
IDS075 - Guide d'accompagnement pour la mise sur le marché d'un Dispositif Médical
DOI mémoire
https://doi.org/10.34746/nf04-kg53Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteures

Auriane GUILLOTEAU 
Eloïse DE BEAUFORT 
Manolie PAUL 
Marie GIORGI
Contacts
- auriane.guilloteau@laposte.net
- eloisedebeaufort@gmail.com
- manolie.paul@laposte.net
- mariegiorgi@outlook.fr
Citation
A rappeler pour tout usage : A. Guilloteau, E. de Beaufort, M. Paul et M. Giorgi, "Guide d'accompagnement pour la mise sur le marché d'un Dispositif Médical", Université de Technologie de Compiègne (France), Master Ingénierie de la santé parcours Technologies Biomédicales et Territoires de Santé, Mémoire de projet, Décembre 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids075/ ; https://doi.org/10.34746/nf04-kg53
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_03_idsap
Résumé
Face à la crise sanitaire du COVID-19, les établissements de santé ont demandé une multiplication des dispositifs médicaux disponibles pour traiter les patients en insuffisance respiratoire : les ventilateurs artificiels. C’est ainsi que de nombreuses initiatives indépendantes ont vu le jour pour proposer des ventilateurs fabriqués par des ingénieurs ou fabricants indépendants afin de suppléer à un manque dans certains hôpitaux. Pour ce faire, les équipes d’experts avaient besoin de connaitre et appliquer les règlementations nécessaires et obligatoires afin de diffuser et commercialiser leur dispositif ou innovation. Pour cela, ce guide d’accompagnement à la mise sur le marché d’un Dispositif Médical, expose toutes les phases dans l’ordre chronologique à suivre pour un porteur de projet d’innovation biomédicale. L’utilisateur sera alors guidé à partir de l’étude du marché et de la concurrence jusqu’à la surveillance après commercialisation de son dispositif. En suivant judicieusement les étapes explicitées dans des fiches techniques spécifiques, le fabricant s’assure de fournir les documents nécessaires aux autorités compétentes et de produire un dispositif conforme aux réglementations européennes. Une cartographie interactive sous forme de spirale, permet à l’utilisateur d’avoir une vision globale de son processus et d’avancer pas à pas vers la mise sur le marché. L’aboutissement de cet outil sera la mise à disposition de l’innovation dans les établissements de santé et l’utilisation par les professionnels pour leurs patients.
Summary
Facing the health crisis of COVID-19, healthcare institutions have requested to increase the number of medical devices available. For the COVID-19 disease, artificial ventilators are needed to treat patient with SARDS (Acute respiratory distress syndrome). As a result, many independent initiatives have emerged to offer ventilators manufactured by independent engineers or manufacturers. It could be used to make up for a shortage in some hospitals. To do so, teams of experts needed to know and apply the mandatory regulations in order to spread and market their new device. Then, this commercialization guide for a Medical Device, sets out all the phases in chronological order to follow for a biomedical innovation project. It will guide the user from the benchmark study to the post-commercialization surveillance of its device. By judiciously following the steps explained in specific technical sheets, the manufacturer ensures that he provides the necessary documents to the competent authorities and produces a device that complies with European regulations. A spiral interactive cartography allows the user to have a global vision of his process and to progress step by step towards the commercialization. The outcome of this tool will be the availability of use of the device in hospitals for patient care.
Téléchargements



Mémoire complet :
Guide d'accompagnement pour la mise sur le marché d'un Dispositif Médical
Introduction
Dans le contexte de crise sanitaire qu’a rencontré le monde entier depuis janvier 2020, la santé et les dispositifs médicaux (DM) sont plus que jamais au centre de toutes les préoccupations. Il est alors primordial aujourd’hui que chaque acteur du monde de la santé puisse agir à son échelle pour contribuer à la santé de la population et aux avancées de la recherche. C’est dans certains élans de solidarité que de jeunes ingénieurs, chercheurs ou médecins ont su unir leurs forces et leurs compétences pour aider le gouvernement et les populations face à une détresse sanitaire. Il parait alors impensable qu’une personne ayant des ressources (intellectuelles ou matérielles) dans le domaine de la santé ne puisse pas s’en servir et en faire profiter ceux qui ont besoin d’aide ou de support technique. Aujourd’hui, c’est à la crise du COVID-19 que le monde doit faire face, mais il est important de noter que demain, une catastrophe naturelle pourrait causer autant de dégâts sanitaires voire plus et que les ressources nécessaires devront être multipliées. Les ingénieurs de demain devront donc être prêts à affronter un manque de ressources en santé et devront pouvoir agir dans l’urgence.
Dans un contexte de commercialisation et de mise à disposition de dispositifs médicaux, les procédures sont parfois très longues et très réglementées. Ce mémoire est à destination de toute personne ayant des connaissances dans le domaine de l’ingénierie biomédicale et qui aurait comme projet de commercialiser une innovation de rupture ou un dispositif médical. Grâce à ce mémoire, il connaitra toutes les étapes nécessaires à suivre de l’étude de marché à la mise à disposition, en respectant les réglementations européennes et en étant conforme à la norme NF EN ISO 13485 - Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires. Le fabricant ou porteur de projet trouvera ici un guide d’accompagnement à la commercialisation et à la diffusion d’un dispositif médical. Un guide détaillé et précis qui lui permettra d’être méticuleux dans son processus et de n’oublier aucune étape. Il semble important de signaler toute l’utilité de ce guide dans un contexte urgent, en offrant un système opérationnel immédiatement qui nécessite simplement d’être suivi de près par le porteur de projet.
Ce mémoire est alors constitué de trois parties, une première qui traitera du contexte et des cibles du projet, une deuxième qui fournit une notice d’utilisation du guide interactif et une troisième qui exprimera les résultats attendus et espérés pour le porteur de projet. Il répondra alors à la problématique suivante : Comment rendre la commercialisation et la diffusion d’un dispositif médical accessible et simple pour un fabricant ?
Chapitre 1 : Mise en contexte – Mettre sur le marché un dispositif médical
1. Contextualisation de la crise sanitaire, les cibles du projet
Il s’agit ici, de mettre en place un outil d’accompagnement suivant la norme ISO 13485 qui permettrait à tout fabricant, ingénieur, start-up, autoentrepreneur de pouvoir mettre sur le marché un dispositif médical qu’il a fabriqué. Cet outil listera toutes les étapes à suivre par le fabricant pour pouvoir commercialiser rapidement le dispositif qu’il a produit.
1.1 Les cibles et objectifs du projet
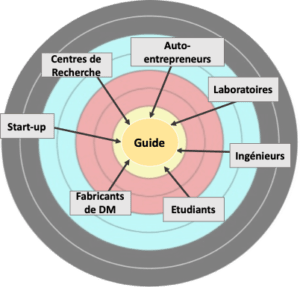
Le fabricant peut être un ingénieur, une start-up, un autoentrepreneur (Figure 1); toute personne avec les capacités techniques et matérielles de fabriquer un dispositif médical dans le besoin d’être guidé. Cet outil s’apparente donc à un guide de commercialisation « pour les nuls ».
Les bénéficiaires de cet outil sont ceux qui vont utiliser le dispositif fabriqué c’est-à-dire, les hôpitaux, les médecins et les patients. Ces personnes seront donc indirectement reliées à l’outil car s’il est efficace, un dispositif sera fabriqué et commercialisé rapidement et sera profitable pour les personnes citées.
Figure 1 : Illustration présentant les cibles du projet, source : auteures
Cet outil a un fort potentiel car il a un important portefeuille de futurs clients et le domaine de l’innovation des dispositif médicaux continue de croître. En effet avant la crise de la covid-19, le marché français des dispositif médicaux était estimé à 30 milliards d’euros [1]. Ensuite, l’OMS et la commission européenne créent un conseil de facilitation de la circulation des dispositif médicaux pour essayer de développer un nouvel accès au marché, encourager la fabrication de nouveaux dispositifs par des particuliers afin de palier à la crise de la covid-19 et anticiper d’éventuelles autres crises à venir qui toucheraient le monde de la santé.
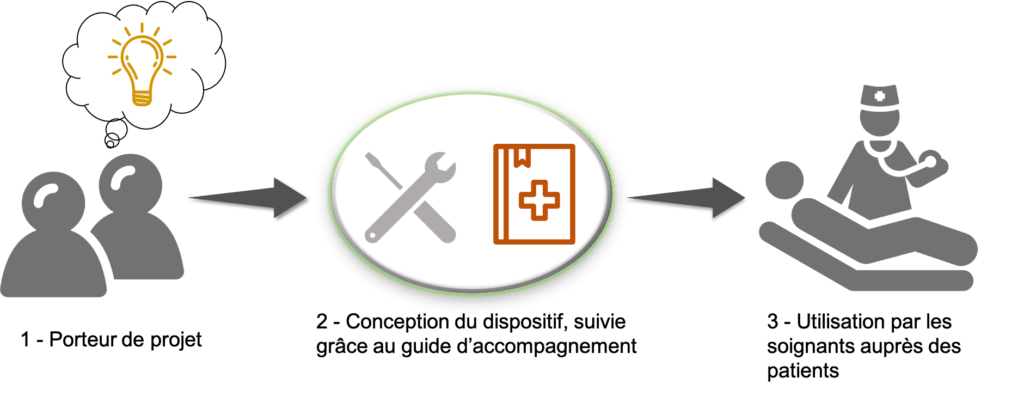
La finalité d’un tel outil est également de remettre au centre des préoccupations le patient. En effet, à première vue le principal concerné par ce guide serait un porteur de projet biomédical ou un ingénieur en capacité de fabriquer un dispositif médical, mais l’aboutissement d’un tel projet profite aux établissements de santé. En effet, une fois le dispositif fabriqué et prêt à être diffuser il pourra être présenter aux services biomédicaux des hôpitaux, puis aux médecins concernés par la spécialité du dispositif médical puis pourra être utilisé auprès d’un patient dans le besoin. Il est donc primordial que le porteur de projet ait toujours comme finalité le patient, celui à qui profitera le dispositif, et qui participera à son diagnostic, son traitement ou son confort de vie. C’est donc comme présenté sur la figure 2 qu’il faut appréhender l’utilisation de ce guide : l’idée nait d’une situation particulière, grâce aux ressources disponibles et à l’outil le dispositif est fabriqué, puis utilisé dans les établissements de santé pour les patients.
Figure 2 : Schéma replaçant l'utilisation de l'outil, source : auteures
1.2 Le contexte actuel en France
En mars 2020, face au manque de dispositifs médicaux disponibles sur le marché en France, les hôpitaux ont cherché tous les moyens possibles pour combler ce manque. Dans le cadre de la crise Covid-19, c’est principalement sur les respirateurs artificiels que s’est porté la demande.
Le gouvernement a donc lancé une demande de production en masse de respirateurs qui pourraient prendre en charge les patients covid, en insuffisance respiratoire. La demande a principalement été prise en charge par l’industriel Air Liquide Medical Systems, fabricant français de ventilateurs artificiels [2].
Cependant, de nombreux appels à projets ont été également lancé par d’autres fabricants, étudiants, ingénieurs ou start-up qui avaient également les capacités matérielles et techniques de fabriquer un MUR : Minimal Universal Respirator (figure 3). C’est un respirateur utilisé dans des situations d’urgence qui permet de suppléer les fonctions respiratoires d’un patient SDRA (syndrome de détresse respiratoire aiguë). C’est un respirateur « de base » qui contient peu de fonctionnalités mais qui suffit à assister le mécanisme de respiration d’un patient en détresse respiratoire [3]. Dans cette situation, de nombreux projets ont vu le jour.
Figure 3 : Photographie d'un MUR, d'après [3]

La problématique ne se tourne donc pas autour de la fabrication du dispositif médical, car chaque projet était lancé par des ingénieurs capables techniquement de produire une telle machine mais consiste à les orienter, une fois le dispositif médical fabriqué, vers la commercialisation et l’obtention du marquage CE.
Finalement, il semble compliqué aujourd’hui pour une start-up ou un jeune ingénieur indépendant de comprendre tout le processus d’avant et après commercialisation, le suivi technique et clinique du dispositif et la méthode d’obtention du marquage CE.
1.3 Une approche de l’outil
Le travail se divise donc en plusieurs parties. En premier lieu, il semble primordial d’analyser et de comprendre la norme ISO 13485 [4], pour en tirer les informations essentielles. Ensuite, il s’agit d’expliciter clairement chaque étape à suivre pour que le fabricant veille à la conformité de son dispositif médical avant de se lancer dans un processus de commercialisation. Il faudra ensuite veiller à l’aptitude de son dispositif médical d’obtenir le marquage CE pour qu’il puisse être commercialisé et utilisé dans les hôpitaux. Enfin, il est important de mettre en place un suivi technique et clinique du dispositif médical, des révisions ou mises à jour, afin de maintenir ce marquage CE et de garder le dispositif médical sur le marché. Il s’agit finalement de permettre au jeune fabricant de comprendre chaque étape du cycle de vie de son dispositif, de sa fabrication à sa réforme.
Cet outil simplifie alors le processus de commercialisation d’un dispositif médical et encourage chaque organisme en capacité de produire un dispositif médical à le mettre sur le marché de manière rapide et efficace.
Il faut réduire la perte de temps à lire les normes et comprendre les réglementations à appliquer, l’outil accompagnera chaque fabricant tout au long de son projet biomédical, pour obtenir le marquage CE et commercialiser son dispositif médical.
Ce projet est donc né dans le contexte de la crise sanitaire actuelle, car il était vraiment compliqué de mettre sur le marché un dispositif médical « home made » tout à fait conforme aux normes qualité. Le projet est aujourd’hui orienté sur le MUR, car c’est un exemple concret sur lequel il y a du recul depuis mars 2020, mais l’outil sera utilisable pour chaque dispositif médical qui doit être commercialisé rapidement, dans un contexte d’urgence sanitaire (ou autre) et qui doit rester sur le marché le temps de sa durée de vie.
2. Les exigences réglementaires indispensables pour un projet biomédical
Il semble avant tout primordial de cibler les différents acteurs qui interviennent tout au long de la démarche de commercialisation d’un dispositif médical :
- Fabricant
- Organisme notifié vis-à-vis de la réglementation
- Autorité compétente : Agence Nationale de Sécurité du Médicament et des produits de santé en France
- Sous-traitant critique : une entreprise qui évalue la sécurité ou les performances du dispositif
- Formateur : responsable des formations à l’utilisation
- Distributeur : organisme qui met à disposition le DM, à l’utilisateur ou à un autre distributeur
- Importateur : organisme qui met sur le marché UE un DM dont le fabricant est hors UE
- Mandataire : le représentant légal d’un fabricant hors UE (obligatoire)
2.1 Le système de management de la qualité
Le système de management de la qualité, ou SMQ sous sa forme abrégée, réunit des règles et des valeurs qui concourent au fonctionnement optimal d’un organisme ou d’une entreprise. Ainsi le fabricant a tout intérêt à s’investir dans un SMQ pour obtenir des performances dans son entreprise.
Ce système est formalisé depuis les années 1990 avec la norme ISO 9001 [5].
L’ISO 9001 donne les exigences d’un SMQ, visant à :
- Fournir en permanence des produits et des services conformes aux exigences du client et aux exigences légales et réglementaires applicables
- Donner de plus grandes opportunités d'amélioration de la satisfaction du client
- Prendre en compte des risques et opportunités associés au contexte et aux objectifs de l'organisme
- Savoir démontrer la conformité aux exigences spécifiées du système de management de la qualité.
Le fabricant va donc devoir répondre aux 7 principes fondamentaux du SMQ, présentés sur la figure 4 [6] :
Figure 4 : Les 7 principes du SMQ, d’après [6]
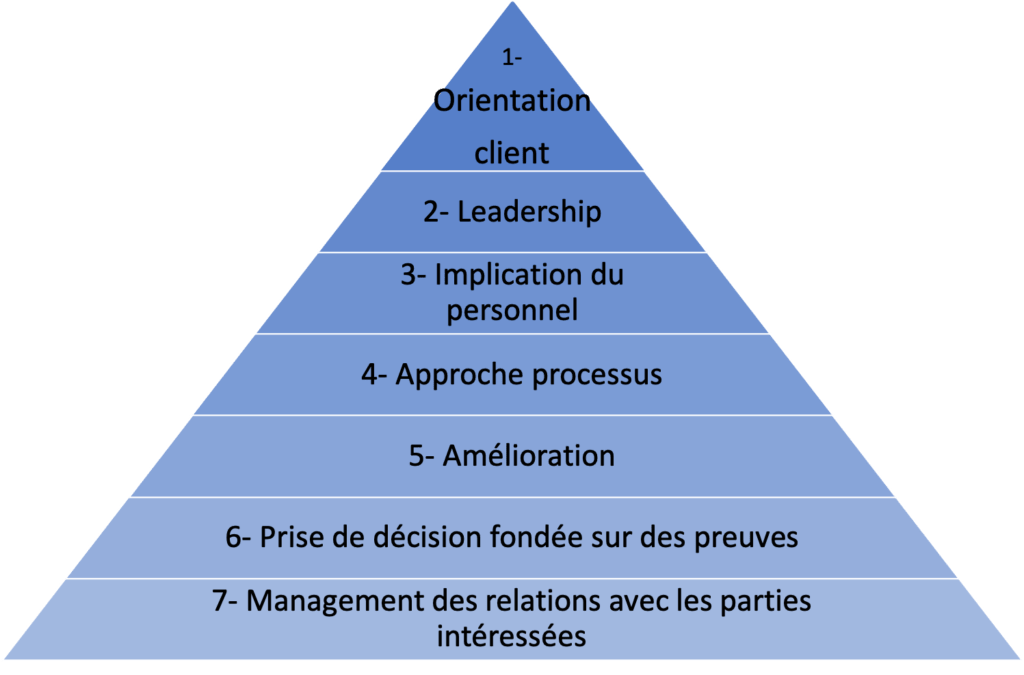
Un SMQ permet de déterminer les processus à réaliser (diagramme des processus), d’avoir une amélioration toujours en continue (PDCA), pouvoir anticiper les risques potentiels et saisir les différentes opportunités (SWOT ou diagramme des décisions). En s’investissant dans un SMQ, le fabricant est gagnant. Sur le long terme il est prouvé que cela apporte de l’efficience, de l’efficacité et de la performance à l’entreprise. Elle sera donc dans la capacité d’être fiable et les clients pourront avoir confiance en leurs produits, choses essentielles à l’heure actuelle [7].
Comment construire un SMQ quand on est un fabricant d’un dispositif médical ?
Pour cela il faut s’inspirer de la norme ISO 13485. C’est une approche par les processus et par les risques propres au domaine médical.
Exigences qualité [8]:
- Activités de management : gestion des RH, revues de direction, suivi et analyse des activités, gestion des non-conformités
- Activités de support : gestion des infrastructures, des équipements, des documents, des achats
- Activités opérationnelles : conception et développement, production, prestations de service, surveillance après commercialisation
Exigences selon deux paramètres fondamentaux :
- La sécurité du dispositif médical : essais de sécurité électrique, biocompatibilité, évaluation biologique, validation de la stérilisation
- Les performances du dispositif médical : évaluation clinique
2.2 La norme ISO 13485
La norme ISO 13485 spécifie les exigences d'un système de management de la qualité pouvant être utilisé par un organisme impliqué dans une ou plusieurs étapes du cycle de vie incluant la conception et le développement, la production, le stockage et la distribution, l'installation ou les prestations associées d'un dispositif médical, ainsi que la conception, le développement ou la prestation d'activités associées (par exemple support technique). Elle permet donc d’offrir un moyen volontaire de se conformer aux exigences du règlement européen [4].
L’outil d’accompagnement doit répondre aux exigences d’un SMQ. Ce SMQ pourra alors être utilisé par une université ou tout établissement de recherche, pour l’accompagner dans les étapes du cycle de vie du dispositif médical : conception, développement, production, distribution et utilisation. Il devra également porter sur la conception, le développement ou la prestation d’activités associées : documentation technique, balance bénéfices/risques etc. Le SMQ devra démontrer la capacité de l’organisme à satisfaire les exigences des hôpitaux et du corps médical sur le dispositif médical choisi et aux exigences réglementaires correspondant à la classe du dispositif médical associé.
Comment obtenir la certification ISO ?
La certification ISO 13485 est définie comme une « présomption » de conformité aux exigences des règlements. Sa raison d’être est d’offrir un moyen volontaire de se conformer aux exigences du règlement européen.
Les articles de l’ISO 13485 permettent de définir les étapes à suivre dans l’outil d’accompagnement.
Dans cette norme, on retrouve 5 articles principaux :
- Système de management de la qualité
- Responsabilité de la direction
- Management des ressources
- Réalisation du produit
- Mesurage, analyse et amélioration
2.3 Du marquage CE à la mise sur le marché
La mise sur le marché des dispositifs médicaux (DM), des dispositifs médicaux implantables actifs (DMIA), et des dispositifs médicaux de diagnostic in vitro (DMDIV) est contrôlé grâce à un marquage CE préalable, « CE » signifie « conformité européenne ». Le fabricant est donc dans l’obligation d’obtenir ce marquage CE pour toute commercialisation de n’importe quel dispositif médical [9]. Avant et après l’obtention de ce marquage de nombreuses étapes présentes sur la figure 5 sont nécessaires.
Figure 5 : Processus de commercialisation d'un DM, auteures d’après [11]
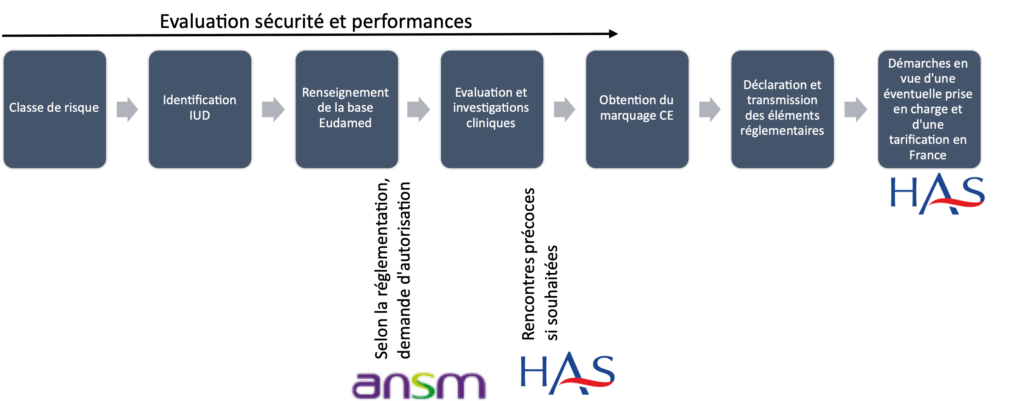
Ce marquage CE est la preuve d’une conformité aux exigences essentielles et que le produit respecte la santé et la sécurité des utilisateurs (stérilité, compatibilité biologique et sécurité électrique).
Pour obtenir le marquage CE, le fabricant est sous sa propre responsabilité pour soumettre les dispositifs à une procédure d’évaluation de conformité aux exigences décrites dans le règlement européen 2017/745 relatif aux dispositifs médicaux. En effet, depuis 1993, c’est la directive européenne 93/42/CEE qui donnait les indications relatives à la mise sur le marché des dispositifs médicaux, mais de nombreux conflits et scandales dans le domaine médical ont mené à un changement et à l’application d’un règlement en avril 2017. Ce nouveau texte vise alors à unifier les acteurs du dispositif médical en étant unifié sur le contexte technologique. Il a pour but d’améliorer la traçabilité et la transparence des organisations et de surveiller les organismes notifiés. Il entrera en vigueur le 26 mai 2021 et en 2024 les dispositifs médicaux ayant un marquage CE selon la directive européenne ne pourront plus être utilisés.
Pour certains dispositifs, une évaluation par un organisme notifié, désigné par l’autorité compétente est nécessaire avant d’apposer le marquage CE. Pour la France, l’autorité compétente est l’ANSM. Le fabricant est tenu de s’adresser à l’ANSM qui évaluera la conformité de ses produits et les certifiera, mais il reste en fin de compte responsable de leur conformité [10].
Avant toute chose, le fabricant doit déterminer la classe de son dispositif médical. Une fois cela fait, il doit répertorier toutes les directives et les exigences associées à la classe de son dispositif médical. En effet, l’ANSM vérifie également si les fabricants ont bien déterminé les directives et les exigences essentielles qui s’appliquent à leur produit.
Le fabricant peut ensuite procéder à la conception (conforme) de son dispositif médical. La réalisation de prototypage afin de réaliser les investigations cliniques. Ces investigations sont fortement réglementées et très contrôlées par l’ANSM et la Haute Autorité de Santé (HAS) [11].
Tout au long de la création de son dispositif, le fabricant doit mener une documentation technique sérieuse sur la conformité de son produit et qui répond aux exigences du marquage CE.
L’évaluation de cette conformité avec les directives européennes comprend différentes activités : fabrication, conception, y compris l’essai du produit, l’inspection visuelle, l’analyse du risque ainsi que l’examen des étiquettes et des instructions relatives à la classe (le risque potentiel).
Les étapes de l’évaluation de la conformité sont notamment les suivantes [12]:
- Examen de la documentation technique relative à la conception, à la fabrication et au fonctionnement du produit
- Essai d’un ou de plusieurs aspects plus précis de chaque produit, ou d’un échantillon des produits, ou d’un exemplaire représentatif du produit
- Évaluation des systèmes liés à la qualité de la production du fabricant
- Vérification continue de la conformité de l’unité de produit.
L’ANSM délivre alors le marquage CE, donnant l’accès aux fabricants au marché européen. Une fois le marquage CE obtenu, il est nécessaire d’y apposer une étiquette CE conforme aux exigences européennes.
2.4 Suivi de la mise sur le marché et dérogation
Une fois sur le marché, le dispositif médical est placé sous la responsabilité du fabricant qui le commercialise et cela durant tout le cycle de vie de celui-ci. Il doit donc en surveiller le contrôle pour vérifier qu’aucun problème ne survient lors de l’utilisation de son dispositif médical. Ainsi il est responsable de la prise de mesures préventives ou correctives. Des audits sont réalisés régulièrement chez le fabricant par l’ANSM. Elle peut d’ailleurs à tout moment retirer le dispositif médical du marché européen.
Cependant, le marquage CE a une date de péremption ! Au bout de 5 ans, le dispositif médical ne pourra plus être vendu ou utilisé. Une nouvelle procédure de renouvellement du marquage CE sera à faire.
Comme démontré ci-dessus, les démarches d’apposition au marquage CE sont longues et fastidieuses. C’est pourquoi, dans le cadre d’une crise sanitaire comme celle de la Covid-19, l’urgence prime à la conformité. C’est ainsi qu’est né un article proposant une dérogation du marquage CE.
Selon l’Article R5211-19 : « Sur demande dûment justifiée, le directeur général de l'Agence nationale de sécurité du médicament et des produits de santé peut autoriser, à titre dérogatoire, la mise sur le marché et la mise en service de dispositifs n'ayant pas fait l'objet des procédures de certification, mais dont l'utilisation présente un intérêt pour la protection de la santé. » [13].
Ainsi cela permet à un fabricant de mettre sur le marché un dispositif ne possédant pas encore de marquage CE, mais il doit rapidement réaliser une documentation afin d’obtenir la certification dans les années qui suivent.
Chapitre 2 : Le guide méthodologique de la commercialisation d'un dispositif médical
1. Mise en contexte : comment utiliser ce guide ?
1.1 Un accompagnement pour le fabricant
Le guide d’accompagnement a pour vocation d’aider toute organisation ou personne en capacité de produire un DM à commercialiser son innovation.
En effet, comprendre précisément chaque item de cette norme semble être un travail fastidieux pour les fabricants et ne correspond pas toujours à leurs aspirations et/ou compétences. Le but de l’outil est d’apporter une aide dans la compréhension de cette norme et de ses exigences, pour que la commercialisation d’un dispositif médical devienne « un jeu d’enfant ». C’est donc un moyen de faire respecter la norme sans devoir s’en préoccuper. C’est moins effrayant et décourageant pour le fabricant.
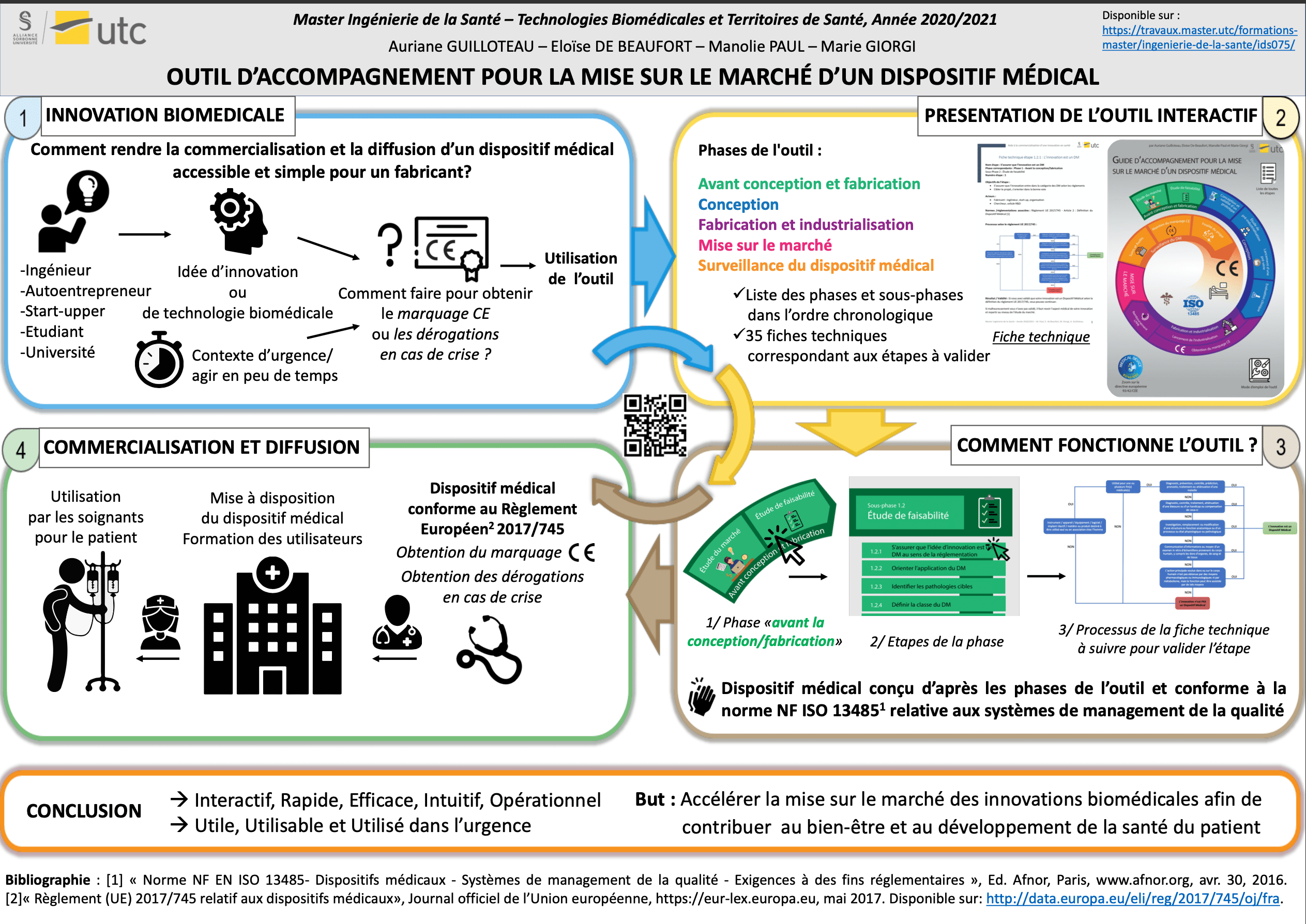
C’est également un gain de temps considérable pour l’utilisateur qui n’a qu’à suivre le fil directeur indiqué par une cartographie qui présente toutes les phases à valider, dans l’ordre chronologique, de l’étude de marché à la commercialisation.
Comme explicité précédemment, la mise sur le marché d’un dispositif médical est un travail qui nécessite la validation de nombreux critères et qui doit se faire progressivement en suivant un ordre bien précis. C’est pour cela que l’outil propose une cartographie en spirale représentant 4 grandes phases, divisées en sous-phases puis chaque sous-phase en étapes. C’est un outil intuitif, qui guidera réellement l’utilisateur dans son projet d’innovation ou de fabrication d’un DM. Cela lui permettra d’agir efficacement dans un contexte urgent.
En utilisant cet outil, le fabricant pourra suivre consciencieusement chaque étape à valider, et s’avancer peu à peu vers la mise sur le marché de son dispositif. L’objectif est double : fournir au fabricant un moyen de démontrer la conformité de son dispositif médical et à un organisme notifié d’évaluer la conformité du fabricant aux exigences requises pour les dispositif médicaux.
1.2 Le fonctionnement de l’outil
Cet outil se construit en 3 parties différentes et sous différentes formes en fonction de l’utilisateur. Une spirale permet de cartographier le processus dans son ensemble, pour visualiser les 4 phases indispensables à parcourir pour le fabricant :
- Avant la conception/fabrication
- Conception
- Fabrication et industrialisation
- Surveillance
Ces 4 phases se divisent ensuite en sous-phases, qui comportent chacune un nombre d’étapes défini pour compléter et valider la phase. A chaque étape, se dresse une fiche technique, qui permet d’aider l’utilisateur à comprendre l’étape qu’il est en train d’effectuer et de faire un lien avec son dispositif médical. Il est donc primordial que l’utilisateur ait bien en tête son dispositif médical ou son idée d’innovation pour permettre de concrétiser au maximum son travail et d’avancer dans son processus.
Voici sur la figure 6, le détail de toutes les étapes recensées qui représentent un processus complet, de l’étude de marché à la surveillance post-commercialisation d’un dispositif médical :
Figure 6 : Sommaire de l'outil, source : auteures

Chaque étape fait référence à un article de la norme ISO 13485 ou à un item de la documentation technique. Ce qui permet à l’utilisateur de fournir toutes les informations nécessaires aux exigences, sans même se plonger dans les documents réglementaires.
Cependant, toutes les étapes sont référencées en fonction des normes auxquelles elles se réfèrent, il faudra donc tout de même s’y référer de temps en temps pour vérifier la conformité. Chaque méthode indiquée aux étapes est non-exhaustive, l’utilisateur devra à la fin de chaque phase tester son dispositif grâce aux outils d’autodiagnostic des normes correspondantes.
L’outil interactif se divise donc en trois axes principaux (figure 7) : la spirale qui sert de fil directeur avec un code couleur bien précis pour les 4 phases du projet, des fiches récapitulatives de l’étapes dans laquelle se trouve l’utilisateur et un lien de chaque étape vers une fiche technique à effectuer et à valider.
Figure 7 : Aperçu de l'outil et de son fonctionnement, source : auteures
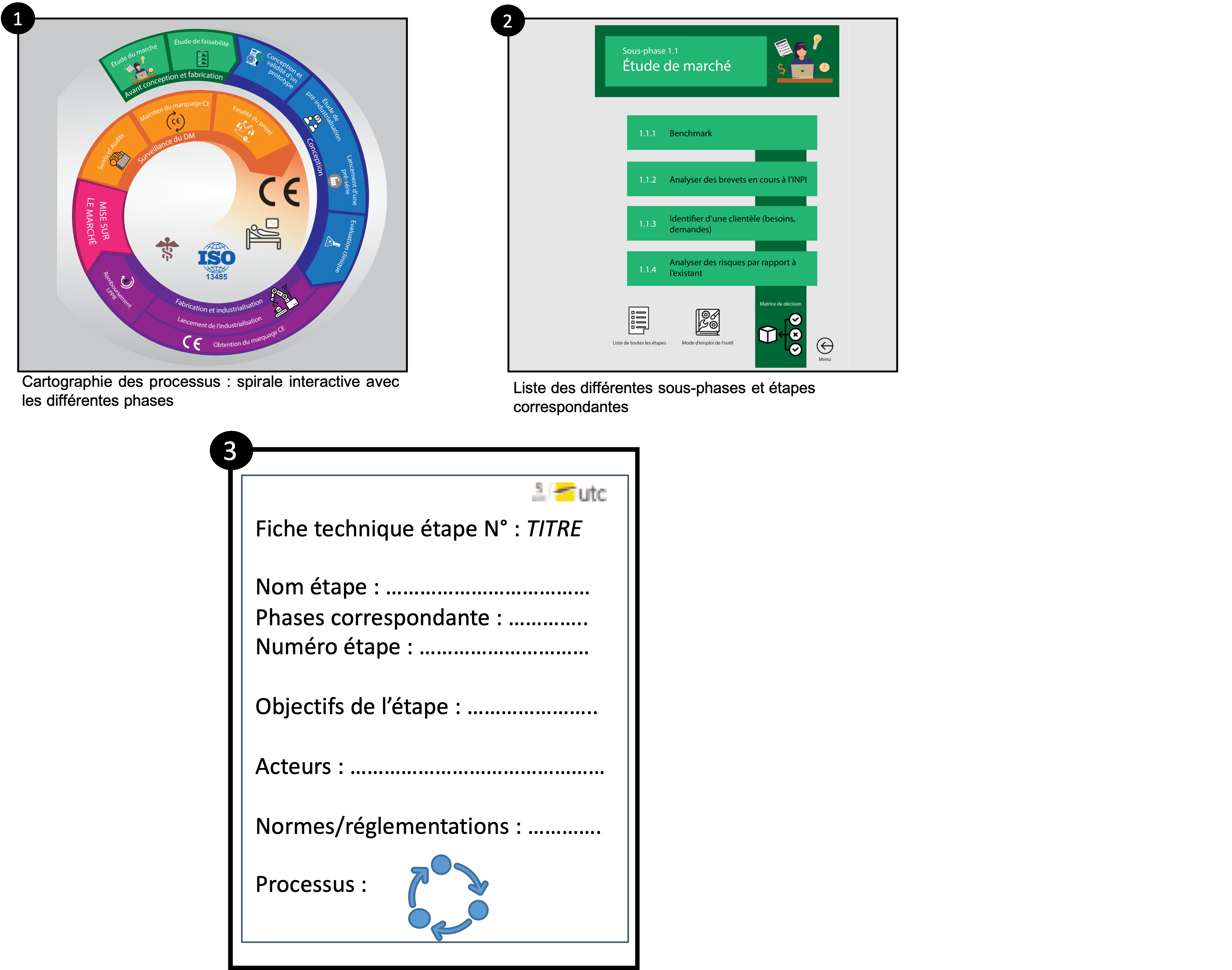
L’outil peut se présenter sous deux formes selon l’envie et le besoin de l’utilisateur :
- Un livret sous format pdf imprimable avec les fiches techniques : l’utilisateur peut écrire et remplir les différents critères à la main
- Un pdf interactif avec la présentation de la cartographie des processus : l’utilisateur peut cliquer sur les phases, qui mènent aux sous-phases, puis aux fiches techniques
1.3 Le mode d’emploi de l’outil
Pour guider l’utilisateur, un mode d’emploi (figure 8) est disponible et explique toutes les fonctionnalités des boutons et les endroits actifs pour cliquer. Il est accessible à n’importe quelle page et à tout moment, de manière à ce que l’utilisateur ne soit pas perdu dans son processus et puisse se repérer à tout moment dans les différentes fonctionnalités de l’outil.
Il est accessible en cliquant sur le bouton :

Figure 8 : Aperçu du mode d'emploi de l'outil, source : auteures

1.4 Une auto-évaluation au fil de l’avancement
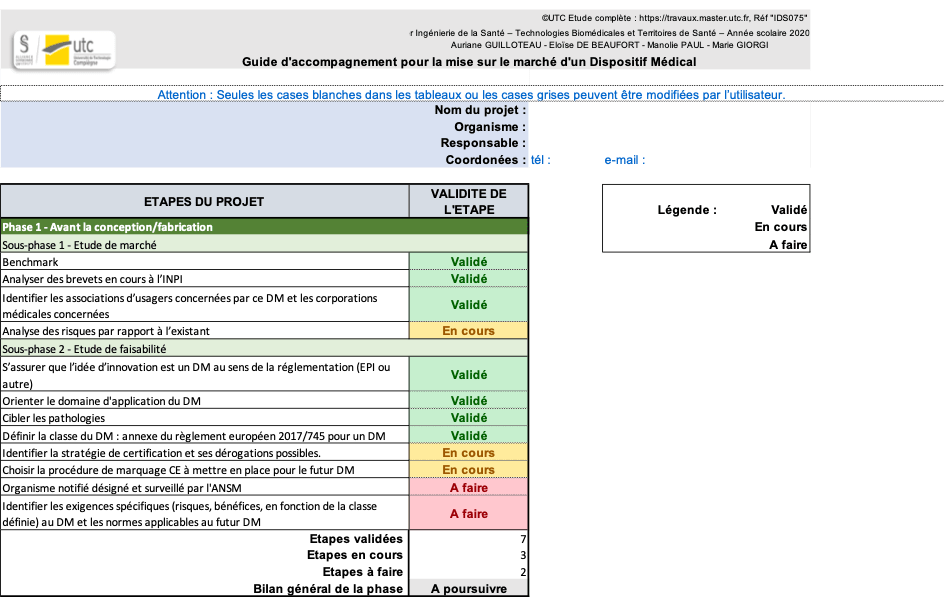
Grâce à la validation de chaque étape, le fabricant avance dans les différentes phases et sous-phases. Pour valider une phase il faut passer par une matrice de décision. C’est un tableau qui permet à l’utilisateur de s’auto-évaluer sur les étapes qu’il vient d’effectuer.
A chaque phase ou sous-phase critique, il devra remplir cette matrice pour pouvoir continuer sereinement. Voici, sur la figure 9 un exemple de la matrice de décision, qui se présente à l’issue de la phase 1 :
Figure 9 : Matrice de décision de la phase 1 : Avant conception/fabrication, source : auteures

Pour chaque étape, le fabricant doit noter si elle est « validée », « en cours » ou « à faire ». Cela lui permet d’évaluer son état d’avancement dans la phase et de vérifier qu’il n’a pas oublié une étape. Cette matrice doit accompagner le fabricant à chaque étape pour qu’il puisse se repérer dans le processus et décider quand il faut passer à la phase suivante.
Toutes les matrices de décision sont également disponibles dans un classeur Excel, comme présenté sur la figure 10. De cette manière l’utilisateur peut remplir les phases et avoir directement une idée de son état d’avancement dans la phase. Le classeur calculera le nombre de phases « validées », « en cours » et « à faire ». Ainsi, cela donnera un état d’avancement en fonction du nombre dans chaque case, « à poursuivre » ou « passage à la phase suivante ». Ce classeur est disponible avec l’outil.
Figure 10 : Apercu de l'outil Excel des matrices de décision, pour la phase 1 - Avant conception/fabrication
Si le fabricant valide chaque étape, il sera certain d’être en accord avec les réglementations en vigueur et d’avoir rempli tous les critères nécessaires pour l’organisme notifié et les autorités compétentes. Il pourra alors voir son DM commercialisé et utilisé auprès du corps médical.
2. Une spirale interactive pour guider l’utilisateur
La cartographie des processus prend la forme d’une spirale interactive (figure 11), elle se construit un peu comme le jeu de l’oie. Elle regroupe toutes les phases et sous-phases du processus de commercialisation rapide d’un DM. Elle est conçue via PowerPoint en créant des liens entre les phases/sous-phases/fiches techniques. Plus précisément, si l’utilisateur clique sur une case de la spirale donc sur une sous-phase, cela l'emmène à la liste des étapes de cette sous-phase, qu’il doit compléter. Ensuite si la personne clique sur une étape, elle est dirigée vers la fiche technique correspondante à l’étape. Chaque livrable est composé d’une notice d’utilisation pour comprendre sa logique et son fonctionnement.
Sur cette spirale, est directement accessible un document intitulé « Zoom sur la directive 93/42/CE » qui correspond à une description des annexes II à VII de cette directive. En effet, elle permet à l’utilisateur de se renseigner sur la procédure de marquage CE à choisir en fonction de la classe de risque de son DM, elle correspond donc à un complément de la fiche 1.2.6 « Choix de la procédure de marquage CE ». Cependant cette directive est valable seulement jusqu’en 2024, il faudra se fier au nouveau règlement européen.
Figure 11 : Aperçu de la spirale, source : auteures

L’avantage majeur de cette spirale est qu’elle est très intuitive. Elle permet de guider l’utilisateur dans la réalisation et la validation des taches, étape par étape afin d’éviter les oublis ou les erreurs. Elle est également attrayante par sa forme et ses couleurs donc encourage l’utilisation. C’est un outil rapide à utiliser car il regroupe toutes les informations essentielles et évite à l’utilisateur d’aller chercher des informations sur d'autres documentations comme les réglementations ou les directives. Toutes les informations sont regroupées dans la spirale afin que l’utilisateur n’utilise qu’un outil, celui-ci.
L’inconvénient de la spirale est qu’elle donne simplement une vision globale des phases à réaliser. Si l’utilisateur veut observer le processus complet avec toutes les étapes il faut se rendre à la page « Sommaire » qui donne une vision générale des phases, sous-phases et étapes correspondantes.
Cependant si l’utilisateur souhaite voir toutes les étapes détaillées, un livret est mis à disposition et répertorie toutes les fiches techniques. Ce support pdf permet à l’utilisateur de pouvoir s’approprier le déroulement des étapes de commercialisation et de pouvoir annoter des commentaires sur ce livret.
3. Un suivi minutieux des étapes grâce aux fiches techniques
Comme vu précédemment, chaque “sous-phase” comprend un certain nombre d’étapes. Les fiches techniques ont pour but d’appuyer ces étapes, en offrant à l’utilisateur toutes les données essentielles à la réalisation de l’étape en question. Elles ont été conçues pour simplifier la compréhension à la commercialisation et à l’obtention du marquage CE. Ainsi chaque étape est clarifiée et classée pour permettre à l’utilisateur de fournir un minimum d’effort de recherche méthodologique.
La fiche technique est donc un condensé d’informations triées et pertinentes. Elles comportent différents points :
- Objectifs de l’étape : clarifier le but de cette étape et ce qu’elle apporte à l’utilisateur
- Acteurs : quels sont les acteurs qui vont intervenir dans cette étape (internes ou externes au projet)
- Normes / réglementations associées : les réglementations qui vont intervenir dans cette étape
- Processus : le processus à suivre pour valider les objectifs de l’étape
- Schéma : un schéma bilan pour simplifier la compréhension
- Résultat / Validité : un point pour s’assurer que l’étape est validée et que l’utilisateur peut passer à la suivante
- Point pour la documentation technique pour le marquage CE (Annexe II du règlement européen 2017/745)
- A quels points de la norme ISO 13485 répond cette étape
- Bibliographie : pour indiquer les sources ou donner des liens intéressants à l’utilisateur
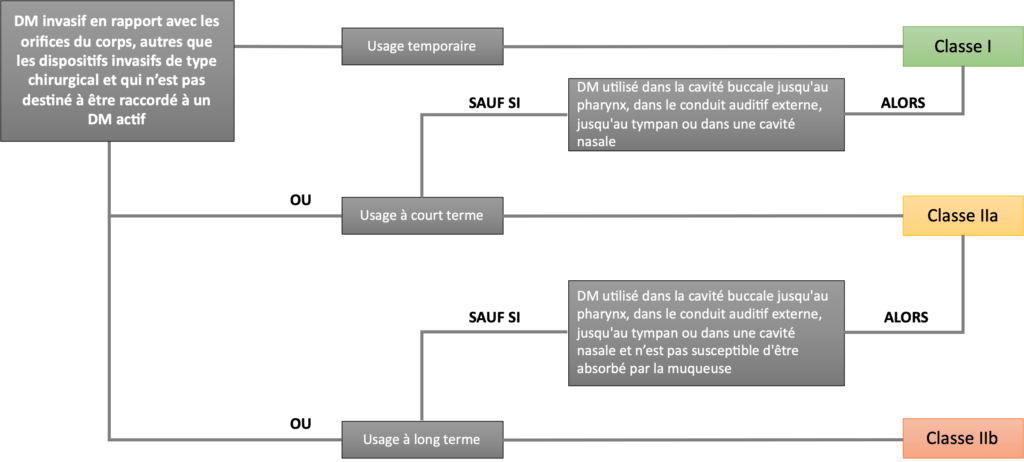
Il est présenté dans chaque fiche technique un processus de réalisation. Cela a pour but de guider très précisément l’utilisateur. Ainsi il était essentiel de la présenter de manière claire : elles comportent donc un schéma “bilan” pour faciliter la compréhension du lecteur (quand cela est pertinent). Prenons un exemple simple. La fiche “Définir la classe du DM.”, située dans la sous-phase 2 de la phase 1 propose un processus pour trouver la classe de risque du DM (figure 12). Un schéma avec “choix” a été choisi pour sa pertinence.
Figure 12 : Processus à suivre pour valider l'étape avant la conception/fabrication qui permet de déterminer la classe de risque du DM, nommée "Définir la classe du DM", source : auteures
Les deux derniers points des fiches techniques concernent les points auxquels répond l’étape pour la documentation technique pour le marquage CE (Annexe II) et la norme ISO 13485. Ainsi tout au long de la réalisation des étapes, l’utilisateur forme sa documentation pour le marquage CE et la norme ISO 13485.
Elles sont conçues pour regrouper différents points de vue, différentes sources, des différents processus ou stratégies. Elles n’indiquent que des conseils méthodologiques et ne sont en rien des documents “officiels”.
Au total, 35 fiches techniques sont réalisées et présentées sous format PDF en mode portrait avec un numéro qui correspond à la phase et à la sous-phase dans laquelle se trouve l'étape.
4. Risques anticipés, alternatives envisagées dans l’utilisation du guide
4.1 Les risques anticipés
Il a été explicité le fonctionnement de ce guide qui, si son fonctionnement reste optimal, permettra au fabricant d’obtenir un suivi du cycle de son dispositif médical de manière intuitive et interactive.
Cependant, il est important de prendre en compte d’éventuels risques de mise en œuvre de cet outil. En effet, il est nécessaire d’identifier tous les types d’aléas susceptibles de ralentir ou de “bloquer” l’évolution du guide par toutes sortes de raisons, afin de les anticiper et pourvoir agir sur ces problèmes pour ne pas être surpris.
Pour cibler ces risques il existe différentes méthodes telles que la méthode AMDEC (Analyse des Modes de Défaillance, de leurs Effets et de leur Criticité) ou encore le logigramme ARA (Actions, Risques, Alternatives) représenté sur la figure 13.
Voici les aléas éventuels qui ont pu être identifiés avec leurs alternatives :
Figure 13 : Logigramme pour évaluer les risques dans l'élaboration du guide, source : auteures
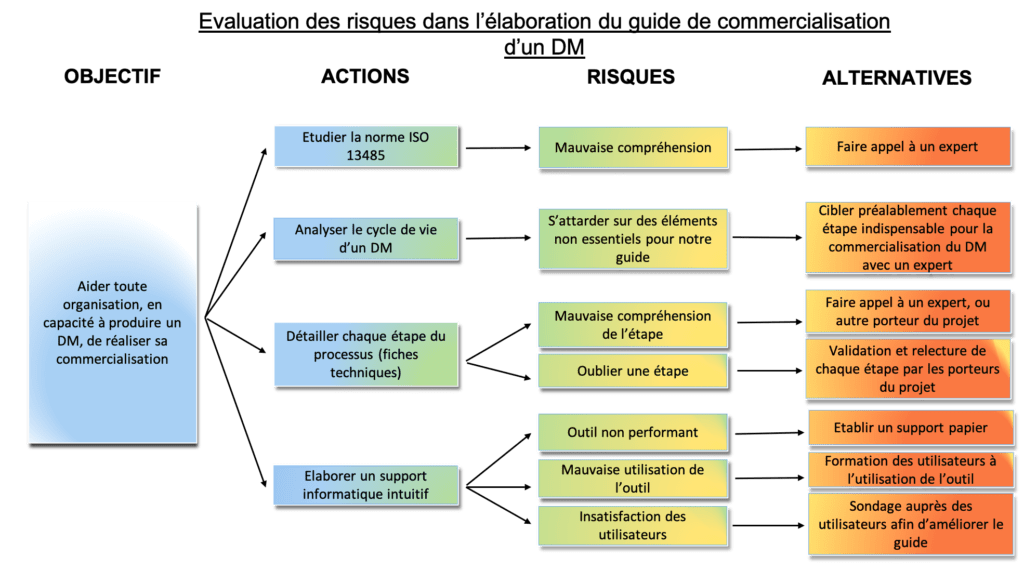
4.2 Les alternatives envisagées
Concernant les alternatives, comme constaté à travers le logigramme ARA, il est indispensable de prendre en compte un éventuel défaut technique, informatique qui pourrait nuire au format PDF du guide. De même, si le format final n’est pas suffisamment interactif, ne plaît pas aux utilisateurs, ou nécessiterait des modifications, un deuxième format est à envisager.
Pour perfectionner l’outil, certaines améliorations informatiques sont à envisager. Tout d’abord, créer des liens directs des références bibliographiques des fiches techniques vers le site internet ou le document concerné. Il serait également judicieux de créer des liens vers les outils d’autodiagnostic des normes afin de vérifier la conformité directement au moment de l’étape correspondante.
Toutes les étapes qui ont été présentées, pourront également être disponibles dans un livret papier afin d’anticiper un problème quelconque mais également de permettre aux fabricants, qui le souhaitent, de disposer d’un support papier. De cette manière, le guide pourra répondre à toutes les exigences souhaitées de la part des utilisateurs, afin de satisfaire leurs besoins.
Il faut tout de même prendre en compte qu’un livret papier sera très conséquent et imposant, au risque de perdre des données ou ne plus se retrouver dans le suivi du cycle du dispositif médical. Il sera plutôt disponible dans un usage de dernier recours.
Une perspective intéressante serait de pouvoir créer un logiciel, ou site internet, qui présente le guide de manière à avoir un impact plus professionnel, et une présentation qui soit plus guidée et intuitive. Pour cela, il faut un budget plus conséquent, et un recul plus approfondi sur l’outil, cette idée reste toutefois envisageable dans un futur proche.
Chapitre 3 : Les résultats attendus, l’utilisation prévue pour les porteurs de projet d’innovation biomédicale
1. Expression des résultats attendus par un exemple concret : le M.U.R
Pour démontrer la pertinence de l’utilisation de ce guide d’accompagnement, il parait judicieux d’appliquer ce processus à la conception et diffusion d’un M.U.R.
Tout d’abord le fabricant ou porteur de projet devra appréhender la spirale interactive en fonction du MUR et de la situation économique/sanitaire actuelle. Voici la démarche globale attendue pour un porteur de projet qui voudrait fabriquer et diffuser un M.U.R dans un contexte de crise sanitaire. Pour appliquer cette étude de manière efficace, le processus sera suivi pour la phase 2 : la phase de conception du dispositif, qui se divise en 3 sous-phases (figure 14).
Figure 14 : Aperçu de la phase de conception du dispositif, source : auteures
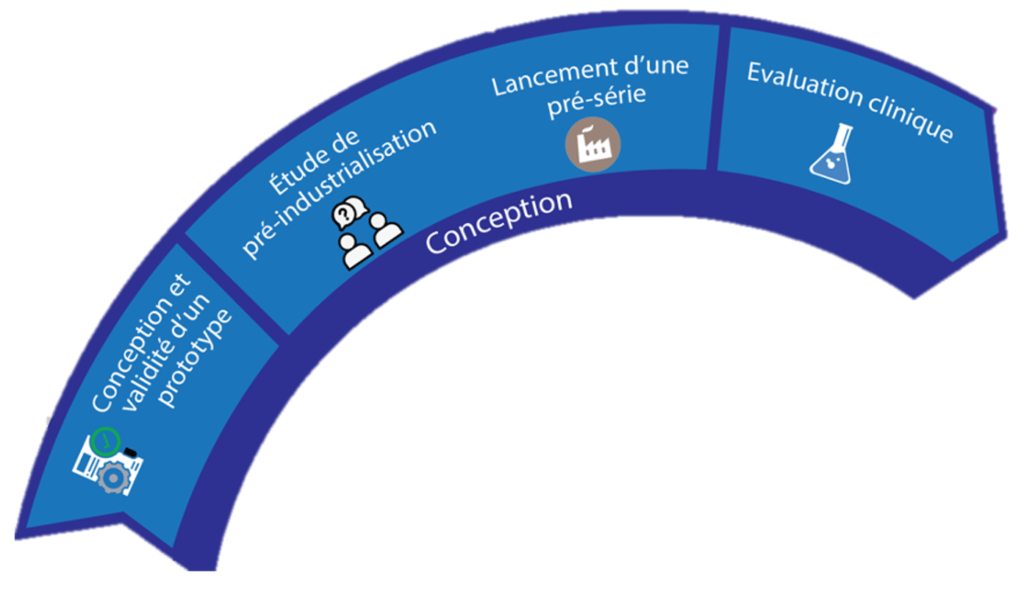
Grâce à la spirale et son code couleur, l’utilisateur prend connaissances des étapes qui l’attend et peut donc anticiper certaines de ses actions en fonction de la suite. Ici, la première sous-phase est la conception et la validité d’un prototype, en cliquant sur le rectangle correspondant, l’utilisateur est directement redirigé vers les étapes qui correspondent aux différentes fiches techniques (figure 15).
Figure 15 : Aperçu de la sous-phase 1, conception et validité du prototype, source : auteures

A présent, l’utilisateur voit les différentes étapes qu’il doit suivre pour valider cette sous-phase :
- Développer un prototype tout en remplissant le dossier de fabrication : méthode de prototypage
- Effectuer les essais de fonctionnalités : vérifier que le ventilateur fonctionne et qu’il répond aux attentes de l’utilisation prévue (échanges d’air avec l’extérieur)
- Analyser les risques : anticiper les risques envisageables à l’utilisation d’un tel ventilateur au niveau des professionnels de santé, du patient et de l’environnement
- Analyser le rapport bénéfices/risques : étudier les bénéfices et réaliser une balance qui permet de conclure sur un rapport positif des bénéfices/risques
- Effectuer les tests cliniques : penser à l’utilisation médicale du ventilateur, des applications médicales, l’aspect thérapeutique et les éventuels effets secondaires à anticiper
Pour le M.U.R, l’utilisateur doit donc fabriquer un premier prototype ou une maquette qui sera à destination de tous les tests qui suivent et qui sera soumis à une analyse des risques. Conformément à la fiche technique n°2.1.1 (Développer un prototype), voici le processus à suivre pour mettre en place un prototype (figure 16).
Figure 16 : Processus à suivre pour élaborer un prototype du MUR, source : auteures

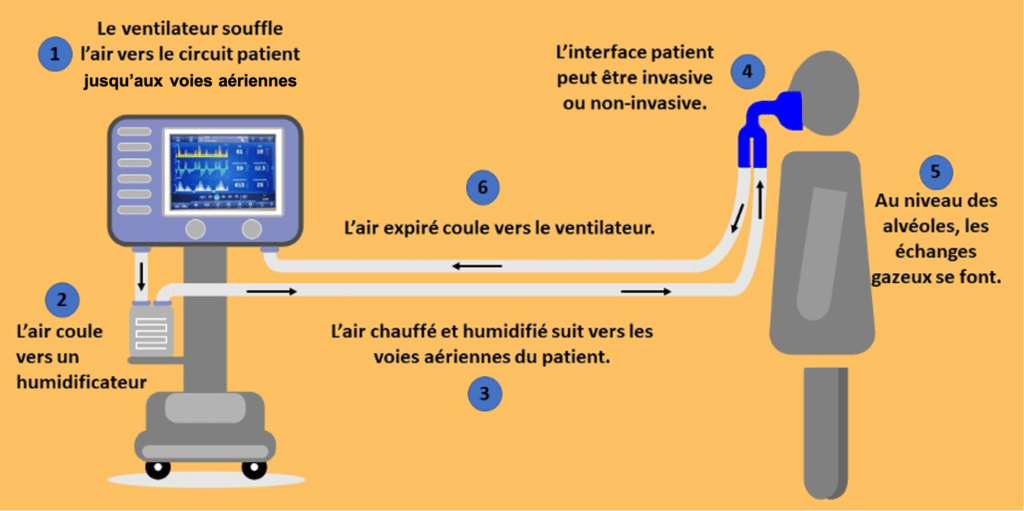
Il est également précisé au niveau de cette étape qu’il faut définir l’utilisation prévue du dispositif et établir l’interaction avec le patient. La classe de risque aura été définie lors de la phase 1 (Avant la conception/fabrication) à la sous-phase 2 (Etude de conception), fiche technique n°1.2.4. Pour le ventilateur artificiel, il parait indispensable de comprendre le circuit d’échange d’air entre la machine et le patient (figure 17).
Figure 16 : Schéma présentant les échanges d'air entre la machine et le patient, source : auteures
A l’issu de ces premières étapes, le premier prototype du MUR devrait être prêt à être utilisé pour les différents essais qui vont suivre. Par exemple, la figure 18 présente le prototype présenté par l’Université de Rice au Texas [14]. L’utilisation prévue est de presser un BAVU (Ballon à valve unidirectionnelle) pour apporter une ventilation minimale à une personne en insuffisance respiratoire. Un petit écran se situe au-dessus du respirateur et sert simplement à afficher le volume courant, le rythme respiratoire et le ratio inspiration/expiration. Trois boutons sont également visibles, un pour allumer le respirateur, un autre pour régler tous les paramètres et le dernier en cas d’arrêt d’urgence. Les composants sont très basiques, trouvables partout (servomoteurs, des cartes arduinos) et la compression se fait grâce à un rail d’engrenages qui avance et recule.
Figure 18 : Photo du ventilateur ApolloBVM, source : d’après [14]

Une fois que le prototype fonctionne selon les attentes de l’utilisation requise, le fabricant peut passer à l’étape suivante afin de s’assurer de la validité de son prototype en fonction des différentes exigences référées à la figure 19, exigées lors de la rédaction de la documentation technique (DT) par les autorités compétentes.
Figure 19 : Tableau présentant les exigences à respecter sur le prototype du MUR, source : auteures

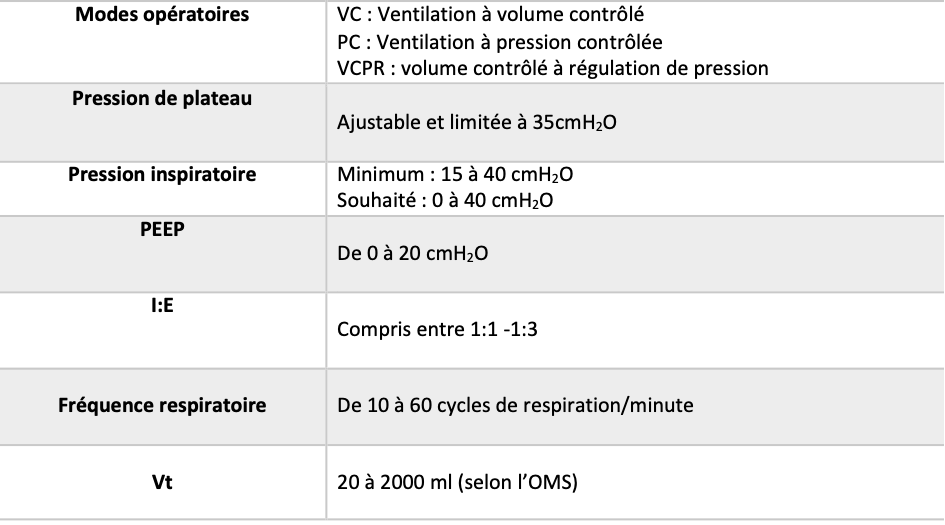
Afin d’être conforme et de respecter les différentes exigences ci-dessus, il est primordial de vérifier que les exigences minimales du respirateur sont intégrées dans le prototype. Pour un ventilateur de respiration minimale, certains paramètres de ventilation sont à respecter [15] (figure 20).
Figure 20 : Tableau présentant les paramètres de ventilation minimale à respecter pour un MUR, source : auteures
Pour conclure, l’efficacité du guide d’accompagnement sera avérée si chaque porteur de projet arrive à valider les phases au fur et à mesure de son processus. Ici, le fabricant du M.U.R doit pouvoir à la fin de cette deuxième phase, lancer une présérie pour effectuer des tests supplémentaires de sécurité, fonctionnalités et des essais cliniques et finir par industrialiser son produit. La finalité sera alors d’obtenir une dérogation de l’ANSM, mise en place exceptionnellement dans le contexte de crise sanitaire actuelle, pour contourner temporairement le marquage CE. Une fois que la dérogation est obtenue, le dispositif pourra être mis sur le marché et utilisé dans les hôpitaux au chevet des patients.
2. Un outil opérationnel et adaptable à tous les contextes
En prenant l’exemple du M.U.R, cela permet d’évaluer l’efficacité du guide dans un contexte urgent. En effet, grâce à la vision globale que l’utilisateur a de son processus, cela lui permet de se situer dans le temps et d’avancer pas à pas. Si son projet est bien ancré et prêt à être mis en place, il n’a plus qu’à suivre des indications inscrites sur les fiches techniques en se référant aux différentes ressources bibliographiques disponibles et de compléter les livrables qu’il doit fournir aux autorités compétentes.
Il s’agit donc d’être minutieux dans le processus sans prendre le temps de lire chaque item des normes ou des textes réglementaires associés. Il est donc évident que l’utilisation de cet outil sera un gain de temps considérable pour les porteurs de projets. Malgré le grand nombre d’étapes et fiches techniques à lire, l’utilisateur peut aller droit au but en lisant uniquement le processus et en validant les étapes dans les matrices de décision. D’autre part, l’utilisateur est autonome dans son processus et est libre de modifier les étapes ou de les adapter plus précisément au dispositif qu’il est en train de développer. Cette autonomie et l’auto-évaluation proposée à la fin de chaque phase, permet à chacun de s’approprier l’outil et de l’adapter aux différentes situations. En effet, il est précisé à chaque étape si elle est obligatoire réglementairement ou fortement recommandée. Dans un contexte urgent, l’utilisateur est donc libre de passer certaines étapes ou d’y revenir plus tard.
Cet outil est qualifié de « guide », il a donc pour unique vocation de guider un porteur de projet ou un fabricant indépendant dans son processus de mise sur le marché. Libre à chacun de se focaliser sur une seule phase du cycle de vie de son produit ou de suivre le processus en entier. Les phases sont accessibles à tout moment de manière à pouvoir anticiper les étapes et sélectionner les plus importantes. Il est vraiment important de pouvoir avoir cette vision globale du processus pour appréhender les étapes au mieux et s’approprier l’outil. Par ailleurs, il reste indicatif et n’a pas de certification officielle, il sera donc indispensable pour tout utilisateur de toujours faire référence aux différentes exigences réglementaires pour fournir les dossiers aux autorités compétentes afin que le dispositif obtienne le marquage CE.
3. Des processus à valider par des professionnels
Afin d’obtenir des résultats pertinents suite à l’utilisation de ce guide, les fiches techniques doivent être validées par des professionnels dans des domaines particuliers. En effet, les résultats attendus sont la conformité à la norme ISO 13485, la rédaction de la documentation technique et des différents rapports de la gestion bénéfices/risques et des tests effectués.
En suivant les étapes de ce guide, l’utilisateur sera conforme aux différentes réglementations et normes en vigueur des dispositifs médicaux :
- NF EN ISO 13485 : Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires
- NF EN ISO 14971 : Dispositifs Médicaux – Application de la gestion des risques aux Dispositifs Médicaux [16]
- NF EN ISO 60601-1 : Appareils électromédicaux Partie 1 - Exigences Générales pour la sécurité de base et les performances essentielles [17]
- NF EN ISO 14155 : Investigation clinique des dispositifs médicaux pour sujets humains — Bonnes pratiques cliniques [18]
- Règlement Européen 2017/745 relatif aux dispositifs médicaux [19]
- Directive Européenne 93/42/CE relative aux dispositifs médicaux
Au fil de l’avancement de l’utilisateur dans le processus proposé par la spirale et par les différentes fiches techniques à suivre, les différentes réglementations relatives aux dispositifs médicaux sont évoquées. L’objectif principal est donc qu’à la fin de chaque phase le dispositif en conception soit conforme aux exigences qui lui correspond.
A la fin de la phase 1 : le projet est viable et concret, dans la sous-phase étude de faisabilité l’utilisateur s’assure que son dispositif est un dispositif médical au sens de la réglementation et commence à engager ses responsabilités auprès des autorités compétentes (ANSM en France). A ce moment, le fabricant doit s’être suffisamment renseigné sur les aspects réglementaires afin de les appliquer à la phase suivante, lors de l’élaboration et de la conception d’un premier prototype.
La phase 2 se divise en deux étapes clés du processus général. Tout d’abord, la conception d’un prototype qui doit être validé dans les fonctionnalités et la sécurité selon la norme 60601-1. Une fois que le prototype est validé, une présérie est lancée après avoir obtenu les autorisations pour les essais pré-cliniques et cliniques. A la fin de cette deuxième sous-phase, l’utilisateur propose un prototype conforme à la norme ISO 14155.
La phase 3 est déterminante pour l’avenir du dispositif médical en cours de fabrication. En effet, c’est ici que le fabricant devient conforme au marquage CE, qu’il envoie la documentation technique de son dispositif et qu’il est en contact direct avec l’organisme notifié. C’est donc la phase qui concrétise réellement le projet et qui permettra, au moment de sa validation, de mettre sur le marché le dispositif médical. A la fin de cette phase, le fabricant attend les retours des organismes notifiés et si son dispositif est validé il pourra procéder à la mise sur le marché, donc à la mise à disposition dans les hôpitaux.
La phase 4 est une phase complémentaire de tout ce processus qui consiste à continuer à surveiller le dispositif qui est en cours d’utilisation. A ce moment, le dispositif est utilisé par des professionnels de santé auprès de patients. Au niveau réglementaire, une surveillance est réalisée par l’ANSM, des contrôles qualité sont effectués dans les établissements et certains audits sont à prévoir. Le dispositif médical est donc suivi tout au long de sa vie jusqu’à sa réforme.
Enfin, la dernière fiche technique propose à l’utilisateur de faire un retour d’expérience sur son projet et sur le processus qu’il vient d’effectuer s’il devait à nouveau mener un projet de cette envergure ou participer à une étape du cycle de vie du dispositif médical. Cela permet donc de mieux appréhender les futurs projets en anticipant les difficultés qu’il a rencontré et les risques qu’il pourrait rencontrer à nouveau.
Finalement, un utilisateur qui a suivi méticuleusement toutes les étapes du guide doit donc avoir mis sur le marché un dispositif médical qui répond à toutes les exigences le concernant et doit voir son dispositif utilisé dans les établissements de santé, par les professionnels pour les patients dans le besoin.
Conclusion
En 2020 la commercialisation d’innovations ou de dispositifs médicaux en rupture est une procédure qui a été compromise suite à cette crise sanitaire qui touche le monde entier. Il était plus que nécessaire, d’apporter un soutien aux fabricants, ingénieurs et chercheurs dans les étapes indispensables à suivre pour la mise sur le marché de leur projet. Cet outil a été élaboré avec précision, en détaillant soigneusement chaque point essentiel dans ce processus d’accompagnement à la commercialisation, tout en répondant aux réglementations européennes et en étant conforme à la norme NF EN ISO 13485.
L’objectif était d’obtenir un outil fiable, intuitif, innovant et simple d’utilisation afin de proposer le plus rapidement possible tout dispositif pouvant sauver des vies en danger dans les établissements en manque de ressources médicales nécessaires. Ce guide est un appui considérable sur le cycle à suivre pour déployer une innovation médicale sur le marché. Cela permet de diminuer les longues procédures, les pertes de données, les combats administratifs (quelle norme pour quel cas, quel organisme pour quel dispositif, etc) tout est simplifié et fait pour aider le monde médical à survivre face à une urgence sanitaire mondiale.
Il est évident que des améliorations sont encore possibles et nécessaires pour assurer la pérennité de ce guide d’accompagnement. C’est pourquoi une amélioration continue est à prendre en compte avec tous les détails, retours, informations des utilisateurs qui seront à prendre en compte. L’évolution des normes, des règlements, des règles de commercialisation sont également très évolutifs, il faut donc être vigilent sur le renouvellement permanent de ce guide.
L’intervention de professionnels garantit la concrétisation de ce projet. En effet, le groupe Metalians (SFAM – CFT – MDP), expert en design et commercialisation de dispositifs médicaux, a montré un grand intérêt dans cette innovation. Il s’est proposé de revoir chacune des étapes de ce guide, pour confirmer ou modifier les points mis en avant dans le bon processus de commercialisation des dispositifs médicaux.