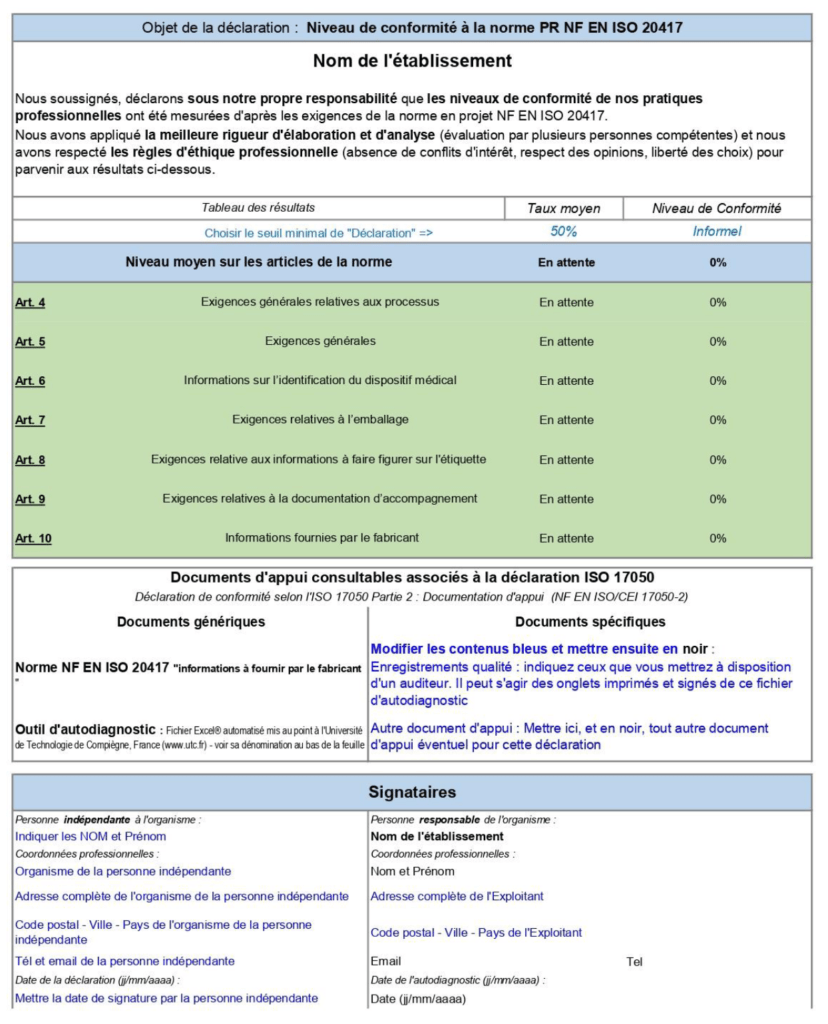
Pour terminer l'onglet « Déclaration ISO 17050 » permet à l'utilisateur en fonction de ses résultats de faire une auto déclaration. En cas de résultats satisfaisants, l'utilisateur fait présomption de conformité au règlement européen (spécifiquement concernant les informations à fournir) avant sa certification auprès d'organisme notifié (voir figure 9).
Interprétations des résultats
Après l'évaluation, une attention particulière doit être accordée aux documents ayant un niveau de conformité « plutôt faux » qui nécessitent des mises à jour. Dans le cas où le document n'existe pas, le fabricant doit très rapidement s'en procurer.
L'outil se positionne donc ainsi comme instrument d'aide pour les fabricants, à élaborer des plans d'action pour la gestion des informations et documents à fournir pour pouvoir mettre et accompagner le produit sur marché.
Conclusion
Parvenu au terme de ce travail de travail, il est noté que la prise en main cette norme est un facteur essentiel de la maitrise du flux informationnel et documentaire dans le processus de mise sur le marché d'un dispositif médical. A travers la cartographie et l'outil autodiagnostic proposé dans ce projet, les fabricants ont la possibilité de bien s'approprier très rapidement des informations et documents à fournir afin de se conformer aux exigences règlementaires selon la norme PR NF EN ISO 20417. Les entreprises de pourront donc assurer la traçabilité des flux d'informations afin de garantir la sécurité du patient