
IDS043 - Stratégie de passage au Règlement Européen 2017/745 d’un dispositif médical de classe I déjà commercialisé
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

Rihab OMRANI 
Chrispy PETNTANG
Contact
- Rihab OMRANI : ryhabomrany@gmail.com
- Chrispy PETNTANG : petntangchrispy@gmail.com
- Silyana SALMI : silyanasalmi@yahoo.fr
Citation
A rappeler pour tout usage : Rihab OMRANI, Chrispy PETNTANG, Silyana SALMI "Stratégie de passage au règlement 2017/745 d'un dispositif médical de classe I déjà commercialisé", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids043/
Résumé
Ce Mémoire d’Intelligence Méthodologique (MIM) a été rédigé par un groupe d’étudiants de l’Université de Technologies de Compiègne en collaboration avec une startup dans le cadre des évolutions réglementaires pour les aider dans la transition de la directive 93/42/CEE au Règlement Européen 2017/745 relatif aux Dispositifs Médicaux.
Ce MIM présente le projet réalisé et le plan d’action proposé comme solution dans cette phase transitoire de l’entreprise, et les outils développés pour aider les fabricants de dispositifs médicaux de classe I dans la maîtrise des écarts entre le Règlement 2017/745 et la directive 93/42/CEE.
Abstract
This Memorandum of Methodological Intelligence (MIM) was written by a group of students from the University of Technology of Compiegne in collaboration with a startup in the context of regulatory developments to help them in the transition from Directive 93/42/EEC on Medical Devices to European Regulation 2017/745 on Medical Devices.
This MIM presents the project carried out and the action plan proposed as a solution in this transitional phase of the company, the tools developed to help manufacturers of Class I medical devices in controlling the gaps between Regulation 2017/745 and Directive 93/42/EEC.
Remerciements
Nous tenons à témoigner toute notre reconnaissance à chaque personne qui a contribué à la réalisation de ce travail. Nous remercions en particulier Mme Pautard, responsable qualité à l’entreprise Blueback , qui nous a fait confiance et nous a guidé durant tout ce projet. Nous la remercions de nous avoir donné l'opportunité de travailler sur son dispositif médical et de proposer nos propres solutions et d'agir avec flexibilité.
Nous tenons à remercier M Jean-Matthieu PROT, notre suiveur UTC qui a sacrifié son temps pour nous aider et nous fournir les conseils nécessaires à la bonne conduite du projet.
Nous remercions notre collègue Yihan Chen pour sa participation active pour l'élaboration de ce travail.
Téléchargements



Outil d'aide à la compréhension du Règlement Européen 2017/745

Maîtriser vos écarts par rapport au Règlement Européen 2017/745
Introduction
C’est dans le cadre de notre formation de master Ingénierie de la Santé (IdS) parcours Dispositif Médical et Affaires Réglementaires (DMAR) à l’Université de Technologie de Compiègne (UTC) que nous sommes amenés à réaliser un projet dans le but de mettre en pratique et consolider les connaissances acquises. Ce document reflète notre travail réalisé avec la startup BLUEBACK pour l’assister dans sa démarche de passage de son DM (classe I) au nouveau Règlement Européen 2017/745 relatif aux dispositifs médicaux[1].
Ce projet initié par cette startup s’inscrit dans le cadre d’un grand changement du point de vu règlementaire autour du dispositif médical qui va impacter les distributeurs, les fabricants, les consommateurs, les concepteurs et les patients.
Notre objectif est de fournir un plan d’action réaliste des actions à entreprendre après identification des exigences pour se conformer au dit règlement avec des dates critiques en fonction des actions prioritaires. Nous proposerons un plan d’action à court (à mettre en œuvre d’ici Mai 2020) et un plan d’action sur le moyen terme (à mettre en œuvre d’ici 2024) pour que le dispositif se conforme aux exigences du nouveau Règlement Européen.
I- Contexte général : secteur des DM, mise sur marché des DM, réglementation
1.1 Secteur des dispositifs médicaux
Le Marché du dispositif médical (DM) en Europe représente près de 110 Milliards d’euros de chiffre d’affaire dans les ventes, et prêt de 700 000 emplois en 2017 [2].En France on compte environ 1500 entreprises pour un marché des dispositifs médicaux représentant près de 30 Milliards d’euros de chiffre d’affaires en 2017[3]. 93% de ces entreprises sont des TPE (Très Petites Entreprises) et des PME (Petites et Moyennes Entreprises)[3]. Les dispositifs médicaux couvrent une gamme de produits très variée du pousse-seringue, à l’IRM en passant par les dispositifs de monitorage personnalisés. Pour des raisons évidentes de sécurité, ce secteur est soumis à diverses réglementations tant au niveau national et international. C’est ainsi qu’a été établi le règlement Européen 2017/745 relatif aux dispositifs médicaux qui vient abroger la directive 90/385/CE relative aux dispositifs médicaux implantables actifs[4] et la directive 93/42/CE relative aux dispositifs médicaux[5].
Ce règlement ainsi que ses exigences définissent des obligations pour chaque opérateur économique (fabricant, mandataire qui est la personne physique ou morale présente au sein de l’UE ayant reçu un mandat écrit d’un fabricant situé hors de l’UE, distributeur qui est toute personne physique ou morale autre que le fabricant ou l’importateur et qui met à disposition sur le marché le dispositif médical, importateur qui est toute personne physique ou morale présente dans l’UE et qui met sur le marché un dispositif provenant d’un pays tiers)
1.2- Mise sur le marché d’un DM de classe I selon la directive 93/42/CE
Le marquage CE d’un dispositif se fait selon une procédure propre à chaque classe de dispositifs médicaux.
- La première étape d’un marquage CE est de démontrer que le dispositif est un DM en regardant sa définition ainsi que l’usage revendiqué par ce DM.
- La deuxième étape est de définir la classe de son DM en se basant sur les règles de classifications de l’annexe IX (de la directive 93/42). Les dispositifs de classe I présentant moins de risques que les autres classes, la directive prévoit juste au fabricant de faire une auto-déclaration [5].
Les DM de classe I avec fonction de mesurage suivent un chemin différent pour le marquage CE. Pour ces DM, il s’agit de suivre trois grands axes à savoir :
- Satisfaire aux exigences essentielles
- Créer une documentation technique
- Mettre en place un système de gestion des risques
L’intervention d’un organisme notifié est nécessaire mais cet ON se limite à l’évaluation de la conformité des produits aux exigences métrologiques [5].
1.3- Augmentation du niveau d’exigences réglementaires
Le nouveau règlement introduit 58 nouvelles définitions, dont dispositif falsifié, étiquette, notice d’utilisation, identifiant unique, nanomatériau, surveillance après commercialisation… De plus, il existe 4 nouvelles règles de classification ajoutées par le règlement (règle 19 à 22 de l’annexe VIII).
Un grand accent a été mis sur l’aspect ressources humaines chez les fabricants et les mandataires en les obligeant systématiquement à disposer d’une « personne chargée de veiller au respect de la réglementation »[1]. Cette personne devra avoir « un diplôme, un certificat ou un autre document de certification formelle sanctionnant des études universitaires en droit, en médecine, en pharmacie, en ingénierie ou dans une autre discipline scientifique pertinente, ou une formation reconnue équivalente par l'État membre concerné, et une expérience professionnelle d'au moins un an dans le domaine de la réglementation ou des systèmes de gestion de la qualité en rapport avec les dispositifs médicaux ou justifier de quatre ans d’expérience ». Ceci représente un budget supplémentaire qui n’est pas forcément supportable par tous.[6]
1.4- Le Blueback Physio
Cette idée de produit est née en 2015 des antécédents médicaux d’enfance d’un des fondateurs et ils ont voulu ainsi contribuer à cette course à l’innovation en créant le « Blueback physio ». Ce DM a été créé avec l’aide d’une professionnelle (kinésithérapeute) qui a participé en émettant les besoins qu’elle pouvait avoir et les fonctionnalités qui lui seront réellement utiles.
Ce dispositif est défini comme « un appareil destiné à être utilisé chez l’homme pour l’investigation d’une structure anatomique ou d’un processus physiologique, ceci à des fins médicales »[1] selon le règlement (UE) 2017/745. Il a pour rôle de caractériser les contractions du muscle abdominal transverse (muscle le plus profond de l’abdomen) lors des séances de rééducation chez un spécialiste. Il est utilisé par des kinésithérapeutes, des médecins, des sages-femmes et a comme pathologies cibles : la Lombalgie, la Pubalgie, les Pathologies respiratoires, les prises en charge Post-partum et le renforcement musculaire ; il n’est pas à but curatif, mais aide le professionnel à prendre des décisions sur ce qu’il observe.

« Le Blueback Box » est constitué d’une application mobile disponible sur playstore et utilisable sur tablette, d’un boitier (le Blueback Physio) et des accessoires.
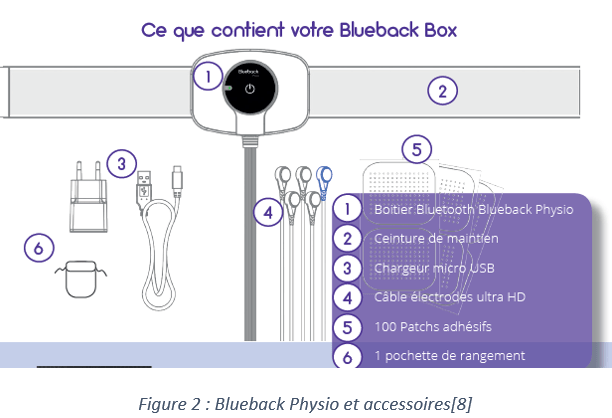
1.5- Environnement concurrentiel de la startup
Le Blueback physio, dispositif commercialisé par la startup Blueback, bien qu’étant innovant, se fait concurrencer par plusieurs autres produits et technologies.
- Le stabilizer biofeedback
Le concurrent majeur du Blueback physio est « le stabilizer biofeedback » (coussin chattanooga) conçu par des physiothérapeutes. C’est un coussin placé sur la colonne vertébrale et qui mesure le changement de pression dans le bassin abdominal à l’aide d’un manomètre. Ce dispositif est utilisé dans une position particulière et effectue une mesure indirecte. Il coûte environ 60 €.
- Le PHENIX liberty
La société Vivaltis propose toute une gamme d’appareil et d’options en fonction du type de mesure qu’on veut faire au niveau du produit PHENIX. Dans cette gamme il y a une partie qui fait du biofeedback pour le périnée avec une sonde périnéale qui est directement insérée dans le vagin de la personne. Avec cette sonde et les algorithmes développés par la société, ils sont en mesure de mesurer la contraction des muscles abdominaux. Ceci est donc une solution qui se rapprocherait du Blueback physio bien que n’étant pas identique.
- L’échographe
L’échographe permet de déterminer l’épaisseur du muscle et ne renseigne pas avec fiabilité (image non dynamique) mais avec cette technologie on peut voir la contraction réelle du muscle transverse abdominal à l’aide d’une sonde abdominale. Depuis 2015 les kinésithérapeutes peuvent acheter des échographes pour faire certains diagnostics du moment où ils ont suivi la formation. Le désavantage ici est le poids de l’appareil et le fait qu’il faut une formation spécifique pour pouvoir l’utiliser. Contrairement au Blueback physio, utilisable facilement par le professionnel de santé et dont la courbe d’évolution de l’activité musculaire du patient est facilement interprétable, le patient pouvant observer son évolution lui-même. Le plus du Blueback ici est donc l’aspect dynamique et la liberté de mouvement du patient.
- L’Electromyogramme (EMG)
Cette technique permet de mesurer l’activité électrique du muscle. Les informations recueillies par ce procédé ne sont pas vraiment faciles à interpréter et ne caractérisent pas vraiment le muscle (au sens médical pour un professionnel) car les mesures sont locales et faites au niveau du capteur. Pour s’approprier un électromyogramme à aiguille aujourd’hui, il faudrait débourser entre 7000 € et 20 000 €. Tandis que le Blueback physio quant à lui coûte environ 2000 €. On constate ici une grande disparité de prix et on peut conclure aux vues de ceci qu’investir sur un Blueback physio serai plus intéressant pour les kinésithérapeutes.
II- Les apports du règlement (UE) 2017/745 (évolutions majeures pour la mise sur marché des DM de classe I)
L’entreprise Blueback Physio a obtenu un marquage CE sur la directive 93/42/CEE. Le nouveau règlement Européen relatif aux dispositifs médicaux sera appliqué en Mai 2020 et abrogera la directive 93/42/CEE. Le dispositif étant de classe I, aucune période transitoire ne leur est accordée. Ils doivent impérativement être conforme d’ici 26 Mai 2020. Il est question d’identifier les nouvelles exigences afin d’anticiper sur les tâches prioritaires.
2.1 Classification DM selon règlement 2017/745
Les règles de classification sont explicitement détaillées dans l’annexe VIII. Selon le guide MEDDEV 2.1/5 relatif aux DM avec fonction de mesurage, un DM avec fonction de mesurage mesure quantitativement un paramètre physiologique ou anatomique, ou une quantité ou une caractéristique qualifiable d'énergie ou de substances livré au corps humain ou retiré de celui-ci. Et, une seconde condition doit être remplie par ces types de DM à savoir , le résultat affiché en unités légales ou autres unités acceptables au sens de la Directive 80/181/CEE ou comparé à au moins un point de référence indiqué en unités légales ou d'autres unités acceptables conformément à la directive précitée [9].
Étant donné que le Blueback physio mesure un paramètre anatomique (activité du muscle transverse abdominal), mais l’affiche sur la tablette sous forme de graphe (grandeur subjective), le DM n’est pas considéré comme DM avec fonction de mesurage. Ainsi, le Blueback physio qui était de classe I selon la Directive reste sous cette classe.
Le Blueback physio fonctionnant impérativement avec comme accessoire un logiciel, il est à son tour classé dans le Règlement.
Selon les règles d’application (Annexe VIII, §3.3) « Le logiciel commandant un dispositif ou agissant sur son utilisation relève de la même classe que le dispositif. » [1]. Donc le logiciel fournit par l’entreprise est de classe I.
2.2 Exigences générales en matière de sécurité et de performances
a. Dispositifs actifs et dispositifs raccordés à des dispositifs actifs
Les fabricants de dispositifs actifs doivent vérifier l’état de la source d’énergie pour garantir la sécurité du patient, s’assurer qu’il n’y a pas d’inférences électromagnétiques ; sécuriser le dispositif en limitant l’accès et disposer des moyens pour limiter et/ou réduire les risques en condition de premier défaut.
b. Les systèmes électroniques programmables
Cette partie fait mention des logiciels qui sont un des nouveaux DM du règlement. N’étant pas pris en compte par la directive, les exigences relatives à ces logiciels sont toutes nouvelles. Il s’agit ici lors de leur fabrication et de leur conception de tenir compte des logiciels utilisés en combinaison avec des plateformes mobiles de la taille et du contraste de l’écran, des caractéristiques spécifiques de ces plateformes et des facteurs extérieures liés à leur bonne utilisation comme la luminosité. Le règlement demande au fabricant d’énoncer les règles minimales pour le matériel informatique, les différentes caractéristiques du/des réseau(x) informatique(s) et des différentes mesures de sécurité informatique (protection du logiciel contre les accès non autorisés).
c. Dispositifs destinés à des profanes
Un point particulier est à prendre en compte dans le cas de ces dispositifs. En effet, le fabricant devra tenir compte de l’utilisateur et lui donner toutes les informations nécessaires à la compréhension du dispositif ; il doit s’assurer au maximum que l’utilisateur n’aura aucun problème pour l’utiliser en le formant au préalable. Le fabricant doit prévoir une procédure qui permettra à l’utilisateur profane de vérifier les performances du DM et le comportement à tenir en cas de résultat non valide.
d. La notice d’utilisation
Les notices doivent désormais comporter un lien dirigeant vers le résumé des caractéristiques de sécurité et de performances, une totale description des bénéfices cliniques escomptés du dispositif, une description sur le choix du logiciel et des accessoires (adaptés). Le fabricant doit aussi prévoir une « mention à l'intention de l'utilisateur et/ou du patient indiquant que tout incident grave survenu en lien avec le dispositif devrait faire l'objet d'une notification au fabricant et à l'autorité compétente de l'État membre dans lequel l'utilisateur ou le patient est établi ».
Le fabricant doit énoncer les exigences minimales concernant le matériel informatique, les caractéristiques des réseaux informatiques et les mesures de sécurité informatique (protection contre les accès non autorisés) garantissant le fonctionnement du logiciel prévu. (§17.4)
Il est également impératif au fabricant de renseigner les contre-indications, les risques liés à d’éventuels surdosages et les effets secondaires indésirables ou une mauvaise utilisation du dispositif.
Si le dispositif est destiné à un profane, le fabricant doit indiquer dans la notice les circonstances dans lesquelles le profane devrait se rapprocher d’un professionnel de santé. Il faut savoir que les notices d’utilisation ne sont pas obligatoires pour les dispositifs de classe I. Cependant, le fabricant s’il le désire peut le mettre à disposition de l’utilisateur.
2.3 Modification du système de management de la qualité et de la Documentation technique
Deux outils d’assistance ont été développé pour aider le fabricant à prendre en main les parties du Règlement relatives au système de management de la qualité et de la documentation technique, et de maîtriser les écarts entre la Directive 93/42/CEE et le Règlement 2017/745. Ces outils feront l’objet d’une description dans cette partie.
a. Mise en conformité du SMQ (article 10 -annexe IX chapitre 1 et 3 ou annexe XI partie a)
Le système de gestion de la qualité démontre l’ensemble des actions mises en place par l’entreprise dans un but d’amélioration de la qualité de production et d’organisation. Il comprend l’ensemble des exigences, éléments et dispositions adoptés par le fabricant. Le fabricant doit tenir à disposition des autorités compétentes la documentation technique du dispositif médical pour une durée de dix ans après sa mise sur le marché.
Au sein du règlement 2017/745, il est précisé qu’avant la mise sur le marché d’un dispositif médical de classe I, le fabricant peut se référer à l’une de ces annexes pour mettre en place un système de gestion de la qualité :
- L’annexe IX(Évaluation de la conformité sur la base d’un système de gestion de la qualité) chapitre I(système de gestion de la qualité)et III(dispositions administratives)
OU
- L’annexe XI(Évaluation de la conformité sur la base de la vérification de la conformité du produit) partie A (assurance de la qualité de la production)
Afin d’avoir une vue directe sur les exigences attendues par le fabricant et pour l’évaluation de la conformité selon les deux annexes, un outil d’appropriation sur PowerPoint a été créé.
La figure 03 présente un extrait de l'outil qui a été développé afin d'assister le fabricant dans sa démarche de mise en conformité du système de gestion de la qualité.
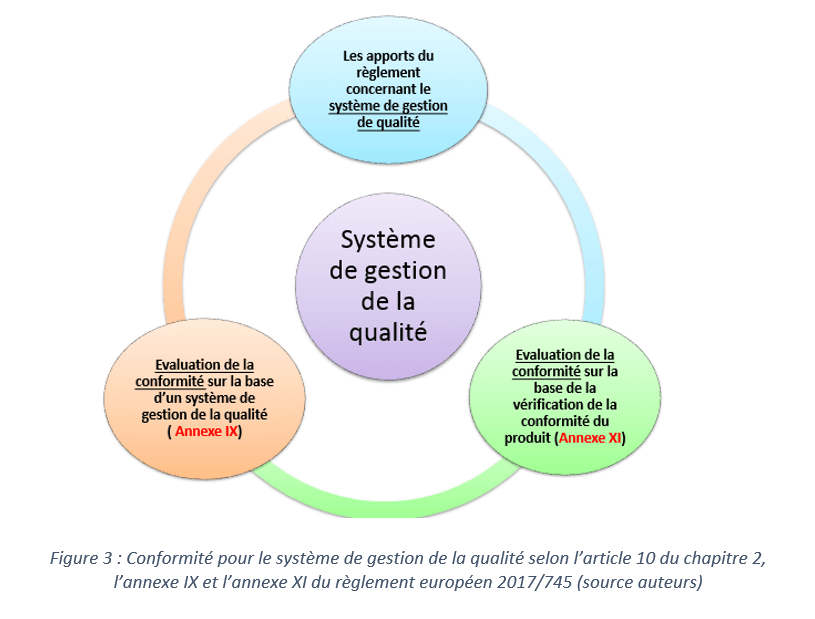
Il a pour but de résumer les nouveautés apportées par le règlement 2017/745 par rapport aux exigences auxquelles la société Blueback répondait déjà sous la directive 93/42/CE pour leur dispositif médical (classe I).
D’autre part, après un résumé général de l’onglet concerné, une check-list permet au fabricant de voir toutes les nouvelles exigences du système de gestion de la qualité auquel il doit pouvoir répondre pour être conforme au nouveau règlement 2017/745. Cette check-list comporte 3 niveaux de véracité : « FAIT » ; « NON FAIT » ; « EN COURS ». En fonction des réponses fournies, des taux de conformités vont être attribués ce qui permettra au fabricant de suivre son état d’avancement.
b. Contrôle de la documentation technique(annexe II)
La documentation technique sert à démontrer la conformité d’un dispositif médical aux exigences essentielles du règlement 2017/745. Elle permet de présenter le dispositif, son mode de fonctionnement et les étapes de sa conception et sa fabrication. L’évolution ou la modification des fonctionnalités du dispositif, du processus de fabrication et de conception ou des éléments concernant la surveillance après commercialisation implique impérativement une mise à jour du dossier technique. Le fabricant doit tenir à disposition des autorités compétentes la documentation technique du dispositif médical pour une durée de dix ans après sa mise sur le marché.
Dans le règlement 2017/745, les annexes II et III sont consacrées pour la présentation du contenu de la documentation technique et la documentation technique de la surveillance après la commercialisation.
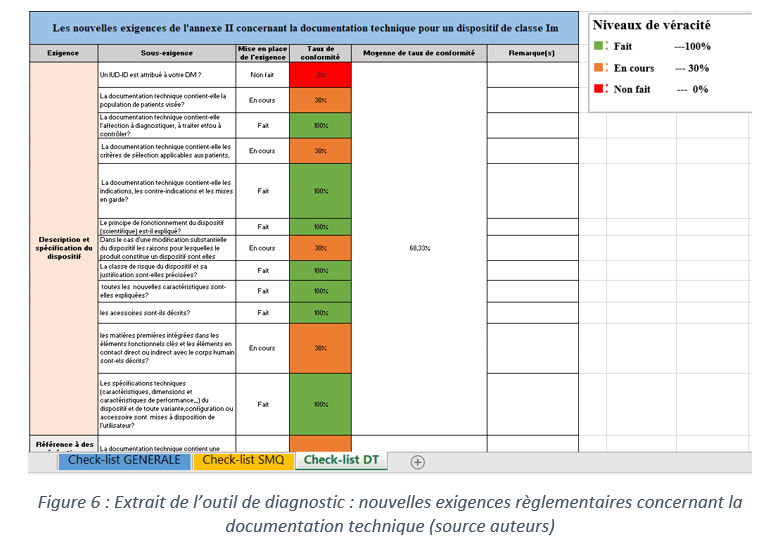
La figure ci-dessous présente les éléments qui doivent figurer dans la documentation technique selon l’annexe II du règlement européen :
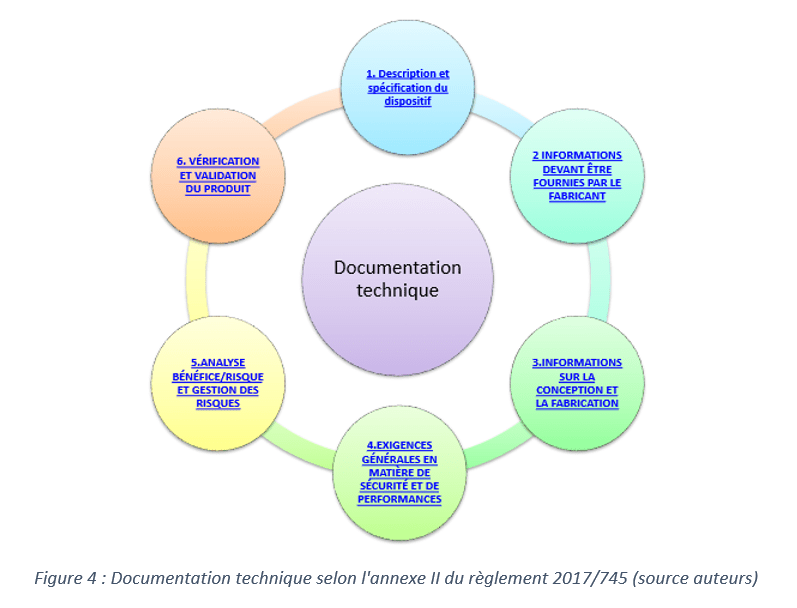
Pour présenter d’une manière simplifiée le contenu de cette documentation technique, un outil d’appropriation sur PowerPoint a été créé. Il a pour but de résumer les nouveautés apportées par le règlement 2017/745 par rapport à la directive 93/42 pour des dispositifs médicaux de classe I.
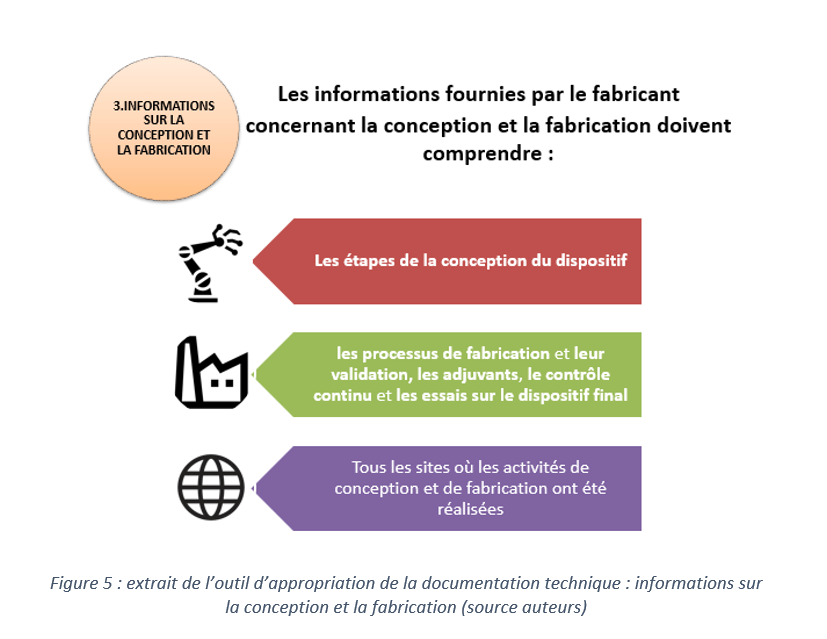
Afin que l’outil d’appropriation soit plus complet un outil d’autodiagnostic, sous forme d’un tableau sur Excel, est fourni permettant à l’entreprise de voir toutes les nouvelles exigences concernant la documentation technique et de suivre ainsi son état d’avancement.
Celui qui réalise le diagnostic aura le choix entre 3 niveaux de véracité : FAIT, NON FAIT et EN COURS. En fonction des réponses fournies, des taux de conformités vont être attribués.
2.4 Évaluation clinique
Les exigences générales à remplir pour poursuivre une évaluation clinique sont indiquées dans l’annexe 14 partie A du Règlement 2017/745. L’évaluation clinique doit prendre en compte les données favorables et défavorables ; elle doit être objective et approfondie. Elle dépend aussi bien de la nature, de la classification du dispositif médical et de sa destination (définie par le fabricant).
Tout au long de l’évaluation clinique, le fabricant établit et fait une mise à jour régulière d’un plan d’évaluation clinique qui doit contenir :
- Les exigences en matière de sécurité et de performances (renforcées par les données cliniques) ;
- L’analyse des données cliniques pertinentes et les conclusions sur la sécurité et les performances ;
- La population visée, les indications et les contre-indications ;
- La destination du produit ;
- Une description précise des résultats cliniques avec mention des bénéfices cliniques pour le patient ;
- Les mesures utilisées pour évaluer qualitativement et quantitativement la sécurité du patient (sécurité clinique) ;
- Une description des risques résiduels et surtout des effets secondaires ;
- Une description des paramètres validés pour la détermination du rapport bénéfice/risque ;
- Les données cliniques disponibles ainsi que tous les écarts relevés des preuves cliniques dans la littérature ;
- Toutes données pouvant être utile au traitement des questions pas encore résolues.
Un rapport sur les évaluations cliniques est établi par le fabricant où il insère les preuves et résultats cliniques. Ce rapport ainsi que les méthodes d’essai (non cliniques), et les documents pouvant démontrer la conformité aux exigences essentielles en matière de sécurité et de performances sont introduites dans la documentation technique.
Les évaluations cliniques pour les fabricants de dispositifs de classe I se font suivant l’un des quatre axes ci-dessous :
- Évaluation critique des publications scientifiques
- Évaluation critique des résultats de toutes les investigations cliniques faites
- Combinaison des méthodes précédentes
- Prise en compte de toutes les alternatives de traitements déjà disponibles sur le marché
L’entreprise qui effectue ses évaluations cliniques sur la base des évaluations critiques des publications scientifiques, devra prouver l’équivalence avec les dispositifs dont elle utilise les données et prouver le respect des exigences générales en matière de performance. Cette équivalence devra être faite sur trois plans :
- Équivalence biologique :
L’entreprise doit démontrer qu’elle utilise les mêmes matériaux, que les durées de contact des substances avec la peau sont similaires et que les produits de dégradation et les autres substances (relargables) sont similaires.
- Équivalence technique :
L’entreprise démontre la conformité de « la conception, les conditions, les spécifications, les propriétés physicochimiques (la traction, viscosité etc…), les propriétés de surface, longueur d’onde, les algorithmes et les méthodes d’installation de son dispositif »[1] avec celui dont elle a fait équivalence ; et elle s’assure qu’ils ont les mêmes performances cliniques et une même méthode opératoire.
- Équivalence clinique :
« L’état clinique utilisé du dispositif, la destination, la sévérité et stade de la maladie sont les mêmes. La population visée (l’âge, l’anatomie et la physiologie) est la même, également le même type d’utilisateur. Les performances critiques sont équivalentes au regard de l’effet clinique prévu » [1].
Pour respecter les échéances du règlement, il faut rassembler des preuves cliniques avant le 26 Mai 2020 [10]. Pour cela, il est urgent que les fabricants utilisent des données post-commercialisations qui sont déjà disponibles ou faire une étude post commercialisation afin de soumettre les preuves cliniques nécessaires dans la documentation technique.
2.5 La surveillance après la commercialisation
Le nouveau règlement européen ne vise pas uniquement à renforcer les exigences relatives à l’évaluation de la conformité des dispositifs médicaux avant leurs mises sur le marché. En effet, ce règlement s’intéresse aussi à la mise en place d’un système de surveillance après la commercialisation ayant pour but de collecter et d’analyser des données concernant la qualité, la sécurité et les performances des dispositifs tout au long de leurs cycles de vie.
En fonction du type du dispositif et de sa classe de risque, les fabricants doivent établir et mettre à jour le système de surveillance après commercialisation qui fait partie du système de gestion de la qualité.
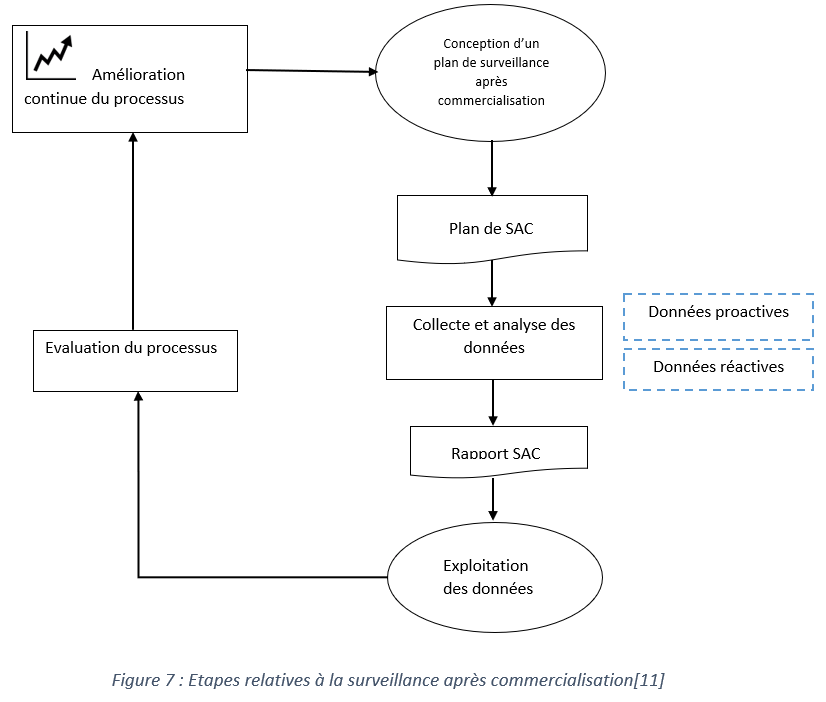
2.5.1 Procédure de surveillance après commercialisation
Le logigramme ci-dessous représente les étapes importantes relatives à la surveillance après la commercialisation à mettre en place par le fabricant :
2.5.2 Documentation technique relative à la surveillance après la commercialisation
Le fabricant doit établir une documentation technique relative à la surveillance après la commercialisation conformément aux exigences de l’annexe II. Cette documentation doit comporter :
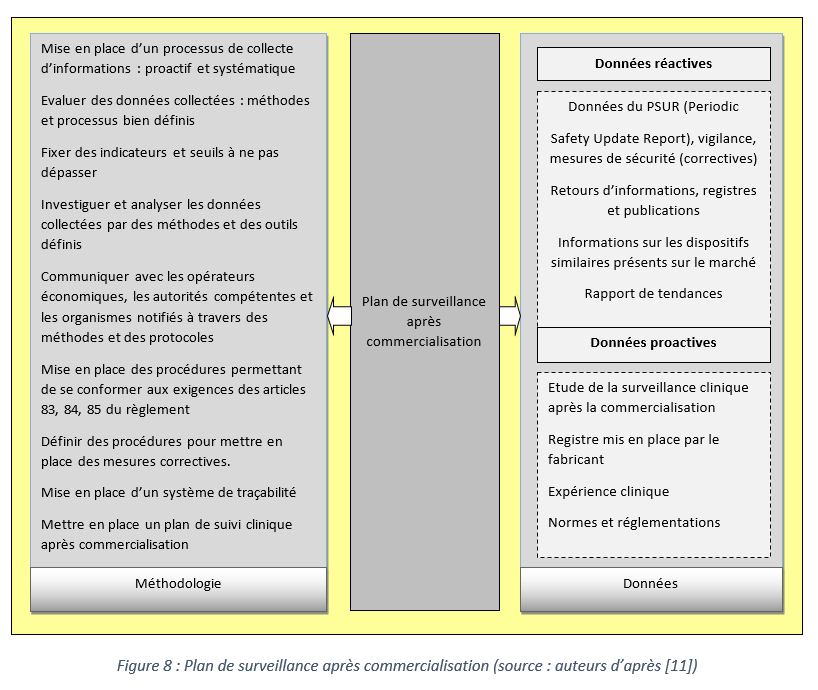
a- Un plan de surveillance après commercialisation
La cartographie ci-dessous résume les actions à mettre en place dans un plan de surveillance après commercialisation ainsi que les types de données nécessaires :
b- Un rapport périodique actualisé de sécurité(PSUR) ou un rapport sur la surveillance après la commercialisation (PMSR)
Selon le chapitre VII, article 85 du règlement européen des dispositifs médicaux, le fabricant des dispositifs médicaux de classe I doit fournir un rapport de surveillance après commercialisation (PMSR). Ce rapport doit contenir les résultats et les conclusions concernant les données de surveillance après commercialisation, ainsi que les actions correctives mises en place pour les dispositifs déjà commercialisés. Ce rapport fait partie de la documentation et il doit être mis à jour par le fabricant si nécessaire et mis à la disposition des autorités compétentes si elles le demandent.
Le PSUR (Periodic Safety Update Report) est une extension du rapport de surveillance post-commercialisation (PMSR). Il est destiné aux dispositifs à risque modéré et élevé (IIa, IIb et III). Il contient les résultats et les conclusions des données relatives à la surveillance après la commercialisation et il doit inclure les justifications et les descriptions nécessaires de toutes les mesures préventives et correctives prises par le fabricant pour les dispositifs sur le marché.
Selon l’article 86 du règlement 2017/745 le PSUR doit contenir :
- « les conclusions à utiliser dans le cadre de la détermination du rapport bénéfice/risque » ;
- « Les principales constatations du SCAC » ;
- « Le volume des ventes du dispositif et une estimation de la taille et d'autres caractéristiques de la population utilisant le dispositif et, si possible, la fréquence d'utilisation du dispositif. »
Le tableau ci-dessous présente le type de rapport à fournir ainsi que sa périodicité selon la classe de risque du dispositif médical.
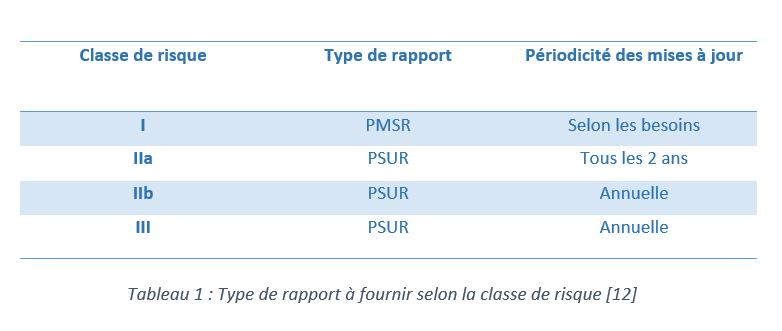
Dans ce cas particulier, on s’intéresse au cas des dispositifs de classe I. Il est donc indisponible de rédiger, mettre à jour et à dispositions des autorités compétentes, si besoin, un PMSR.
III- Stratégie (plan d’action)
3.1 Démarche de conformité
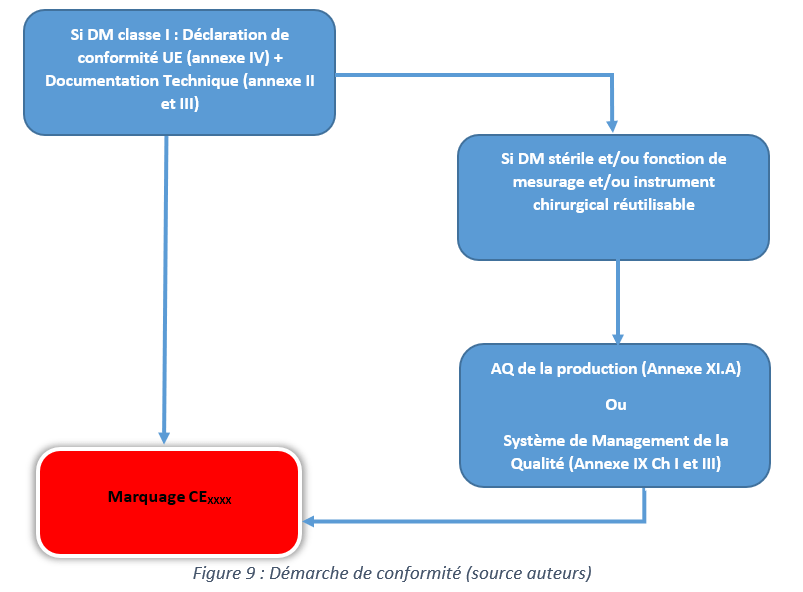
La figure 9 explique la démarche à suivre pour avoir un marquage CE selon l’article 52. Ainsi, les fabricants de dispositifs de classe I doivent avoir une documentation technique conforme au règlement 2017/745 et avoir un système de management de la qualité conforme à l’ISO 13485 qui fait présomption de conformité ; ou suivre les procédures de l’annexe IX (Ch I et III)ou de démontrer qu’il maîtrise la production (annexe XI.A).
3.2 Plan d’action à court terme (Mai 2020)
Cette partie traite des exigences à mettre en place avant Mai 2020 pour maintenir le dispositif sur le marché. Malgré le deuxième corrigendum pour le règlement qui a été publié pour soulager les organismes notifiés et aider les fabricants en leur accordants des délais pour se conformer, il y a des points majeurs à anticiper. Les dates de transition selon la situation de l’entreprise sont les suivantes : [13]
- DM de classe I : mai 2020
- Nouveaux DM de classe IIa, IIb et III : mai 2020
- DM de classe Is, Im, IIa, IIb et III déjà CE : fin du certificat ou mai 2024 au plus tard
- DM de classe Icr (Instrument chirurgical réutilisable), Is, Im : mai 2024
- DM de classe I changeant de classe : mai 2024
Les nouveauté décrites ci-dessous sont détaillées dans la check-list présente dans notre outil d’autodiagnostic.

3.3 Plan d’action Moyen terme (Mai 2022)
La base de données Eudamed ayant été repoussé de deux ans, les fabricants ont jusqu’en Mai 2022 pour attribuer des IUD à leurs dispositifs et les enregistrer dans la base de données Européenne Eudamed. Cependant, les fabricants doivent vérifier les disponibilités d’une éventuelle version dégradée de la base de données d’ici le 26 Mai 2020.
Conclusion
Ce projet proposé par l’entreprise Blueback dans le cadre de la mise en conformité de leur dispositif de classe I selon le règlement 2017/745 nous a permis de relever et d’analyser les nouvelles exigences réglementaires. Ces exigences ont été regroupées dans un outil d’autodiagnostic facile à exploiter permettant aux fabricants d’évaluer leurs taux de conformité aux exigences spécifiques.
De même, un plan d’action a été proposé à l’entreprise sur le court et moyen terme en se basant sur les échéances recensées dans le règlement.
Par ce projet, nous avons eu des connaissances plus approfondies dans le domaine de la règlementation et des compétences managériales plus développées. Il a été question de mener à bien le projet avec une équipe pluridisciplinaire en coopération avec l’entreprise Blueback en s’échangeant les connaissances et l’expérience. Ceci nous a permis de concrétiser les connaissances théoriques acquises concernant le Règlement des dispositifs médicaux.
Références bibliographiques
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE.) ». Ed. JO L 117, http://data.europa.eu/eli/reg/2017/745/oj, 05-mai-2017.
[2] J. Doe, « Medical Devices », Internal Market, Industry, Entrepreneurship and SMEs - European Commission, 21-nov-2018. [En ligne]. Disponible sur : https://ec.europa.eu/growth/sectors/medical-devices_en. [Consulté le : 09-janv-2020].
[3] Syndicat National de l’Industrie des Technologies Médicales, « Snitem - Panorama de la filière industrielle des dispositifs médicaux en France 2019 », calameo.com, 13-janv-2020. [En ligne]. Disponible sur : https://www.calameo.com/read/0006105424e05e637bda8. [Consulté le : 22-janv-2020].
[4] « Directive 90/385/CEE du Conseil, du 20 juin 1990, concernant le rapprochement des législations des États membres relatives aux dispositifs médicaux implantables actifs ». Ed. JO L 189, http://data.europa.eu/eli/dir/1990/385/oj, 20-juill-1990.
[5] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux ». Ed JO L 169, http://data.europa.eu/eli/dir/1993/42/oj, 12-juill-1993.
[6] C. Mangeol, « Enjeux et exigences de la nouvelle réglementation européenne des dispositifs médicaux », Thèse, Aix Marseille Université, Marseille, 2019. [en ligne] Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-02140163/document.
[7] Blueback, « Brochure BluebackPhysio ». 2018. Disponible sur : https://www.blueback.fr/produit/blueback-physio/
[8] Blueback, « Notice Blueback physio V7 ». 2019. Disponible sur : https://www.blueback.fr/produit/blueback-physio/
[9] Commission Européenne, « Medical devices with a measuring function ». Juin1998. [En ligne] Disponible sur : https://www.gpc.center/download/817.
[10] P. RENARD, « Le temps presse pour les fabricants de DM de classe I », DeviceMed.fr, 28-mai-2019. [En ligne]. Disponible sur : https://www.devicemed.fr/dossiers/reglementation/le-temps-presse-pour-les-fabricants-de-dm-de-classe-i/19342. [Consulté le : 24-déc-2019].
[11] M. Jacquemart, « La surveillance après commercialisation dans le domaine des dispositifs médicaux et l’impact du nouveau règlement (UE) 2017/745 », Thèse, Université Toulouse III PAUL SABATIER, Toulouse, 23 Janvier2019. [en ligne] Disponible sur : http://thesesante.ups-tlse.fr/2604/1/2019TOU32002.pdf?fbclid=IwAR0S4xM95LvMUOESvYN72ABE-AR9086kJQ1pvp7xmFzAY5Rbyu-4RnvcBRE
[12] A. Groell, H. Zkeik, et K. Benaceur, « IDS005 - Surveillance après commercialisation des dispositifs médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositif Médical et Affaires Règlementaires (DMAR), Mémoire de projet, www.travaux.master.utc.fr, puis « IDS » réf n° IDS005, janv. 2019.
[13] Guillaume Promé, « Corrigendum du règlement DM et mesures transitoires », Qualitiso, 05-mars-2019. [En ligne]. Disponible sur : https://www.qualitiso.com/periode-transition-dm-classe-i/. [Consulté le : 16-déc-2019].