
IDS148 - Guide pour faciliter la mise sur le marché d’un Dispositif Médical selon la règlementation européenne
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Contact
Kévin GOUFFAUD : kevin.gouffaud@gmail.com
Citation
A rappeler pour tout usage : Kévin GOUFFAUD, « Guide pour faciliter la mise sur le marché d'un Dispositif Médical Implantable et des instruments associés selon la règlementation européenne », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS148, juillet 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids148/
Résumé
Avec une population mondiale de plus en plus vieillissante, le secteur des dispositifs médicaux est un secteur en croissance. Cependant, après plusieurs incidents majeurs comme le scandale des prothèses mammaires PIP, les autorités européennes ont mis en place une nouvelle réglementation 2017/745 sur les dispositifs médicaux permettant la mise sur le marché européen de dispositifs médicaux sûrs pour les patients et les utilisateurs.
En 2022, la transition entre l’ancienne Directive 93/42/EEC et la nouvelle réglementation sur les dispositifs médicaux n’est pas achevée. Dans ce contexte, ce mémoire de stage a pour but de guider les acteurs du DM à comprendre et faciliter la mise sur le marché européen d’un dispositif médical implantable et de ces instruments chirurgicaux en orthopédie. La rédaction du dossier technique indispensable au marquage CE sera approfondie.
Mot(s)-clé(s)
Transition règlement, Union européenne, Règlement des DM 217/745 (RDM), Directive des DM 93/42/EEC (DDM), Dispositif médical implantable (DMI), Instrument chirurgical, Orthopédie, Marquage CE, Dossier technique, Organisme notifié, Système Management Qualité (SMQ), ISO 13485
Abstract
With an increasingly aging world population, the medical device market is a growing business. However, after several major incidents such as the PIP breast implants scandal, the European authorities have put in place a new Medical Device Regulation 2017/745 allowing the placing on the European market of medical devices that are safe for patients and users.
In 2022, the transition between the old Medical Device Directive 93/42/EEC and the new Medical Device Regulation 2017/745 is not completed. In this context, this report aims at guiding the medical device actors to understand and facilitate the European marketing of an implantable medical device and of these surgical instruments in orthopedics. The drafting of the technical file essential to the CE marking will be detailed.
Keyword(s):
Regulation transition, European union, Medical Device Regulation 2017/745 (MDR), Medical Device Directive 93/42/EEC (MDD), Implantable Medical Device (IMD), Surgical instrument, Orthopedics, CE marking, Technical file, Notified body, Quality Management System (QMS), ISO 13485
Téléchargements

Remerciements
Ces remerciements sont un peu particuliers pour moi, car ce ne sont pas mes premiers remerciements pour un stage de fin d’études.
Je voudrais avant tout remercier Monsieur Christophe EGLES, professeur d’université pour avoir toujours cru en moi depuis ces nombreuses années et su m’apporter un soutien sans faille dans les moments difficiles. Je ne te remercierais jamais assez pour m’avoir guidé vers la formation de l’UTC.
Je souhaite remercier Madame Mireille LEMERY, vice-présidente qualité et affaires règlementaires d’Amplitude pour m’avoir accueilli au sein de son équipe, avoir pris de son précieux temps pour échanger avec moi et relu mon mémoire de stage.
Je souhaite remercier Monsieur Jean-Mathieu PROT, enseignant-chercheur et Madame Isabelle CLAUDE, maître de conférences pour m’avoir donné l’opportunité de suivre le Master 2 Ingénierie de la Santé et de m’avoir accompagné dans mes choix durant ces deux semestres.
Je souhaite remercier Monsieur Gilbert FARGES pour m’avoir inculqué la qualité au-delà d’un simple cours, mais d’une philosophie à conserver pour l’ensemble de sa carrière professionnelle.
Je souhaite remercier le service des affaires réglementaires :
- Philippe DOURIAUX, ex-responsable et Emmanuelle VERDIER, responsable du service
- Cindy PINEAU, Géraldine FAYET, Ange-Manuella NJIOYA NGONGANG, Leïla RHAYAT, Anthony CHAUVAIN, Céline DUJET pour leur accueil chaleureux, leur pédagogie et le temps pour me donner les meilleures conditions pour apprendre, découvrir et échanger.
Une mention spéciale à l’équipe Suivi Post-Marché (Roxane COLOMBANI, Sandra BERGERON) et la Qualité (Blandine BRIAT) pour leur bonne humeur communicative.
Je remercie toutes les personnes de près ou de loin qui ont été en interaction avec moi durant mon stage chez Amplitude.
Je souhaite remercier Sophie, ma colocataire, pour le soutien mutuel qu’elle a pu m’apporter pour rédiger ce mémoire.
Pour conclure mes remerciements, je dédie ce mémoire à mes parents, ma sœur, mes grands-parents maternels et mes grands-tantes qui m’ont encouragé, donnés la force de me relever et d’aller de l’avant. Un grand merci à eux. Je vous dois beaucoup.
Abréviations
- Is : Dispositif médical de classe I stérile
- Im : Dispositif médical de classe I avec une fonction de mesure
- Ir : Dispositif médical de classe I type instrument chirurgical réutilisable
- ANSM : Agence National de Sécurité du Médicament et des produits de santé
- AR : Affaires règlementaires
- DM : Dispositif Médical
- DT : Dossier technique/ Documentation technique
- MDD : Medical Device Directive 93/42/CEE ou Directive des Dispositifs Médicaux 93/42/CEE
- MDR : Medical Device Regulation 2017/745 ou Règlementation des Dispositifs Médicaux 2017/745
- PMS : Suivi post-marché
- PTH : Prothèse Totale de Hanche
- PTG : Prothèse Totale de Genou
- QVP : Qualification et validation des procédés
- Re-certification : recertification et certification
- R&D : Recherche & Développement
- SMQ : Système Management Qualité
- Snitem : Syndicat national de l’industrie des technologies médicales
- UDI : Unique Device Identifier
- UE : Union Européenne
- UTC : Université de Technologie de Compiègne
Introduction
Le 29 mars 2010, l’Agence National de Sécurité du Médicament et des produits de santé (ANSM), anciennement l’AFSSAPS, décide de suspendre l’ensemble des activités de la société Poly Implant Prothese (PIP). Cette déclaration provient du scandale qui a été révélé par l’ANSM en 2009 sur des prothèses mammaires. Le gel de remplissage des prothèses avait été remplacé par un gel non adapté pour les dispositifs médicaux (DM) et différents de celui inscrit dans le dossier de conception de la prothèse [1].
D’autres scandales, comme les prothèses totales de hanche (PTH) métal-métal ont mis en évidence un manque de contrôle des autorités après la commercialisation, mais également une gestion des risques insuffisants lors de la production des DM [2].
Dans ce contexte, l’Union européenne a décidé de faire évoluer la règlementation en mettant en place deux nouveaux règlements : le règlement 2017/745 pour les DM [3] et le règlement 2017/746 pour les DM de diagnostic in vitro (DM-DIV) [4]. Ces mesures doivent combler les faiblesses révélées par les affaires précédentes. Ainsi, ces nouveaux règlements européens viennent remplacer les anciennes directives 93/42/CEE sur les DM [5], 90/385/CEE sur les DM Implantables Actifs (DMIA) [6] et 98/79/CE sur les DM de diagnostic in vitro [7].
Le règlement étant applicable actuellement et les directives étant abrogés, l’Union Européenne (UE) a prévu une période de grâce qui doit s’achever le 25 mai 2024 pour donner le temps nécessaire aux entreprises à se conformer au règlement [3]. Dans ces conditions, pour accompagner les entreprises du DM à obtenir un nouveau marquage CE de leurs DM, des outils et des guides doivent être mis en place pour faciliter la mise sur le marché des DM selon la règlementation 2017/745.
Ce mémoire de stage est composé de plusieurs parties. La première consiste à rappeler le contexte socio-économique des DM, particulièrement en chirurgie orthopédique. Les pathologies principales des membres inférieurs, les implants et les instruments chirurgicaux en orthopédie seront explicités. En deuxième partie, un rappel sur la transition règlement-directive et les différences entre le règlement 2017/745 et la directive 93/42/CEE sur les DM seront abordés. En troisième partie, un exemple de dossier technique sera présenté en donnant plusieurs pistes de réflexion sur la stratégie règlementaire et les choix à envisager. Un outil d’aide au suivi de la rédaction d’un dossier technique sera proposé.
1. Le contexte socio-économique des dispositifs médicaux orthopédiques
1.1 Les dispositifs médicaux et l’orthopédie, un secteur en croissance
Selon le règlement européen 2017/745 (Art. 2) [3], un DM peut être défini comme :
Un « dispositif médical, tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
- investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
- communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus,
et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. »
Cette définition est équivalente dans le Code de la Santé Publique (Art. L5211-1 et R5211-1) [8]. Les DM représentent un nombre varié de produits de santé : lit médicalisé, seringue stérile, pansement, IRM, pompe à insuline, prothèse totale de hanche (PTH) … Le but de la filière étant de proposer des solutions pour les professionnels du médical et du paramédical pour soigner les patients en toute sécurité.
D’après l’International Trad Center, le marché mondial des DM a rapporté 260 milliards $ en 2019 [9]. Les implants et les DM orthopédiques représentent un quart du marché des DM. En 2020, le marché des DM orthopédiques est évalué à 40,6 Milliards $ dont 16,8 Milliards $ pour les DM de reconstructions articulaires. En prévision, la croissance des DM en chirurgie orthopédique est estimée à plus de 5% pour la période 2021-2027 ce qui porterait la valeur du secteur à plus de 60 milliards $ [10].
L’ensemble des sociétés du DM ont eu un chiffre d’affaires de 30,7 milliards € sur le marché français avec un tiers du chiffre d’affaires réalisé à l’exportation. L’exportation est un axe privilégié par les entreprises pour leur croissance (4,3% de croissance annuelle). Les trois marchés principaux privilégiés sont l’Europe, l’Amérique du Nord et l’Asie.
Cette augmentation s’inscrit dans les changements sociétaux actuels et à venir. En effet, le vieillissement de la population occidental entraîne un besoin supplémentaire dans les DM dû à une prise en charge plus fréquente que les populations jeunes. Dans le cas de l’orthopédie, les traumatologies associées au sport et aux accidents risquent d’augmenter comme la rupture du col du fémur chez les personnes âgées [10]. La sédentarité et le surpoids pourraient également entraîner une augmentation du nombre de poses de prothèse totale ou partielle de genoux ou une fusion de vertèbre due à un affaissement de disque intervertébral.
Figure 1 : Les chiffres clés du secteur des dispositifs médicaux en France [11]
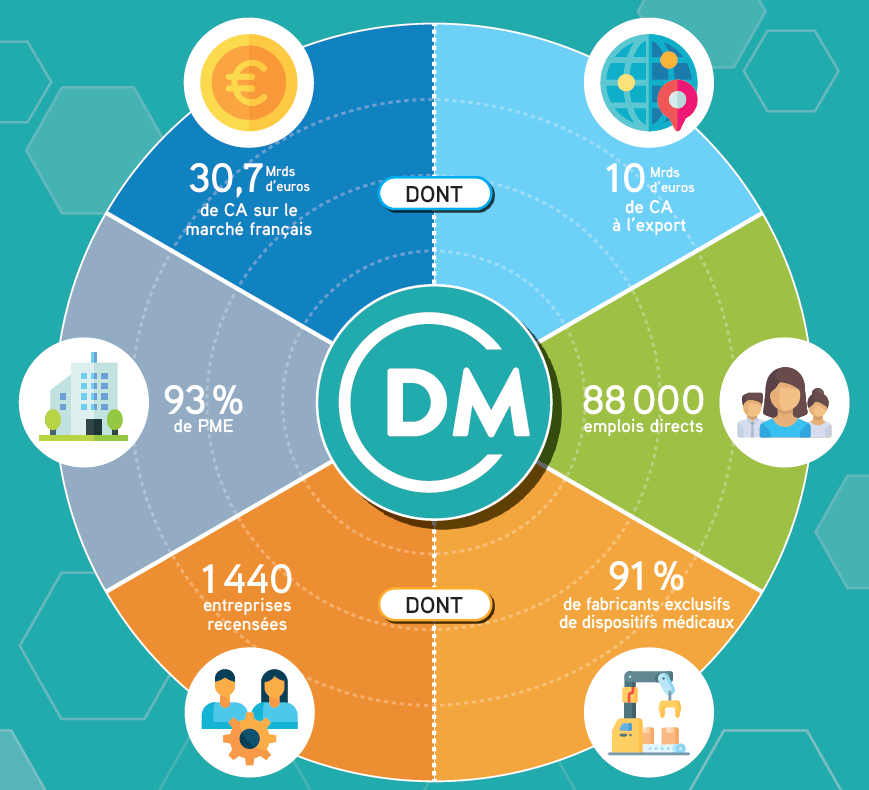
À l’échelle de la France, en 2021, le secteur des DM représente un bassin d’emploi de 88 000 emplois. Les PME correspondent à 93% des entreprises du DM soit 14440 entreprises recensées selon le Snitem (Figure 1).Les sociétés du secteur sont inégalement réparties sur le territoire. Ainsi, la première région d’accueil est l’Ile de France, 34% des entreprises. La seconde région est l’Auvergne-Rhône-Alpes avec 18% des sociétés [11].
Également, le secteur peut s’appuyer sur un tissu industriel composé de sous-traitants dans les domaines de la mécanique (22%), la plasturgie (17%) et la conception (12%). Les sous-traitants sont aux deux tiers des PME et emploient environ 15 000 personnes.
La région Auvergne-Rhône-Alpes profite particulièrement de ce bassin industriel en étant la première région en nombre de sous-traitants [11].
Cependant, la croissance du secteur des DM pourrait être perturbée suite à la crise du Covid-19 et de la guerre en Ukraine. Ces deux crises impactent la production et le prix des matières premières qui a pour conséquence l’augmentation des prix des DM [12]. De plus, le secteur connaît une pénurie de recrutements pour différents services comme les affaires règlementaires qui impactent le temps de mise sur le marché des DM durant la transition directive-règlement.
Dans le cas de l’industrie du DM, les entreprises ont pu être favorisées ou pénalisées. En effet, des actes chirurgicaux ont été reportés ou déprogrammés comme en orthopédie. Ainsi, les industriels de la chirurgie ont vu leur croissance réduite. En France, le secteur doit s’adapter à une baisse tarifaire du remboursement des implants articulaires comme la prothèse totale de genoux (PTG) ou la PTH [13].
1.2 Amplitude Surgical, un challenger de la chirurgie orthopédique
Ce stage a été réalisé dans la société française du DM, Amplitude Surgical. La société a été créée en 1997 par Olivier Jallabert, directeur général actuel. Cette entreprise est une SAS (Société par Action Simplifié) d’environs 450 collaborateurs située dans la région Auvergne-Rhône-Alpes.
Figure 2 : Répartition géographique des filiales (en rouge) et des distributeurs (en orange) (Source : Auteur d’après Amplitude Surgical)
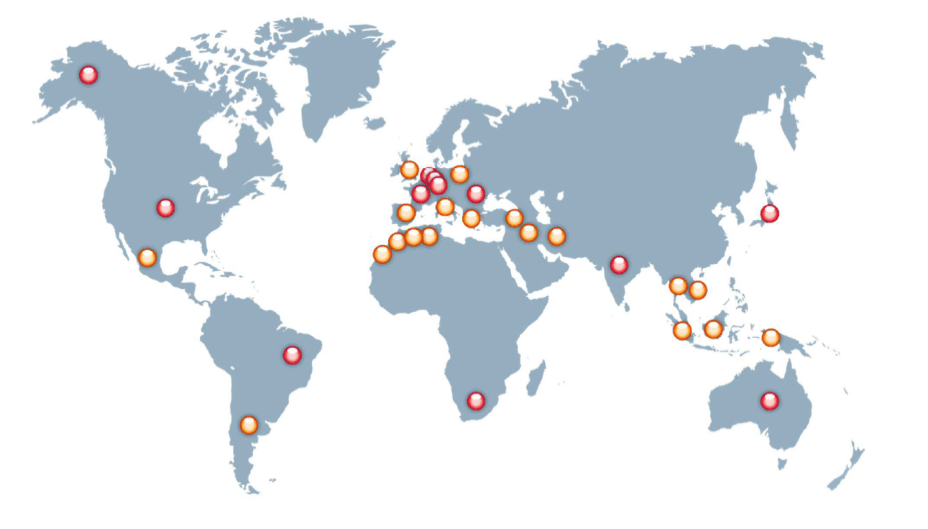
Cette société appartient au Groupe Amplitude Surgical composé d’Amplitude, de Novastep et de filiales dans plusieurs pays comme le Brésil, l’Australie, l’Afrique du Sud, les États Unis d’Amérique, la Suisse, l’Allemagne, la Belgique... (Figure 2). Les activités du Groupe Amplitude sont la production et la distribution de dispositifs médicaux dans le domaine de la chirurgie orthopédique des membres inférieurs.
Amplitude est organisé en deux sites. Le premier site basé à Valence dans la Drôme concentre toutes les activités administratives, règlementaires, marketing, export, conception et logistique (Figure 3). Un deuxième site localisé à Neyron dans l’Ain traite de la partie commerciale et marketing. Le stage que j’ai effectué était dans les locaux de Valence au sein du service des Affaires Règlementaires (AR).
Figure 3 : Site d’Amplitude Surgical localisé à Valence, France (Source : Auteur)

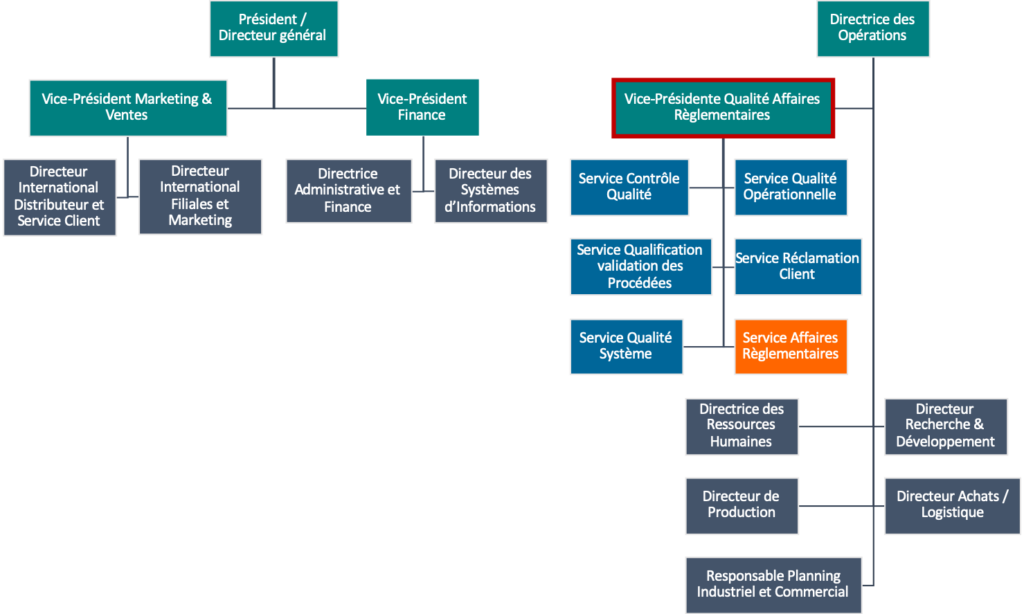
Hiérarchiquement, le service est sous la direction de la vice-présidente Qualité Affaires règlementaires (Annexe I). En qualité de Personne Chargée de Veiller au Respect de la Règlementation (PCVRR), la vice-présidente possède un statut particulier au sein de la société en rendant compte simultanément à la directrice des opérations et au directeur général. Le rôle de ce poste est d’assurer que l’ensemble de l’entreprise et des sous-traitants respectent les exigences règlementaires et les standards qualités revendiquées.
Annexe I : Organigramme d’Amplitude (Source : Auteur d’après Amplitude Surgical)
Le service des AR est en interaction avec différents services :
- Service Recherche & Développement (R&D) : Le service a pour mission principale de concevoir, améliorer et industrialiser les produits Amplitudes. La R&D est composée de plusieurs pôles selon les gammes de produits (PTH, PTG, ligaments artificiels, station d’aide à la chirurgie). Le bureau des méthodes est également présent pour assurer l’industrialisation des DM.
- Service Qualification et Validation des Procédés (QVP) : Le service est chargé de veiller au respect des normes revendiquées, valider la biocompatibilité, le nettoyage, le conditionnement et la stérilisation des produits Amplitude.
- Service Suivi clinique : Le but du service est de collecter les données cliniques de la bibliographie ou des études cliniques spécifiques aux dispositifs de la société pour s’assurer que la balance bénéfice/risque est favorable pour le patient.
- Service Réclamation clients : Son rôle consiste à suivre les incidents (matériovigilance, incidents de DM similaire …) pouvant survenir après la mise sur le marché d’un DM et à suivre les actions appropriées pour l’amélioration des produits et services
- Service Marketing : Le Marketing relève les attentes des chirurgiens et des professionnels de santé pour développer de nouveau DM en concertation avec le service R&D. Les pays de distribution sont également définis par le service. Le service joue également un rôle de formateur pour les produits Amplitude
- Service Export : Le rôle du service consiste à délivrer les différentes commandes en fonction du pays concerné. Les commandes peuvent être bloquées lorsque les conditions règlementaires, contractuelles et normatives ne sont pas respectées pour le pays distribué.
La stratégie d’Amplitude est de produire des dispositifs médicaux « haut de gamme » pour compenser des pathologies articulaires des membres inférieurs comme la hanche, le genou (ou le pied pour Novastep).
Grâce à ces différentes filiales et ces distributeurs présents dans plus 30 pays, Amplitude peut mettre sur le marché ses produits en utilisant la même stratégie adoptée en France, son premier marché historique. Amplitude est à contrepied de ces concurrents en se focalisant en priorité sur le besoin des chirurgiens.
En effet, Amplitude Surgical a une stratégie cherchant à travailler en collaboration avec des chirurgiens orthopédistes de renom selon les pays, en développant des produits répondant à leurs attentes et à celles de leurs patients.
Cependant, cette stratégie pourrait à terme engendrer un conflit avec la croissance de l’entreprise en proposant des produits personnalisés aux chirurgiens, de « niches ».
La société devrait supporter, à long terme, une augmentation du volume de DM produits et un élargissement de la gamme. En pratique, cette stratégie pourrait être risqué, voir intenable économiquement. En effet, le développement d’un nouvel instrument, outils d’aide à la chirurgie, ou d’implants chirurgicaux représente un coût important en R&D et frais administratif pour sa mise en vente.
Dans le contexte pandémique actuel, Amplitude continue ses investissements grâce à ses investisseurs confiants dans sa rentabilité. En effet, Amplitude est une société rentable depuis plus de 15 ans. En 2021, la croissance de l’entreprise est de 4,5% [14].
Cette entreprise ne recherche pas l’innovation de rupture comme les grands groupes du secteur des dispositifs médicaux implantables orthopédiques, mais de s’inspirer des innovations en apportant sa propre vision du produit (PTH, PTG). Pour résumer, au sens marketing, Amplitude est un « Challenger ». La stratégie induite par Amplitude, a permis de se classer comme 2ème fabricant et fournisseur de PTG sur le sol français. Amplitude se place également dans les fabricants principaux des PTH sur le territoire français [14].
À l’international, Amplitude cherche à prendre une part de marché significative par rapport aux autres concurrents dont les Groupes américains comme la filiale DePuy Synthes de Johnson & Johnson, Zimmer Biomet, Stryker Corporation, …. En s’appuyant sur son expérience historique de fabricant français et européen, Amplitude souhaite développer son activité aux États-Unis pour toucher le premier marché mondial [10], [14].
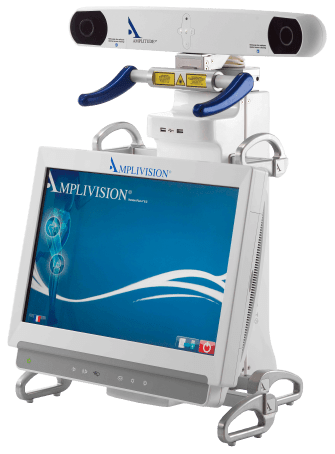
Pour se faire, Amplitude peut s’appuyer sur des produits « phares » comme E.T.O.I.L.E.® [15], i.M.A.G.E.® [16], Amplivision® [17] (Figure 4).
Figure 4 : Les dispositifs médicaux E.T.O.I.L.E.®, i.M.A.G.E.® et Amplivision® de l’entreprise Amplitude, de gauche à droite [18].


- Plateforme d’instrumentation E.T.O.I.L.E.®
Le système E.T.O.I.L.E.® signifie Extension de Table Orthopédique avec Instruments Liés à Exposition [15]. Cet instrument est utilisé dans le cadre d’une chirurgie pour la pose d’une PTH. L’intérêt de ce système est d’aider le chirurgien dans l’abduction de la hanche, lors de la voie chirurgicale antérieur de Hueter. Cette voie a l’avantage d’être mini-invasif et de préserver les muscles entourant la hanche. Le temps de rééducation est ainsi réduit [19].
- Système d’instrumentation sur mesure à usage unique i.M.A.G.E.®
Le système i.M.A.G.E.® est un ensemble d’instruments à usage unique basés sur l’anatomie du patient [16]. En effet, le but est de proposer une pose en première intention d’une PTG en prenant en compte les spécificités du patient. Pour cela, le patient doit réaliser un scan de l’ensemble des articulations de la hanche, du genou et de la cheville pour obtenir une reconstruction 3D. Puis, Amplitude réalise des positionneurs adaptés au patient grâce à un frittage laser. Les positionneurs facilitent et améliorent la coupe et la pose de la PTG. La planification, le temps d’intervention et les pertes sanguines sont ainsi réduits.
- Système de chirurgie assistée par ordinateur Amplivision®
Le système de chirurgie assistée par ordinateur Amplivision® est une alternative au gabarit personnalisé comme le système i.M.A.G.E.®.
L’intérêt est de cartographier, dans un premier temps, la forme de l’articulation (genou ou hanche) à l’aide de marqueurs, puis de poser la PTH ou la PTG de la marque Amplitude en ajoutant une précision supplémentaire dans les angles de l’articulation, l’alignement et la longueur de la jambe. Cette précision est rendue possible par l’utilisation de marqueurs infrarouges qui permettent une cartographie de la surface de l’articulation ou cibler un point d’intérêt chirurgical de l’articulation.
Dans le but de simplifier le travail du chirurgien et d’améliorer la vie du patient, Amplitude souhaite poursuivre dans les pas d’Amplivision® en proposant une solution de robot chirurgical permettant une découpe précise et personnalisé anatomiquement. Cette démarche aurait un intérêt sur la qualité de vie du patient implanté [20]. Ce robot permet d’élargir les technologies proposées aux chirurgiens et de donner des possibilités complémentaires aux chirurgiens. D’autre part, Amplitude prévoit de construire un nouveau bâtiment de production à côté du siège social pour maîtriser davantage ses coûts opérationnels, accroître sa capacité de production et assurer une qualité adéquate aux exigences de la règlementation [21].
Amplitude, installée dans la Drôme, possède une localisation intéressante sur plusieurs points :
- L’accès direct au port de Marseille par l’autoroute facilite l’exportation des DM et l’importation de pièces ou de matières premières. De plus, la proximité avec la frontière suisse, italienne et allemande permet de distribuer, dans un temps réduit, les principaux pays européens occidentaux.
- Amplitude peut s’appuyer sur un bassin industriel présentant de nombreux sous-traitants, PME et TPE, fournissant des produits finis ou des pièces indispensables à la production des produits d’Amplitude. De plus, Monsieur Olivier JALLABERT, président et fondateur d’Amplitude, souhaite qu’Amplitude s’appuie sur les savoir-faire régionaux à défaut nationaux pour développer et produire les pièces des DM. La proximité avec les partenaires industriels permet une meilleure réactivité et une meilleure adaptation des sous-traitants à la demande d’Amplitude. D’autre part, cette proximité facilite les audits internes des fournisseurs et permet de contrôler que les exigences du SMQ selon la norme ISO 13485 sont respectées [22]. SOFAB Orthopédie est un exemple de cette stratégie.
Cette entreprise produit des pièces du DM pour Amplitude. Le chiffre d’affaires de SOFAB Orthopédie est de 5 M€ dont 80% venant d’Amplitude. Cette collaboration étroite avec Amplitude a d’ailleurs débouché sur la participation au capital de la société SOFAB Orthopédie à hauteur de 50% [23]. Actuellement, la participation d’Amplitude chez SOFAB Orthopédie est de 100%.
- Le Site de Valence se situe dans une zone artisanale adaptée au transport de produit et ayant un accès aux voies rapides rejoignant les autoroutes en direction de Grenoble, Lyon ou Aix-en-Provence/Marseille.
Malgré cette stratégie efficace, Valence est une ville moyenne de province. Le besoin en main d’œuvre qualifié, ingénieurs et administratifs, devrait croître en parallèle de la croissance de la société. L’attractivité de Valence pourrait être un facteur limitant. En effet, Lyon, Grenoble, Aix-en-Provence et Marseille sont des villes avec une forte activité industrielle et Amplitude pourrait se faire cannibaliser des profils dont la société a besoin pour sa croissance. Cependant, l’envie des cadres à vivre en province pourrait jouer en faveur d’Amplitude en proposant une alternative aux métropoles citées précédemment.
1.3 Les pathologies des membres inférieurs, les techniques opératoires et les implants orthopédiques
Les membres inférieurs sont composés de plusieurs articulations : la hanche, le genou et la cheville. Une articulation est définie par l’extrémité de deux os recouverts par une couche de cartilage. Le cartilage sert à amortir les contraintes mécaniques entre les deux os. De plus, le cartilage aide à la mobilité de l’articulation. Pour éviter la luxation de l’articulation, des ligaments sont attachés de part et d’autre de l’articulation au niveau de l’os. Les tendons jouent un rôle dans la mobilité de l’articulation. Les tendons sont situés à chaque extrémité des muscles et s’attachent sur les os. Lors de la contraction du muscle, le tissu musculaire se contracte et raccourcit provoquant un mouvement de flexion.
Figure 5 : Schéma anatomique du genou et de la hanche avec ou sans arthrose. En haut, une articulation du genou avec (droite) ou sans arthrose (gauche). En bas, une articulation de la hanche avec (droite) ou sans arthrose (gauche) [24], [25].
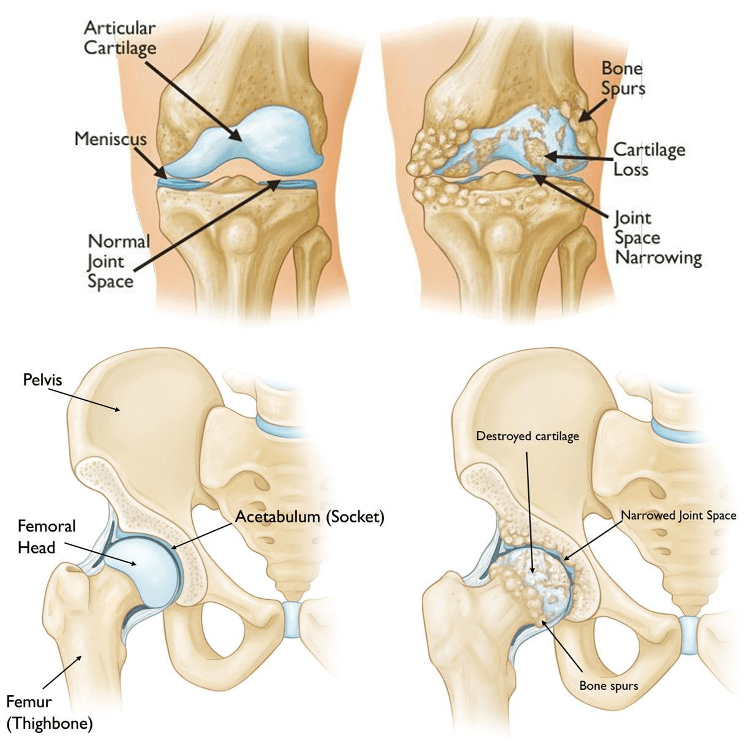
Dans le cas de la hanche et du genou, ces articulations sont sollicitées tout au long de la vie par le mouvement essentiel de la marche ou le passage de la position assise-debout. Selon la génétique du patient, son activité ou lors d’un traumatisme, l’articulation peut être endommagée au niveau de cartilage (Figure 5). Le cartilage ne pouvant plus jouer son rôle de mobilité, les mouvements de l’articulation deviennent plus douloureux pour le patient et limités. . Cette dégénérescence du cartilage est appelée coxarthrose pour l’articulation de la hanche et gonarthrose pour l’articulation du genou. Le patient est encouragé à prolonger au maximum ces activités. Cependant, lorsque le cartilage devient trop endommagé et que les douleurs ne peuvent plus être contrôlées par la prise de médicaments antidouleur ou par des infiltrations d’acide hyaluronique, l’intervention chirurgicale peut être proposée au patient [24]–[27].
Dans ce mémoire, l’intervention chirurgicale développée sera l’arthroplastie de la hanche. L’arthroplastie existe également pour le genou (Figure 6).
Figure 6 : Schéma d’une PTG (gauche) et d’une PTH (droite) [24], [25].
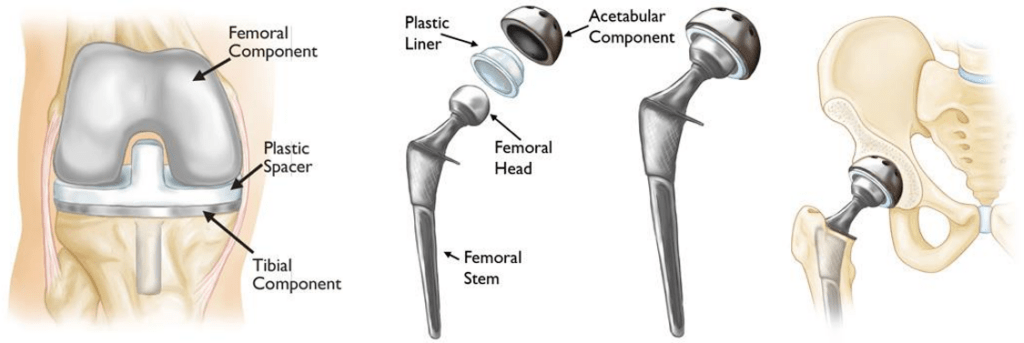
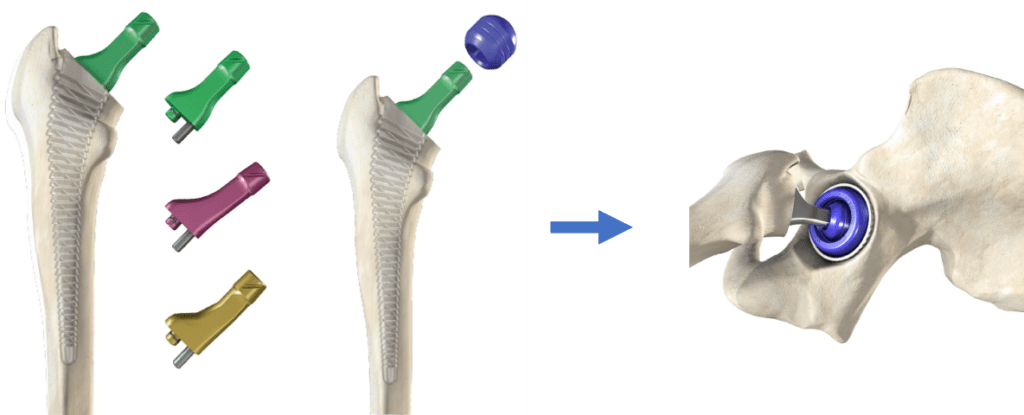
L'arthroplastie de la hanche est une technique chirurgicale bien connue, principalement pratiquée en cas d'arthrose ou de traumatisme. Le but de cette chirurgie est de remplacer l’articulation de la hanche par une PTH. Une PTH est composée d’une tige fémorale, d’une tête fémorale et d’une cupule.
La tige vient s’insérer dans le fémur puis la tête est fixée à la tige. D’autre part, la tête est coiffée par la cupule qui vient se loger dans le cotyle osseux, cavité qui vient accueillir la tête fémorale en absence de prothèse. La première pose de PTH s’appelle une chirurgie de première intention. Lorsque l’implant ou l’os subit des dégradations, une deuxième chirurgie peut être effectuée. Dans ce cas, une prothèse de révision est utilisée. Dans les cas extrêmes, des prothèses de reconstruction sont utilisées lorsque le patient est atteint d’ostéoporose ou en cas de défects osseux. La différence générale qui sépare ces prothèses est leur caractère invasif. Une tige de première intention est courte, comparée à une tige de révision. De plus, des vis sont ajoutées pour améliorer la fixation de la prothèse aux os du patient.
Lorsque le cartilage du cotyle n’est pas détérioré, le remplacement de la tête fémorale peut être suffisant. L’arthroplastie est alors dite partielle.
Bien sûr, la mise en place des implants demande un ensemble d’instruments adaptés à la préparation osseuse et à l’implantation lors des étapes chirurgicales.
L’arthroplastie de la hanche de première intention est composée de 5 étapes chirurgicales majeures :
- Planification préopératoire : La morphologie du patient est évaluée. Des repères anatomiques sont effectués au niveau du col. Le type et la taille des implants sont sélectionnés.
Figure 7 : Les étapes de la préparation fémorale (Source : Auteur d’après Amplitude Surgical).
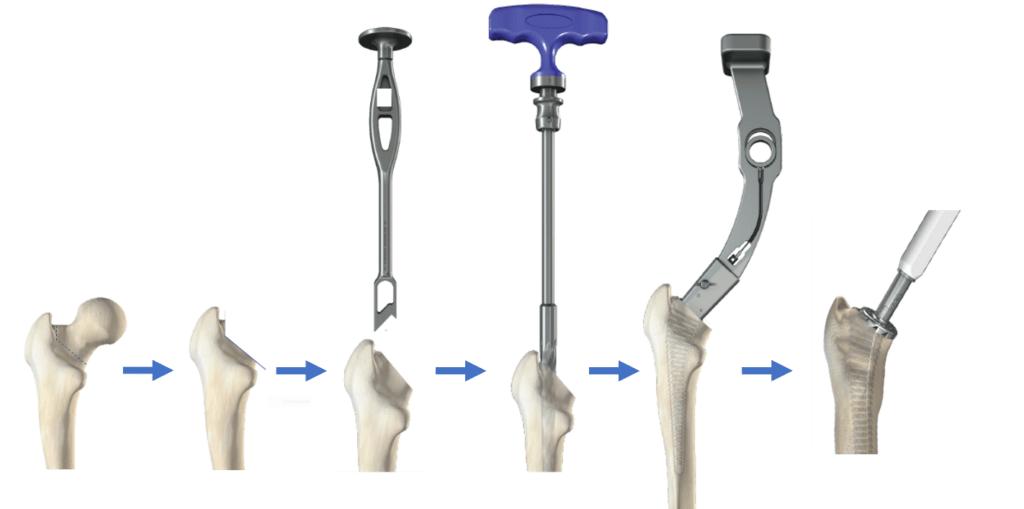
- Préparation fémorale : Cette étape consiste à préparer le fémur à l’implantation des composants fémoraux. L’articulation de la hanche du patient est ouverte puis le chirurgien procède à la coupe du col fémoral. Dans un deuxième temps, le canal fémoral est foré et alésé. Puis une râpe est insérée pour assurer l’espace nécessaire à l’implantation de la tige fémorale dans le canal fémoral. L’alésoir utilisé possède des dimensions similaires à celle de la tige. Lorsque la râpe est mise en place, un alésage peut être réalisé en se fixant sur l’extrémité la râpe pour faciliter la pose du col et retirer l’excédent d’os (Figure 7).
Figure 8 : Préparation de la cupule à l’aide d’une fraise à cotyle (Source : Auteur d’après Amplitude Surgical).
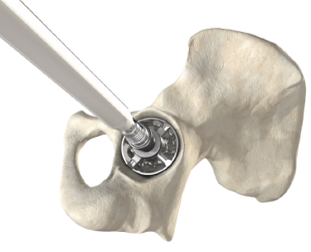
- Préparation de la cupule : Cette partie a pour but de préparer l’implantation de la cupule. La cavité du cotyle est fraisée à l’aide de la fraise à cotyle pour être aux dimensions de la cupule choisie (Figure 8).
Figure 9 : Utilisation de col d’essai, de tête d’essai et de cupule d’essai (Source : Auteur d’après Amplitude Surgical).
- Essais : Les essais ont pour but d’ajuster le modèle du col et de la tête fémorale pour obtenir une tension musculaire et un centrage satisfaisant de la hanche. Le décalage entre la tige fémorale et les composants de la tête sont ajustés pour optimiser l’adaptation de la PTH à l’état musculosquelettique du patient. Le chirurgien procède également à des tests de stabilité, de mobilité et d’amplitude des mouvements de l'articulation avec des pièces d’essais (cupule, col et tête) avant l'implantation des implants définitifs (Figure 9).
Figure 10 : Impaction de la tête fémorale, impaction de la cupule, impaction de la tête avec la cupule (gauche à droite) (Source : Auteur d’après Amplitude Surgical).
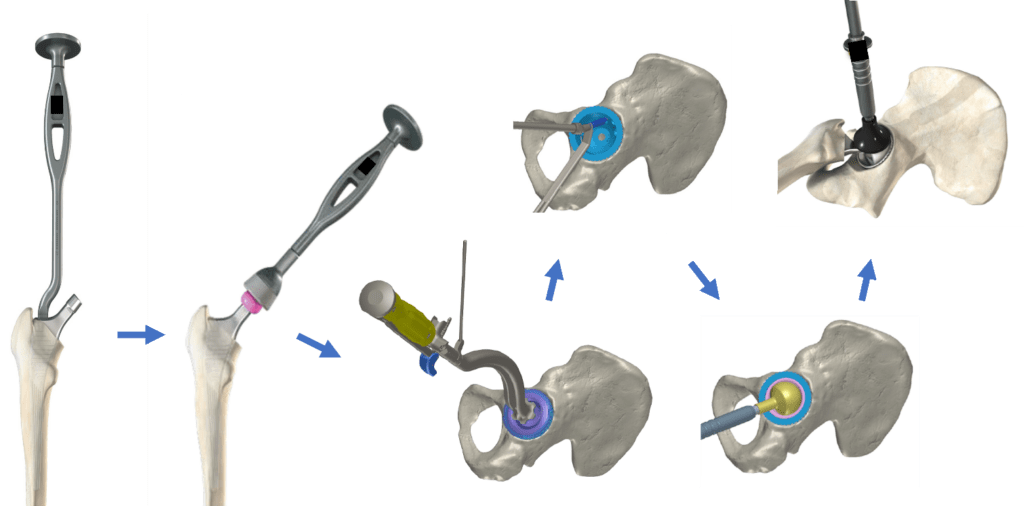
- Implantation des implants : L’implantation des implants s’effectue par impaction sur l'os préparé ou par assemblage des composants. L'implantation peut nécessiter l'utilisation de ciment orthopédique pour assurer la fixation à long terme de l'implant sur l'os (Figure 10).
2. La mise sur le marché européen d’un dispositif médical
2.1 Le besoin de règlementaire européen pour les dispositifs médicaux
Les DM n’ont pas toujours été règlementés. En effet, les premières indications sur les DM portaient sur les prothèses de hanches qui devaient être homologuées pour chaque pays. Le dossier technique (DT) était rudimentaire et n’avait pas de notion de gestion de risque. Les autres DM pouvaient être mis sur le marché sans homologation. Depuis que la fin de la Seconde Guerre mondiale, les pays européens sont dans une démarche d’échange commercial.
Les premiers pays à se regrouper formeront la Communauté Européenne du Charbon et de l’Acier (CECA) puis s’élargir avec la Communauté Européenne Economique (CEE). Dans une envie d’échange sans frontière douanière et économique, les Etats Membres de la CEE s’engagent en 1986 avec l’Acte Unique Européen (AUE) à « créer un espace sans frontières intérieures pour permettre la circulation des personnes, des biens, des capitaux et des services » [28]. Dans la continuité de ce texte, la CEE instaure plusieurs directives comme la directive 93/42/CEE sur les DM [5], la directive 90/385/CEE sur les DM implantables actifs [6] et la directive sur les DM pour le diagnostic in vitro [7] qui régissent des exigences communes.
Ainsi, les DM ont pu être soumis à des règles harmonisées pour l’ensemble des Etats Membres. Les directives ne sont pas applicables directement et doivent être traduites dans des textes lois de chaque pays. Si la philosophie du texte reste inchangée, certaines particularités entre les pays peuvent apparaître comme la date de révision de la notice d’utilisation. Les directives mises en vigueur obligent l’obtention du marquage CE pour la mise sur le marché européen d’un DM. Le marquage CE est renouvelable tous les 5 ans et permet une libre circulation au sein de l’Espace Economique Européen.
Malgré la présence dans la directive 93/42/CEE de normes harmonisées, telles que la gestion des risques ISO 14971, ou de la classification des DM selon l’ordre de dangerosité potentielle croissante, plusieurs scandales ont révélé des failles dans la réglementation :
- Les prothèses mammaires PIP (2009) : La société française utilisait pour le remplissage des prothèses mammaires des gels de grade non implantable. Les prothèses contenaient des huiles de silicone provenant de l’industrie du caoutchouc. De plus, la société a caché sciemment à l’autorité de santé, l’utilisation de ces huiles [1].
- Les prothèses articulaires Ceraver (2011) : Ceraver Italie a mis sur le marché des prothèses implantables en apposant le marquage CE sur leur produit sans informer les autorités du changement de revêtement effectué sur l’implant. Les prothèses ont pu être implantées chez des patients grâce à la corruption de chirurgiens [29].
- Les PTH métal-métal (2010) : Les prothèses métal-métal sont composés d’alliage métallique contenant en autres éléments du chrome et du cobalt Plusieurs études ont fait mention de la libération d’ions dans le corps des patients implantés. Ce phénomène se produit par les effets de frottements entre la tête fémorale et la cupule. Les ions chrome et cobalt sont alors libérés dans le corps. Une inflammation peut se produire dû à la présence de macrophage qui vont venir détruire l’os (cytolyse) autour de l’implant. Le phénomène détériore l’os et provoque de la douleur chez le patient. La société DePuy avec ces PTH ASR sera condamnée lors d’une "class action" aux États-Unis en 2010. Cependant, d’autres sociétés continueront la commercialisation de PTH métal-métal. De plus, l’ANSM ne réagira pas à cette décision [30].
Ces scandales répétés ont mis en évidence le manque de réactivité, de suivi des incidents, de contrôle des fabricants et de traçabilité des DM. Pour renforcer la sécurité des DM et la protection des patients, deux nouvelles règlementations européennes ont été mises en place pour pallier les faiblesses associées aux scandales précédents. La règlementation européenne 2017/745 sur les DM remplace les directives 93/42/CEE et 90/385/CEE et la directive 98/79/CE est remplacé par le règlement 2017/746 sur les DM pour le diagnostic in vitro [3]–[7].
Le passage de directive à règlementation permet à l’Union Européen de se doter d’exigences harmonisées pour l’ensemble des pays européens.
2.2 Les conséquences du passage à la règlementation 2017/745 et la transition directive - règlementation
Comme la réglementation 2017/745 le permet, les directives sont abrogées et l’ensemble des DM doivent respecter le règlement en application à partir du 26 mai 2021. Cependant, le règlement prévoit une période de grâce (Article 120) du 26 mai 2021 au 26 mai 2024. Cette période a pour but de donner aux fabricants le temps nécessaire pour obtenir un nouveau marquage CE des DM ayant obtenu précédemment un marquage CE avec l’ancienne directive. En attendant le passage des DM avec un marquage CE sous règlement, les DM déjà présents sur le marché européen peuvent continuer à être commercialisés sous certaines conditions. Pour faciliter la compréhension des parties suivantes, un DM sous MDD est un DM qui a obtenu son marquage CE sous la directive alors qu’un DM sous MDR est un DM ayant un marquage CE sous la nouvelle réglementation (Figure 11).
Figure 11 : Calendrier initial de la mise en application du règlement 2017/745 et 2017/746 [31]. La date de mise en application de la règlementation sur les DM est modifiée au 26 mai 2021.
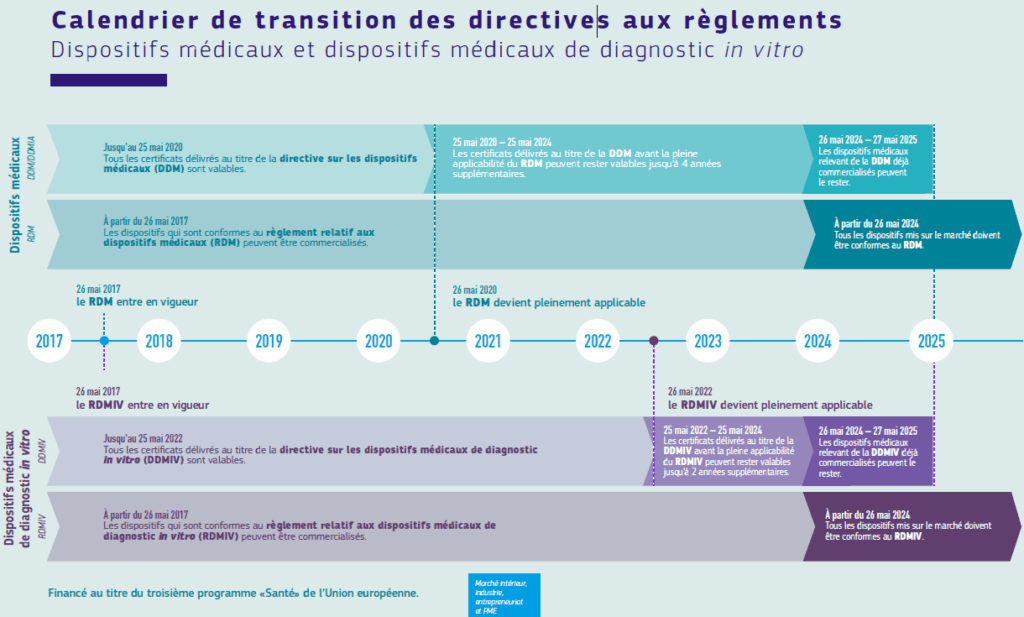
Actuellement, en 2022, en pleine transition directive-règlement, plusieurs difficultés sont rencontrées par les différents acteurs du marquage CE. En effet, la demande de marquage CE est ralentie par plusieurs facteurs qui provoquent un goulet d’étranglement dans la soumission des dossiers :
- Le manque d’Organismes notifiés (ON) : L’ON est « un organisme d’évaluation de la conformité désigné en application du présent règlement » qui est accrédité et inspecter par l’ensemble des autorités compétentes européennes [3]. Sous la directive, le nombre d’ON était d’environ 80. Lors de la mise en vigueur du règlement, de nombreux ON n’ont pas pu se mettre au niveau des critères réglementaires ou n’ont pas souhaités se faire accréditer. En 2022, le nombre d’ON est de 30 selon la base de données européenne NANDO [32]. Ce chiffre reste insuffisant au regard du nombre de certifications à effectuer pour l’ensemble des fabricants européens du DM qui représente 33 000 entreprises [33].
- Un nombre de renouvellements de certificats et de nouvelle certification MDR insuffisant : En avril 2022, l’ensemble des DM ayant obtenu une certification pour la réglementation représente 10% des certificats pour l’obtention du marquage CE [34]. Ainsi, la majorité des DM, 90% des certificats, sur le marché européen, ne possède pas de certificats sous la nouvelle réglementation [34]. De plus, de nouveaux DM émergents tous les ans ce qui vient s'ajouter à la liste des DM voulant obtenir un marquage CE sous la nouvelle réglementation. Sachant que la fin de la transition est prévue pour le 26 mai 2024, les ON n’ont pas la charge suffisante pour certifier l'ensemble des dispositifs médicaux. Un facteur aggravant des ON notifiés MDR comme le BSI a été de privilégier des marquages CE MDD au lieu de marquages CE MDR au début de la période de transition ce qui a provoqué des retards dans la gestion des dossiers. Actuellement, en 2022, les syndicats des DM, les ON et les autorités de santé européennes sont vigilantes sur l'avancement de ces certifications. Ce manque de certification et de re certification pourrait entraîner, à long terme sur le marché européen, une pénurie de DM dans certains domaines médicaux. Malheureusement, la Commission européenne ne souhaite pas modifier la date de la fin de transition à ce jour [35].
- Un coût élevé pour les fabricants : Certains fabricants ont tendance à ne pas renouveler leur certification sous MDD pour reculer les frais à engager pour payer une certification MDR. En effet, le prix d'une certification MDR est d’environ 100% plus cher qu'une certification MDD.
- Une prise de conscience tardive des fabricants : Des fabricants n'ont pas encore pris conscience de l'importance de la réglementation et des enjeux qui y sont associés. En particulier, les fabricants de DM de classe I qui ne connaissent pas les procédures de certification sous MDD des classes supérieures. Les DM de classe I sous MDD n'ont pas besoin d'être audité par un ON le marquage et s’autocertifie. Ces sociétés, par leur méconnaissance de la réglementation, ont tendance à sous-estimer le temps nécessaire à la constitution d'un DT sous MDR pour son évaluation auprès d'un ON.
- Un retard dans le traitement des dossiers par les ON : Plusieurs causes sont à mettre en évidence dans le retard du traitement des dossiers par les ON. La première est le décalage observé entre la demande de l'ON et ce qui est demandé par le règlement 2017/745. Certains ON ont tendance à demander certaines spécificités qui ne sont pas présentes dans la réglementation 2017/745. De plus, la mise en forme du DT est différente selon les ON ce qui peut ajouter une confusion aux fabricants et ajouter certains retards dans le traitement des dossiers. La deuxième cause peut également s'expliquer par des dossiers qui ne sont pas rédigés correctement par les fabricants et qui prolongent d'autant la procédure pour l'évaluation du dossier en vue d'un marquage CE voir le refus d’évaluer le dossier [34].
- Le manque de ressources humaines dans le secteur des DM : Les fabricants rencontrent également un manque de ressources humaines dans la constitution des DT et pour mettre en place leur système de management de la qualité. Le nombre de chargés d'affaires réglementaires et assurance qualité est insuffisant. De plus, un chargé d'affaires réglementaires est un poste qui demande une certaine expérience pour pouvoir constituer un DT sans rencontrer de difficultés. Le nettoyage, la stérilité, ou la biocompatibilité, ont pris de l’importance sous la nouvelle réglementation. Ainsi, les ingénieurs en validation et qualification des procédés sont plus impliqués dans la constitution du DT. Certains procédés spéciaux demandent également une expertise associant la toxicologie, la biocompatibilité et la science des matériaux ce qui reste marginal dans les formations professionnelles ou universitaires. Ce manque de ressources est actuellement en train d'être résolu par la mise en place d'alternance par les formations spécialisées dans les affaires réglementaires dans le but d'améliorer la capacité des profils juniors à être opérationnel.
Un autre point aggravant a été le retard du côté des fabricants et des ON dû au Covid-19. Malgré, que l’UE ait mis en place un report de la date de transition d’un an, passant du 26 mai 2020 au 26 mai 2021. La mesure semble insuffisante pour les acteurs pour que chacun puisse obtenir le marquage CE de ces DM dans les temps [35].
Les DM qui auront été commercialisés avant le 26 mai 2021, dans les hôpitaux, pourront être utilisés jusqu’au 26 mai 2025. D’autre part, le marquage CE n’est pas un certificat permanent. Les DM qui ont été recertifiés sous MDD avant le 26 mai 2021 auront leur certificat valable jusqu’à 4 ans supplémentaires (26 mai 2024 maximum).
La nouvelle réglementation est plus détaillée comparée à la directive. En comparant les deux textes réglementaires, le nombre d'articles est de 123 pour la réglementation alors qu'elle n'est que de 23 pour la directive. La réglementation peut également s'appuyer sur 17 annexes contre 12 annexes pour la directive. La définition d'un dispositif médical était élargie (Article 2) permettant d'incorporer des DM assimilés à vocation non médicale comme les lentilles de contact. La classification des dispositifs médicaux a été renforcée en passant certains dispositifs médicaux à une classe supérieure. La notion de sécurité et de risque est plus présente dans la réglementation en demandant davantage de preuves cliniques et pré-clinique sur les dispositifs médicaux. Le bénéfice-risque est ainsi privilégié pour le patient. Les documentations marketing (notice d’utilisation, brochure) sont contrôlées pour éviter toute dérive publicitaire. Une nouveauté du règlement 2017/745 est de renforcer le suivi clinique et la surveillance après commercialisation des DM. Également, la traçabilité est davantage mise en avant avec l’identifiant unique (UDI) et l’utilisation de la plateforme EUDAMED. Sur cette plateforme, dans un but de transparence, le patient peut s’informer sur les dispositifs grâce à un résumé spécifique décrivant les caractéristiques de sécurité et de performances cliniques du produit (SSCP).
2.3 La stratégie règlementaire pour la mise sur le marché européen pour un Dispositif Médical
Lorsqu’un fabricant souhaite mettre un DM sur le marché européen, une stratégie règlementaire doit être définie en prenant en compte les différents services. Cette activité doit être réalisée de manière collaborative pour une meilleure prise en compte des exigences de chaque service. En absence de concertation entre les services, la mise sur le marché d’un DM pourrait provoquer des frais supplémentaires en comptant les frais administratifs, mais aussi le temps de travail supplémentaire causé par un pays supplémentaire. En premier lieu, les pays de commercialisation doivent être définis ainsi que les pays qui seraient visés par le marketing dans un second temps ou un troisième temps.
À savoir, un programme de certification commune existe pour certains pays, permettant un accès à leur marché économique. Le Canada, les États-Unis, le Brésil, le Japon et l’Australie font partie d’un groupe appelé MDSAP, Medical Device Single Audit Program [36]. L’avantage du MDSAP est de réduire le nombre d’audits règlementaires dans le but de mettre un DM sur le marché. Ainsi, les exigences sont respectées pour un nombre étendu de pays tout en réduisant le temps et les ressources pour traiter un dossier.
Cette possibilité permet également d’accélérer la disponibilité d’un DM pour les patients. Dans le cas de la règlementation européenne et du MDSAP, il est demandé d’avoir un système de management de la qualité (SMQ). Pour le cas, de la règlementation européenne, un SMQ respectant la norme harmonisée ISO 13485 :2016 est recommandé, mais non obligatoire (Article 10.9) [3], [22]. Par exemple, si le DM doit être commercialisé en France et en Australie, un certificat valide pour la norme ISO 13485 sera obligatoire. En effet, le MDSAP reconnaît la norme ISO 13485 pour preuve d’un SMQ. Selon la situation, un SMQ d’un fabricant devrait s’inspirer de la norme sans pour autant se faire certifier (cas des classe I). Les exigences demandées par la réglementation européenne et couvertes par la norme ISO 13485 sont :
- Garantir la conformité des produits aux exigences règlementaires
- Documenter le SMQ
- Définir les objectifs qualité
- Définir les responsabilités et autorités
- Définir les responsabilités de la direction
- Assurer la gestion des ressources
- Sélectionner et le contrôler les fournisseurs et sous-traitants
- Maîtriser la réalisation du produit de la conception à la production
- Surveillance, mesure et analyse des produits et du SMQ
- Gestion de la communication interne et externe
- Gérer des actions préventives et correctives
- Identifier les dispositifs et assurer la traçabilité
Si le fabricant possède un certificat de norme ISO 9001 [37], la transition vers la norme ISO 13485 peut être simplifiée [38]. La norme ISO 13485 a été créée sur les fondements de la norme ISO 9001 qui se veut être une norme sur le SMQ dans un secteur d’activité indifférencié [38].
Cet argument peut être intéressant pour les sous-traitants qui seraient réticents à se conformer à la norme ISO 13485.
De plus, le choix d’un sous-traitant ayant déjà un certificat pour la norme ISO 13485 peut être bénéfique pour le fabricant et éviter une négociation entre le fabricant et le sous-traitant pour connaître celui qui prendra en charge les coûts de mise aux normes. Ce problème peut être rencontré dans les PME qui n’ont pas toujours une démarche qualité pouvant être coûteuse pour une petite structure.
Un SMQ repose sur un système de gestion documentaire efficace et une amélioration continue des procédures. Dans ces conditions, un logiciel de gestion documentaire peut être intéressant pour faciliter la révision des documents, la signature ou la disponibilité des dernières versions du document. Cette méthode permet de centraliser l’ensemble des documents SMQ et d’accélérer la recherche d’une procédure lors d’un audit MDSAP ou 2017/745 portants sur le SMQ.
Une autre exigence demandée est la désignation d’une Personne Chargée de Veiller au Respect de la Règlementation (PCVRR). Le rôle de cette personne n’est pas à prendre à la légère. Si une personne devient PCVRR, la personne s’engage à ce que l’entreprise respecte la règlementation 2017/745. La PCVRR s’engage à des poursuites légales en cas de manquement de l’entreprise. Ce poste est accessible à une personne ayant une formation scientifique/règlementaire bac+4 avec une année d’expérience ou 4 ans d’expérience dans les AR. Dans le cas d’une société de moins de 50 salariés, les entreprises peuvent faire sous-traiter le statut. Néanmoins, la sous-traitance peut-être risquée due à l’éloignement du PCVRR par rapport à l’entreprise. D’autre part, ce statut demande une affirmation et une éthique sans faille lorsqu’un incident survient ou que la pression hiérarchique est forte pour céder sur un aspect règlementaire.
Figure 12 : Les grandes étapes pour la mise sur le marché d’un DM et son suivi au cours de sa commercialisation [39].

Dans le cas, où un fabricant souhaiterait mettre sur le marché européen un DM, le DM doit posséder un marquage CE. Pour cela, le fabricant doit fournir une déclaration de conformité comportant les certificats de SMQ et d’évaluation du DT (Figure 12).
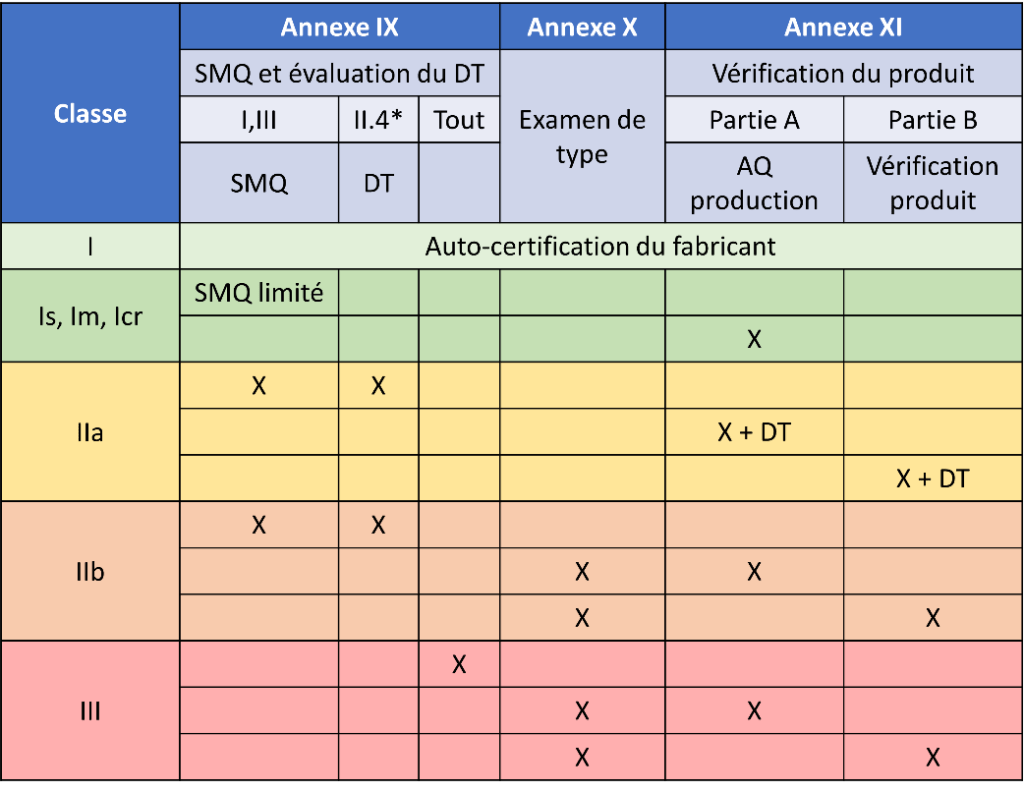
Pour obtenir une certification 2017/745 par un ON, différents choix peuvent être réalisés selon la stratégie employée et la classe du DM à certifier (Figure 13).
Les différentes procédures pour la certification du règlement 2017/745 sont présentées dans les quatre annexes suivantes :
- Annexe IX Évaluation de la conformité sur la base d’un SMQ et de l’évaluation de la DT : Cette voie est la plus complète pour obtenir un marquage CE. Cette annexe correspond à l’annexe II de la directive.
- Annexe X Évaluation de la conformité sur la base de l’examen de type : Cette évaluation repose sur un échantillon représentatif de la production. L’équivalence sous la directive est l’annexe III.
- Annexe XI Évaluation de la conformité sur la base de la vérification de la conformité du produit : La partie A évalue l’assurance qualité de la production. Tandis que la partie B vérifie la fabrication de chaque DM. Cette annexe peut être comparée avec l’annexe V (Partie A) et l’annexe IV (Partie B) de la directive.
- Annexe XIII Procédure pour les dispositifs sur mesure
À connaître, si le DM implantable possède une substance médicamenteuse, la procédure de certification devra suivre la procédure pour les classes III, mais également l’annexe IX.II.5.2.
Figure 13 : Les différentes procédures de certification pour le marquage CE [40]. * L’évaluation de la documentation est différente selon la classe du DM. Les IIa sont évalués par catégorie de DM. Les IIb sont évalués par groupe de DM (utilisation et technologies identiques). Chaque DM de classe IIb implantable et III est évalué. Pour le cas des Is, Im ou Ir, une évaluation du SMQ est limitée aux procédés spéciaux comme la stérilisation.
Au sein de la DT, dans les preuves attendues pour la gestion des risques, des données cliniques sur le DM sont à fournir. Si le DM à certifier est considéré comme innovant, l’absence de données cliniques peut engendrer le besoin d’une investigation clinique. Une investigation clinique est coûteuse financièrement et peut prendre plusieurs années pour obtenir suffisamment de données cliniques sur le DM. À savoir que les DM de classe III ou les DM implantables doivent obligatoirement procéder à une investigation clinique. Néanmoins, quelques cas particuliers permettent d’éviter l’investigation clinique comme un DM sous MDR ayant un marquage CE ou une modification d’un DM déjà mis sur le marché ou démontrer l’équivalence d’un DM similaire sur la base de sa DT.
Lorsque la stratégie règlementaire a été mise en place, le fabricant doit prendre contact le plus tôt possible avec l’ON. En juin 2022, certains ON ne peuvent pas prendre de nouveaux clients dus à un manque de ressource de leur part.
Le choix de l’ON doit être fait judicieusement en fonction des codes de certification des ON (capacité des ON à certifier certaines parties/annexes du règlement 2017/745), de la classe du DM, de la capacité de certification de l’ON, du coût de certification et des délais de traitement. Un ON est un organisme désigné par les autorités de santé pour évaluer la conformité des DM selon la règlementation 2017/745.
Les ON doivent également procéder à des audits de suivi annuel, des audits de renouvellement des certificats (tous les 3 ans pour le SMQ et tous les 5 ans pour le marquage CE du DM) et des audits inopinés (au moins un tous les 5 ans). La plateforme NANDO peut aider le fabricant à choisir l’ON en fonction des évaluations demandées [32]. En effet, tous les ON ne peuvent pas évaluer toutes les parties du DT. Un ON (Auditing Organization) comme le BSI, le G-MED ou le TÜV Rheinland possédant l’accréditation pour la certification MDSAP peut être également un gain de temps.
Selon la règlementation (Article 50), les ON doivent publier le prix des audits et des certifications. Néanmoins, certains ON ne propose pas clairement leur tarif. De plus, le prix des audits ou des certifications peut être un tarif fixe comme pour l’ON Kiwa ou un tarif horaire comme le BSI. Ces choix de tarifications peuvent jouer sur le coût global du marquage CE. Dans ces conditions, les fabricants doivent se pencher sur la rentabilité de l’ON choisit [41].
Bien que l’ensemble des ON se doivent de suivre la réglementation, des changements sont observés dans la façon de traiter les dossiers entre les ON en interprétant différemment les exigences du règlement. Dans le cadre d’un changement substantiel, ayant un impact sur le risque patient, le signalement auprès du BSI entraîne un marquage CE systématiquement sous MDR. A contrario, Kiwa demande seulement une notification de changement substantiel comme le permet l’article 120. Globalement, dans l’intérêt du fabricant, le fabricant doit s’assurer de sélectionner un ON facilitant la mise sur le marché des dispositifs.
2.4 Les points essentiels à la rédaction d’un Dossier Technique d’un Dispositif Médical
Comme vu précédemment, la constitution d’un DT est une composante essentielle dans l’obtention d’un marquage CE pour un DM (hors classe I).
Même si, la structure d’un DT reste libre dans sa forme. Des ON (GMED, BSI,…) mettent en ligne des guides pour uniformiser la forme et faciliter les preuves et les documentations demandées [42], [43].
De plus, le MDCG fournit aux fabricants des guides d’aide à la compréhension du règlement et des précisions sur des cas particuliers ou des points spécifiques. Globalement, le DT est composé de deux parties. La première consiste à identifier si le produit est un DM et la classe de risque qui lui est associé. Dans la deuxième partie, le but est de montrer que l’ensemble des risques lors de la production et de la mise sur le marché sont contrôlés et surveillés.
- La classification des DM
Tout d’abord, le DT doit décrire les caractéristiques techniques qui définissent le DM. Cette étape se doit de définir également les accessoires ou les autres DM en interaction avec le DM à certifier. Dans un second temps, la classification du DM doit être identifiée. La classification est définie par plusieurs critères (Annexe VIII Chapitre 3) : le temps d’utilisation continu du DM, le caractère invasif, le besoin d’une énergie extérieur au DM (DM actif), le caractère thérapeutique ou de diagnostic, les sites anatomiques critiques d’emploi du DM (Système circulatoire central et système nerveux central). Les DM actifs selon la règlementation ont une définition qui peut être déroutante. La notion d’actif signifie qu’un DM a besoin d’une énergie extérieure à celle d’un humain ou à l’effet de pesanteur pour fonctionner. Les DM ayant besoin d’électricité pour fonctionner sont considérés comme actifs. Inversement, un percuteur ayant besoin d’être frappé sera considéré comme non actif. Lorsque plusieurs règles peuvent s’appliquer pour un DM, la classe de risque la plus élevé est retenue (Figure 14). Cette étape est complexe, car chaque règle peut avoir des exceptions dans sa classification. Le lecteur doit rester vigilant et lire l’ensemble des règles.
Figure 14 : Exemple de questions logiques pour définir la classification d’un DM orthopédique (Source : Auteur). Deux exemples de DM orthopédique sont présentés. La fraise à calcar est un instrument chirurgical réutilisable après stérilisation permettant de fraiser l’os résiduel de la tête fémorale. L’instrument a besoin d’un moteur électrique extérieur pour fonctionner.

- L’approche par les risques
Le but premier du DT est de montrer à l’auditeur que l’ensemble des risques associés aux DM est maîtrisé et réduit lorsqu’il ne peut être éradiqué. La gestion des risques s’appuie sur la norme harmonisée 14971 :2019 [44]. L’ensemble des procédés industriels doivent être évalués. La criticité de chaque risque doit être clairement définie en suivant un barème cohérent. De plus, la gestion des risques ne repose pas que sur le calcul de criticité, mais fait appel à la cohérence des données avec celle rencontrée lors des réclamations clients ou le retour des données cliniques (effets secondaires, contre-indications …). Un point important dans la gestion des risques est l’évaluation des risques biologiques du DM. Différents critères sont évalués comme la biocompatibilité selon la norme ISO 10993-1:2020 [45], les procédés de nettoyage ou de stérilisation. D’autres normes sont revendiquées pour garantir une sécurité supplémentaire des DM. De plus, les matières premières du DM cancérigène, mutagène et toxique pour la reproduction humaine, CMR, doivent être indiquées comme l’alliage de Chrome Cobalt.
- Les documents informatifs pour le DM
Chaque document informatif (brochure patient, technique opératoire, notice d’utilisation, carte implant, étiquette) doit respecter une règle fondamentale est de ne pas utiliser de superlatifs, de rester factuel en s’appuyant sur des données cliniques. Cette partie demande une relecture minutieuse des documents que le marketing pourrait transmettre au patient ou au chirurgien pour s’assurer de la conformité des documents. De plus, la stratégie réglementaire peut impacter ces documents. En effet, selon le règlement, les documents fournis doivent traduit dans la langue de chaque pays où le DM est distribué. La traduction des documents doit être effectuée par un organisme traducteur certifié. Malgré, la certification des organismes, les traducteurs peuvent sur interpréter certains mots ou modifier un texte ayant une importance règlementaire. Une vérification par le service des AR doit être réalisé pour éviter de fautes de traduction critique. Pour cela, un glossaire doit être construit avec les mots règlementaires pour s’assurer que l’équivalent de chaque mot traduit soit similaire selon la règlementation 2017/745. Du point de vue de la stratégie règlementaire, la définition des pays doit être la plus représentative des besoins du marketing. À défaut d’avoir anticipé les pays de mise sur le marché, la rédaction du DT pourrait être freinée dans l’attente des traductions pour les pays demandés. D’autre part, le service des AR doit également vérifier d’autres critères : la cohérence entre le rapport d’évaluation clinique et les indications, contre-indications et la population cible ; la cohérence des normes identifiées entre la notice et la DT ; les pictogrammes règlementaires et normatifs ; les unités de mesure…
- L’évaluation clinique du DM
Comme expliqué dans la partie 2.3, un rapport clinique doit venir soutenir les résultats pré-cliniques en laboratoire. Ce rapport doit contenir des données cliniques. Différentes possibilités sont envisageables : analyser l’état de l’art, s’appuyer sur des données cliniques recueillies lorsque le DM était sous MDD, utiliser des données venant de DM similaire. Le dernier cas, le plus complexe à mettre en œuvre pour les fabricants est l’investigation clinique. Grâce à ces données cliniques, la balance bénéfice/risque peut être évaluée.
- La Surveillance Après Commercialisation et le Suivi Clinique Après Commercialisation
Une nouveauté par rapport à la directive est de surveiller son DM tout au long de sa commercialisation.
Pour cela, la stratégie de surveillance porte sur la surveillance après commercialisation (SAC, chapitre VII) et le suivi clinique après commercialisation (SCAC, annexe XIV.B). Ces documents ont pour intérêt de ne pas évaluer la sécurité et la performance à un instant t, mais d’avoir une réactualisation périodique des données cliniques ou de la matériovigilance. Le SAC et le SCAC sont des documents qui doivent être autoporteurs de sens et font appel à des sources de données similaires. Si le SCAC est orienté sur la revue des données clinique, le SAC s’intéresse aux réclamations clients, aux incidents de DM équivalents. Dans le SAC, un rapport périodique actualisé de sécurité doit être mis à jour tous les ans pour les DM de classe III, IIb et tous les deux ans pour les DM de classe IIa. Pour construire au fil du temps ces documents, une veille des incidents doit être mise en place auprès des autorités de santé. La veille des incidents accessible pour un francophone/anglophone est la newsletter de l’ANSM (France), la base de données DAEN de la TGA (Australie) et la base de données MAUDE de la FDA (Etats-Unis). D’autres bases de données existent, mais parfois la barrière de la langue peut être un frein à la consultation des incidents de certains pays.
- La traçabilité avec la carte implant, l’UDI et la plateforme EUDAMED
Dans le cas des DM implantables, chaque patient doit recevoir une carte implant par DM implantable.
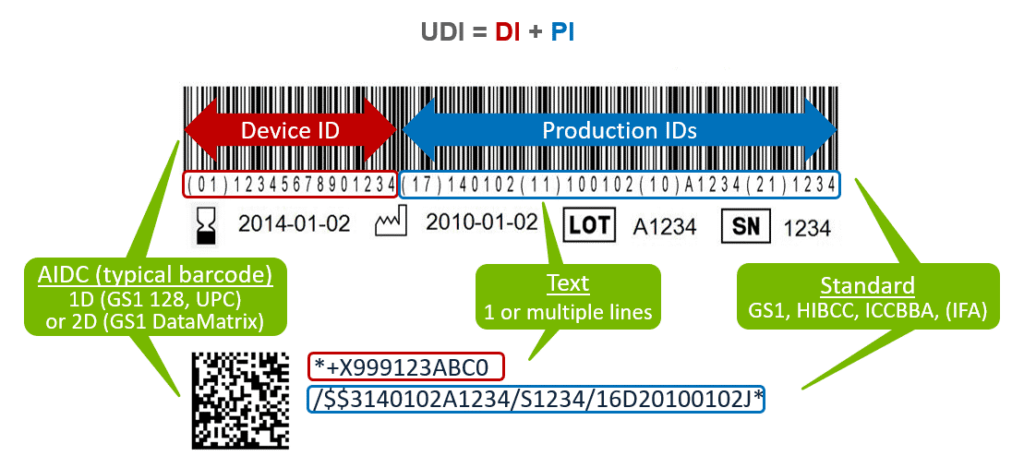
Pour renforcer la traçabilité des DM, la règlementation souhaite mettre en place une base de données, EUDAMED, permettant aux fabricants d’enregistrer leur entreprise et leurs DM (Article 33). Un numéro d’identification, Unique Device Identifier (UDI) est demandé sur la plateforme. Cette UDI est également reconnue par la FDA et utilisable sur la plateforme GUDID. L’UDI est apposé exclusivement par le fabricant sur le DM et sur le conditionnement du DM (Figure 15). L’UDI est composant de deux parties (UDI = UDI-DI + UDI-IP) :
- UDI-DI : Cette partie fixe représente l’Identifiant Dispositif, intégrant l’identifiant du fabricant et commun à tous les DM possédant une même référence commerciale.
- UDI-PI : L’Identifiant Production représente la partie variable de la référence UDI du DM.
Cette partie se compose de plusieurs informations telles que le numéro de lot, le numéro de série, la date d’expiration (pour les dispositifs fournis stériles), la date de fabrication.
Des organismes tels que le GS1 ont la capacité d’attribuer les UDI.
Figure 15 : Exemple d’un UDI composé de l’UDI-DI et de l’UDI-PI [46].
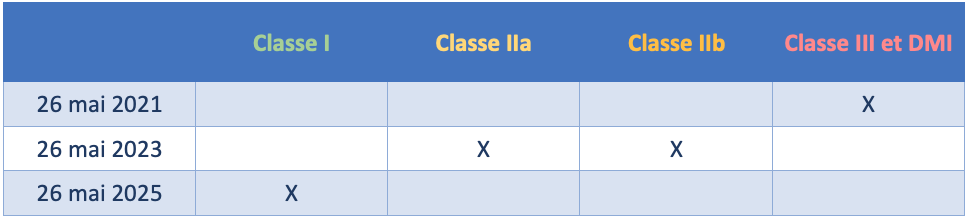
Un autre code est également utilisé et ne doit pas être confondu avec le précédent. Le Basic UDI-DI est le principal identifiant d'un modèle de DM. Cet identifiant est la donnée principale de la base de données EUDAMED et de la documentation réglementaire (certificats MDR, déclaration de conformité MDR, notice, certificats de libre vente, DT et résumé des caractéristiques de sécurité et des performances cliniques). Un numéro est utilisé pour les DM possédant la même destination, la même classe de risque et les mêmes caractéristiques. Le Basic UDI-DI est présent sur la notice, mais pas sur les autres étiquetages. Pour rationaliser les numéros, un système de classement des DM peut être mis en place en suivant les classements EMDN et GMDN des DM. Cependant, ces classements par fonction des DM sont loin d’être optimisés pour le classement des Basic UDI-DI. La base de données EUDAMED est en cours de déploiement. Tous les modules ne sont pas disponibles. Ce retard mis en application de la plateforme a été pris en compte dans le règlement (Article 123.3.d). Des dates ont été définies pour que chaque DM possède un UDI (Figure 16).
Figure 16 : Date de mise en application des UDI pour chaque classe de DM. (Source : Auteur)
2.5 Un outil pour le suivi de la rédaction du Dossier Technique
L’établissement du DT est une mission qui peut devenir complexe dans la manière de suivre son avancement. En effet, si la rédaction du DT et la mise en conformité pour le marquage CE reste un pilotage pour le service des AR, un autre service comme la R&D peut avoir la charge du projet de développement d’un nouveau DM. Cette situation peut entraîner une confusion pour les intervenants entre les contraintes du développement et les exigences règlementaires. La présence de plusieurs services sollicités pour la rédaction et la relecture du DT demande une rigueur dans le suivi de la rédaction du projet. De plus, une confusion sur l’état d’avancement peut-être présente due à un manque de vision globale sur le projet Pour y remédier, un outil de suivi à la rédaction du DT peut être utilisé pour centraliser et clarifier l’état d’avancement de chaque partie/annexe du projet. Chaque acteur peut ainsi connaître l’avancement du projet et anticiper certains points de blocage éventuel comme le manque de ressources humaines, l’attente de relecture ou également l’absence d’un rédacteur. Cet outil se focalise sur le suivi de chaque chapitre du DT et les annexes associées.
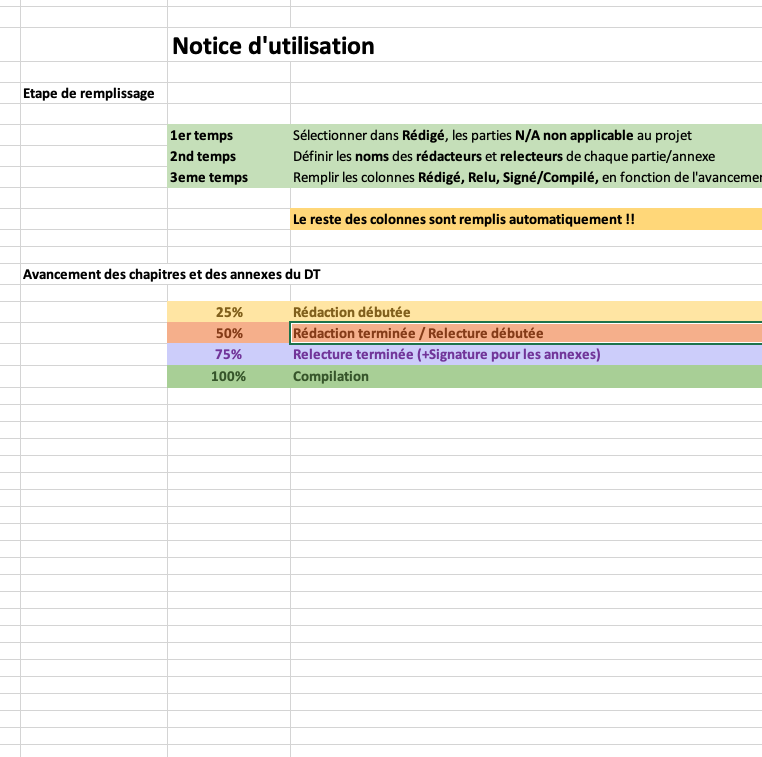
À l’échelle d’un manager dans un service d’AR, l’outil permet au responsable de connaître l’état d’avancement global du projet. Si l’utilisation de l’outil est généralisée à l’ensemble des DT de l’entreprise, le manager peut ainsi obtenir une vision d’ensemble des projets en cours, faciliter sa prise de décision dans l’allocation de ses ressources et communiquer les résultats à sa hiérarchie plus efficacement. L’outil est un fichier Excel comportant quatre onglets. Le premier est la notice d’utilisation (Figure 17).
Figure 17 : Notice d’utilisation de l’outil (Source : Auteur)

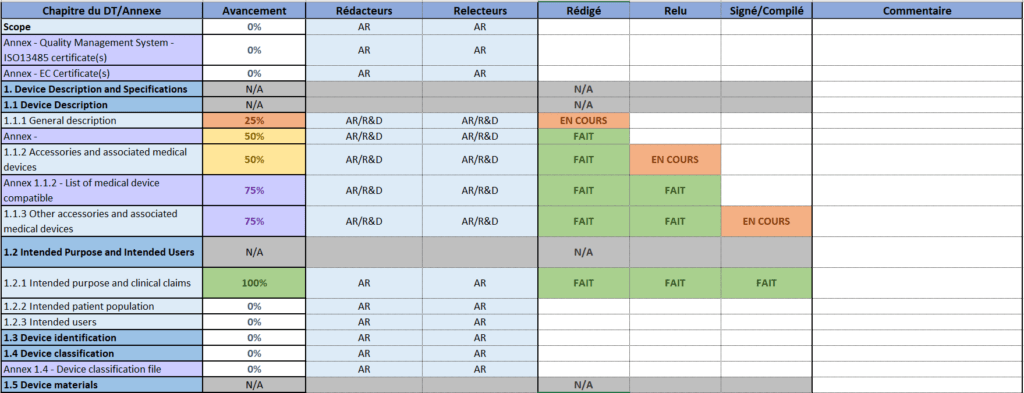
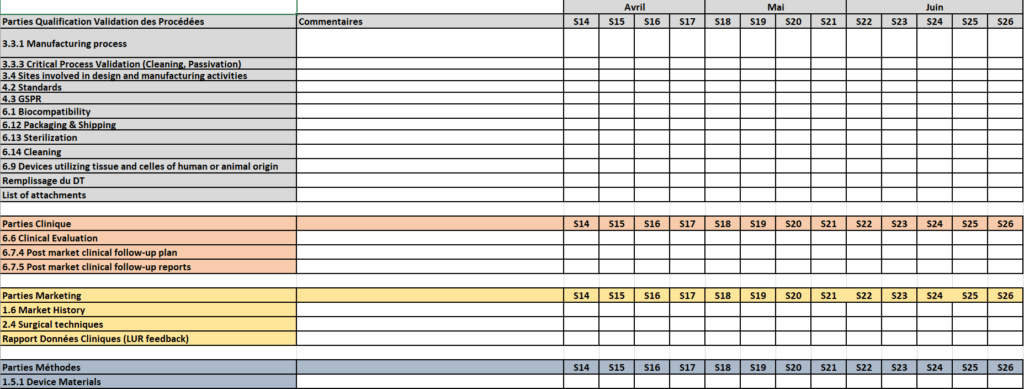
Le deuxième onglet est composé (Figure 18) :
- Des chapitres et des annexes correspondant à l’attente de l’ON pour la rédaction du DT. L’organisation des chapitres est particulièrement adaptée à l’ON du BSI. Cependant, le contenu des chapitres reste universel pour les exigences du règlement 2017/745.
- L’état d’avancement du projet est défini en pourcentage. 25% montrent que la rédaction du chapitre ou de l’annexe a été commencée par le rédacteur. 50% signifient que le chapitre ou l’annexe a été entièrement rédigé et prêt à être relu. 75% correspondent aux relectures et aux corrections finalisées.
100% signifient que le chapitre ou l’annexe a été rédigé, relu et compilé. Dans le cas des annexes, les documents doivent être également signés par l’ensemble des acteurs concernés. Un pourcentage total est disponible pour connaître de manière relative l’avancement de la rédaction du DT (chapitres et annexes). L’avancement est calculé automatiquement en complétant les autres parties de l’onglet.
- Les personnes chargées de la rédaction et de la relecture des différentes parties. Le but est d’identifier les services qui doivent rédiger et relire le chapitre ou l’annexe. Dans un deuxième temps, connaître les représentants pour la partie concernée. Le modèle présente les services responsables de manière arbitraire.
- Le suivi de la rédaction, de la relecture ou de la compilation de la partie ou de l’annexe est mis en avant avec un choix déroulant : Non applicable, En cours de réalisation ou Fait.
- Des commentaires peuvent être ajoutés pour chaque partie ou annexe.
Figure 18 : Suivi de la rédaction du DT par chapitre et annexe (Source : Auteur)
Le troisième onglet est tourné sur la planification temporelle du projet. Chaque partie est présente et associée à un diagramme de Gantt pour connaître les dates et les deadlines. Cette vue permet de donner un aperçu des prochaines échéances sur le projet que ce soit pour la rédaction d’un chapitre ou la compilation des données (Figure 19).
Figure 19 : Planification de la rédaction du DT par service (Source : Auteur)
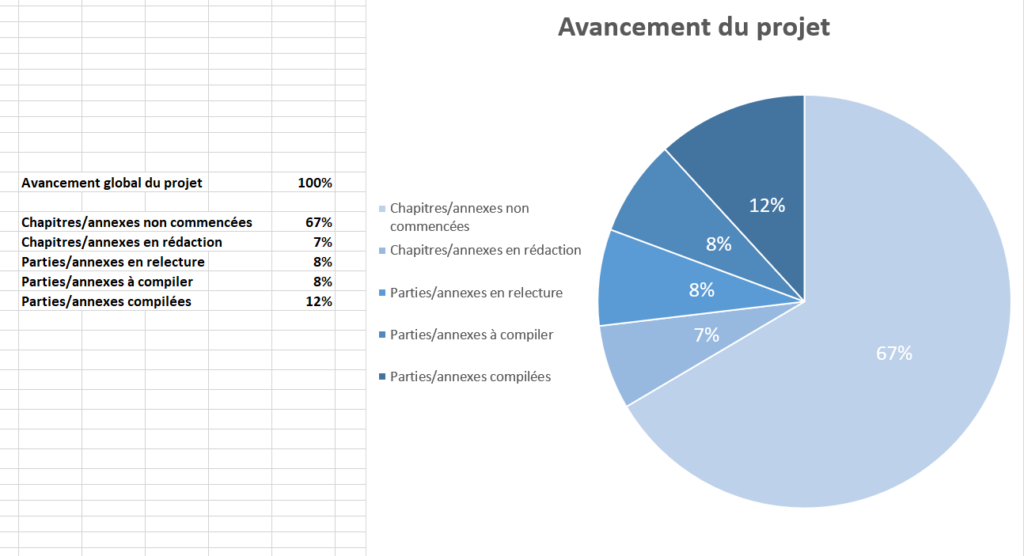
Le quatrième onglet est un bilan de l’avancement du projet. Le but de cette feuille Excel est de donner les points clés d’avancement du projet (Figure 20). Le manager peut visualiser à l’aide d’un graphique camembert :
- Le nombre de parties non commencées
- Le nombre de parties/annexes en rédaction
- Le nombre de parties/annexes en relecture
- L’avancement de la compilation du DT
- L’avancement global du projet
Figure 20 : Suivi global du projet (Source : Auteur)
Conclusion
Le secteur des DM est un domaine régi par de nombreuses exigences réglementaires. Actuellement, dans la transition de la directive vers la règlementation 2017/745, la démarche pour obtenir un marquage reste compliquée. Chaque fabricant doit certifier d’un SMQ et certifier que son DM est conforme à la règlementation par l’audition d’un DT pour la plupart des DM. Dans ces conditions, le besoin de guide pour comprendre l’ensemble des exigences est impératif. Dans ce guide, un outil a été proposé pour faciliter la rédaction de la DT. La règlementation souhaite améliorer la protection des patients en renforçant la traçabilité des DM, la surveillance du marché et la gestion des risques. Malgré, la volonté de la majorité des fabricants de se conformer à la règlementation européenne, de nombreux freins sont présents comme le manque de clarté dans la démarche d’audition des ON ou un nombre réduit d’ON par rapport au secteur des fabricants européens. Le parlement européen montre de plus en plus son inquiétude sur la faisabilité de la mise en application de la réglementation sur l’ensemble des dispositifs dans les temps impartis. La Commission européenne pourrait répondre à cette question, fin juillet 2022 selon Qualitiso.