
IDS121 - Evaluation et suivi clinique des dispositifs médicaux
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, , nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- Hamza BENCHERRIF :
- Christina GBELAY :
- Charline LEVEL :
- Catherine DALLA RIVA :
- Hamza AIT SAID :
Citation
A rappeler pour tout usage : Hamza BENCHERRIF, Christina Roxanne GBELAY, Hamza AIT SAID, Charline LEVEL, Catherine DALLA RIVA, « Evaluation et suivi clinique des dispositifs médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS121, https://doi.org/10.34746/3hfj-jb17, janvier 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids121
Résumé
Le 26 Mai 2017, le nouveau règlement européen 2017/745 est entré en vigueur et a chamboulé les opérateurs du secteurs, en particulier les fabricants. En effet, ce nouveau règlement, apparu suite à différents scandales sanitaires et notamment l’affaire des implants PIP, a pour vocation de renforcer le cadre règlementaire autour des dispositifs médicaux. De fait, les produits déjà en service doivent être certifiés CE selon ces nouvelles exigences pour pouvoir continuer d’être commercialisés au sein de l'Union Européenne.
L’évaluation clinique est un des points les plus concernés par cette évolution règlementaire et les fabricants doivent faire face à l’apparition de nouvelles exigences dans leur stratégie d’évaluation clinique. Ce processus continu peut prendre différentes formes selon la classe du dispositif, son degré d’innovation et les données disponibles dans la littérature. Dans la course à la certification, l’optimisation de l’évaluation clinique permet au fabricant de gagner du temps et de disposer d'un avantage concurrentiel.
Ce mémoire propose une synthèse des grandes étapes du processus d’évaluation clinique d'un dispositif médical, ainsi que la documentation et les acteurs associés à chaque étape. Un guide pratique destiné aux fabricants est associé à ce projet et permet d’obtenir relativement rapidement une proposition de stratégie clinique adaptée à chaque dispositif.
Abstract
On May 26, 2017 the new European regulation 2017/745 entered into force and upset the operators of the sector, in particular the manufacturers. Indeed, this new regulation, which appeared following various health scandals and in particular the CASE of PIP implants, aims to strengthen the regulatory framework around medical devices. In fact, products already in service must be CE certified according to these new requirements in order to continue to be marketed in Europe.
Clinical evaluation is one of the points most affected by this regulatory evolution and manufacturers must face the emergence of new requirements in their clinical evaluation strategy. This continuous process can take different forms depending on the class of the device, its degree of innovation and the data available in the literature. In the race for certification, optimizing the clinical evaluation saves the manufacturer time in the certification process and therefore money.
To do this, this brief offers a synthesis of the main stages of clinical evaluation, as well as the documentation and actors associated with each step. With this in mind, a practical guide for manufacturers is associated with this project and makes it possible to obtain a clinical strategy proposal adapted to each device relatively quickly.
Téléchargements


Remerciements
Nous souhaiterions tout d’abord remercier Jean-Matthieu PROT, notre suiveur de projet, de nous avoir
accompagné tout au long de ce semestre. Ses recommandations bibliographiques, ses conseils et
indications, nous ont été précieux et nous ont guidé tout au long de cette démarche. Merci d’avoir pris
le temps de répondre à nos interrogations.
Nous aimerions remercier également Isabelle Claude et Gilbert Farges, pour avoir assisté à nos trois soutenances, relu notre rapport et nous avoir conseillé pour progresser dans notre travail.
Merci à Béatrice Koening pour nous avoir formé sur les techniques de mise en page d’une bibliographie, puis d’avoir pris le temps de relire et corriger la nôtre.
Un grand merci aussi aux différents intervenants, qui sont venus, dans le cadre de notre formation, nous partager leurs connaissances et leurs retours d’expériences. Ces informations nous ont été d'une grande utilité pour développer et améliorer notre mémoire d’intelligence méthodologique.
Un grand merci pour finir à Dunstan Becht, étudiant ingénieur, qui nous a aidé en apportant ses connaissances en informatique pour la mise en forme de notre guide interactif à destination des fabricants.
Liste des abréviations
- ANSM : Agence Nationale de la Sécurité du Médicaments et des dispositifs médicaux
- CNIL : Commission Nationale de l’Informatique et des libertés
- DM : Dispositif Médical
- DMDIV : Dispositif Médical de Diagnostic In Vitro
- DMIA : Dispositif Médical Implantable Actif
- IUD : Identification Unique d’un Dispositif
- MIM : Mémoire d’Intelligence Méthodologique
- MDCG : Medical Device Coordination Group (Groupe de coordination des dispositifs médicaux)
- OE : Opérateurs Economiques
- ON : Organismes Notifiés
- PDS : Planification Dynamique Stratégique (Outil qualité)
- PSUR : Rapports Périodiques Actualisés de Sécurité
- QQOQCP : Qui, Quoi, Où, Quand, Comment, Pourquoi (Outil qualité)
- RDM : Règlement Des Dispositifs Médicaux
- SCAC : Surveillance Clinique Après Commercialisation
- SAC : Suivi Après Commercialisation
- UE : Union Européenne
Introduction
La médecine est en constant progrès et ce grâce à l’évolution des techniques et des instruments de soins, avec comme objectif constant la sécurité du patient. Ces avancées sont le fruit de nombreuses recherches et investigations cliniques. Cependant, ces expérimentations n’ont pas toujours été exécutées dans des conditions éthiques. Par exemple, avant 1945, certaines expériences dirigées par des médecins nazis (inoculation de germes mortels, injections intraveineuses de phénol, etc) ont été réalisées sur des prisonniers, en l'absence des protocoles scientifiques et des codes déontologiques actuellement admis et reconnus par la communauté scientifique et médicale internationale. Ces expériences ont exposé les cobayes humains à des conditions cruelles avec des apports scientifiques contestables, voire inutiles.
A l’issue de la Seconde Guerre Mondiale, le procès des Médecins de Nuremberg a eu lieu afin d’incriminer ces médecins accusés de crimes de guerre et crimes contre l'humanité et juger leurs actes. Ce procès historique a donné lieu à la déclaration d’Helsinki et au code Nuremberg. Le principe général de la déclaration est qu'une recherche médicale, impliquant des êtres humains, ne peut être conduite que si l’importance de l’objectif, dépasse les risques et inconvénients pour les personnes impliquées. Le code de Nuremberg à quant à lui été le point de départ de la reconnaissance des dangers excessifs que peuvent apporter les progrès scientifiques et de l’importance fondamentale de les encadrer avec des règles [2], [3].
L’évaluation clinique fait partie de la recherche clinique et correspond à l’évaluation de données cliniques obtenues suite à l’utilisation d’un médicament ou d’un dispositif médical, pour s’assurer de sa pertinence en termes de performance et sécurité pour les patients et utilisateurs [4]. Aujourd’hui encore, cette vigilance est primordiale. Assurée en majeur partie par l’ANSM, elle se traduit par une évaluation des dispositifs médicaux tout au long de leur cycle de vie. Il y a 10 ans environ, l’affaire des implants mammaires PIP a démontré une fois de plus les enjeux de la mise sur le marché d’un dispositif et de sa surveillance après commercialisation. En 2016, l’ANSM publiait une synthèse d’incidents sur ces prothèses indiquant environ 10 000 signalements d’incidents par an [5].
Sur le site de l’ANSM, des déclarations de défaut de qualité ou de risques d’effets secondaires sont renseignés par les fabricants, les patients, les professionnels de santé ou encore les distributeurs. Cette année par exemple, deux gammes de produit du fabricant NUVASIVE SPECIALIZED ORTHOPAEDICS Inc. (NSO) ont vu leur marquage CE suspendu pour cause d’insuffisance de conformité des dispositifs aux exigences règlementaires [6].
Au cœur de l’actualité ces derniers mois, les vaccins contre le Covid 19 sont aussi soumis à une vigilance
stricte de la part de l’ANSM [7]. C’est pour limiter les risques, garantir la performance des dispositifs et renforcer la sécurité des patients que le 26 mai 2021, le nouveau règlement européen 2017/745 est entré en application. Cette
nouvelle réglementation s’applique spécifiquement aux dispositifs médicaux et vise en une harmonisation des règlementations au sein de l’Europe [8].
Cette nouvelle règlementation implique, de la part des fabricants, de réévaluer leurs dispositifs mis sur le marché afin de certifier leur conformité aux exigences générales requises. Cependant, les changements dans les procédures plongent les fabricants dans une grande confusion. Les processus d’évaluation clinique font parties des exigences qui ont été renforcées, de fait il est indispensable d’en connaitre les évolutions afin que le fabricant puisse espérer obtenir un nouveau marquage CE. Composé de près de 175 pages et faisant référence à d’autres normes, le règlement est un document conséquent et en prendre connaissance prend du temps, et nécessite une certaine expertise.
Contexte
L’entrée en application du nouveau règlement 2017/745 vient remplacer les anciennes directives 90/385 et 93/42, et contrairement à ces dernières, il s’applique directement sans transposition, dans tous les États membres de l’Union Européenne.
Aussi, le règlement introduit quelques nouveautés concernant les exigences cliniques par rapport à l’état de l’art, particulièrement dans l’article 61, et notamment :
- La possibilité de consultation d’un groupe d’expert par le fabricant
- L’obligation d’établir un contrat entre deux fabricants pour la démonstration de l’équivalence
- Le recours incontournable aux investigations cliniques pour les dispositifs médicaux implantables et ceux en classes III avant commercialisation
- Une définition précise du Suivi Clinique Après Commercialisation (SCAC) et de son contenu
- L’obligation pour les groupes de produits n’ayant pas de destination médicale d’effectuer une évaluation clinique
Abordons plus en détails chacune d’entre elles :
1. La consultation d’un groupe d’experts par le fabricant :
Pour tous les dispositifs de classe III et IIb destinés à administrer dans l'organisme et/ou à retirer de l'organisme un médicament, le fabricant aura la possibilité de consulter un groupe d’experts avant d'effectuer son évaluation clinique, y compris son investigation clinique.
Ce groupe est un ensemble des experts de premier plan dans leur domaine, nommés par la commission européenne sur la base de leur expertise scientifique, clinique et technique. Leur sélection est faite par la Commission européenne, en consultation avec le groupe de coordination en matière de dispositifs médicaux (GCDM/MDCG). Ce groupe permettra de guider les fabricants dans leur stratégie de développement clinique et de préparation d'investigation cliniques.
2. Un contrat entre fabricants pour la démonstration de l’équivalence :
L’évaluation clinique suivra « une procédure définie et méthodologiquement fondée sur :
- Une évaluation critique des publications scientifiques pertinentes, actuellement disponibles concernant la sécurité, les performances, les caractéristiques de conception et la destination du dispositif, à condition que :
- L’équivalence du dispositif faisant l'objet de l'évaluation clinique, en ce qui concerne la destination, et du dispositif auquel se rapportent les données soit démontrée, et
- Le respect des exigences générales pertinentes en matière de sécurité et de performances soit dûment établi.
- Une évaluation critique des résultats de toutes les investigations cliniques disponibles en tenant dûment compte de la question de savoir si les investigations ont été conduites au titre des articles 62 à 80, de tout acte adopté en vertu de l'article 81 et de l'annexe XV ; et
- La prise en compte des alternatives de traitement actuellement disponibles à cette fin, s'il en existe. » Autrement dit la prise en compte de l’état de l’art.
Mais en termes de démonstration de l’équivalence le règlement introduit surtout les nouvelles exigences suivantes :
- Les deux fabricants doivent avoir conclu un contrat qui accorde explicitement au fabricant du second dispositif un accès total et permanent à la documentation technique, et
- L'évaluation clinique d'origine doit avoir été effectuée conformément aux exigences du règlement, et le fabricant du second dispositif doit en apporter la preuve manifeste à l'organisme notifié (ON).
3. Le recours incontournable aux investigations cliniques pour les dispositifs implantables et de classe III.
Dans le cas des dispositif médicaux implantables et de classe III, les investigations cliniques seront obligatoires avant mise sur le marché.
Cette obligation de conduire des investigations cliniques, avant mise sur le marché, pourra être évitée par les fabricants si les conditions suivantes sont réunies :
- Le dispositif a été conçu en modifiant un dispositif déjà commercialisé par le même fabricant,
- Le fabricant a démontré que le dispositif modifié est équivalent au dispositif commercialisé et cette démonstration a été approuvée par l’organisme notifié.
- L’évaluation clinique du dispositif commercialisé suffit à démontrer la conformité du dispositif modifié avec les exigences pertinentes en matière de sécurité et de performances.
- Les dispositifs qui ont été légalement mis sur le marché ou mis en service conformément à la directive 90/385/CEE ou à la directive 93/42/CEE et pour lesquels l'évaluation clinique :
- Est fondée sur des données cliniques suffisantes, et
- Est conforme à la spécification commune par produit qui est applicable pour l'évaluation clinique de ce type de dispositif, lorsqu'il en existe une ; ou des sutures, agrafes, produits d'obturation dentaire, appareils orthodontiques, couronnes dentaires, vis, cales, plaques, guides, broches, clips ou dispositifs de connexion et pour lesquels l'évaluation clinique est fondée sur des données cliniques suffisantes et est conforme à la spécification commune par produit qui est applicable, lorsqu'il en existe une.
4. Exigences relatives à la surveillance après commercialisation :
Le Suivi Clinique Après Commercialisation (SCAC) est décrit comme un processus continu de mise à jour de l'évaluation clinique qui s’inscrit dans le plan de surveillance après commercialisation établi par le fabricant.
Pour le fabricant, mettre en place un SCAC consiste à collecter et évaluer de manière proactive les données cliniques résultant de l'utilisation d’un dispositif marqué CE et commercialisé.
Les objectifs du SCAC sont :
- De confirmer la sécurité et les performances pendant toute la durée de vie prévue du dispositif
- D’assurer le caractère constamment acceptable des risques identifiés
- De détecter les risques émergents sur la base d'éléments de preuve concrets
C’est la partie B de l’annexe XIV du règlement qui définit et décrit le contenu du SCAC.
Le plan de SCAC comprend au moins :
- Les méthodes et les procédures générales du SCAC à appliquer, telles que la collecte de l'expérience clinique acquise et des retours d'information des utilisateurs ainsi que la consultation de la littérature scientifique et d'autres sources de données cliniques.
- Les méthodes et les procédures spécifiques du SCAC à appliquer, par exemple l'évaluation des registres appropriés ou des études de SCAC.
- Une justification de l'adéquation des méthodes et des procédures précédentes.
- Une référence aux parties pertinentes du rapport d'évaluation clinique et du dossier de gestion des risques.
- Les objectifs spécifiques fixés pour le SCAC.
- Une évaluation des données cliniques relatives à des dispositifs équivalents ou similaires.
- Une référence aux spécifications communes, normes harmonisées ou aux guides utilisés par le fabricant, concernant le SCAC.
- Un calendrier détaillé et justifié pour les activités de SCAC (par exemple, analyse des données issues du SCAC et rapport).
5. Cas des groupes de produit n'ayant pas de destination médicale :
En ce qui concerne les groupes de produits sans destination médicale (qui ont des technologies et une utilisation très proche d’un dispositif, mais sans revendication liée au diagnostic ou au thérapeutique. NB : auparavant ils faisaient partie de la réglementation des produits cosmétiques et grand public), le point 9 de l’article 61 précise que ces produits sont soumis aux mêmes exigences que les dispositifs médicaux en matière d’évaluation clinique, d’investigations cliniques, de SAC et de SCAC [1], [9].
La mise en pratique de ces principes énoncés ci-dessus fera l’objet du chapitre suivant.
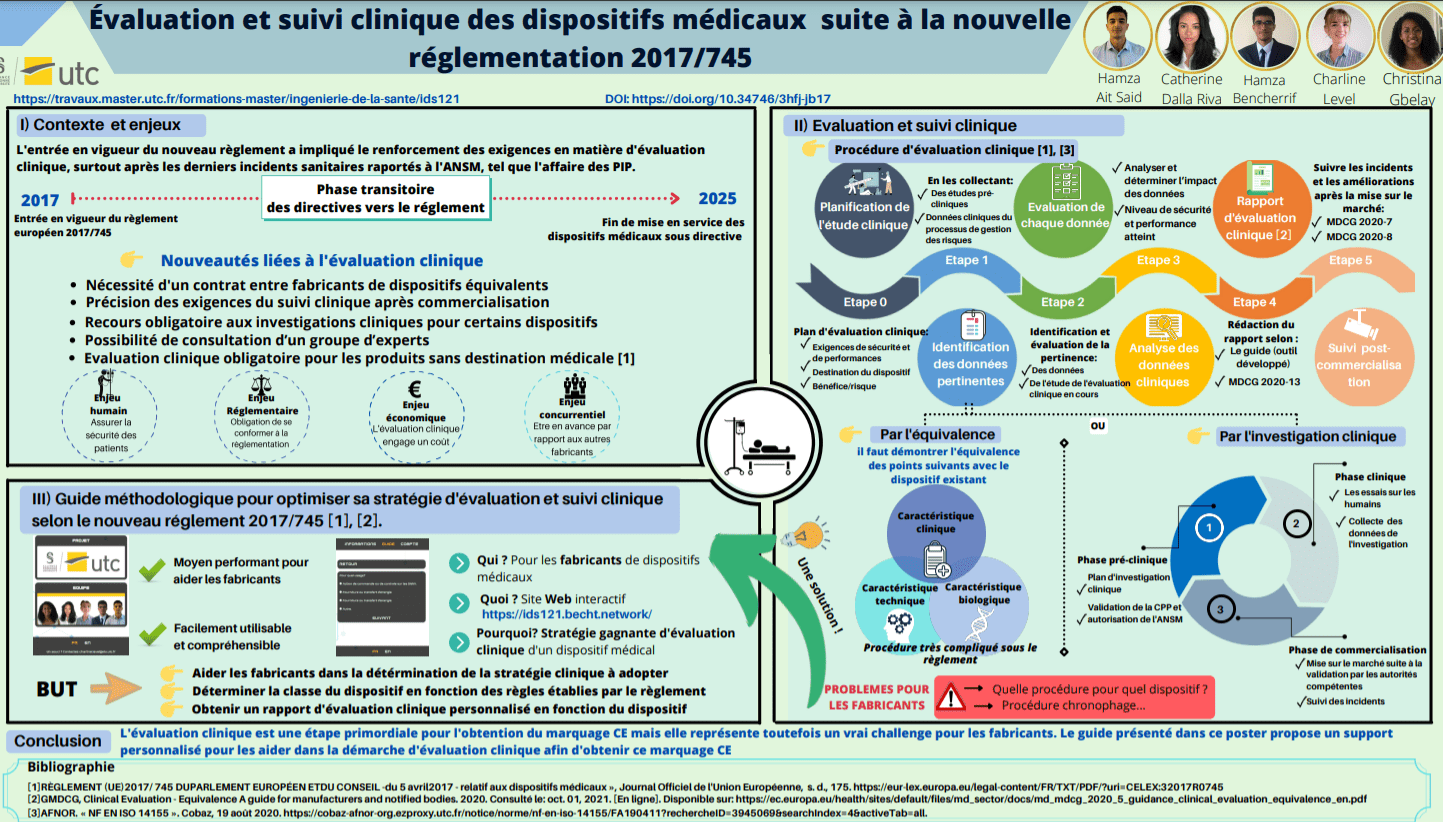
Outre ces nouveaux aspects théoriques de l'évaluation clinique apportés par le règlement, on peut constater également, grâce au tableau 1 présenté ci-dessous, une augmentation des textes évoquant l'évaluation clinique.
Tableau 1 : Evolution de la description des obligations cliniques entre directive et règlement (Source : Auteurs d’après [7])
Ainsi, la mise en place du nouveau règlement avait pour objectif de renforcer les exigences pour assurer la performance et la sécurité des dispositifs, notamment au niveau de l’évaluation clinique. Ce tableau 1 comparatif le confirme. Des précisions relatives au processus d’évaluation clinique ont été ajoutées, ainsi que pour le suivi post commercialisation. Cette précision des exigences a pour objectif de limiter les abus en proposant un cadre plus strict.
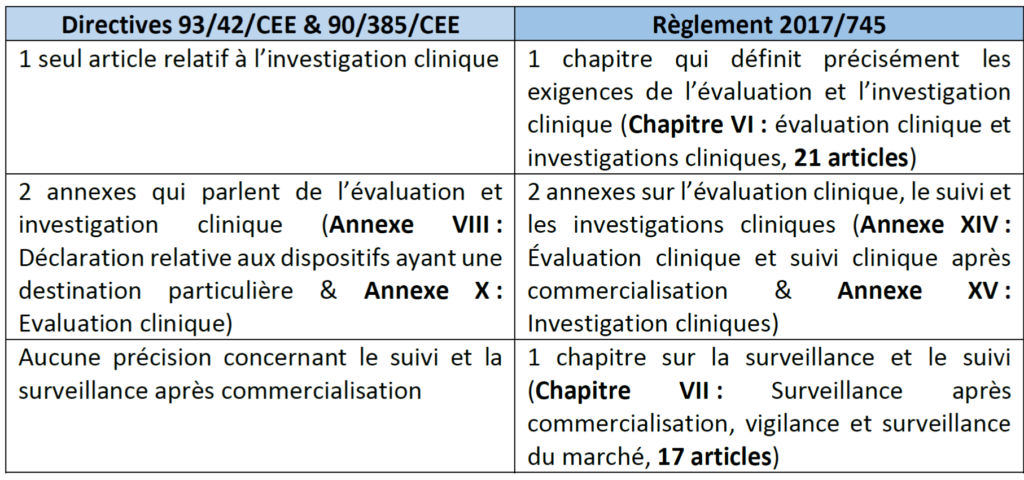
Actuellement, les fabricants et autres opérateurs économiques du secteur se trouvent dans une phase de transition (figure 1) au cours de laquelle ils doivent certifier la conformité de leurs dispositifs aux nouvelles exigences du règlement 2017/745, qu’ils soient nouvellement mis sur le marché ou déjà certifiés sur les anciennes directives.
Figure 1 : Schéma chronologique simplifié de la phase transitoire induite par l'entrée en vigueur du nouveau règlement européen (source : d’après [10])
Aussi, malgré les différents éléments mis en place pour faciliter l’utilisation de ces réglementations, la prise en main des exigences reste un processus chronophage pour les opérateurs du secteur. En effet, pour la mise sur le marché ou la réévaluation des dispositifs déjà sur le marché, les fabricants doivent s’appuyer sur les exigences énoncées dans le règlement.
Seulement, après consultation dudit règlement, il apparaît que les informations peuvent parfois être difficiles à appréhender. Effectivement, les différentes réglementations, que ce soit pour l’évaluation clinique, le marquage CE, le suivi clinique après commercialisation et toute la documentation technique, sont réparties dans différents chapitres et articles, mais aussi dans les annexes. De fait, la détermination de la meilleure stratégie d’évaluation clinique peut prendre du temps à être élaborée. Et ce temps, les fabricants ne l’ont pas.
L’objectif de ce projet est de fournir un support aux fabricants afin de les accompagner dans ces démarches et rester concurrentiels dans cette course à la mise sur le marché.
Pour ce faire, il est d’abord nécessaire de préciser les enjeux, les étapes de déroulement et les acteurs impliqués dans ce processus d’évaluation. Au vu de ces informations, il sera alors possible de définir les processus à mettre en place par le fabricant pour assurer cette évaluation clinique et ce, lors de phases clés du cycle de vie du dispositif [11].
Sous la forme d’un guide, les fabricants auront accès à un processus de détermination des stratégies cliniques à mettre en œuvre selon les différents critères de leur dispositif : type de données collectées, niveau de risque du dispositif… Ce qui leur permettra d’obtenir une stratégie clinique adaptée.
Également, grâce à ce guide stratégique, les fabricants pourront évaluer le niveau de risque de leur dispositif et ainsi déterminer sa classe de risque.
Enjeux
Le principal enjeu de l’évaluation clinique, et du renforcement des exigences associées, est un enjeu humain. C’est un enjeu crucial pour assurer la qualité des soins, puisque les dispositifs ont pour objectif de soigner, diagnostiquer ou encore prévenir d’une pathologie de la façon la plus performante possible et sécuritaire pour les patients. Autrement dit, le dispositif entre indirectement et directement en contact avec le patient. Assurer la sécurité humaine, ainsi que la performance de l’instrument utilisé est alors la priorité, le rapport bénéfices/risques doit être donc en faveur du bénéfice pour le patient. C’est d'ailleurs sur cette base de rapport bénéfices/risques que l’ANSM délivre ou non une autorisation d’essais et d’investigation clinique.
Le marché du dispositif est important et génère de grands bénéfices financiers pour les différents fabricants et opérateurs économiques du secteur. C’est pourquoi l’enjeu économique est aussi un enjeu important à prendre en compte. En effet, plusieurs dépenses sont à prévoir pour accomplir l’évaluation clinique et notamment lorsqu'on doit avoir recours à une investigation clinique. En effet, le coût de cette dernière comprend entre autres : le nombre de patients dans l’investigation, la durée de l’étude, la durée de recrutement des patients, la pathologie étudiée ainsi que les surcoûts hospitaliers et heures de travail. Ainsi, un promoteur, ayant à sa charge un dispositif nécessitant une investigation clinique, devra anticiper le budget adéquat de façon optimale. Selon une professionnelle du domaine, la fourchette de prix est de l'ordre de 50000/80000 euros et peut aller jusqu'à des centaines de milliers d'euros, en fonction du dispositif médical.
Ce marché est toutefois très règlementé et la mise sur le marché de chacun des dispositifs ne peut avoir lieu que en respectant les exigences requises. Ainsi, l’enjeu réglementaire fait partie des enjeux principaux de cette nouvelle règlementation et oblige les fabricants à s’aligner aux exigences réglementaires en termes d’évaluation clinique. Il est, entre autres, essentiel pour l’obtention du marquage CE sous le règlement 2017/745. La règlementation influe grandement sur la durée nécessaire à la mise sur le marché des nouveaux dispositifs, de fait, la prise en main rapide de ces nouvelles exigences devient un enjeu concurrentiel pour les fabricants. C’est pourquoi, les différents outils proposés par les entreprises de conseil, les organismes notifiés et le guide élaboré dans le cadre de ce projet, deviennent des supports prisés du secteur.
Chapitre 1 : Principes généraux sur l’évaluation clinique selon le Nouveau Règlement Européen 2017/745
1) Distinction entre évaluation clinique et investigation clinique
1.1) L’évaluation clinique
Décrite comme un processus continu, l’évaluation clinique, permet de recueillir, évaluer et analyser des données cliniques se rapportant à un instrument médical et d’évaluer si les preuves sont suffisantes, pour prouver la conformité aux exigences essentielles pertinentes en matière de sécurité, sûreté et de performance lors de l’utilisation du dispositif, conformément aux instructions d’utilisation du fabricant.
Ces exigences générales sont données par le règlement 2017/745 dans le chapitre VI et l'annexe XIV, et sont relatives à la sécurité et à la performance du dispositif. Ces exigences sont, pour la plupart, largement inspirées du MEDDEV 2.7.1, progressivement remplacé ou mis à jour dans les guides publiés par le MDCG [12].
Ce guide, établi à l’intention des fabricants et des organismes notifiés, résume les processus nécessaires à la mise en place des évaluations cliniques de façon à constituer un document de référence.
C’est le fabricant qui définit le niveau de preuves cliniques nécessaires pour confirmer le respect de ces exigences dans le plan d’évaluation clinique défini avant sa production.
C’est la classification du dispositif qui détermine le niveau d’exigence requis. La classe attribuée au dispositif dépend de son niveau de risques, de sa durée d’utilisation sur le patient, et de sa destination. Pour chaque dispositif, l’évaluation clinique doit être intégrée dans sa documentation technique [13], [14].
1.2) L’investigation clinique
Selon le Règlement (UE) 2017/745, l’investigation clinique est définie comme « toute investigation systématique impliquant un ou plusieurs participants humains destinés à évaluer la sécurité ou les performances d’un dispositif ».
En effet, à compter du 26 mai 2021, tous les projets de recherche visant à évaluer un dispositif médical, ou un dispositif utilisé à des fins non-médicales listé à l’annexe XVI du règlement (UE) 2017/745, seront encadrés par ce dernier et seront nommés investigations cliniques (IC) [15].
L'ISO 14155 définit l’investigation clinique comme une investigation systématique portant sur un ou plusieurs sujets humains, entreprise en vue d'évaluer les performances cliniques, l’efficacité ou la sécurité d'un dispositif médical. Les termes « essai clinique » ou « étude clinique » sont des synonymes d'« investigation clinique » [16].
Malgré les progrès actuels, la médecine ne permet pas encore de tout guérir. Pour évoluer, elle a besoin de trouver de nouveaux traitements plus efficaces, moins toxiques, moins contraignants, pour le patient. La recherche clinique ou investigation clinique, intervient après la recherche fondamentale et représente toutes les recherches faites sur l’homme. Elle a pour but d’améliorer les connaissances sur les maladies, améliorer leur dépistage, améliorer les traitements mais aussi améliorer la prise en charge des patients à court, moyen et long terme [17].
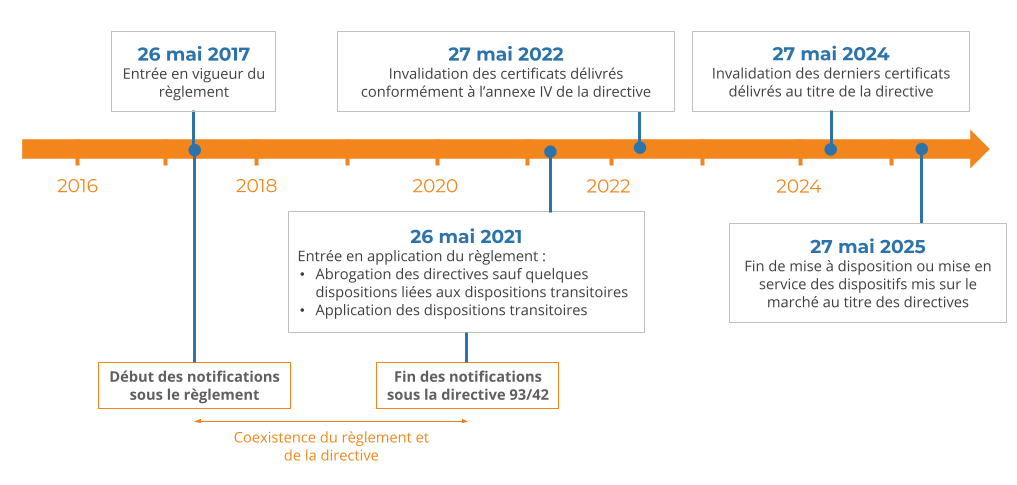
Les projets de recherche clinique sont basés sur la participation volontaire des personnes à des essais cliniques qui leurs sont proposés par le médecin. On distingue les recherches interventionnelles, des recherches non-interventionnelles, aussi appelées études observationnelles (figure 2).
Les recherches interventionnelles impliquent une participation directe de la personne et peuvent modifier la prise en charge habituelle. Cela peut aller de la simple réalisation d’une prise de sang supplémentaire à la mise en route d’un traitement spécifique en fonction du degré de contrainte et du risque pour le patient.
Figure 2 : Types de recherches cliniques impliquant la personnes humaines et caractéristiques associés (source auteurs, d’après [18]).
2) Quand faire une évaluation clinique et comment ?
2.1) Quand faire l’évaluation clinique ?
Pour commencer, il apparaît primordial de clarifier les différentes étapes de cette évaluation et de préciser à quel moment du cycle de vie du dispositif médical elle intervient (figure 4). Le Groupe de Coordination en matière de Dispositifs Médicaux (MDCG) rappelle que l’évaluation clinique est un processus qui est continu tout au long de la vie du dispositif (figure 3). Il est toutefois possible d’identifier 3 séquences en particulier dans laquelle elle intervient :
- La 1ère évaluation clinique a lieu dès la conception du dispositif. A ce moment, elle va permettre d’identifier les données en termes de performance et de sécurité clinique, d’équivalence avec d’autres dispositifs déjà sur le marché, analyse des lacunes par rapport aux données trouvées dans la littérature nécessaire à la future mise sur le marché du dispositif. Au cours de cette séquence, le fabricant va mettre en place le plan d’évaluation clinique. Dans ce document, le fabricant doit énoncer clairement la thématique du projet de recherche et les éléments mis en place pour y répondre. Ils seront pris en compte pour la définition de la procédure d’évaluation clinique.
- La 2ème évaluation a lieu lors du marquage CE initial. L’objectif est de collecter des données cliniques suffisantes pour confirmer le respect des exigences en matière de performance et de sécurité clinique. Au cours de cette séquence, les données qui doivent être systématiquement contrôlées après la mise sur le marché, vont être identifiées. Cette analyse va permettre d’évaluer les risques résiduels. Ces critères désignent typiquement les possibilités de complications, les incertitudes, le niveau de rareté, …
- C’est finalement lors de la mise à jour par le fabricant qu’à lieu la 3ème étape de l’évaluation clinique. C’est le fabricant qui définit et justifie la fréquence de mise à jour nécessaire pour le dispositif. Au cours de ces étapes, il se dit notamment :
- D’analyser les risques significatifs du produit,
- De considérer les innovations dans le secteur au niveau clinique, technologique ou autres qui sont liés au dispositif,
- D’évaluer le niveau de performance et de sécurité du dispositif d’après les données cliniques disponibles et le nombre de dispositifs équivalents disponibles sur le marché.
L’évaluation clinique peut être mise à jour selon un processus dit « actif » à la suite :
- D’un potentiel changement requis dans l’évaluation,
- Ou dans le cas où aucune information de ce type ne s’applique :
- Mise à jour annuelle pour les dispositifs de classe III, considérés comme à risque, ou encore pour des dispositifs pour lesquels les risques ne sont pas encore tout à fait bien établis
- Mise à jour tous les 2 à 5 ans si le dispositif ne présente pas de risque significatif [19].
Figure 3 : Représentation simplifiée du processus continu d'évaluation clinique d'un dispositif médical (Source : Auteurs)
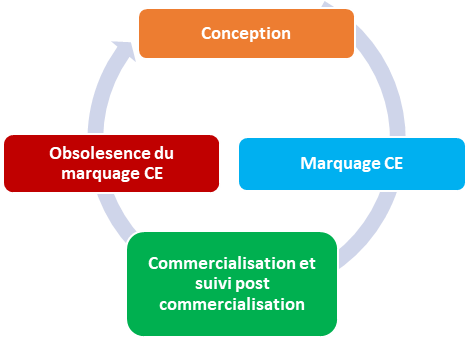
Comme évoqué plus haut, l’évaluation clinique est un processus continu tout au long du cycle de vie de dispositif, elle va donc nécessiter une mise à jour continuelle de la documentation technique afin de conserver le marquage CE.
Figure 4 : Schéma des différentes phases clés de l'évaluation clinique au cours du cycle de vie du dispositif (source : d’après [19])
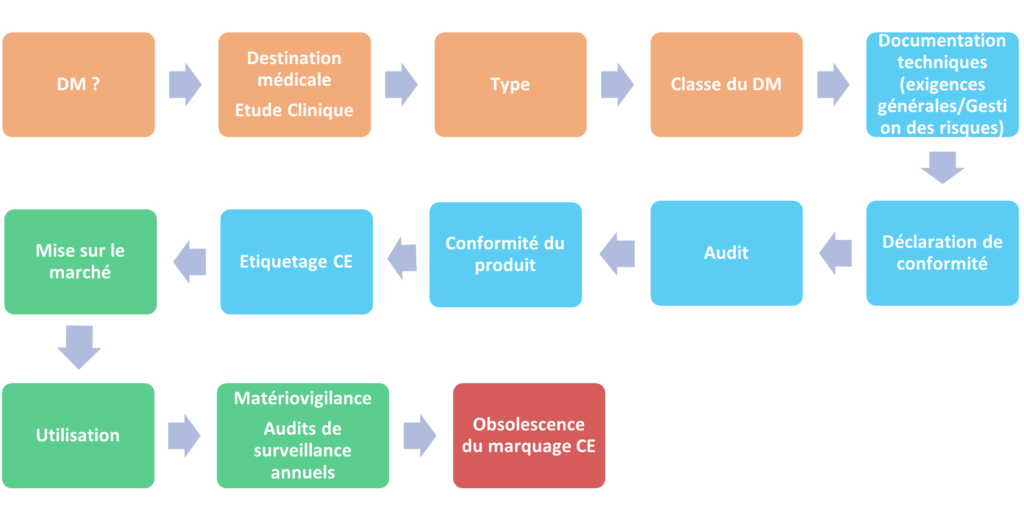
2.2) L’intérêt de l’évaluation clinique
A la lecture des exigences essentielles générales de la directive 93/42/CEE (modifié par la directive 2007/47/CE), il peut être noté qu'elles sont toutes autour du patient et de l’utilisateur pour garantir leur santé ainsi que leur sécurité (voir tableau 2). Notez bien que la sécurité et les performances d’un dispositif doivent être assurées pendant tout son cycle de vie. Pour assurer sa conformité à ces exigences, l’évaluation clinique des dispositifs reste indispensable pour garantir la sécurité et avoir un rapport bénéfice/risque favorable.
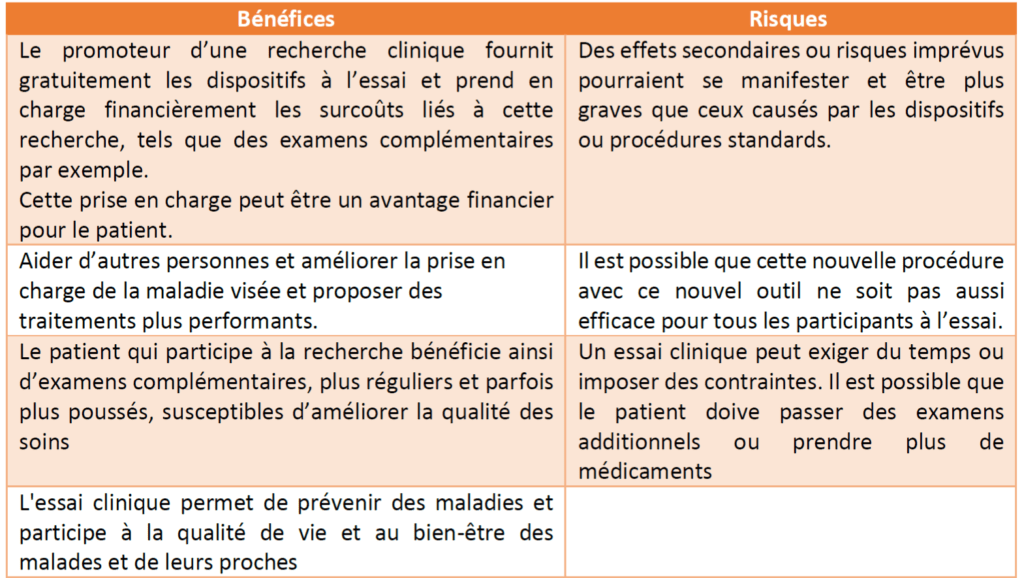
Les essais cliniques sont soigneusement conçus pour engendrer le moins de risques et le plus d’avantages possibles pour chaque participant. Cet équilibre est désigné sous l’appellation de « rapport bénéfice/risque » et correspond à l’élément central dans tous les processus d’évaluation clinique. Les bénéfices et les risques possibles sont propres à chaque essai et il est important de les anticiper au maximum et d’en informer les responsables le plus rapidement possible. Le tableau 2 ci-dessous présente des exemples non-exhaustifs des bénéfices et risques pouvant survenir au cours d’une étude.
Tableau 2 : Exemples de bénéfices et risques liés aux essais cliniques (Source : Auteurs)
En recherche clinique sur les dispositifs médicaux, ainsi que sur les médicaments, de nombreuses mesures sont prises pour limiter les risques encourus par les patients. Par exemple dans le cas des médicaments, de nombreux tests pré-cliniques et des programmes d’expérimentation animale sont conduits avant toute administration à l'homme. Ensuite, les plans de recherche prévoient d’exposer d’abord de très faibles nombres de volontaires en bonne santé à de faibles doses de produit, avant d’augmenter les doses, puis le passage à un plus grand nombre de patients [20], si les résultats sont concluants. Ces précautions sont appelées « pré-requis » de la recherche. Des précisions à ce sujet seront abordées dans la partie relative au déroulement d’un essai clinique.
3) Acteurs mobilisés dans l’évaluation clinique
Une évaluation clinique est un processus complexe, qui nécessite l’intervention de différents acteurs avec un champs d’expertise et une implication différente (figure 5). Ainsi, ils vont rythmer le parcours de lancement d’un essai clinique.
Parmi eux, le promoteur initie l’essai. Il peut être un individu, une entreprise, une institution ou un organisme qui prend la responsabilité de mettre en place, de gérer et/ou de financer un essai clinique. Selon les textes de loi, le promoteur est "la personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain, qui en assure la gestion et vérifie que le financement de la recherche est prévu". Il s’assure que la recherche se déroule selon les Bonnes Pratiques Cliniques et il est considéré Interlocuteur unique du CPP et de l’Autorité Compétente.
Le promoteur a plusieurs missions :
- Initier le projet scientifique et concevoir le protocole
- Choisir l'investigateur
- Assurer et contrôler la qualité
- Faire le lien avec le Comité de Protection des Personnes et l'autorité compétente
- Contracter une assurance couvrant les conséquences éventuelles de cette recherche
- Déclarer au Ministère et à l’Autorité compétente les éventuels événements indésirables survenus au cours de la recherche
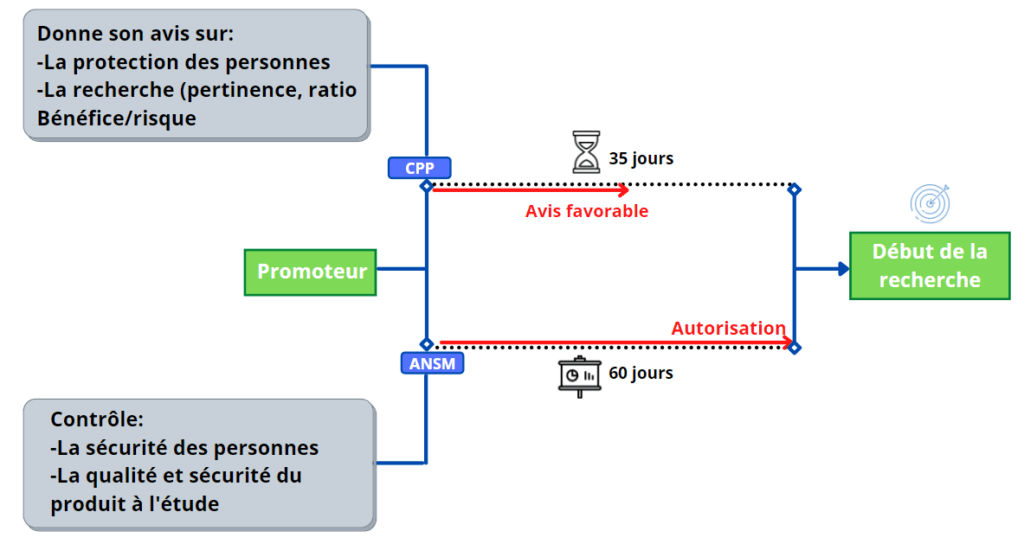
Le Comité de Protection des Personnes (CPP) possède un rôle clé et incontournable dans le déroulement d’un essai. En effet, sa validation est nécessaire pour lancer un essai clinique en France. La mission du CPP est de vérifier que la protection des personnes est bien assurée en évaluant un protocole (avant sa réalisation), selon :
- La pertinence de la recherche
- L’information délivrée au participant
- Le rapport bénéfice/risque
- La qualification des médecins investigateurs
- L’adéquation entre les objectifs et les moyens mis en œuvre.
L’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM), est l'autorité compétence en France en matière de dispositifs médicaux, à cet égard elle assure la surveillance du marché, valide la pertinence et l’intérêt médical du dispositif. Elle évalue :
- La sécurité d’emploi
- La qualité
- Le bon usage des médicaments et produits de santé.
Elle attribue notamment l’autorisation de mise sur le marché (AMM) pour les médicaments. Elle a la responsabilité de délivrer aux promoteurs les autorisations pour tous les essais cliniques interventionnels qui comportent une intervention possiblement à risque sur la personne, non justifiée par sa prise en charge habituelle. Cela concerne les essais cliniques interventionnels sur les médicaments, les dispositifs médicaux, les dispositifs médicaux de diagnostic in vitro, les produits biologiques ainsi que les essais cliniques ne portant pas sur des produits de santé se déroulant en France. L’agence peut demander des informations complémentaires ou des modifications des protocoles d’essais.
N.B : Le CPP et l’ANSM sont chargés d’autoriser le début de l'investigation clinique après l’évaluation des dossiers.
L’ANSM est l’autorité compétente en matière d’essais cliniques conduits en France. Elle peut suspendre ou arrêter un essai à tout moment.
La Commission Nationale Informatique et Liberté (CNIL) veille au respect de la confidentialité des informations recueillies notamment dans le cadre d’une recherche. Elle donne son autorisation sur la mise en place de tout fichier informatisé concernant un essai et les patients de cet essai. Elle est donc fondamentale au bon déroulement d’un essai clinique.
L’investigateur est un individu médicalement qualifié, responsable de la conduite de la recherche sur un centre choisi, appelé « centre investigateur ». Il inclut les patients dans l'étude et veille à la sécurité des personnes. Ses principales missions lors d’un essai sont :
- Superviser la recherche dans un service (centre investigateur) : il est le seul qui est déclaré au Comité de Protection des Personnes
- Responsable du déroulement de la recherche (engagement de responsabilités)
- Désigner, au début de l'étude, ses collaborateurs et leur fonction dans la recherche (formulaire de délégation de fonctions transmis au promoteur [21].
Figure 5 : Chronologie de l'implication successive des différents acteurs d'un essai clinique (Source : d’après auteurs)
Chapitre 2 : Processus d'évaluation clinique
1) Etapes d'évaluation clinique
NB : Les références énoncées dans cette partie font référence au règlement 2017/745.
L'évaluation clinique peut être divisée en 6 étapes distincts (figure 6).
Figure 6 : Etapes définies du plan d’évaluation clinique (Source : d’après auteurs)
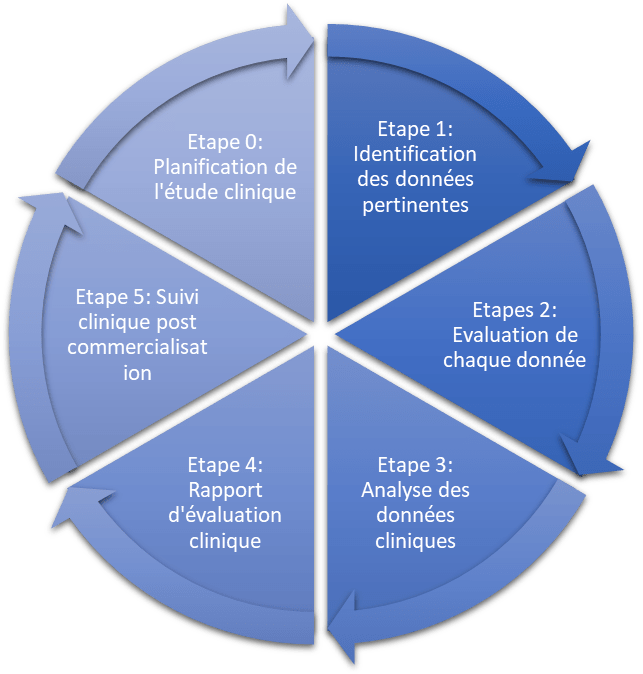
- Étape 0 : définir le domaine d’application et le plan d’évaluation clinique
Avant de démarrer une étude, le fabricant doit en définir la portée en s’appuyant sur les exigences essentielles, qui doivent être abordées d’un point de vue clinique selon la nature et l’historique du dispositif. C’est ce qu’on appelle aussi le cadrage.
La portée sert de base à d’autres étapes, y compris l’identification des données pertinentes (Etape 1). Le fabricant établit une description de l’instrument évalué et un plan d’évaluation clinique. Une évaluation clinique doit être critique. Par conséquent, il doit identifier, évaluer et analyser les données favorables et défavorables aux hypothèses de l’étude. En fonction du stade du cycle de vie du produit, des considérations relatives à la mise en place d’un plan d’évaluation doivent comprendre certains aspects spécifiques.
- Etape 1 : identifier les données pertinentes
Ces données sont collectées par le fabricant et proviennent :
- Des investigations pré-commercialisation ;
- Des données cliniques obtenues grâce au processus de gestion des risques : rapports de vigilance, recherches issues de la littérature, plaintes reçues ;
- Des études précliniques réalisées, avec les rapports des tests incluant la vérification et la validation des données.
Si les données ne proviennent pas du fabricant, elles peuvent aussi être extraites de la littérature, par exemple lors d’études réalisées pour des dispositifs équivalents ou provenant d’articles relatifs aux connaissances actuelles d’un domaine.
- Etape 2 : évaluer chaque données (validité et poids scientifique)
L’objectif de cette étape est :
- D’identifier les données présentes dans chaque étude,
- D’évaluer la qualité méthodologique de l’étude et sa valeur scientifique,
- D’évaluer la pertinence de l’étude vis-à-vis de l’évaluation clinique en cours,
- De pondérer systématiquement la contribution de chaque étude par rapport aux autres données retenues
Cette évaluation de données est souvent réalisée selon un plan précis et déterminée à l’avance (au cours de l’étape 0) en fonction de critères relatifs à la qualité, à la validité scientifique, à la pertinence de l’évaluation clinique, de l’intérêt clinique général du produit.
- Etape 3 : analyser les données vis-à-vis des performances, de la sécurité, des informations fournies par le fabricant et des risques résiduels afin d’envisager des conclusions d’étude. Afin de démontrer la conformité, les évaluateurs se doivent d’utiliser des méthodes précises, d’effectuer une analyse complète, afin de déterminer si d’autres études cliniques ou d’autres mesures sont nécessaires. Cette étape va aussi permettre de déterminer les exigences à indiquer dans le CRF (Case Report Form). Ce document a pour objectif de contenir toutes les informations requises par le protocole concernant les personnes impliquées dans un projet de recherche biomédicale.
NB : Ces 3 étapes seront abordées plus bas dans le chapitre 3.
- Etape 4 : rapport d’évaluation (check list à vérifier avant de libérer le rapport) selon le plan de Suivi après commercialisation (SAC)/Suivi clinique après commercialisation (SCAC)
Le rapport contient tous les documents relatifs à cette évaluation, ainsi que toutes les informations relatives à l’étude qui doivent être explicités clairement. Ce rapport contient aussi des références à la littérature.
Des groupes d'experts issus de l’organisme notifié devraient s'acquitter de la tâche consistant à fournir un avis sur les rapports d'évaluation sur l'évaluation clinique des organismes notifiés dans le cas de certains dispositifs à haut risque. Ces groupes d’experts doivent être désignés en amont, lors de la planification de l’étude [19], [22]. Ils vont ainsi valider les données récoltés et les processus scientifiques mise en place, ainsi que le respect des règlementations et de l’éthique.
L’étape 5 concernant le suivi clinique après commercialisation (SCAC) sera abordée dans la suite du mémoire au chapitre 3.
2) Types d’évaluation clinique
2.1) Par le biais de la littérature
La première phase de l’évaluation clinique est l’évaluation par la littérature, afin de collecter les données existantes et d’établir l’état de l’art de la technologie proposée par le fabricant. Ces premières recherches vont être essentielles pour établir la suite de la procédure d’évaluation clinique. Elles répondent à des exigences particulières qui sont définies dès la rédaction du plan d’évaluation clinique et vont être inscrites dans le rapport final. En effet, parmi les documents à fournir on retrouve un protocole ainsi qu'un rapport de recherche documentaire.
A titre d’exemple, le GMED propose un sommaire récapitulant les différents éléments attendus dans la constitution de cet état de l’art. Tout d’abord, il va être indispensable de définir le contexte clinique, en précisant les domaines médicaux et pathologies associées ; mais aussi les évolutions et les conséquences possibles, population cible et facteurs d’incidences. Ensuite, un résumé de la stratégie clinique envisagée basée sur des recherches bibliographiques avec les connaissances actuelles (questions de recherches, résultats d’études, publications, …). Pour finir, tous les documents consultés doivent être mentionnés, les articles retenus comme ceux exclus, avec dans chaque cas une justification. Les données récoltées vont être évaluées par pondération selon leur pertinence et leur validité scientifique [22].
Au vu des données collectées, le fabricant va pouvoir s’orienter soit vers une procédure d’équivalence, soit vers une investigation clinique si le dispositif est innovant ou à haut niveau de risque.
2.2) Par le biais de l’équivalence avec un dispositif existant
Dans certains cas, un dispositif de technologie similaire existe déjà sur le marché, dans ce cas, le fabricant n’a pas la nécessité de réaliser de nouvelles études cliniques. Si l’équivalence est démontrée et que les données de ce dernier sont suffisantes, le fabricant peut donc gagner du temps dans son processus de certification. Cependant, le nouveau règlement européen renforce les exigences pour démontrer cette équivalence. La démonstration de l’équivalence doit se faire sur trois plans : techniques, clinique et biologique (figure 7) (Annexe XIV.3) [1].
Figure 7 : Schématisation simplifiée des trois points de démonstration d'équivalence d'un dispositif médical (Source : d’après auteurs)
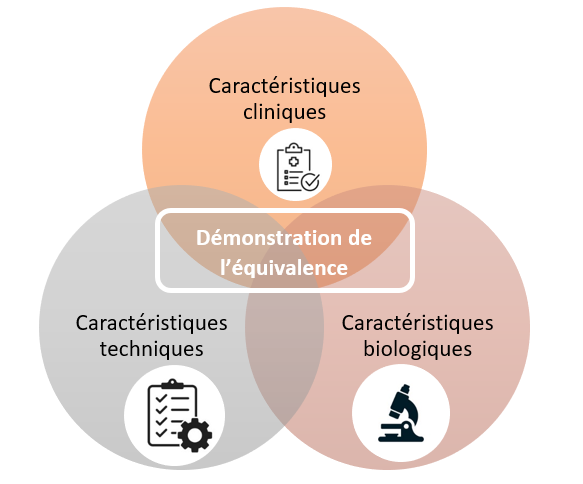
Au niveau des caractéristiques techniques, certaines conditions sont donc à prendre en compte :
- L'appareil doit être de conception similaire.
- Il doit être utilisé dans des conditions d'utilisation similaires et posséder des spécifications et des propriétés similaires, notamment des propriétés physico-chimiques telles que l'intensité de l'énergie, la résistance à la traction, la viscosité, les caractéristiques de surface, la longueur d'onde et les algorithmes logiciels.
- Il doit posséder des principes de fonctionnement et des exigences de performance critiques similaires.
Les caractéristiques cliniques doivent aussi être équivalente et ce à différents niveaux :
- L’état clinique (c’est-à-dire des sévérités et stades similaires de la maladie, la même indication clinique),
- Le même usage prévu,
- La même zone anatomique cible,
- Une population similaire (cela peut concerner l'âge, le sexe, l'anatomie, la physiologie, éventuellement d'autres aspects),
- Les performances (il s’agit ici des performances cliniques pertinentes telles que l’effet clinique attendu, les destinations prévues spécifiques, la durée d’utilisation, etc.).
Cette équivalence se retrouve finalement aussi au niveau des caractéristiques biologiques :
- Mêmes matériaux ou substances en contact avec les mêmes tissus humains ou fluides corporels pour un même type ou une même durée de contact et des caractéristiques de libération des substances similaires, y compris les produits de dégradation et substances relargables.
- Des exceptions peuvent être prévues pour les dispositifs en contact avec la peau intacte et les composants mineurs de dispositif ; dans ces cas, l’évaluation de la sécurité biologique (par exemple en conformité avec les normes EN ISO 10993) et l’analyse des risques doivent prendre en considération le rôle et la nature des matériaux similaires, aussi bien que d’autres aspects nécessaires à une démonstration complète de l’équivalence.
- Une justification expliquant la situation doit être fournie pour toute différence.
Pour confirmer l’équivalence sur ces trois points, différentes évaluations peuvent donc être réalisées. Concernant, l'évaluation des caractéristiques biologiques, il peut être nécessaire de montrer à partir d'études histopathologiques que la même réponse de l'hôte est obtenue in vivo dans l'application et la durée de contact envisagée et la durée de contact envisagée. Aussi, lorsqu'une caractérisation chimique détaillée des matériaux en contact avec le corps est nécessaire, l'ISO 10993-18 Annexe C peut être utilisée pour montrer l'équivalence toxicologique, mais ceci n'est qu'une partie de l'évaluation des critères biologiques. Les procédures d'approvisionnement et de fabrication peuvent affecter négativement les profils d'impuretés ; les méthodes analytiques choisies pour caractériser les dispositifs médicaux doivent prendre en compte de manière appropriée les connaissances concernant les profils d'impuretés attendus (les tests peuvent devoir être répétés lorsque les méthodes de production ou l'approvisionnement sont modifiés). Pour les tests sur des animaux, les différences entre espèces peuvent limiter la valeur prédictive du test ; le choix du test et sa valeur prédictive doivent être justifiés.
Pour l’évaluation des caractéristiques cliniques, les seules données cliniques considérées comme pertinentes sont les données obtenues quand le dispositif équivalent est un dispositif médical marqué CE, utilisé conformément à sa destination tel que documenté dans la notice d'utilisation.
Au niveau technique, les caractéristiques vont être évalués dès le processus de fabrication qui doit être reconnu comme équivalent. Si un traitement spécial a été appliqué (par exemple une modification de surface, un procédé qui modifie les caractéristiques du matériau) ; il pourrait entraîner des différences dans le produit final au niveau technique et biologique notamment. Cela doit être, ainsi, pris en compte pour la démonstration de l'équivalence et documenté dans le rapport clinique d’évaluation.
Les différences entre le dispositif en cours d'évaluation et le dispositif présumé équivalent doivent donc être identifiées, pleinement divulguées et évaluées ; des explications doivent être fournies pour lesquelles les différences ne devraient pas affecter de manière significative les performances cliniques et la sécurité clinique du dispositif en cours d'évaluation.
Certains paramètres vont être pris en compte, tels que le degré d’innovation et le niveau de risque du dispositif. En effet, cela va influer sur les exigences de démonstration de l’équivalence. Par exemple, Lorsque l’équivalence concerne des dispositifs de classe III et implantables déjà commercialisés et non fabriqués par le fabricant lui-même et que cette équivalence permet au fabricant de ne pas conduire d’investigations cliniques, le fabricant doit fournir le contrat conclu entre les deux fabricants, qui accorde explicitement au fabricant du dispositif faisant l’objet de l’évaluation clinique un accès total et permanent à la documentation technique, ainsi que la preuve manifeste que l’évaluation clinique d’origine a été effectuée conformément aux exigences du règlement (UE) 2017/745.
Lorsque l’équivalence concerne des dispositifs autres que des dispositifs de classe III et implantables déjà commercialisés et non fabriqués par le fabricant lui-même et que cette équivalence permet au fabricant de ne pas conduire d’investigations cliniques, le règlement (UE) 2017/745 n’exige pas de contrat entre les fabricants pour réglementer l’accès à la documentation technique. Cependant, le fabricant doit avoir un niveau d’accès suffisant aux données relatives aux dispositifs avec lesquels il revendique l’équivalence et doit documenter cet accès [9], [21], [23].
Parmi les documents attendus pour cette démonstration, le fabricant doit notamment fournir des tableaux comparant les mesures des spécifications et propriétés cliniquement pertinentes, les rapports d'études pré cliniques présentant des justifications d’équivalence cliniques et non-cliniques.
Ainsi, sous le nouveau règlement, il est désormais très compliqué pour les fabricants de démontrer cette équivalence. En effet, il doit confirmer que son produit est équivalent en tout point à un produit déjà sur le marché. Deux problèmes se posent alors au fabricant ; d’une part, il doit avoir accès à toutes les données du produit vendu par le concurrent et d’autre part, vendre un produit exactement équivalent à un autre déjà vendu n’a pas de grand intérêt commercial. La procédure d’équivalence tend donc à disparaitre dans les années à venir et encourage les fabricants à se tourner vers la réalisation d’études cliniques (toujours dans un but de renforcer la sécurité des dispositifs médicaux pour les patients et utilisateurs).
2.3) Par le biais de l’investigation clinique (incluant les types)
2.3.1. Description
L’objectif majeur d’une investigation clinique est d’établir la conformité du dispositif aux exigences du règlement européen afin d'en assurer les performances et la sécurité pour le patient et ainsi, pouvoir le commercialiser au sein de l’Union Européenne.
La conduite d’une investigation n’est pas la même selon si elle porte sur un dispositif marqué ou non marqué CE, ou sur un dispositif déjà commercialisé pour une autre destination médicale.
L’Agence Nationale de la Sécurité du Médicament et des produits de santé (ANSM) propose différents outils, sous forme de tableaux, pour aider les fabricants à savoir la procédure à suivre dans chaque cas. Toutefois, les tableaux contiennent beaucoup d’informations, et ne sont pas dans des formats faciles à lire ou facilement imprimables [24].
Le champ d’application des produits pouvant être soumis à ce type d’évaluation clinique est aussi précisé par cette réglementation. Ainsi, le règlement s’applique aux investigations cliniques des produits suivants :
- Les dispositifs médicaux et leurs accessoires,
- Les produits listés dans l’annexe XVI du règlement et n’ayant pas de destination médicale (lentilles de contact, substances destinées à un comblement du visage, …) ;
- Les dispositifs produits à partir de tissus ou cellules d’origine animale, ou de leur dérivé, non viables ou rendus non-viables ;
- Les dispositifs fabriqués à l’aide de dérivés de tissus ou de cellules d’origine humaine non viables ou rendus non viables ;
- Les dispositifs incorporant comme partie intégrante un dispositif médical de diagnostic in vitro (DMDIV) ;
- Les dispositifs incorporant comme partie intégrante une substance qui, utilisée séparément, serait considérée comme un médicament, notamment un médicament dérivé du sang ou du plasma humains, dont l’action est accessoire à celle du dispositif ;
- Les dispositifs destinés à l’administration d’un médicament ;
- Les dispositifs qui contiennent comme partie intégrante des tissus ou cellules d’origine humaine non viables ou leurs dérivés.
Il existe toutefois plusieurs situations pour lesquelles les investigations cliniques ne sont pas obligatoires. Passer outre cette investigation permet souvent au fabricant de gagner du temps, car la procédure d’investigation est souvent celle qui demande le plus de temps et le plus de documentation.
Dans le cas où les investigations seraient menées dans différents États membres de l’Union Européenne, la mise en place de la base de données EUDAMED permettra la mise en place d’une procédure d’évaluation unique. Cette base de données ne sera complètement effective qu’à partir de 2022.
En attendant, les demandes d’investigations cliniques devront être effectuées par mail à l’ANSM [1].
2.3.2) Étapes des investigations cliniques
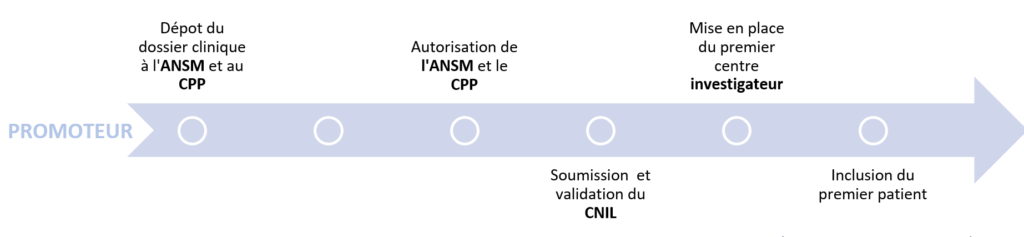
De manière générale, pour démarrer un nouvel essai clinique, le promoteur de l’étude doit soumettre le projet de recherche à l’avis d’un Comité de Protection des Personnes (CPP) et à l’autorisation de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) ce qui est démontré dans la figure 8. Pour tout projet de ce type, la notion de bénéfice/risque détermine la faisabilité du projet et doit être considéré avec grand attention.
Figure 8 : Processus et délais d'autorisation de lancement d'une investigation clinique (Source : d’après auteurs)
La norme ISO 14155 :2020 définit les bonnes pratiques à suivre pour mener à bien un protocole d’investigation clinique. Elle précise les différentes phases d’un essai : pré-clinique, clinique et de commercialisation. Elle précise les responsabilités de chacun des acteurs impliqués dans ce processus et la documentation relative à ses essais.
- Phase pré-clinique
Avant de lancer une investigation, le fabricant, souvent accompagné de médecins ou cliniciens, rédige un plan d’investigation clinique précis. Il définit le protocole d’étude, les sites d’investigations choisis, les investigateurs recrutés, les critères d’inclusions des patients (stade de pathologie, sexe, âge, …), le nombre de patients et les résultats espérés. Il doit aussi prévoir toute la documentation informative à destination des patients. En effet, il est indispensable d’informer les patients inclus dans l’étude et de s’adapter aux différentes populations cibles (gériatriques, pédiatriques, handicapées, …).
Chaque patient est libre d’intégrer un protocole de recherche clinique et de se retirer à tout moment sans aucune conséquence. Une liste de documents et d’informations précises à fournir au patient est explicitée dans la norme ISO 14155. Pour formaliser ce principe, le patient doit signer un consentement éclairé avant de commencer l’étude. Pour les populations pédiatriques, le représentant légal devra lui aussi signer un formulaire de consentement éclairé. Par éclairé, il est sous-entendu que les patients ont parfaitement compris les actes qui vont être réalisés, les procédures et les risques possibles.
Une fois le dossier mis en place, le fabricant doit le soumettre aux autorités compétentes afin de pouvoir l’initier (figure 9) :
Figure 9 : Diagramme simplifié de l'outil DGS proposé par l'ANSM (Source : Auteurs d'après [15])
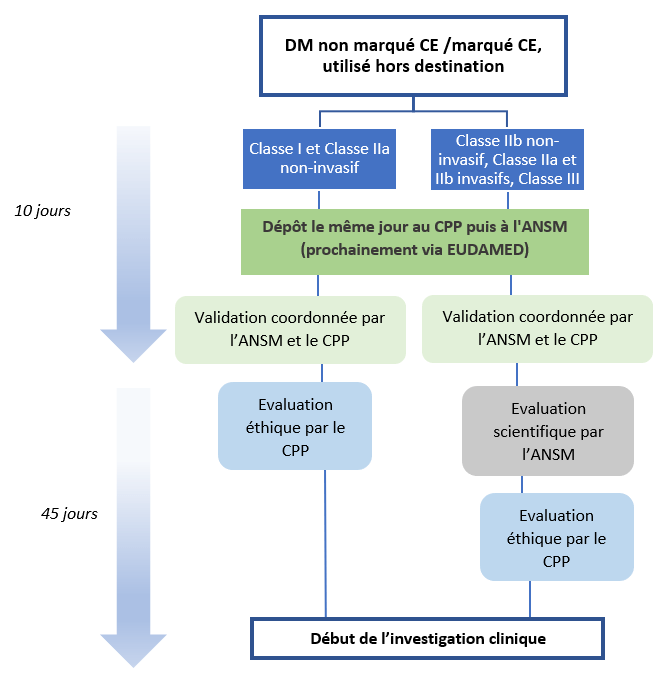
*DM : Dispositifs médicaux
*CPP : Comité de Protection des Personnes
* ANSM : Agence Nationale de sécurité du Médicament et des produits de santé
Cette procédure est assez longue et s’adapte au niveau de risques du dispositif à évaluer.
2. Phase clinique : Planification de l’investigation clinique
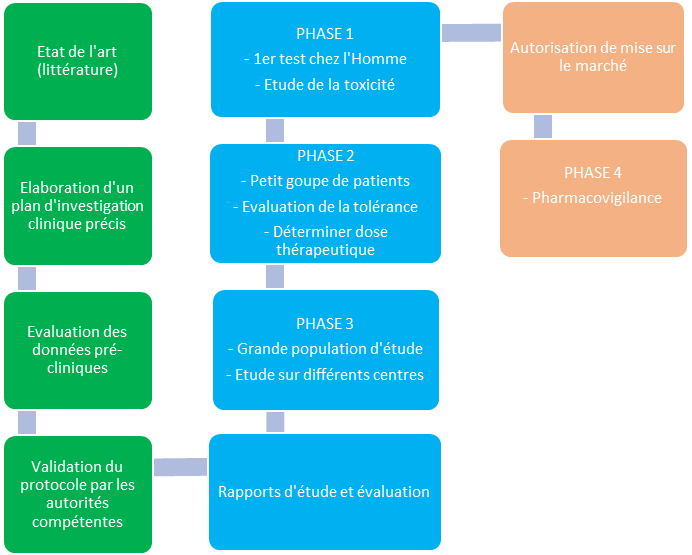
L’investigation clinique est sectionnée en 3 phases distinctes (figure 10) :
- La phase 1 correspond aux premiers tests réalisés sur l’Homme pour évaluer les risques du produit. Elle est menée sur un petit groupe de patients sélectionnés selon des critères d’inclusions définis. Tous les paramètres physiologiques d’intérêts seront évalués.
- La 2ème phase a pour objectif de déterminer la dose minimale efficace basé sur le rapport tolérance/efficacité. Le dispositif sera alors testé sur une population de malades plus grande.
- La phase 3 évalue l’intérêt du dispositif sur une population plus large. Les patients inclus dans ces études sont sélectionnés selon des critères précis et peuvent être répartis en sous-groupe de façon aléatoire (population témoin, différents dosages, …).
3. Commercialisation
Si les résultats sont concluants et validés par les autorités compétentes, le produit pourra obtenir une autorisation de mise sur le marché.
La dernière phase d’étude a lieu après la commercialisation du produit et correspond au suivi clinique du dispositif afin d’identifier d’autres risques résiduels ou évènements indésirables.
Figure 10 : Schéma des différentes phases d’un essai clinique (Source : Auteurs d'après [25])
Chapitre 3 : Documentation et données utiles pour l’évaluation clinique
1) Processus nécessaire à la collecte des données cliniques
Les données cliniques, issues d’une évaluation clinique d’un dispositif médical, représentent des résultats permettant d’apporter des informations relatives à la sécurité et à la performance de l’utilisation dudit dispositif. La collecte des données cliniques représente l’ensemble des moyens mis en œuvre pour parvenir à récupérer les données cliniques pouvant être exploitables pour mener à bien l’évaluation clinique [26].
Aussi, avant tout collecte de donnée, il faut s’assurer que les éléments suivants sont réunis :
- Être parvenu à avoir l’autorisation et l’appui des autorités compétentes (ANSM, CPP, CNIL...) et des parties prenantes (professionnels de santé, participants...), notamment en leur transmettant une vision et une mission leur procurant de l’intérêt dans votre démarche
- Être en mesure de garantir la formation des collecteurs de données et leur apporter le soutien nécessaire (ressources...)
- Établir des processus permettant la vérification de la qualité des données [27]
Figure 11 : Schéma général du cycle de vie des données (Source : d'après [23])
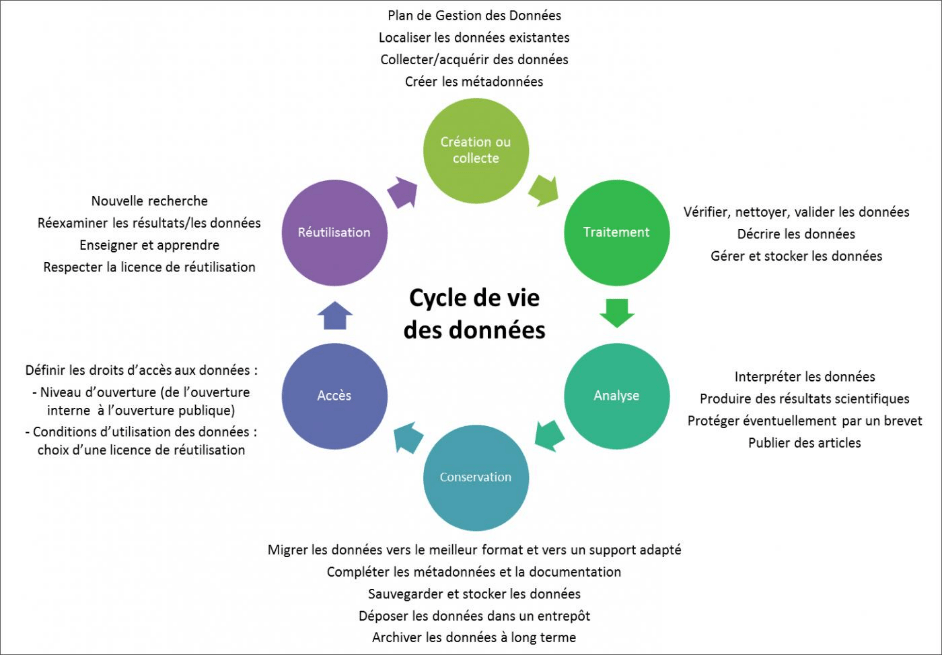
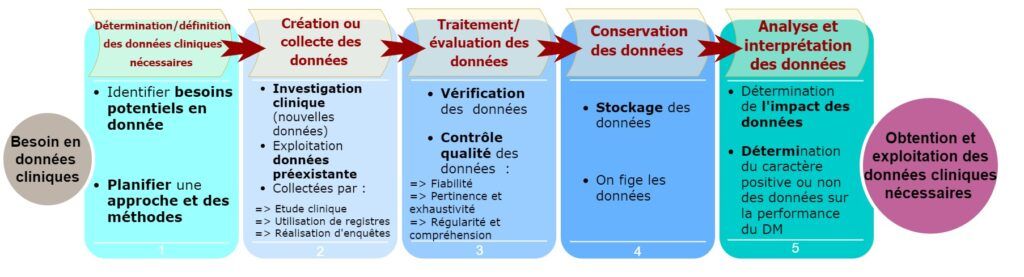
La définition et la collecte des données englobent un ensemble d’activité à effectuer pour s’assurer du bon déroulement de cette démarche (figure 12) :
- Détermination/définition des données cliniques nécessaires
- Identifier les besoins potentiels en donnée
- Planifier une approche et des méthodes
La définition des données consiste essentiellement à déterminer le type de données cliniques qui sera nécessaire de générer, la méthodologie adéquate pour leur gestion (acquisition traitement, analyse ect), les résultats attendus (qu’il faudra apporter), les délais, mais aussi le coût.
Le(s) moyen(s) de collecte de données clinique est/sont fonction du niveau de preuve clinique nécessaire, lui-même fonction principalement de la classe de risque du dispositif et de sa finalité, il en est de même pour la proportion/la quantité de données clinique adéquate [28].
Ces preuves cliniques sont définies par le nouveau règlement « comme étant le volume et la qualité de données suffisantes pour évaluer en connaissance de cause, si le dispositif est sûr et offre le ou les bénéfices cliniques attendus lorsqu'il est utilisé conformément à la destination prévue par le fabricant. » [1].
Il est difficile de définir quantitativement le niveau de preuve attendu pour les différents types de dispositifs.
La quantité de données nécessaires est prévue dans un protocole documentaire émis en amont. Toutes les recherches et articles vont être listés dans le rapport d’évaluation clinique, en précisant ce qui ont été retenus ou non et les raisons de ce choix [11].
Cette bibliographie sera évaluée par un expert de l’organisme notifié, qui va évaluer si le niveau de données est suffisant en termes de quantité et de qualité scientifique pour confirmer le bénéfice du dispositif et son niveau de sécurité. C’est-à-dire le service attendu, définit par le fabricant dans le plan d’évaluation clinique. C’est grâce à l’analyse de gestion des risques et l’identification d’indicateur d’analyse de ces risques, que l’expert pour comparer le rapport bénéfice/risque.
Dans le cas, d’un essai clinique une centaine de patients inclus est souvent considérés comme le minimum acceptable pour justifier l’intérêt de l’étude. En fonction du niveau de risque du dispositif, ce nombre de patients est variable est définit dès le plan d’évaluation clinique. Il peut évoluer au vu des résultats observés tout au long de l’étude.
- Création ou collecte des données
- Réalisation d’essai clinique permettant d’obtenir de nouvelles données cliniques
- Exploitation des données cliniques d’autres solutions similaires au dispositif étudié
La collecte de données peut notamment se faire par des études cliniques, par l’utilisation de registres ou encore par la réalisation d’enquêtes, dans le cas des dispositifs innovants ou des dispositifs à haut risque pour le patient.
Ces données peuvent provenir du fabricant lui-même ou être relevé de sources extérieures, elles peuvent être de nature qualitative ou quantitative.
- Traitement/évaluation des données
- Vérification des sources de données et de la fiabilité des données
- Les données doivent être, revues, contrôlées et validées [29]
La qualité des données collectées est fondamentale lorsque l’on effectue une évaluation clinique, de ce fait les données récoltées sont soumises régulièrement à de nombreux contrôles qualité. Cette qualité des données dépend notamment de 3 critères/facteurs :
- Leur fiabilité : les données cliniques, pour être exploitables, doivent présenter un haut degré de fiabilité. En effet, ce sont ces données qui vont permettre de déduire si le dispositif sur et pour lequel on collecte les données, remplit les critères nécessaires à sa commercialisation (notamment par rapport à son rapport bénéfice/risque) et peut donc être utilisé sur des patients, sans risques inconsidérés. Pour s’assurer de la fiabilité des données, il faut notamment s’assurer de la fiabilité des sources dans lesquelles ont été extraites ces données.
- La pertinence et exhaustivité : il faut s’assurer que les données cliniques collectées répondent ou apportent des éléments aux différentes interrogations ayant mené à l’évaluation clinique (le rapport bénéfice/risque est-il acceptable ?...). Aussi, il faut s’assurer de collecter un nombre suffisant de données pour apporter une significativité à l’exploitation des données. La quantité de données collectées doit s’avérer suffisante pour permettre d’établir une étude statistique pertinente. Ces données sont souvent accompagnées d’un niveau de confiance calculé. Ce niveau de confiance est déterminé selon différents critères (durée d’utilisation, produits utilisés, …). La Food and Drug Administration (FDA) propose par exemple des formulaires d’accompagnement pour les fabricants afin de les guider dans le processus d’évaluation [32]. Il reste toutefois difficile de donner clairement une quantité de donnée précise car le niveau de preuve est fonction du type de dispositif et de sa nature, ainsi que de la prévalence sur le marché. Par exemple, pour certaines sociétés qui n’ont aucune concurrence et donc aucun antécédent sur le marché seulement 5 articles peuvent suffire. Pour d’autres secteurs, tels que l’orthopédie par exemple, qui concernent des dizaines des milliers de patients tous les jours, une centaine d’articles doivent être pris en compte.
- La régularité et la compréhension : il faut être en mesure de collecter les données à une fréquence convenant à l’usage prévu. Il faut vérifier également que les données collectées soient compréhensives par les personnes qui vont les exploiter, analyser ect, il peut donc être nécessaire de les retranscrire dans un langage qui soit compréhensible, sans ambiguïté, à leur destinataire [27].
Une fois la vérification effectuée, les données sont validées si elles répondent correctement au besoin et sont suffisamment fiables.
- Conservation des données
- Stockage des données
On fige les données pour permettre leur réutilisation ultérieure [31]
- Analyse et interprétation des données
- Détermination de l’impact des données
L’analyse statistique et l’interprétation des données vont permettre de déterminer si le niveau de sécurité et la performance du dispositif, et ainsi permettre de déduire la pertinence de la commercialisation dudit dispositif.
Figure 12 : Schéma récapitulatif du processus de définition et collecte des données cliniques (Source : d’après auteurs)
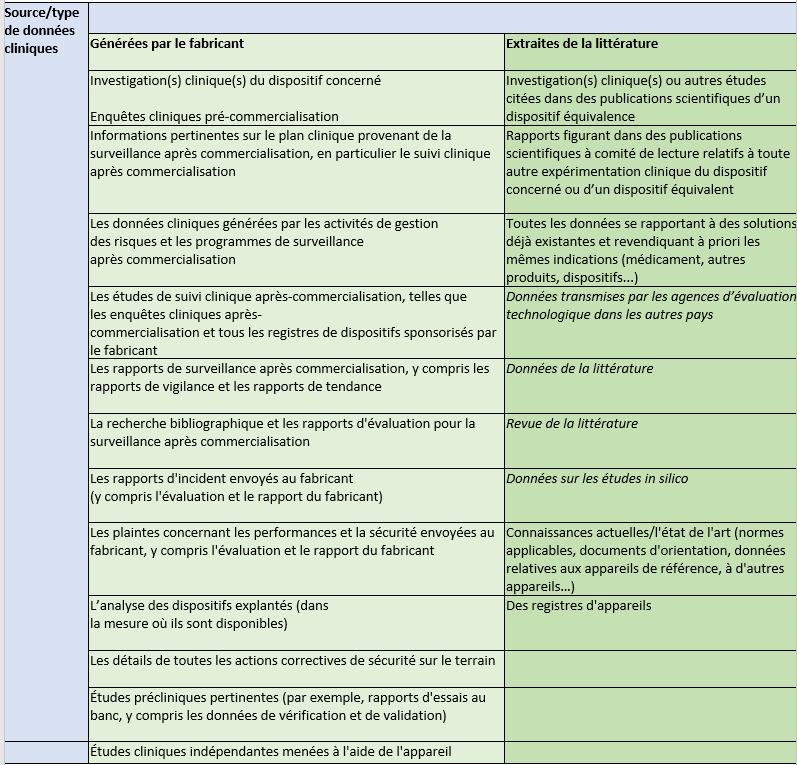
2) Exemples de sources et données cliniques pouvant être utilisées pour l’évaluation clinique
Lors de la collecte des données cliniques, le fabricant a donc différentes stratégies possibles pour obtenir ces données. Le tableau 3 ci-dessous présente un récapitulatif non exhaustive des sources et/ou type de donnée clinique pouvant être utiliser pour mener à bien l’évaluation clinique [19], [26], [32], [33] :
Tableau 3 : Tableau récapitulatif des différentes sources et types de données cliniques collectées par le fabricant (Source : d’après auteurs)
3) Documentation spécifique à avoir pour la réalisation d'une investigation clinique
Comme exposé dans les parties précédentes, l’investigation clinique est un processus très chronophage. Plusieurs documents sont essentiels à avoir avant le début de l’investigation clinique. Le tableau 4 ci-dessous, extrait de la norme NF EN ISO 14155 (2020) présente une partie des documents à fournir, accompagné d’annotations et de précisions sur leur utilisation. L’annexe XV, Chapitre III du règlement européen [1] présente de façon exhaustive le reste des documents nécessaires avant, pendant et après l’investigation clinique [16].
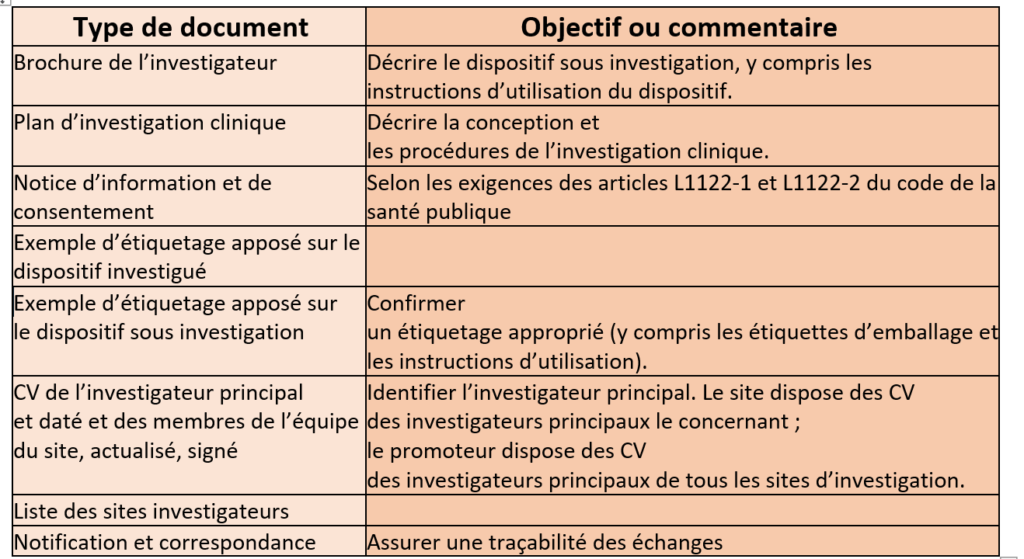
Tableau 4 : Documents à réunir par le fabricant dans le cadre d'une investigation clinique (Source : d’après auteurs)
4) Documentation nécessaire pour le suivi clinique
Le suivi clinique correspond à la collecte de données cliniques après la commercialisation, c’est-à-dire pendant l’utilisation réelle du dispositif. Il s’effectue selon deux voies de surveillance : sur site par le fabricant et par l’autorité compétente via les retours terrains et les inspections.
4.1) Surveillance après commercialisation
4.1.1. Par les fabricants
Ce système a donc pour objectif de collecter, enregistrer et analyser de façon active les données relatives à la qualité, la performance et la sécurité du dispositif tout au long de son cycle de vie. Les données sont récoltées via un système de vigilance, mis en place par le fabricant. Par ces plateformes, les usagers et patients peuvent notifier les incidents et qui sont transmis et documentés par les fabricants. Ainsi, quand cela est nécessaire des actions de prévention, correctives peuvent être appliquées et assurer le suivi du produit sur le marché. Cette surveillance est donc basée sur une évaluation continue du rapport bénéfice/risque, de la gestion de risques associés, des informations techniques, l’évaluation clinique en fonction des remontées terrains.
Cette surveillance n’a pas seulement pour objectif de sanctionner les fabricants, elle va aussi permettre de notifier les améliorations d’utilisation, de performances et de sécurité par la mise en place de mesures correctives et préventives pour garantir la sécurité et la performance du dispositif tout au long de sa vie. Toutes ces données sont inscrites dans un rapport périodique de sécurité (PSUR) réalisé par le fabricant (annexe III). Selon la classe du DM les spécifications sont différentes [1].
Ces rapports pourront être directement enregistrés dans EUDAMED, une fois que la plateforme sera pleinement active [35].
4.1.2. Par l’ANSM
En plus de cette surveillance établie par le fabricant, une matériovigilance est mise en place par l’ANSM. Grâce aux déclarations/signalements réalisées par l’ANSM, experts du comité technique, utilisateurs, établissements de soins, patients ou le fabricant lui-même, les incidents liés à un dispositif vont pouvoir être identifiés. Suivant le niveau de risque suspecté, une étude pourra être mise en place et conduire à la prise de décision (suspension, recommandations, interdiction…) à appliquer pour éviter que d’autres incidents se reproduisent. Pour cela, le fabricant doit déclarer une personne de sa société responsable de la matériovigilance et qui devient, alors, le correspondant avec l’ANSM [35].
4.2. Documentation associée
Pour répondre à cette exigence de suivi clinique du règlement, certains documents sont nécessaires et doivent être fournis par le fabricant en cas d’audit :
- Le plan de surveillance après commercialisation
Le plan de surveillance après commercialisation comprend une surveillance de toutes les données et informations en lien avec l’utilisation du dispositif mis en service, tels que les réclamations, le nombre de matériovigilance, le nombre de produit vendus, …
- Le rapport de surveillance après commercialisation
- Une planification du SCAC (Suivi Clinique Après Commercialisation)
Il existe des templates, tel que le MDCG 2020-7 « Post-market clinical follow-up (PMCF) Plan template », afin de guider les fabricants dans la rédaction de ce document, en accord avec les exigences du règlement européen [12], [36].
Le plan ou protocole du SCAC est sans cesse réactualisé en fonction de l’évolution des pratiques, qui questionne sa performance et son intérêt médical. Cette planification a été rendu obligatoire par l’entrée en vigueur du règlement.
- Un rapport d’évaluation du SCAC, inclus dans le rapport d’évaluation clinique, lui-même inclus dans la documentation technique
Ce rapport contient notamment les synthèses des événements indésirables et événements indésirables graves, une synthèse des résultats et des conclusions de l’analyse des données collectées. Le rapport doit aussi exposer la justification des mesures correctives et préventives établies à la suite des conclusions. Ces preuves permettant de mettre en avant le rapport bénéfice/risque, le volume de vente, la fréquence d’utilisation du dispositif. Pour les classes les plus à risque, il doit être mis à jour une fois par an et il est inclus dans la documentation technique du produit. Ce rapport est accessible aux organismes notifiés (article 8) et il permet aux autorités, aux utilisateurs et aux patients d'avoir les informations concernant l’évolution du dispositif à la suite de sa commercialisation. Ce rapport doit être rédigé par, ou en collaboration avec un clinicien.
De plus, le Suivi Après Commercialisation (SAC) (Annexe III et Article 85) compris avec le Suivi Clinique Après Commercialisation (SCAC) doit être ajouté après la commercialisation du dispositif ainsi que le Rapport Périodique Actualisé de Sécurité (PSUR) (Article 86). Pour finir, la durée de vie du dispositif doit être définie par l’ensemble des données précliniques et cliniques et précisé dans le plan de suivi post commercialisation.
- Le suivi des actions préventives et correctives décidées à la suite de SCAC
Le rapport du PSUR comporte une synthèse de l’évaluation et l’analyse des données collectées dont différents éléments relatifs aux dispositifs, comme :
- Des données administratives,
- Date de commercialisation du dispositif sur le marché et le nombre d’unité vendues,
- Taux de plaintes,
- Nombres d’évènements identifiables,
- Nombre de rapports notifiés à l’autorité compétente,
- Risques imprévus,
- Nombre de rappels de produits.
Ce type de rapport est obligatoire pour des dispositifs de classe IIb et III. La périodicité des rapports est définie dans le protocole de suivi de commercialisation et dépend du niveau de risque et de la proportion de lots vendus.
Conclusion
Depuis toujours, la santé est au service du patient. Les progrès technologies et l’avancée des connaissances a permis d’améliorer la prise en charge de nombreuses pathologies grâce au développement régulier de nouveaux traitements et dispositifs médicaux.
La sécurité des patients est la priorité. Pour s’en assurer différentes procédures sont mise en place. L’évaluation clinique est un processus continu tout au long de la vie du dispositif qui permet de rassemblées toutes les données relatives à un dispositif issus de la littérature, des tests pré-cliniques et des investigations cliniques. Depuis 2016, différents documents tendent à préciser ces exigences tels que le MEDDEV et sa nouvelle version le MDCG. Le nouveau Règlement européen 2017/745 a intégré ces exigences et les officialise, ce qui contraint les fabricants à fournir des preuves cliniques suffisantes selon la classe de leur produit. L’évaluation est une procédure qui peut apparaitre complexe pour le fabricant et qui s’avère donc être un frein à l’innovation si le fabricant ne la maitrise pas bien. Les cabinets de conseils et autorités compétentes proposent différents supports pour guider les fabricants dans cette démarche.
Cependant, chaque dispositif requiert un plan d’évaluation clinique adapté, qui découlera sur un rapport final qui pourra être validé par l’organisme notifié. C’est pourquoi, il semblait nécessaire de proposer aux industriels un outil interactif, s’adaptant aux besoins du fabricant. Cet outil se devait être accessible facilement, pratique et fiable. Surfant sur les tendances actuelles, ce guide est proposé au format numérique. Toutes les étapes de l’évaluation clinique y sont précisées, telle que l’état de l’art, la démarche d’équivalence ou l’investigation clinique. La documentation est aussi précisée. Le marché du dispositif est un marché en constante progression et très compétitif. De fait, l’optimisation de cette procédure est un moyen de gagner du temps et donc, d'améliorer ses gains.
Références Bibliographiques
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux », Journal officiel de l’Union européenne, JO L 117 du 5.5.2017, avr. 2017. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj
[2] B. Halioua, « Du procès au code de Nuremberg : principes de l’éthique biomédicale | article | Espace éthique/Ile-de-France », Espace éthique/Région Ile-de-France, 19 mai 2014. https://www.espace-ethique.org/ressources/article/du proces-au-code-de-nurembergprincipes-de-lethique-biomedicale (consulté le nov. 23, 2021).
[3] Association Médicale Mondiale, « Déclaration d’Helsinki de L’AMM – Principes éthiques applicables à la recherche médicale impliquant des êtres humains », WMA - The World Medical Association-, 15 février 2017. https://www.wma.net/fr/policies-post/declaration-dhelsinki-delamm-principes ethiques-applicables-a-la-recherche medicale-impliquant-des-etres-humains/ (consulté le nov. 23, 2021).
[4] G. Promé, « Évaluation clinique des Dispositifs Médicaux », Qualitiso, 5 janvier 2019. https://www.qualitiso.com/evaluation-clinique-dispositif-medical/ (consulté le nov. 21, 2021)
[5] ANSM, « Dossier thématique - Synthèse des données d’incidents déclarés », ANSM Santé, 22 décembre 2020. https://ansm.sante.fr/dossiers-thematiques/implants-mammaires-pip-pre-remplis-de-gel-de-silicone/synthese-des-donnees-dincidents-declares-chez-les-femmes-porteuses-dimplants-pip (consulté le déc. 13, 2021).
[6] ANSM, « Information de sécurité - Suspension du marquage CE des gammes », ANSM Santé, 11 mai 2021. https://ansm.sante.fr/informations-de-securite/suspension-du-marquage-ce-des-gammes-de-dispositifs-medicaux-magec-et-precice (consulté le déc. 13, 2021).
[7] ANSM, « Point de situation sur la surveillance des vaccins contre la Covid-19 - Période du 29/10/2021 au 11/11/2021 », ANSM Santé, 19 novembre 2021. https://ansm.sante.fr/actualites/point-de-situation-sur-la-surveillance-des-vaccins-contre-la-covid-19-periode-du-29-10-2021-au-11-11-2021 (consulté le déc. 13, 2021).
[8] ANSM, « Entrée en application du nouveau règlement européen relatif aux dispositifs médicaux », ANSM Santé, 26 mai 2021. https://ansm.sante.fr/actualites/entree-en-application-du-nouveau-reglement-europeen-relatif-aux-dispositifs-medicaux (consulté le oct. 11, 2021).
[9] N. Lionel Ekedi, « Évaluation clinique des dispositifs médicaux selon la voie de la littérature », Thèse de doctorat, Faculté de Pharmacie - Université de Grenoble Alpes, 2018. Consulté le : 1 octobre 2021. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-01882924/document
[10] EcoMundo, « Dispositifs médicaux : La nouvelle réglementation européenne, applicable en 2021 », Ecomundo.eu, 29 août 2019. https://www.ecomundo.eu/fr/blog/dispositifs-medicaux-reglement-2020 (consulté le déc. 13, 2021).
[11] lne GMED, « L’évaluation clinique à l’ère du règlement (UE) 2017/745 », mars 2021. Consulté le : 23 octobre 2021. [En ligne]. Disponible sur : http://lne-gmed.com/wp-content/uploads/2021/03/Newsletter_GMED-Evaluation_clinique-20210330.pdf
[12] « Guidance - MDCG endorsed documents and other guidance », Santé publique - European Commission. https://ec.europa.eu/health/md_sector/new_regulations/guidance_en (consulté le nov. 19, 2021).
[13] C. Mangeol, « Enjeux et exigences de la nouvelle réglementation européenne des dispositifs médicaux », Thèse de doctorat, Faculté de pharmacie - Aix Marseille Université, 2019. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-02140163/document
[14] ANSM, « Essais cliniques : procédure standard pour la constitution et le traitement des demandes pour les médicaments », ANSM Santé. https://ansm.sante.fr/page/essais-cliniques-procedure-standard-pour-la-constitution-et-le-traitement-des-demandes-pour-les-medicaments (consulté le nov. 21, 2021).
[15] ANSM, « Dispositifs médicaux - Demander une autorisation pour une investigation clinique », ANSM Santé, 29 septembre 2021. https://ansm.sante.fr/vos-demarches/industriel/demander-une-autorisation-pour-un-essai-clinique-pour-des-dispositifs-medicaux-categorie-1 (consulté le nov. 23, 2021).
[16] « norme ISO 14155 - Investigation clinique des dispositifs médicaux pour sujets humains — Bonne pratique clinique », Ed. ISO, https://www.iso.org/fr/home.html, juillet 2020. Consulté le : 21 novembre 2021. [En ligne]. Disponible sur : https://www.iso.org/cms/render/live/fr/sites/isoorg/contents/data/standard/07/16/71690.html
[17] Inserm, « La recherche clinique », Inserm, 7 juin 2021. https://www.inserm.fr/our-research/clinical/la-recherche-clinique/ (consulté le janv. 20, 2022).
[18] S. Gallien, « Autorisations règlementaires : CPP, comité d’éthique, ANSM, CNIL », avr. 2019. [En ligne]. Disponible sur : https://www.infectiologie.com/UserFiles/File/formation/desc/2019/seminaire-avril-2019/jeudi-04-04-2019/recherche-9-jeudi-04-dr-gallien.pdf
[19] A. Bignon, « Evaluation clinique des dispositifs médicaux : Évolutions du MEDDEV 2.7.1 Rev.4 », juill. 2016. [En ligne]. Disponible sur : https://www.dm-experts.fr/wp-content/uploads/2016/07/BioM-ADVICE-Note-danalyse-TechReg-07-2016-Evaluation-clinique-des-DM-MEDDEV-2.7.1-Rev.4-1.pdf
[20] S. Lee, « Avantages, risques et frais liés aux essais cliniques », Société canadienne du cancer. https://cancer.ca/fr/treatments/clinical-trials/clinical-trial-benefits-risks-and-costs (consulté le nov. 24, 2021).
[21] S. Makhlouf, « Les différents intervenants dans la recherche clinique et Relation entre ces différents acteurs », nov. 2016. [En ligne]. Disponible sur : https://www.recherchecliniquepariscentre.fr/wp-content/uploads/2016/11/DIU-COMMUN-Les-diff%C3%A9rents-acteurs-de-la-RC_04-11-16_S.Makhlouf.pdf
[22] lne GMED, « Évaluation clinique des dispositifs médicaux résumé des caractéristiques de sécurité et des performances cliniques règlement (UE) 2017/745 », juill. 2020. [En ligne]. Disponible sur : https://lne-gmed.com/wp-content/uploads/2020/10/Guide_GMED-Evaluation_clinique_des_DM-RDM-FR.pdf
[23] MDCG, « MDCG 2020-5 Clinical Evaluation - Equivalence A guide for manufacturers and notified bodies », avr. 2020. Consulté le : 1 octobre 2021. [En ligne]. Disponible sur : https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2020_5_guidance_clinical_evaluation_equivalence_en.pdf
[24] DGS et ANSM, « Classification et process d’évaluation des investigations cliniques (DM) selon le règlement 2017/745 (RDM) et adaptations nationales », mai 2021. [En ligne]. Disponible sur : https://ansm.sante.fr/uploads/2021/05/21/2021-05-21-tableau-classification-ic-20210517.pdf
[25] Inserm, « Les essais cliniques (Recherches interventionnelles portant sur un produit de santé) », Inserm, 9 août 2017. https://www.inserm.fr/nos-recherches/recherche-clinique/essais-cliniques-recherches-interventionnelles-portant-sur-produit-sante/ (consulté le déc. 13, 2021).
[26] G. Promé, « Données cliniques », Qualitiso, 18 janvier 2018. https://www.qualitiso.com/dictionnaire/donnees-cliniques/ (consulté le nov. 07, 2021)
[27] Fédération Mondiale de l’Hémophilie (FMH), « Collecte de données de qualité ». Consulté le : oct. 23, 2021. [En ligne]. Disponible sur : http://www1.wfh.org/publications/files/pdf-1595.pdf
[28] S. Sorrel, « Evaluation clinique des dispositifs médicaux : le cas des « legacy devices » », DeviceMed, 4 février 2020. https://www.devicemed.fr/dossiers/reglementation/evaluation-clinique-des-dispositifs-medicaux-le-cas-des-legacy-devices/21909 (consulté le nov. 07, 2021).
[29] S. Sohier et O. Capronnier, « Comprendre les enjeux de la recherche clinique : notions de méthodologie », DeviceMed, 21 juillet 2020. https://www.devicemed.fr/dossiers/sous-traitance-et-services/etudes-cliniques/comprendre-les-enjeux-de-la-recherche-clinique-notions-de-methodologie/23836 (consulté le nov. 07, 2021).
[30] « Clinical Trials Guidance Documents », FDA.gov. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trials-guidance-documents (consulté le déc. 16, 2021).
[31] Bibliothèque du Ceris, « Gérer ses données de recherche », Institut Pasteur, 26 février 2019. https://www.pasteur.fr/fr/ceris/bibliotheque/gerer-ses-donnees-recherche (consulté le nov. 07, 2021).
[32] Haute Autorité de santé, « Evaluation des DM connectés : Spécificités méthodologiques d’évaluation clinique d’un DM connecté (DMC) », Saint Denis La plaine, janv. 2019. Consulté le : 10 octobre 2021. [En ligne]. Disponible sur : https://www.has-sante.fr/upload/docs/application/pdf/2019-02/rapport_methodologiques_devaluation_clinique_dun_dmc.pdf
[33] MDCG, « MDCG 2020-6 Regulation (EU) 2017/745 : Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC », avr. 2020. Consulté le : 16 octobre 2021. [En ligne]. Disponible sur : https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2020_6_guidance_sufficient_clinical_evidence_en.pdf
[34] G. Promé, « SCAC : le Suivi Clinique Après Commercialisation », Qualitiso, 24 novembre 2020. https://www.qualitiso.com/scac-suivi-clinique-apres-commercialisation/ (consulté le nov. 07, 2021).
[35] A. Romain, « Parcours du dispositif médical en France », nov. 2017. [En ligne]. Disponible sur : https://www.has-sante.fr/upload/docs/application/pdf/2009-12/guide_pratique_dm.pdf
[36] MDCG, « MDCG 2020-7 Post-market clinical follow-up (PMCF) Plan Template A guide for manufacturers and notified bodies », avr. 2020. Consulté le : 1 octobre 2021. [En ligne]. Disponible sur : https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2020_7_guidance_pmcf_plan_template_en.pdf
[37] A. Groell, H. Zkeik, et K. Benaceur, « Surveillance après commercialisation des dispositifs médicaux », Université de Technologie de Compiegne, Mémoire de projet réf n°IDS005 https://doi.org/10.34746/nq28-4w94, janv. 2019. Consulté le : 16 décembre 2021. [En ligne]. Disponible sur : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids005/
Glossaire
Les définitions ci-dessous sont issues du règlement européen 2017/745 [1].
Dispositif médical : tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- Diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d’une maladie.
- Diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci.
- Investigation, remplacement ou modification d'une structure ou fonction anatomique ou d’un processus ou état physiologique ou pathologique.
- Communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus, et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens.
Bénéfice clinique : l'incidence positive d'un dispositif sur la santé d'une personne physique, se traduisant par des résultats cliniques significatifs, mesurables et pertinents pour le patient, y compris les résultats en matière de diagnostic, ou une incidence positive sur la prise en charge de la santé du patient ou sur la santé publique.
Évaluation clinique : un processus systématique et planifié visant à produire, collecter, analyser et évaluer en continu les données cliniques relatives à un dispositif afin de vérifier la sécurité et les performances, y compris les bénéfices cliniques, de celui-ci lorsqu'il est utilisé conformément à la destination prévue par le fabricant.
Investigation clinique : toute investigation systématique impliquant un ou plusieurs participants humains destinée à évaluer la sécurité ou les performances d'un dispositif.
Données cliniques : des informations relatives à la sécurité ou aux performances obtenues dans le cadre de l'utilisation d'un dispositif et qui proviennent de différentes sources (investigations, publications scientifiques, surveillance après commercialisation, suivi clinique après commercialisation).
Annexes
Annexe 1 : Développement du projet : Planification Dynamique Stratégique
L'annexe 1 illustre la planification dynamique stratégique (PDS) effectuée et utilisée en début de projet afin de clarifier, pour l'ensemble de l'équipe de projet, la direction à entreprendre pour réaliser le projet.
Annexe 2 : Déroulement du projet : Analyse des risques associés au projet
Le tableau ci-dessous représente l'analyse des risques de notre projet. Cette analyse a permis d'identifier les points critiques et ainsi d'anticiper des solutions pour neutraliser ou réduire les risques repérés.
Annexe 3 : Présentation de l’outil : Note de clarification
La note de clarification présentée dans l'annexe 3 permet d'apporter les informations principales concernant notre projet sur l'évaluation et le suivi clinique des dispositifs médicaux et plus particulièrement de l'outil réalisé dans le cadre de ce projet.
| Note de clarification Projet sur “L’évaluation et le suivi des dispositifs médicaux” |
Origine du projet
Dans le cadre de l’UV IDCB “Ingénierie de projet”, par groupe de 5 élèves un choix a été fait entre différents sujets en lien avec notre parcours et portant sur des thèmes actuels impactant le domaine du biomédical. Notre choix s’est ainsi porté sur l’évaluation et le suivi des dispositifs médicaux, thème phare du nouveau règlement relatif aux dispositifs médicaux EU 2017/745.
Sujet et contexte du projet
Le sujet de notre projet porte sur “L’évaluation et le suivi clinique des dispositifs médicaux”. Ce sujet s’inscrit dans une période de transition de la réglementation relative aux dispositifs médicaux. En effet, les directives précédentes qui portaient sur les dispositifs médicaux, ont été abrogées et unifiées pour donner place à 2 règlements relatifs aux dispositifs médicaux. Ainsi, la notion d’évaluation et de suivi clinique a évolué avec cette transition, laissant les fabricants de dispositifs médicaux dans une certaine incompréhension. Avec ce projet, l’objectif est d’apporter un éclaircissement aux fabricants sur les nouveautés concernant l’évaluation et le suivi clinique.
Produit du projet
Le produit final du projet est un guide interactif et personnalisé, permettant à chaque fabricant d’obtenir, à partir d’un dispositif donné, une évaluation du niveau de risques du produit et de la stratégie clinique recommandée sous le nouveau règlement.
Produit associant des livrables matériels et immatériel
Ce guide est associé à un Mémoire d’Intelligence Méthodologique (MIM) permettant de de comprendre plus en détail les enjeux relatifs à l’évaluation clinique sous le nouveau Règlement européen 2017/745.
Aussi, un poster résumant les éléments importants du MIM et présentant les grands axes de l’outil sera aussi réalisé.
Contraintes à respecter
L’une des plus grosses contraintes a été de respecter les délais impartis selon les différents jalons du projet. En effet jongler entre les différents projets professionnels mais aussi les projets universitaires de chacun a été le plus gros défi de ce projet.
Aussi, prendre connaissance de tous les documents et toutes les exigences relatives à l’évaluation clinique afin de fournir un travail de qualité accessible à l’ensemble de la communauté biomédicale n’a pas été une mince affaire.
Pour finir, le guide mis en place représentait peut-être un projet trop important pour le délai qu’il restait. En effet, pour s’assurer de rendre un outil exhaustif, complet et pratique pour tous, un peu plus de temps aurait été bénéfique.
Conséquences et bénéfices attendus
Le principal bénéfice attendu est un gain de temps et donc d’argent, pour les fabricants qui vont pouvoir optimiser leur planification d’évaluation clinique. En effet, les fabricants ont accès sur une même plateforme à toutes les exigences et dispositions à prendre pour chacun de leur dispositif.
Une autre conséquence de ce projet, c’est un bénéfice non négligeable pour les membres du groupe avec une valorisation auprès de nos futurs employeurs. Grâce à ce projet, ils pourront avoir un aperçu de nos connaissances, de notre intérêt et de notre implication dans le domaine des dispositifs médicaux.
Et enfin bien évidemment, la validation de l’Unité d’Enseignement est un bénéfice certain pour le groupe.
Donnée d'entrée du projet
Les données d’entrées de ce projet sont, tout d’abord le règlement européen 2017/745 et son chapitre relatif à l’évaluation clinique, mais aussi tous les documents relatifs aux nouvelles exigences concernant l'évaluation clinique tels que le MDCG.
Le projet se base aussi sur le manque d’informations des fabricants sur ce sujet malgré tous les documents apparemment mis à disposition.
Acteurs du projet
Ce projet regroupe différents acteurs, qui ont œuvrés tous ensemble pour arriver à bout de ce projet.
Tout d’abord, la maîtrise d’œuvre a été assuré par :
Les maîtres d’ouvrage ou les personnes pour qui est destiné ce projet sont principalement les fabricants de dispositifs médicaux ou autres opérateurs économiques responsables de l’évaluation clinique d’un produit.
Les parties prenantes directs sont donc :
- Opérateurs économiques (fabricants, industriels, distributeurs, mandataires, importateurs)
- Organismes notifiés
- Agence Nationale de la Sécurité du Médicaments et des Produits de Santé (ANSM)
Les parties prenantes indirects dont :
- Patients, qui vont bénéficier de dispositifs médicaux plus sûr, dont la sécurité et la performance est contrôlée ;
- Professionnels de santé, qui vont manipuler ces dispositifs dans leur routine de soin.
Déroulement du projet
Le projet s’est organisé autour de 3 jalons, au cours desquels nos avancements sur le projet ont été présentés au jury de suivi afin de recueillir des critiques constructives permettant d’améliorer nos rendus.
Les deux premiers jalons ont été majoritairement concentrés sur la prise de connaissance des informations et du contexte du sujet conduisant à la rédaction du mémoire. C’est au cours du dernier jalon que l’outil a été développé.
Améliorations envisagées
Pour le moment, l'outil n'est pas totalement complet. Certaines améliorations sont encore à envisagées, telles que :
- Proposer des templates personnalisées et personnalisables
- Assurer l'exhaustivité des exigences