
IDS107 - Mise en place d'un Système de Management de la Qualité dans une start-up d'Intelligence Artificielle.
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteure

Woodeline
Contact
Citation
A rappeler pour tout usage : W. P-L « Mise en place d'un Système de Management de la Qualité dans une start-up d'Intelligence Artificielle », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de Stage, réf n° IDS107, juillet 2021, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids107/
Résumé
A l’ère où les start-ups spécialisées dans le numérique sont en pleine expansion, le monde des affaires règlementaires connaît lui aussi un grand tournant. Le 26 mai 2021, le nouveau Règlement Européen 2017/745 [1] est devenu effectif, impliquant alors de nombreux changements pour l’ensemble des parties prenantes dans le monde des dispositifs médicaux. Des fabricants aux organismes notifiés, l’ensemble des acteurs du domaine a dû faire de nombreux ajustements afin d’être en conformité avec les exigences règlementaires. . Les Annexes 9 à 11 du règlement détaillent les recommandations concernant l’évaluation de la conformité du système de management de la qualité. Afin de les appliquer au mieux la norme NF EN ISO 13485 :2016 détaille les exigences à des fins règlementaires.
Ce projet de fin d’étude avait pour but d’implémenter un Système de Management de la Qualité selon ce référentiel. Ce mémoire a pour but d’exposer les différents challenges que peuvent rencontrer les entreprises de petite envergure dans leur désir d’allier d’innovation et qualité. Dans cette optique, un système documentaire (nomenclature, fiche processus, cartographie des processus, procédure), une stratégie clinique et une veille règlementaire ont été élaborées.
Abstract
At a time when digital start-ups are booming, the world of regulatory affairs is also undergoing a major shift. On May 26, 2021, the new European Regulation 2017/745 [1] became effective, implying many changes for all stakeholders in the medical device world. From manufacturers to notified bodies, all actors in the field had to make many adjustments in order to comply with the regulatory requirements.
The goal of this end-of-study project at WeDiagnostiX was to implement a Quality Management System according to the NF EN ISO 13485:2016 standard. The purpose of this thesis is to expose the different challenges that small companies can face in their desire to combine innovation and quality. In this perspective, a documentary system (nomenclature, process sheet, process mapping, procedure), a clinical strategy and a regulatory watch have been developed.
Keywords : European Regulation 2017/745 - Quality Management System - NF EN ISO 13485:2016 - Innovation - Quality - Documentary system - Clinical strategy - Documentary watch
Téléchargements

Mise en place d'un Système de Management de la Qualité dans une start-up spécialisée dans l'Intelligence Artificielle à destination du secteur dentaire.
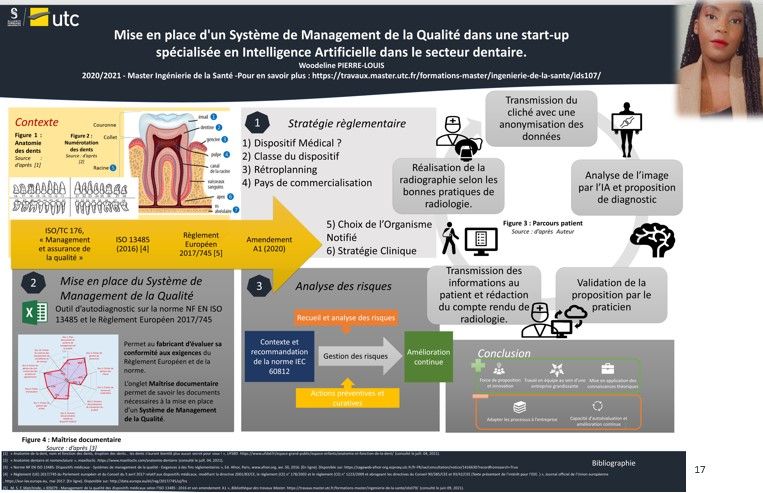
Poster
Mémoire Complet
Mise en place d'un Système de Management de la Qualité dans une start-up d'Intelligence Artificielle
Introduction
Henry Ford disait « La qualité signifie faire bien les choses quand personne ne regarde ».
Au cœur de la transition entre les directives européennes 93/42/CEE relative aux dispositifs médicaux [2], 98/79/CE relative aux dispositifs médicaux de diagnostic in vitro [3] et le nouveau Règlement Européen 2017/745, la course en quête du marquage CE se fait de plus en plus pressante. Bien qu’indispensable, l’intérêt de l’implémentation d’un Système de Management de la Qualité (SMQ) reste très abstrait pour un grand nombre de fabricants. Plus qu’une liste de tâche à accomplir, l’objectif est d’inscrire la qualité au cœur de la politique de l’entreprise. Cependant, les ressources à mobiliser peuvent s’avérer être un frein pour ceux dont l’inventivité ne tarit pas mais dont les connaissances du monde de la qualité restent limitées. « Normes », « processus », « enregistrements », …, autant de termes qui restent abstraits pour des profils purement scientifiques, qui n’ont qu’une idée en tête : mettre sur le marché un dispositif innovant, permettant d’améliorer considérablement les conditions de soins des patients.
Un secteur particulièrement concerné par ces changements est celui du numérique en santé. Aux problématiques préexistantes s’ajoutent des exigences règlementaires en constante fluctuation, et de plus en plus sévère. Le secteur de la e-santé, qui comprend la télémédecine, mais aussi l’intelligence artificielle ou les dispositifs connectés, représentera, selon le cabinet Frost & Sullivan, environ 234 milliards de dollars d’ici 2023 [4].
L’institut Montaigne estime à environ 3 milliards d’euros, le gain d’efficience pouvant être réalisé uniquement grâce aux avancées technologiques réalisées à l’aide de l’Intelligence Artificielle et son apport dans le diagnostic de pathologies [5].
Des chiffres qui présagent un bel avenir à la e-santé ; mais qui laissent en suspens la question de la gestion des données de santé, qui est au cœur du débat public.
Pour aider à la gestion de l’ensemble des problématiques de financement, de cybersécurité, d’aptitude à l’utilisation, la mise en place d’un Système de Management de la Qualité (SMQ) peut être utile pour structurer les différentes activités de l’entreprise. Mais elle peut être un challenge à part entière pour les novices de ce domaine.
Ce stage a été réalisé dans une structure de petite taille spécialisée dans l’Intelligence Artificielle dans le secteur dentaire. Comment initier une démarche qualité au sein d’une start-up ?
Pour répondre à cette question, la méthodologie appliquée pour l’entreprise d’accueil sera présentée dans ce mémoire en présentant premièrement la structure et son environnement, en abordant les différentes problématiques à résoudre et la mise en place des exigences de la norme NF EN ISO 13485 :2016 au sein de l’entreprise. Une présentation des résultats obtenus sera détaillée.
I- Situation et Environnement de la structure d’accueil
1. Transition vers le nouveau Règlement Européen 2017/745
Un dispositif médical est défini comme « tout instrument, appareil, équipement, logiciel, implant, réactif ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l’homme pour l’une ou plusieurs des fins médicales précises suivantes :
– diagnostic, prévention, contrôle, prévision, pronostic, traitement ou atténuation d’une maladie,
– diagnostic, contrôle, traitement, atténuation ou compensation d’une blessure ou d’un handicap,
– étude, remplacement ou modification d’une structure ou fonction anatomique ou d’un processus ou état physiologique ou pathologique,
– communication d’informations au moyen d’un examen in vitro d’échantillons provenant du corps humain, y compris les dons d’organes, de sang et de tissus,
et dont l’action principale voulue dans ou sur le corps humain n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens.
Les produits spécifiquement destinés au nettoyage, à la désinfection ou à la stérilisation de dispositifs médicaux et de dispositifs destinés à la maîtrise de la conception ou à l’assistance à celle-ci, sont considérés comme des dispositifs médicaux [6]. »
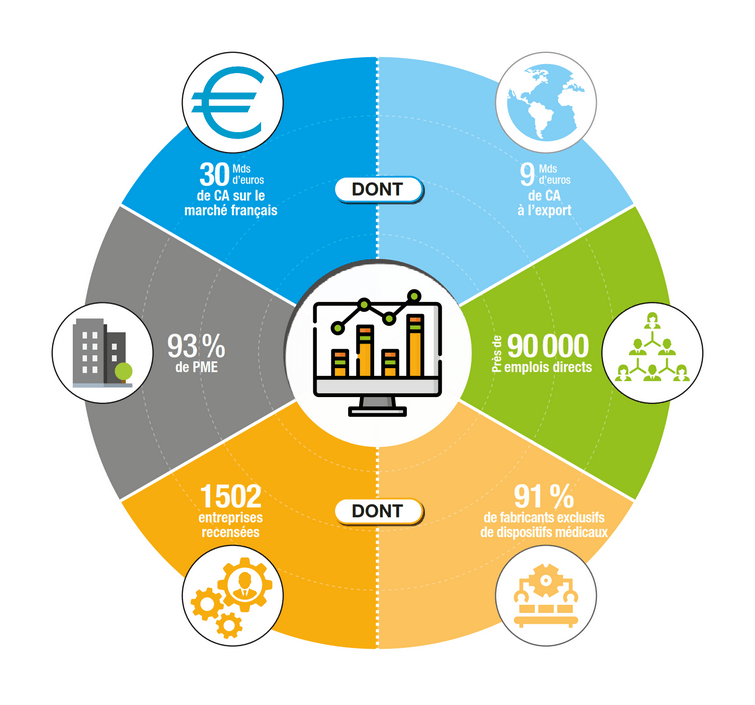
Cette définition montre l’étendue du secteur des dispositifs médicaux, qui connait un essor depuis plus de 70 ans grâce aux avancées technologiques. En France, ce secteur a montré une évolution du chiffre d’affaires de 2 milliards d’euros entre 2016 et 2018, passant alors de 28 à 30 milliards d’euros. Ce dernier a été réalisé par 1502 entreprises recensées dont le secteur d’activité est pour une grande majorité, exclusivement dans les dispositifs médicaux. Il y a parmi elles un grand nombre de petites et moyennes entreprises qui emploient un peu moins de 100 000 personnes [7] (Figure 1).
Figure 1 : Panorama et analyse qualitative de la filière industrielle des dispositifs médicaux en France
Source d’après [7]
Historique règlementaire [8] :
Afin de garantir la sécurité du patient dans ce secteur de grande envergure, la règlementation est en constante évolution.
- C’est en 1976, qu’est apparue la loi sur les dispositifs médicaux aux Etats-Unis. Instaurée par la Food Drug Administration, elle régule la fabrication et la consommation des dispositifs sur le territoire américain.
- Le comité technique ISO/TC 176, « Management et assurance de la qualité » est à l’origine de la première édition des norme ISO 9000 éditées en 1987 [9].
- L’ISO 13485 « Dispositifs médicaux — Systèmes de management de la qualité — Exigences à des fins réglementaires », a elle fait son apparition en 1996. Une seconde version de la norme à été éditée en 2003. Basée sur une structure similaire à celle de l’ISO 9001 : 2008, Systèmes de management de la qualité – Exigences, cette norme traite du management de la qualité en y ajoutant des thèmes spécifiques aux dispositifs médicaux tels que la stérilisation ou encore la traçabilité.
- Les annexes ZA,ZB et ZC font leur apparition dans la version 2012 de la norme afin de relier les recommandations de la normes aux exigences des directives CE 90/385/CEE relative aux dispositifs médicaux implantables actifs), 93/42/CEE relative aux dispositifs médicaux et 98/79/CEE relative aux dispositifs médicaux de diagnostic in vitro. Ces directives énoncent les exigences de performance et de sécurité auxquelles doivent répondre les produits.
- La version effective de la norme ISO 13485 date de mars 2016. Elle aborde notamment l’approche par les risques, le cycle de vie du dispositif ou encore le dossier du dispositif médical. Elle met l’emphase sur les exigences documentaires, plus présentes que dans les versions précédentes.
- Afin de mieux appréhender les recommandations de la norme, le comité technique ISO/TC 210 a écrit un guide nommé : « ISO 13485 :2016 - Medical devices – A pratical guide).
- L’évaluation des risques est une étape indispensable à la conception et à la fabrication du dispositif. Les normes ISO 14971 (2019) : « Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux » et NF S99-170 (2013) : « Maintenance des dispositifs médicaux – Système de management de la qualité pour la maintenance et la gestion des risques associés à l’exploitation des dispositifs médicaux » permettent de mieux appréhender cette notion et d’évaluer l’ensemble des situations dangereuses associées à l’exploitation du dispositif médical. Le guide « Medical devices — Guidance on the application of ISO 14971 » rédigé par le groupe d’experts ISO/TR 24971, permet de mieux comprendre les recommandations de cette norme.
- L’Agence nationale de sécurité du médicament et des produits de santé (ANSM) récence également un grand nombre de ressources documentaires utiles ; tout comme le site Santé Publique de la Commission européenne MEDDEV.
- D’autres normes peuvent s’avérer être très utiles à la compréhension des exigences règlementaires comme par exemple la norme ISO 15223-1 (2012), « Dispositifs médicaux - Symboles à utiliser avec les étiquettes, l'étiquetage et les informations à fournir relatifs aux dispositifs médicaux - Partie 1 : Exigences générales », la norme IEC 62304 (2006) « Logiciels de dispositifs médicaux - Processus du cycle de vie du logiciel » ou encore la norme expérimentale XP S99-223 « Dispositifs médicaux - Gestion du rapport bénéfice/risque ».
- Les annexes de l’amendement A1 de la norme NF EN ISO 13485/A1, publié le 06/01/2020, fait le lien entre la présente norme et les exigences des directives 90/385/CEE, 93/42/CEE, 98/79/CEE et celles des Règlement UE 2017/745 et 2017/746.
- La norme ISO 13485 est applicable dans les structures, de toutes tailles, en contact avec les dispositifs médicaux. Une justification est à fournir si certaines des exigences ne sont pas applicables. Il faut alors prouver que ces exclusions ne compromettent pas la conformité du produit.
- Le 26 mai 2021 le Règlement Européen 2017/745 relatif aux dispositifs médicaux a pris le relais sur la Directive Européenne 93/42/CEE. Retardée d’un an pour cause de la crise sanitaire liée au COVID – 19, cette transition marque un tournant dans le monde des Affaires Règlementaires. Pour les start-ups du numérique, il s’agit d’un changement draconien de la considération des logiciels de santé sur le marché des dispositifs médicaux. Régulièrement, de nouveaux guides, français ou internationaux font leur apparition pour aider les fabricants à se familiariser avec ces nouvelles exigences. Par exemple :
- Guide sur l’application du règlement (UE) 2017/745 relatif aux dispositifs médicaux à destination des établissements de santé [10].
- Guide GMED : Demande de certification en vue du marquage CE - Règlement (UE) 2017/745 [11].
Le respect du règlement permet d’obtenir le marquage CE, obligatoire pour les dispositifs médicaux mis sur le marché de l’Union Européenne.
2. Intelligence Artificielle pour les dispositifs médicaux
En pleine expansion, une discipline se démarque des autres dispositifs médicaux : l’Intelligence Artificielle. « L'intelligence artificielle (IA, ou AI en anglais pour Artificial Intelligence) est définie comme une mise en œuvre d’un certain nombre de techniques visant à permettre aux machines d'imiter une forme d'intelligence réelle. L'IA se retrouve implémentée dans un nombre grandissant de domaines d'application. [13] ».
Cette technologie est régulièrement utilisée afin de comprendre un texte et fournir une réponse adéquate (ex : chatbox), d’aider à la prise de décision, de générer des prédictions basées sur des informations préalablement fournies, de reconnaître des images ou de reproduire des tâches humaines [14].
Pour ce faire, de nombreuses informations sont implémentées dans une base de données afin que l’ordinateur soit en mesure de prédire, de modéliser et d’analyser les différentes situations auxquelles il fait face.
En constante augmentation, de nouvelles innovations en santé font leur apparition, soit en intégrant de l’Intelligence Artificielle (IA) ou en tant que dispositif médical à part entière.
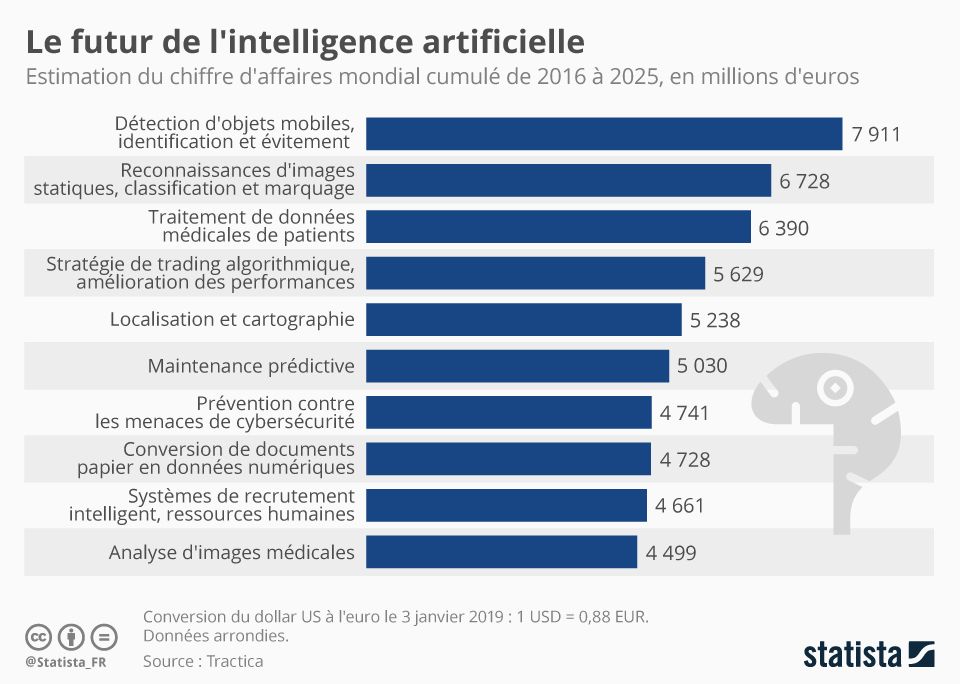
Le secteur le plus touché est celui de l’imagerie. L’IA est utilisée pour l’identification et la segmentation des organes ou encore pour classer les différentes images [15] (Figure 2).
Figure 2 : Estimation du chiffre d’affaires mondial cumulé de 2016 à 2025, en millions d’euros
Source d’après [16]
Très appréciée des praticiens, elle est cependant sujette à une règlementation de plus en plus rigoureuse afin de prouver l’exactitude du résultat fourni et de garantir la santé du patient.
Données de santé
Les exigences en matière de protection des données sont inscrites dans le Règlement (UE) 2016/679 relatif à la protection des personnes physiques à l'égard du traitement des données à caractère personnel et à la libre circulation de ces données [17].
En France, les patients sont des usufruitiers de leurs données de santé [18]. « L'usufruit est le droit d'utiliser un bien et d'en percevoir les revenus, sans en être propriétaire. [19]». Ici, cela signifie que le patient peut disposer de ses données mais non les vendre. Le traitement de ces données doit se faire avec le consentement préalable du possesseur.
Afin de s’assurer que ces exigences sont correctement respectées, la Commission Nationale de l’Informatique et des Libertés (CNIL) émet des recommandations et fournit des guides d’aide à la compréhension comme par exemple Guide des bonnes pratiques de l’informatique, réalisé par l’ANSSI et la CPME [20] ou Guide sécurité des données personnelles de la CNIL [21].
La norme ISO/CEI 27001 « Technologies de l'information - Techniques de sécurité - Systèmes de gestion de sécurité de l'information – Exigences » dans sa version 2013, permet à tout organisme de mettre en place un Système de Management de l’Information conforme aux règlementations. Basée sur la roue de Deming, elle permet de contextualiser, d’identifier et de lister les risques, ainsi que de choisir les actions à mettre en place vis-à-vis de la sécurité et l’intégrité des données de la structure [22].
Figure 3 : La place de la cybersécurité dans la sécurité du patient
Source : d’après [23]
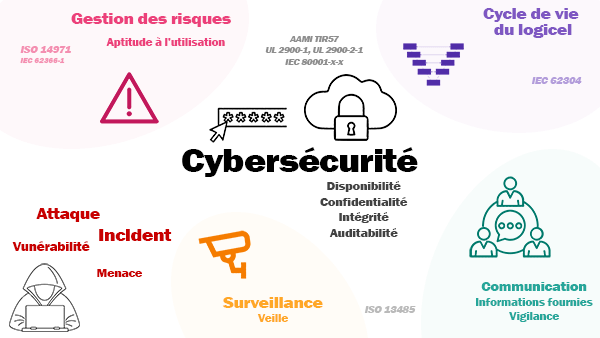
De manière plus spécifique aux dispositifs médicaux, plusieurs ressources permettent de mieux appréhender le sujet :
- Le Medical Device Cybersecurity Guide rédigé par l’International Medical Device Regulator Forum (IMRDF) [24].
- Le relatif à la cybersécurité des dispositifs médicaux rédigé par l’Agence Nationale de sécurité du médicament et des produits de santé [25].
- Les guidances rédigées par la Food Drug Administration (FDA) [26].
3. Le secteur dentaire dans le monde des dispositifs médicaux
La règlementation changeante s’applique à tous les domaines du secteur de la santé. La dentisterie ne faisant pas exception, cette discipline possède des spécificités qui la distingue des autres domaines.
L’être humain possède 32 dents à l’âge adulte. Parmi elles, se trouvent douze molaires, quatre canines, huit incisives, quatre dents de sagesse et huit prémolaires [27].
Figure 4 : Anatomie de la bouche
Source : d’après [28]
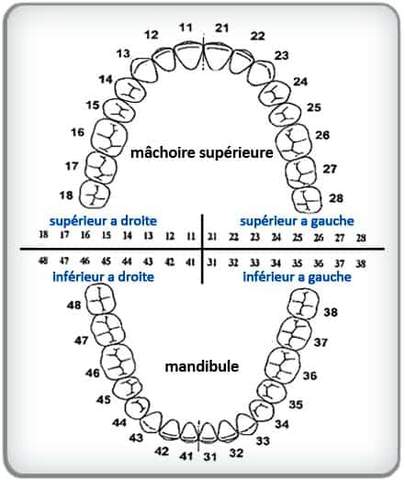
Afin que les professionnels du secteur dentaire identifient les structures anatomiques du patient, deux systèmes de numérotation des dents sont généralement utilisés :
- Le système de numérotation dentaire FDI (utilisé en France et en Europe) qui divise la bouche en 4 semi-arcades où les dents sont numérotées de 1 à 8.
- Le système de numérotation dentaire universelle (Etats-Unis), qui numérote les dents de 1 à 32 en partant de la partie supérieure droite et en finissant au niveau inférieur droit de la mâchoire.
Cette démarche permet de localiser les traitements des dents et de les consigner dans les comptes rendus. Le schéma dentaire a pour but de résumer en une seule représentation, les traitements présents dans la bouche du patient et les pathologies.
L’imagerie : un outil indispensable aux spécialistes du secteur dentaire
Afin de visualiser et dans un soucis de traçabilité, la réalisation de radiographies est un élément indispensable à la dentisterie. « Elle permet de mettre en évidence les lésions des dents et des tissus adjacents qui ne peuvent être décelées lors d’un examen clinique, notamment les débuts de caries, les kystes, les tumeurs et les abcès » [30].
Plusieurs types de radiographies sont réalisés :
- La radiographie interproximale
- La radiographie périapicale
- La radiographie panoramique dentaire
Cet outil présente cependant des limites : la radiographie émet des radiations ce qui implique le fait de prendre des précautions supplémentaires. De plus sa représentation en 2D ne permet pas une visualisation complète de l’anatomie de la bouche.
Dans le cas où une fracture ou une infection est suspectée, une radiographie panoramique est demandée. Une panoramique dentaire ou orthopantomogramme est un « acte de radiologie qui permet d'obtenir une image très large de la dentition : les deux rangées de dents, les os de la mâchoire supérieure et inférieure, ainsi que l'os du maxillaire et la mandibule [31]. »
Pour ce faire, un spécialiste place une languette dans la bouche du patient pour écarter ses dents et réaliser les images. La tête est stabilisée grâce à des tiges afin qu’elle ne bouge pas et l’examen dure en moyenne une vingtaine de secondes. Certaines pathologies peuvent cependant passer inaperçues du fait du caractère plan de l’image [32].
Le parcours patient dans le secteur dentaire
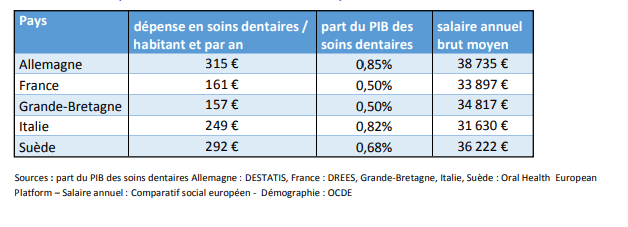
On estime à 17 milliards d’euros les coûts des soins dentaires directs et indirects liés à la dentisterie.
Tableau : Dépenses en soins dentaires en Europe en 2015
Source : d’après [33]
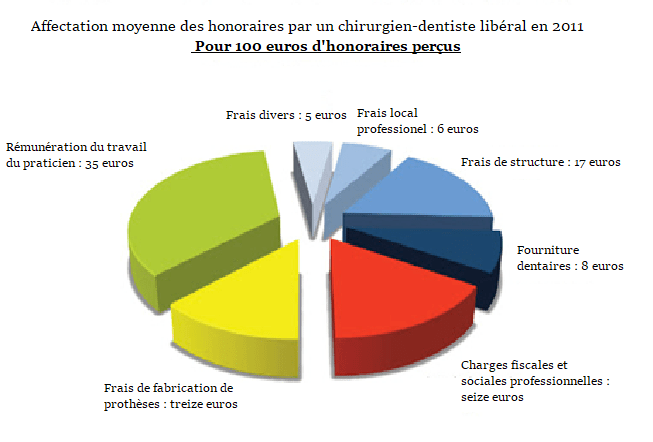
Aujourd’hui, plus de 85% des chirurgiens-dentistes exercent de manière libérale. Plus de dix milliards d’euros ont été dépensés en 2014. Il est important de noter que plus de 60% des revenus du cabinet sont dédiés aux charges du cabinet et qu’une partie du reste doit être affectée aux impôts [33] (Figure 5).
Le temps affecté à chaque patient est donc crucial et une aide au diagnostic peut-être un réel atout dans cet environnement.
Figure 5 : Affection moyenne des honoraires encaissés pas un chirurgien libéral en 2011.
Source : d’après [33]
Déroulé de l’examen
- Un examen approfondi est réalisé afin de connaître la situation dentaire du patient. Aucune préparation n’est nécessaire avant la réalisation de l’examen. Pour des raisons de sécurité, une salle de radiologie doit être affectée à la réalisation de cet examen. Un diagnostic est alors établi.
- Suite à ce diagnostic, un plan de traitement est établi et un devis est présenté au patient.
- Le patient décide alors s’il est d’accord avec le plan établi.
- Suite à son consentement, le traitement commence.
- Si nécessaire, le praticien met en place un suivi de traitement.
II- Missions et/ou observations réalisées
1. Contexte
Figure 6 : Schéma masque dentaire réalisé par une technologie d’Intelligence artificielle.
Source : d’après [6]

Fondée par une équipe de professionnels de la santé et d’experts mondialement reconnus en Intelligence Artificielle, l’entreprise d’accueil a été créée en 2020, afin d’aider à la réalisation d’un compte-rendu précis de la radio panoramique des patients, autrement connue sous le nom d’orthopantomogramme.
Le dispositif étudié utilise une intelligence artificielle permettant de réaliser une première lecture de la radiographie. Il met en évidence les zones potentiellement pathologiques et propose une aide au diagnostic. Cela évite aux patients d’être soumis à des examens intrusifs. Les plans de traitement sont alors mieux acceptés par le patient et les explications sont plus compréhensibles grâce à l’affichage des informations sur sa radiographie.
L’intelligence artificielle est une technologie en constante évolution : elle permet de progresser dans la détection de pathologie et d’appréhender plus facilement de nouvelles anomalies jusque lors méconnues du secteur dentaire.
A ce jour, il n’existe aucune information documentée sur de potentiels concurrents ayant déjà obtenu le marquage CE sur un dispositif similaire. Cependant, en Europe et à l’international, de nombreuses start-ups spécialisées dans l’Intelligence Artificielle à destination du secteur dentaire font leur apparition.
2. Enjeux
L’Union Française pour la Santé Bucco-Dentaire dit ceci : « La réalisation, la lecture et surtout l’interprétation des images dentaires représentent un ensemble cohérent d’une pratique pluri quotidienne pour tous les chirurgiens-dentistes quelle que soit l’orientation de leurs activités.
C’est un acte médical qui suppose, à chaque étape, la maîtrise des bonnes pratiques, en particulier une bonne connaissance des règles de réalisation des incidences, une lecture précise de la radio anatomie et une évaluation juste des aspects pathologiques tous nécessaires à la qualité du service rendu au patient.
Ainsi chaque professionnel pourra agir dans un cadre de qualité et sécurisé pour son patient, mais aussi dans un cadre de compétences maîtrisées tant du point de vue de sa pratique (contraintes / limites) que du point de vue de la gestion de son risque assurantiel » [34].
Figure 7 : Illustration d’une radio Source : d’après [35]
Le remplissage du schéma dentaire sur le logiciel de management des patients du cabinet est une opération fastidieuse et est souvent délaissée par les praticiens du fait de son caractère chronophage. Selon les professionnels du secteur, aujourd’hui, moins de 10% des praticiens dentaires réalisent leur compte rendu dentaire de manière conforme aux bonnes pratiques du métier.
Manque de régularité, ou manque d’exhaustivité, cette activité chronophage n’est pas particulièrement appréciée des professionnels du secteur. Son intérêt est pourtant crucial : L’article L 1111-2 du code de santé publique stipule que « Toute personne a le droit d’être informée de son état de santé ; cette information porte sur les différentes investigations, traitements ou actions de prévention qui lui sont proposées, leur utilité, leur urgence éventuelle, leurs conséquences, les risques fréquents ou graves normalement prévisibles qu’ils comportent, ainsi que les autres solutions possibles. ».
Le compte rendu permet donc un suivi plus approfondi de l’état de santé du patient qui voit l’ensemble de ses données de santé dentaire rassemblé dans son dossier de manière rigoureuse, et permet au praticien d’avoir une vision claire et précise des traitements préalablement effectués. Le code de déontologie médicale oblige d’autant plus les praticiens à garder ces résultats pour une durée minimum de 20 ans [35]. Dans ce contexte, l’intelligence artificielle permet une meilleure traçabilité, efficacité et rapidité du praticien.
Outre la traçabilité, un autre intérêt majeur de la bonne tenue du dossier patient est l’identification odontologique dans le cadre de la médecine légale. En effet, en fonction de l’état dans lequel est trouvé un cadavre, l’identification traditionnelle peut s’avérer périlleuse. Cependant d’autres alternatives sont envisageables notamment grâce aux empreintes digitales, à l’analyse de l’ADN ou encore à l’analyse des données dentaires.
Que ce soit sur un plan judiciaire, pénal, social, religieux ou moral, les intérêts de l’identification sont nombreux [36]. Pour ce faire, il faut cependant que les comptes-rendus de radiologie soient établis de la manière la plus juste et précise possible. Ce compte-rendu peut également être utile et indispensable en cas de litige entre le patient et le praticien.
Les intérêts du dispositif concernent également la prise en charge du patient.
Le temps consacré à chaque patient fait partie des critères de choix du chirurgien par le patient, comprenant également les prix, la disponibilité et la compétence de ce dernier. Une aide à la rédaction du compte-rendu a donc tout son intérêt pour répondre à la problématique de non-remplissage actuel, sans entamer le temps consacré à chaque consultation.
Rapide et efficace, l’utilisation du dispositif nécessite tout de même une confirmation du professionnel de santé. Cela lui permet d’avoir un deuxième avis sur son diagnostic et de lui signaler des anomalies qu’il n’aurait pas détectées à la première lecture.
Certains patients nécessitent une attention particulière, par exemple s’ils sont atteints de diabète, ou s’ils sont sur le point de subir une intervention chirurgicale. Le dispositif permet d’identifier les foyers infectieux et de prévenir le praticien en cas de danger potentiel.
3. Problématique et objectif
L’objectif de ce stage en entreprise était d’implémenter un Système de Management de la Qualité dans l’entreprise d’accueil. Le référentiel choisi est la norme NF EN ISO 13485 :2016 [35].
La norme ISO 21500:2012 rappelle les « Lignes directrices sur le management de projet ». Cette norme énonce les recommandations pour une gestion de projet optimale [38] (Figure 8).
Figure 8 : Aperçu général des concepts de management de projet et de leurs interrelations
Source : d’après [38]
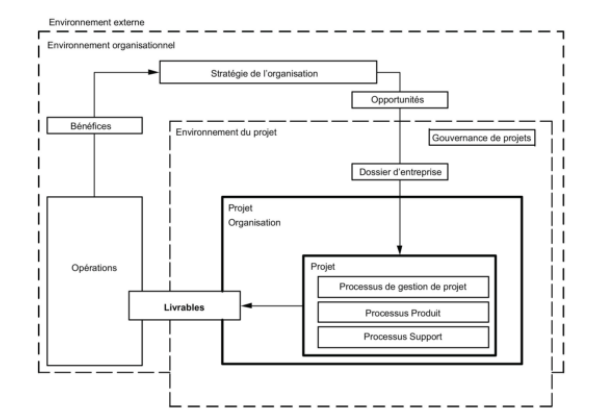
Afin d’appliquer au mieux les recommandations de la norme, une cartographie de projet a été réalisée (Figure 9). Elle permet de visualiser la stratégie adoptée, les opérations à effectuer et l’environnement du projet.
Figure 9 : Approche méthodologique de projet
Source : d’après Auteur
Conformément à cette dernière, un état des lieux des documents préexistants dans l’entreprise a été effectué. L’outil bi-diagnostic (Norme et Règlement) réalisé par les étudiants de l’Université de Technologie de Compiègne s’est avéré être très utile dans l’accomplissement de cette tâche [39] (Figure 10). Cet outil permet d'évaluer sa conformité à la norme ainsi qu'au Règlement UE 2017/745 et d'établir un plan d'actions d'amélioration. Plusieurs onglets composent cet outil : parmi eux, l'onglet Maîtrise documentaire qui permet de faire un état des lieux du système documentaire de l'entreprise vis à vis de la norme.
Figure 10 : Extrait du résultat de l’autodiagnostic initial (t=0)
Source : d’après Auteur, [39]
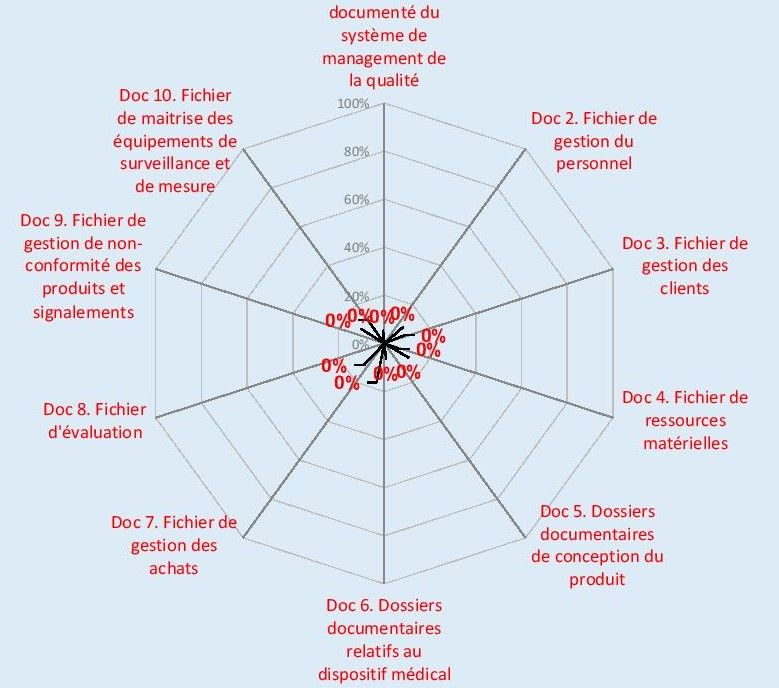
Ce graphique permet une visualisation rapide de la situation de l’entreprise.
Il permet en un coup d’œil de savoir quels sont les résultats escomptés et obtenus. Bien que son format n’assure pas un enregistrement automatique des précédents résultats, cet outil permet d’assurer la traçabilité dans l’évolution de la mise en place d’un Système de Management de la Qualité.
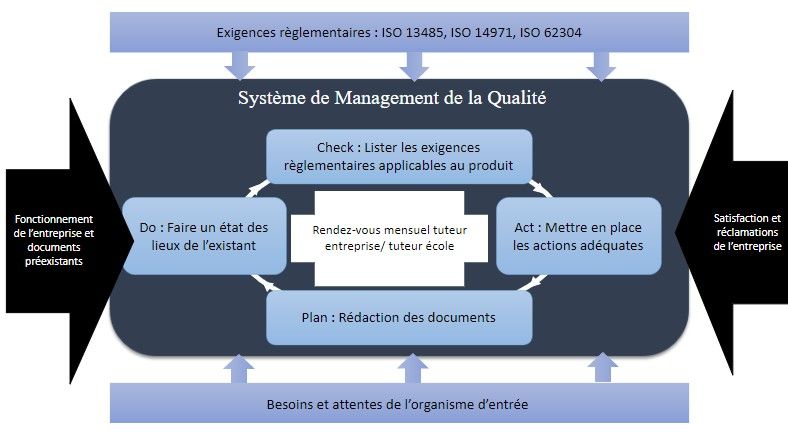
L’entreprise entamant sa démarche de SMQ par l’intermédiaire de ce stage, aucun document n’avait encore été rédigé. Une première réunion a donc été réalisée dans l’optique de déterminer et de quantifier les objectifs de l’entreprise.
Les réunions suivantes ont permis de cibler précisément les utilisateurs, leurs besoins, et le type de patients auxquels s’adresse le dispositif. Un plan d’action et d’amélioration a été définis. Les livrables à fournir à l’issu de ce stage ainsi qu’un rétroplanning ont été établis.
Les acteurs du Système de Management de la Qualité
Figure 11 : Parcours patient
Source : d’après Auteur
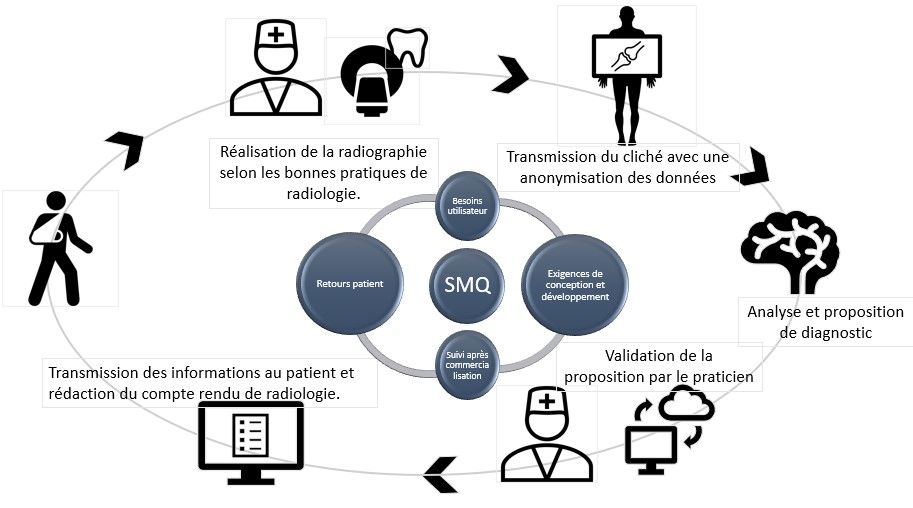
L’objectif principal est d’assurer la sécurité du patient en étant au plus proche des processus métiers.
Une sensibilisation aux enjeux de la qualité est donc nécessaire à tous les niveaux :
- Au sein de l’entreprise, la direction doit établir une politique qualité afin d’assurer l’application des recommandations du SMQ. Cela implique une communication constante entre les différents services est essentielle afin de s’assurer que le personnel est pleinement conscient de son impact dans la démarche qualité.
- L’utilisateur final du dispositif doit lui aussi être inclus afin que ses exigences soient prises en compte et que le dispositif corresponde à ses besoins.
- Le patient quant à lui, contribue au système grâce à ses retours dans le suivi clinique après commercialisation.
Un temps de formation sur la norme NF EN ISO 13485 doit donc être réalisé auprès de toutes les parties prenantes afin d’expliquer les tenants et aboutissants de la qualité.
La revue de direction est une réunion périodique planifiée afin d’assurer la constante amélioration du Système de Management de la Qualité. Durant cette réunion, les différents intervenants peuvent intervenir sur les modifications pouvant mener à une amélioration du SMQ et sur la pertinence des processus en place.
Les réclamations des utilisateurs et les résultats des audits internes sont également examinés afin d’assurer l’amélioration continue au sein de la structure. De cette réunion découlent des actions préventives et correctives ainsi qu’un plan d’action pour les problèmes qui ont été soulevés. Un rapport est ensuite rédigé afin que les éléments de sortie soient passés en revue lors de la prochaine réunion.
Classe du dispositif
Le Système de Management de la Qualité est dépendant du type de dispositif médical concerné. Il est impératif de s’assurer, dans un premier temps, que la technologie répond bien à la définition de dispositif médical. Tout dispositif est caractérisé par son caractère invasif et sa durée de contact avec le corps.
Ces deux critères définissent la classe du dispositif selon des critères préétablis. Connaitre la classe du dispositif permet de s’assurer que les ressources nécessaires à l’implémentation du SMQ sont disponibles.
Pour ce faire, chacune des règles de l’annexe VIII du Règlement a été examinée afin de savoir si le dispositif était concerné. Une première ambiguïté est apparue sur le mot « chirurgie ».
Le Larousse définit la chirurgie comme étant une « discipline médicale spécialisée dans le traitement des maladies et des traumatismes, qui consiste à pratiquer, manuellement et à l'aide d'instruments, des actes opératoires sur un corps vivant […] »[40]. Or le Règlement stipule à la règle 11 du chapitre 2 de l’annexe VIII que :
« Les logiciels destinés à fournir des informations utilisées pour prendre des décisions à des fins thérapeutiques ou diagnostiques relèvent de la classe IIa, sauf si ces décisions ont une incidence susceptible de causer :
- la mort ou une détérioration irréversible de l'état de santé d'une personne, auxquels cas ils relèvent de la classe III, ou
- une grave détérioration de l'état de santé d'une personne ou une intervention chirurgicale, auxquels cas ils relèvent de la classe IIb. [1] »
Le domaine de la dentisterie est donc concerné par cette règle ; mais il reste à déterminer si les exigences sont applicables au dispositif.
Lors de la réflexion autour de cette problématique, une question importante a émergé : L’utilisation du logiciel a-t-elle une incidence susceptible d’entrainer une intervention chirurgicale qui n’aurait pas eu lieu sans son utilisation ?
Le professionnel de santé, utilisateur du dispositif est qualifié pour établir seul un diagnostic précis du patient. Son aptitude est assurée par l’obtention d’un diplôme relatif à sa profession. De plus il est dans l’obligation de vérifier les informations fournies par l’aide au diagnostic fournie parle dispositif. De ce fait, aucune intervention chirurgicale supplémentaire n’est entrainée par l’utilisation du dispositif.
Cette justification, approuvée par des professionnels du secteur est celle qui sera présentée devant les autorités compétentes et les organismes notifiés.
Abaisser la classe de risque du dispositif, revient également à abaisser le niveau d’exigence auquel il doit répondre. Cependant il est important de garder à l’esprit que la sécurité et la santé du patient ne doivent pas être menacées. C’est pour cette raison qu’une justification argumentée sera exigée par les auditeurs. Bien définir la classe de risque de son dispositif est donc une étape préliminaire essentielle pour la suite de son développement.
Choix de l’organisme notifié
Assez rapidement dans la mise en place de la stratégie règlementaire, le choix de l’Organisme Notifié (ON) doit être fait. Il en existe à ce jour qu’un seul en France : il s’agit du GMED. Cependant d’autres ON devrait rapidement voir le jour sur le territoire. Tous les ON présents en Europe peuvent être choisis par l’entreprise en vue de la certification. Parmi les critères de choix, il y a :
- Le prix ; en moyenne aux alentours de 1 300 € la journée d’audit. Un audit sur le SMQ dure environ un jour et demi. A cela s’ajoute les jours de vérification de conformité du dispositif, dont la durée peut varier en fonction du caractère innovatif et atypique de la technologie. Certains frais supplémentaires peuvent s'ajouter si l’intervention d’un spécialiste du domaine est nécessaire, et les frais de dossier. Effectuer une étude de marché comparative peut donc s’avérer être une tâche très importante d’un point de vue financier.
- La stratégie de certification. En effet, tous les organismes notifiés ne possèdent pas les mêmes accréditations. Il est donc primordial de s’assurer que celui qui sera choisi possède donc toutes les compétences nécessaires. Tout particulièrement si l’entreprise souhaite pouvoir mettre le dispositif sur le marché en dehors de l’Europe au même moment (MDSAP, 510(k)…). Dans ce cas, il peut être intéressant de rédiger l’ensemble des documents en anglais et d’en discuter avec les ON. Cela engendrera cependant un ajustement des documents et un coût supplémentaire.
- La disponibilité. La transition entre les deux référentiels a entrainé un embouteillage dans l’emploi du temps des ON. En fonction de la classe du dispositif et de la spécialité concernée, les temps d’attente peuvent être supérieurs à une année entière. Envisager un organisme notifié d’un autre pays peut raccourcir les délais, mais les démarches doivent être réalisées en amont afin de ne pas entraver la mise sur le marché du dispositif.
Les entreprises désireuses d’obtenir le marquage CE ont plusieurs alternatives.
Exemples :
- Dans le cas où il s’agirait d’une petite structure et que les actions à mettre en place ne sont pas nombreuses, l’entreprise peut envisager d’obtenir la certification pour la norme ISO 13485 :2016 en même temps que le marquage CE ou de réaliser le marquage CE seul.
- Si l’entreprise cible un ou plusieurs pays de l’Union Européenne en particulier, elle peut se contenter du marquage CE et décider de s’étendre en dehors de l’Europe dans un second temps.
Ce choix doit être murement réfléchi car un changement peut entrainer un ralentissement de la mise sur le marché et des frais supplémentaires.
Mise en place d’une structure documentaire
Pour des soucis écologiques, économiques et pratiques, les entreprises tendent de plus en plus vers un système documentaire entièrement dématérialisé. Des outils ultra performants permettent une gestion optimale de l’ensemble des documents : les logiciels de gestion électronique des documents. Prêts à l’utilisation, collaboratifs et adaptables, ces outils peuvent accompagner et rassurer les novices dans le domaine de la qualité.
Figure 12 : Interface d’accueil d’un logiciel de gestion documentaire
Source d’après [41]


Cependant, ces plateformes ont un coût. Les prix varient entre 36 €/mois pour les plus élémentaires, à 100 voire 200 €/mois pour les plus complets. Pour les structures de petite taille, cela représente une somme d’argent conséquente, mais d’autres alternatives sont envisageables.
La mise en place d’un serveur d’entreprise accessible par le biais d’une authentification renforcée pour des soucis de confidentialité, peut permettre l’accès et l’utilisation des documents par l’ensemble des membres de l’entreprise.
Parmi ces documents, se trouvent notamment les normes, régulièrement mises à jour en fonction des modifications de versions et dont les exigences sont implémentées dans les procédures de l’entreprise.
Rédaction des documents associés
Afin de mettre en place l’ensemble du système documentaire, plusieurs alternatives sont possibles :
- Le fabricant peut faire appel à une société de consulting qui l’aidera à rédiger ces documents (ex : Strategical, Efor, Nexialist …). Il faut compter environ 20 000 € pour la rédaction des documents concernant Système de Management de la Qualité.
- Il peut également recruter un profil junior (Stagiaire, Jeune diplômé), encadré par un consultant ponctuel.
- Un profil expérimenté seul peut également être envisagé.
Il est important de noter que le nouveau Règlement impose à la société d’avoir une PCVRR.
Une PCVRR est une « Personne Chargée de Veiller au Respect de la Règlementation ».
Elle doit être diplômée ou avoir une expérience suffisante pour la bonne réalisation de cette tâche. Dans le cas de petites structures, elle peut faire partie d’une organisation externe. Ces informations sont à prendre en compte lors du recrutement du personnel du service qualité/affaires règlementaires [42].
Dans le cadre de la mission au sein de l’entreprise, une consultante a encadré la réalisation des documents. Riche de nombreuses expériences en tant qu’auditrice et consultante, son apport a été majeur dans la réalisation des différentes tâches. Grâce à un accompagnement régulier, un changement dans la stratégie de rédaction a pu être effectué afin que les documents correspondent au mieux à la réalité de l’entreprise.
Rédaction des processus
Selon Edwards Deming :« Si vous ne pouvez pas décrire ce que vous faites en tant que processus, vous ne savez pas ce que vous faites. ». Un processus transforme des éléments d’entrée en éléments de sortie par l’intermédiaire d’activités préalablement définies. Il s’agit d’un concept répétable, mesurable et prévisible.
Les processus d’une entreprise peuvent être séparés en trois grandes catégories :
- Les processus de management qui ont pour but d’établir l’organisation globale et d’assurer la politique de l’entreprise.
- Les processus de réalisation qui sont attenants aux produits développés par l’entreprise et qui permettent la maîtrise de la conception et du développement du produit.
- Les processus de support qui permettent de s’assurer que l’ensemble des ressources nécessaires au bon fonctionnement des autres processus est bien disponible.
Des pilotes sont alors affectés à chaque catégorie et à chaque processus afin de s’assurer de leur bonne mise en œuvre.
La réalisation d’une cartographie des processus permet d’avoir une vision globale des interactions et des flux au sein de l’entreprise. La norme ISO 9001 : 2018 fournit un exemple de cartographie où le leadership est au centre. Cette modélisation permet au processus de Management d’être à l’interface de chacun des autres processus et d’assurer une cohésion au sein de l’entreprise.
Une des difficultés rencontrées a été celle de ne pas faire de la sur-qualité : il est important de se rapprocher au mieux de la réalité du terrain et d’adapter le SMQ au personnel de l’entreprise afin que les documents rédigés soient utiles, utilisés et utilisables, et afin que la qualité s’inscrive réellement dans la culture de l’entreprise. Pour cela, l’ensemble des acteurs de l’entreprise doivent être investis dans la bonne marche du SMQ.
Suite à la rédaction de l’ensemble de ces documents, voici les résultats obtenus via l’outil d’autodiagnostic utilisé précédemment :
Figure 13 : Extrait du résultat de l’autodiagnostic à t=4 mois
Source : d’après Auteur, [39]
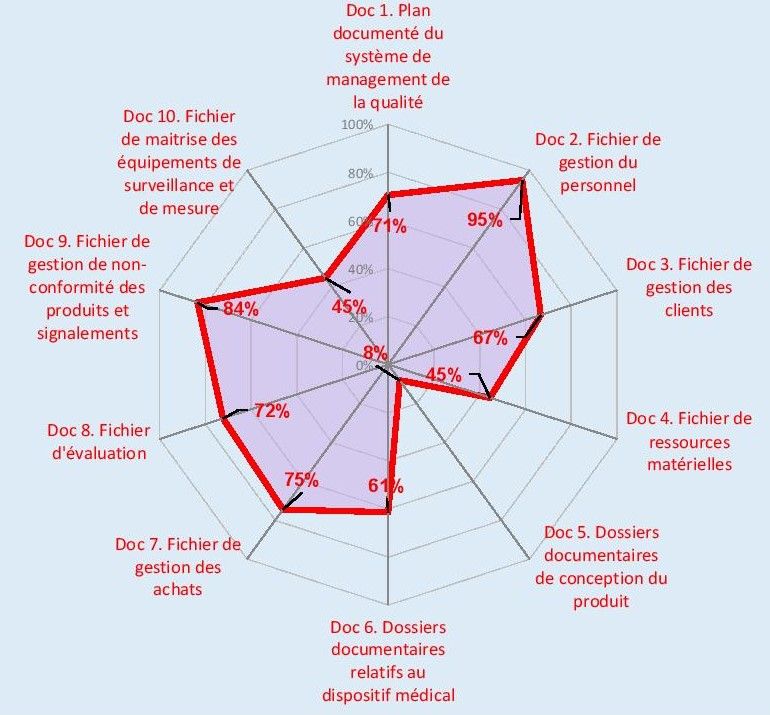
Une disparité dans les résultats est présente et justifiée par les choix adoptés par l’entreprise pour la chronologie de rédaction des documents en fonction des différentes thématiques.
Stratégie clinique
En parallèle de la mise en place du SMQ, une réflexion autour de la stratégie clinique à adopter a été entreprise. L’article 61 du Règlement Européen détaille les exigences en matière d’évaluation clinique et d’investigation clinique. Le point n°10 de cet article permet dans certaines conditions, que « […] le fabricant justifie dûment dans la documentation technique visée à l'annexe II pourquoi il juge adéquate une démonstration de la conformité aux exigences générales en matière de sécurité et de performances qui se fonde uniquement sur les résultats de méthodes d'essai non cliniques, comme l'évaluation des performances, les bancs d'essai et l'évaluation préclinique. »
C’est d’ailleurs une méthode courante dans la démonstration de performances des dispositifs du secteur de l’imagerie. Mais une question très importante subsiste :
Prouver les performances d’une aide au diagnostic revient-il à démontrer la performance du diagnostic fourni ?
C’est une question complexe à laquelle il peut s’avérer difficile sans informations provenant de dispositifs similaires. Les guides « Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software » [12] ainsi que « Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR » permettent de se faire une première idée des études réalisées pour les logiciels de santé. Mais ces derniers ne détaillent pas les recommandations pour les intelligences artificielles.
Un protocole doit donc être monté de toutes pièces par l’entreprise afin de convaincre et rassurer sur les performances du dispositif, soit grâce à une étude clinique impliquant un panel de praticiens, soit grâce à une étude approfondie de l’état de l’art.
Conclusion
En regard des objectifs à atteindre et des problématiques à résoudre, cette expérience a été très enrichissante. En effet, bien que le Système de Management de la Qualité soit inachevé, son initiation a permis à l’entreprise d’entamer sa réflexion autour des processus à mettre en place au sein de la structure.
Cette expérience démontre que plusieurs méthodes permettant de commencer une démarche qualité existent et que les critères de choix vont dépendre de la culture de l’entreprise, de sa taille et du type de dispositif concerné.