
IDS171 - Évaluation des dispositifs médicaux en vie réelle
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs




Contacts
- DE BARROS CORREIA Daniel : daniel.debarroscorreia@outlook.fr
- EL YAMANI Loubna : loubna.elyamani.20@gmail.com
- MOISSET Aure : aure.moisset@gmail.com
- NTOMEGNE TCHIMI Steve Harison : harisonentomegne@yahoo.fr
- SOH KOUDJOU Marie Josée : mariejoks@gmail.com
Citation
A rappeler pour tout usage : DE BARROS CORREIA D., EL YAMANI L., MOISSET A., NTOMEGNE TCHIMI S. H., SOH KOUDJOU M. J., « Évaluation des dispositifs médicaux en vie réelle », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS171, https://doi.org/10.34746/ids171, Décembre 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids171
Résumé
Afin d’assurer une sécurité optimale du patient lors de sa prise en charge, les dispositifs médicaux doivent répondre aux exigences de performance et de sécurité énoncées dans le règlement (UE) 2017/745.
La preuve du respect de ces exigences va le plus souvent être apportée par des essais cliniques randomisés. Bien que ces essais soient les plus utilisés dans le domaine médical grâce au niveau de preuve qu’ils apportent, leurs résultats peuvent toutefois être complétés par des données de vie réelle récoltées en pratique de soins courants. Encore trop peu utilisées dans le domaine des dispositifs médicaux, ces données peuvent, à l’avenir, représenter un intérêt majeur pour les fabricants.
C’est dans ce contexte que s’inscrit ce projet afin d’informer les fabricants de dispositifs médicaux ainsi que les différents acteurs de ce domaine (tels que les industries ou les chercheurs) quant à l’importance de ces données de vie réelle et de la manière dont elles sont déjà utilisées aujourd’hui. L’accent a également été mis sur l’aspect réglementaire concernant le traitement de ces données qui constituent des données dites « sensibles » et doivent de ce fait respecter le Règlement Général sur la Protection des Données (RGPD).
Ce mémoire propose un état des lieux des données de vie réelle des dispositifs médicaux existants, avec une cartographie permettant d’étudier ces différentes données et leur collecte. Cette analyse est accompagnée de problématiques auxquelles ces données peuvent amener des éléments de réponse tout au long du cycle de vie du dispositif.
Abstract
In order to ensure optimal patient safety during care, medical devices must meet the performance and safety requirements set forth in Regulation (EU) 2017/745.
Proof of compliance with these requirements is most often going to be provided by randomized clinical trials. Although these trials are the most widely used in the medical field due to the level of evidence they provide, their results can however be complemented by real-life data collected in routine care practice. Still not widely used in the field of medical devices, these data may, in the future, represent a major interest for manufacturers.
It is in this context that this project was set up to inform medical device manufacturers as well as the various actors in this field (such as industries or researchers) about the importance of this real-life data and the way it is already used today. Emphasis was also placed on the regulatory aspect concerning the processing of this data, which constitutes so-called "sensitive" data and must therefore comply with the General Data Protection Regulation (GDPR).
This dissertation proposes an inventory of the real-life data of existing medical devices, with a mapping to study these different data and their collection. This analysis is accompanied by issues to which these data can provide answers throughout the life cycle of the device.
Remerciements
À l'issue de ce travail nous tenons à adresser nos plus sincères remerciements à Jean Mathieu Prot, responsable de formation, pour nous avoir encadré tout au long de ce projet. Merci d’avoir répondu à toutes nos questions, merci de nous avoir aidé à structurer notre réflexion, nos idées et nos questionnements.
Nous remercions également Isabelle Claude, Gilbert Farges et Julie Follet pour tous les conseils reçus lors de nos présentations de projet, pour votre aide lors de la découverte de nouveaux outils ainsi que les encouragements qui nous ont vraiment permis de progresser et d’élargir nos horizons.
Nous remercions également Béatrice Koenig pour tous les conseils délivrés pour la rédaction correcte d’une bibliographie, pour les corrections apportées, pour votre rigueur et tout le savoir que vous nous avez transmis tout au long de ce projet.
Enfin, nous remercions tous les acteurs impliqués dans la naissance de ce projet. Au travers de conseils prodigués, de réponses lors d’entretiens ou encore de suggestions d’améliorations et de questionnements, pour votre temps et vos retours toujours bienveillants nous vous en remercions profondément. Tout cela nous a permis de construire ce mémoire d’intelligence méthodologique.
Téléchargements



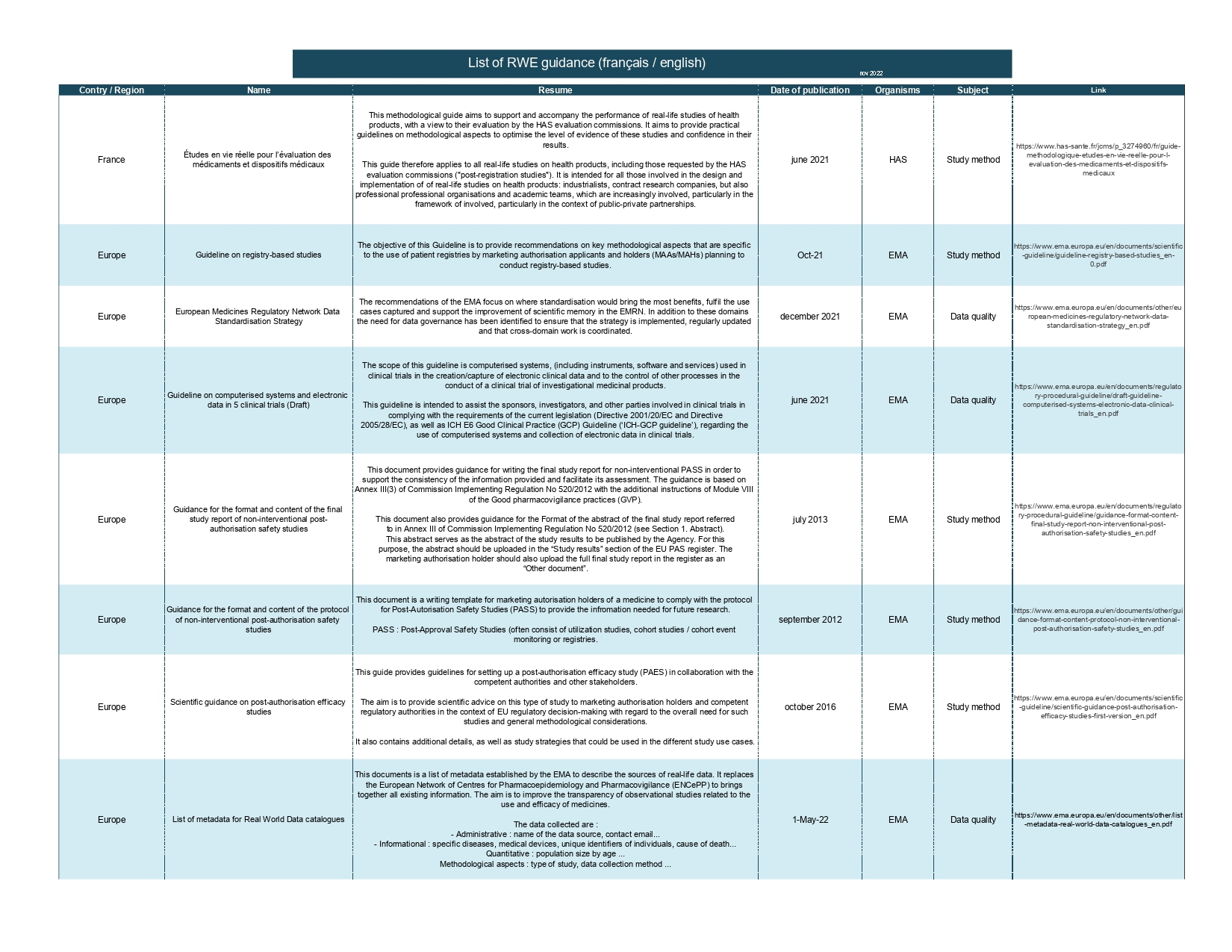
liste de guides et de référentiels portant sur les données de vie réelle en français et en anglais pour approfondir les connaissances sur le sujet
Liste des abréviations
| AIPD | Analyse d’Impact relative à la Protection des Données |
| ASA | Amélioration du Service Attendu |
| ASCO | American Society of Clinical Oncology |
| ASMR | Amélioration du Service Médical Rendu |
| ASR | Amélioration du Service Rendu |
| CCAM | Classification Commune des Actes Médicaux |
| CEPS | Comité Économique des Produits de Santé |
| CHU | Centre Hospitalo-Universitaire |
| CNEDiMTS | Commission Nationale d’Évaluation des Dispositifs Médicaux et des Technologies de Santé |
| CNIL | Commission Nationale de l’Informatique et des Libertés |
| CNP | Conseil National Professionnel |
| CT | Commission de Transparence |
| DM | Dispositif Médical |
| DPO | Data Protection Officer |
| EDSH | Entrepôts de Données de Santé Hospitaliers |
| EPI | Étude Post-Inscription |
| ERC | Essais Randomisés Contrôlés |
| GHS | Groupe Homogène de Séjour |
| HAS | Haute Autorité de Santé |
| IA | Intelligence Artificielle |
| LPPR | Liste des Produits et des Prestations Remboursables |
| NABM | Nomenclature des Actes de Biologie Médicale |
| NGAP | Nomenclature Générale des Actes Professionnels |
| OMS | Organisation Mondiale de la Santé |
| PMSI | Programme de Médicalisation des Systèmes d’Information |
| PROMs | Patient-Reported Outcome Measures |
| RGPD | Règlement Général sur la Protection des Données |
| SA | Service Attendu |
| SCAC | Suivi Clinique Après Commercialisation |
| SMR | Service Médical Rendu |
| SNDS | Système National des Données de Santé |
| SNIIRAM | Système National d’Information Inter-Régimes de l’Assurance Maladie |
| SR | Service Rendu |
| UNCAM | Union Nationale des Caisses d’Assurance Maladie |
Glossaire
Acte professionnel : geste réalisé par un professionnel de santé qui peut avoir recours à du matériel dans un but de diagnostic, de prévention, de traitement ou de rééducation [1].
Biais de confusion : déterminants qui faussent la relation de cause à effet.
Biais de sélection : les sujets réellement observés dans l’enquête ne constituent pas un groupe représentatif des populations étudiées.
Cohorte : groupe de sujets aux caractéristiques communes, qui sont suivis individuellement de manière prospective.
Destination : utilisation à laquelle un dispositif est destiné d'après les indications fournies par le fabricant sur l'étiquette, dans la notice d'utilisation ou dans les documents ou indications publicitaires ou de vente, et comme celles présentées par le fabricant dans l'évaluation clinique [2].
Dispositif médical : tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens [2].
Données à caractère personnel : toute information se rapportant à une personne physique identifiée ou identifiable ; est réputée être « une personne physique identifiable » une personne physique qui peut être identifiée, directement ou indirectement, notamment par référence à un identifiant, tel qu’un nom, un numéro d’identification, des données de localisation, un identifiant en ligne, ou à un ou plusieurs éléments spécifiques propres à son identité physique, physiologique, génétique, psychique, économique, culturelle ou sociale [3].
Données de santé : données relatives à la santé physique ou mentale, passée, présente ou future, d’une personne physique (y compris la prestation de services de soins de santé) qui révèlent des informations sur l’état de santé de cette personne [3].
Essai randomisé en double aveugle : étude où les participants sont attribués à un groupe de manière aléatoire (randomisé) et où les patients comme les médecins ne savent pas à quels groupes les participants appartiennent (double aveugle).
Évaluation clinique : processus systématique et planifié visant à produire, collecter, analyser et évaluer en continu les données cliniques relatives à un dispositif afin de vérifier la sécurité et les performances, y compris les bénéfices cliniques, de celui-ci lorsqu’il est utilisé conformément à la destination prévue par le fabricant [2].
Performance : capacité d'un dispositif à atteindre la destination indiquée par le fabricant [2].
Pseudonymisation : traitement de données à caractère personnel de telle façon que celles-ci ne puissent plus être attribuées à une personne concernée précise sans avoir recours à̀ des informations supplémentaires, pour autant que ces informations supplémentaires soient conservées séparément et soumises à̀ des mesures techniques et organisationnelles afin de garantir que les données à caractère personnel ne sont pas attribuées à une personne physique identifiée ou identifiable [3].
Questionnaire EQ-5D : questionnaire générique pouvant être utilisé lors d’une évaluation clinique. Il permet une mesure standardisée de la qualité de vie des patients liée à leur santé.
Registre : recueil continu et exhaustif de données, dans une population géographiquement définie.
Suivi après commercialisation : ensemble des activités réalisées par les fabricants, en collaboration avec d'autres opérateurs économiques, pour établir et tenir à jour une procédure systématique de collecte proactive de données sur leurs dispositifs mis sur le marché, mis à disposition sur le marché ou mis en service de manière à dresser le bilan de leur utilisation, dans le but de repérer toute nécessité d'appliquer immédiatement une mesure préventive ou corrective [2].
Système National des Données de Santé (SNDS) : regroupement de données médico-administratives pseudonymisées des soins remboursés de la population française. Cet entrepôt de données permet notamment de chaîner les données de l’assurance maladie.
Mémoire - Évaluation des dispositifs médicaux en vie réelle
I) Introduction
Le secteur des dispositifs médicaux (DM) est un domaine très hétérogène dans lequel des produits à la pointe de la technologie comme les organes artificiels ou les équipements d’imagerie médicale et de monitorage côtoient des dispositifs plus simples comme les seringues, les pansements, les lunettes ou encore les lits d’hôpitaux. Au fil des années et de leurs évolutions technologiques, les dispositifs médicaux se sont imposés comme des outils incontournables de l’univers médical. En France, le secteur ne représente pas moins de 2 millions de références avec 1 440 entreprises réalisant un chiffre d’affaires de 30,7 milliards d’€ en 2021 [4].
Pour rappel, le règlement (UE) 2017/745 définit un dispositif médical comme tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé chez l’homme pour une ou plusieurs des fins médicales suivantes :
- Diagnostic, contrôle, traitement ou atténuation d’une maladie, d’une blessure ou d’un handicap,
- Investigation, remplacement ou modification d’une structure, d’une fonction anatomique, d’un processus ou état physiologique ou pathologique,
- Communication d’informations à l’aide d’un examen in vitro d’échantillons issus du corps humain,
- La maitrise de la conception,
Et dont l’action principale voulue n’est pas obtenue par des moyens pharmacologiques, immunologiques ni par métabolisme.
Le développement de ces dispositifs est très fortement lié à la découverte de nouveaux matériaux à l’image des prothèses de hanches, à l’origine en verre puis en téflon, et aujourd’hui, en céramique. L’avènement de la microélectronique et de l’informatique dans les années 70 ouvre le champ des possibles et permet la miniaturisation des dispositifs tout en augmentant leurs fonctionnalités (exemple des défibrillateurs externes pesant à l’origine 18 kg et étant aujourd’hui automatisés et implantables).
Ces dispositifs interviennent tout au long du parcours de soins des patients et ont pour mission de permettre à la population d’avoir la meilleure santé possible. Par conséquent, il est alors primordial de s’assurer qu’ils répondent aux exigences de sécurité et de performances applicables du règlement (UE) 2017/745 dans les conditions normales d’utilisation d’une part, pour qu’ils soient commercialisables, et d’autre part, pour le bien-être des patients et la sécurité des utilisateurs.
De plus, les fabricants doivent évaluer les effets secondaires indésirables, ainsi que le rapport bénéfice/risque du dispositif. La confirmation de ces deux aspects se base sur des données cliniques apportant une preuve clinique suffisante.
Dans ce cadre, le règlement (UE) 2017/745 exige la mise en place d’une évaluation clinique pour s’assurer de la conformité du DM aux exigences de sécurité et de performances pour l’ensemble des dispositifs. Le niveau de preuves nécessaire à démontrer doit être déterminé, justifié par le fabricant, et approprié au dispositif en prenant en compte sa destination et ses caractéristiques [5].
Cette évaluation clinique est réalisée sur l’ensemble du cycle de vie du dispositif en tant que processus actif. Lors de son développement et reposant sur les intentions médicales revendiquées par le fabricant, l’évaluation clinique permet d’identifier les données à générer afin de pouvoir commercialiser le dispositif. Elle est obligatoire et doit être mise à jour de manière systématique en continu dans le cadre de l’obtention du marquage CE.
Malheureusement, les essais cliniques pouvant être réalisés lors de ces évaluations comme source de données cliniques ne sont pas sans failles, des doutes peuvent alors persister quant à la sécurité et à la performance du dispositif. C’est alors qu’entrent en jeu les données de vie réelle qui vont permettre de renforcer la robustesse des résultats de ces essais [6].
Pour prouver la sécurité et l’efficacité d’un dispositif, les Essais Randomisés Contrôlés (ERC) constituent une approche systémique dans la résolution des problèmes cliniques en intégrant les meilleures preuves de recherches disponibles à l’expertise clinique et aux valeurs du patient [7]. Certains modèles de recherches sont plus robustes que d’autres en ce qui concerne la recherche d’efficacité des interventions. Une hiérarchie des preuves est illustrée en figure 1 à travers une pyramide qui a été mise en place afin d’avoir une vue globale sur la puissance des preuves dans les tests d’efficacité d’un traitement ou d’une intervention.
Figure 1 : Hiérarchie des preuves de recherches [8]
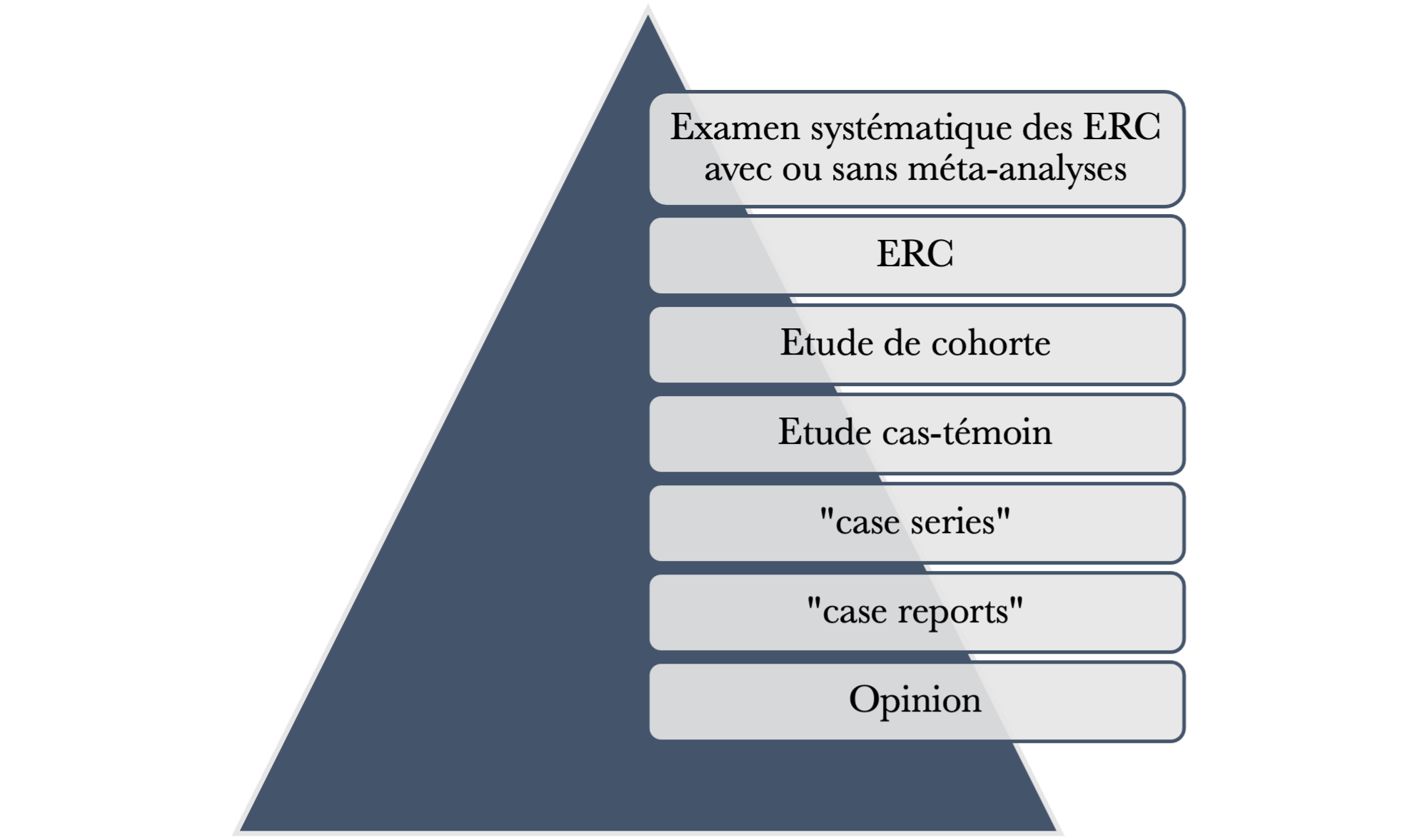
Les preuves les plus faibles sont les méthodes d’observation simples. Plus les preuves se trouvent à la base de la pyramide, plus leur utilisation peut être responsable d’introduction de biais. Les ERC sont considérés comme une méthodologie robuste grâce à leur minimisation de risque de facteurs de confusion pouvant influencer les résultats. Ainsi les résultats produits par ce type d’études se rapprochent plus de la réalité [8], car ces études sont réalisées en suivant un protocole avec des critères très stricts qui ont l’avantage de ne pas présenter de biais de sélection et de biais de confusion limitant ainsi les incertitudes des résultats.
Bien que ces essais soient les plus probants lorsqu’il s’agit d’apporter des preuves d’efficacité, ils comportent encore certaines limites pouvant remettre en cause leurs résultats. Ces études sont entre autres réalisées sur un court temps et sur une population très réduite par rapport à la population qui à terme pourra bénéficier de ce traitement.
Nous pouvons retrouver un certain nombre de limites à propos desquelles les données de vie réelle peuvent être utilisées afin d’apporter une certaine robustesse aux résultats d’efficacité finaux.
Ces données étant nouvelles, il est primordial de définir réellement quelles sont ces données de vie réelle et où nous pouvons les retrouver. Leur intérêt et leur utilisation lors de l’évaluation clinique d’un dispositif médical sera ensuite analysé. Enfin, afin d’assurer la continuité ainsi que la rapidité des résultats, il est important de proposer aux fabricants un outil/ protocole pour les accompagner à la compréhension de l’évaluation des dispositifs médicaux en vie réelle suivant un processus bien défini, ce qui fera l’objet de notre étude par la suite en répondant à ces questions :
- Que sont les données de vie réelle ?
- Comment les recenser ?
- Comment les utiliser au cours du cycle de vie du DM ?
- Quelle est la réglementation autour de ces données ?
II) Les données de vie réelle
A) Définition
Les « Données en vie réelle » ou « Données observationnelles », correspondent à l’ensemble des données issues de la pratique de soins courants. Elles sont produites en dehors de tout cadre expérimental comme les essais randomisés contrôlés et impliquent l’utilisation, l’efficacité, ou la tolérance d’un dispositif médical. Elles sont recueillies lors des soins de routine sur les patients, reflétant ainsi la pratique quotidienne. La nouveauté de l’exploitation de ces données constitue un défi majeur dans l’évaluation des dispositifs médicaux.
Avant de commencer la collecte de ces données, il faut :
- Justifier l’intérêt de l’étude, pour avoir l’autorisation des autorités compétentes (ANSM, CNIL…) et des parties intéressées (participants…),
- Vérifier la crédibilité et la qualité des données et les valider avant leur utilisation.
B) Types de données de vie réelle
Il existe différents types de données de vie réelle présentés comme suit en Figure 2 [9]–[11] :
Figure 2 : Les différents types de données de vie réelle [source : Auteur]
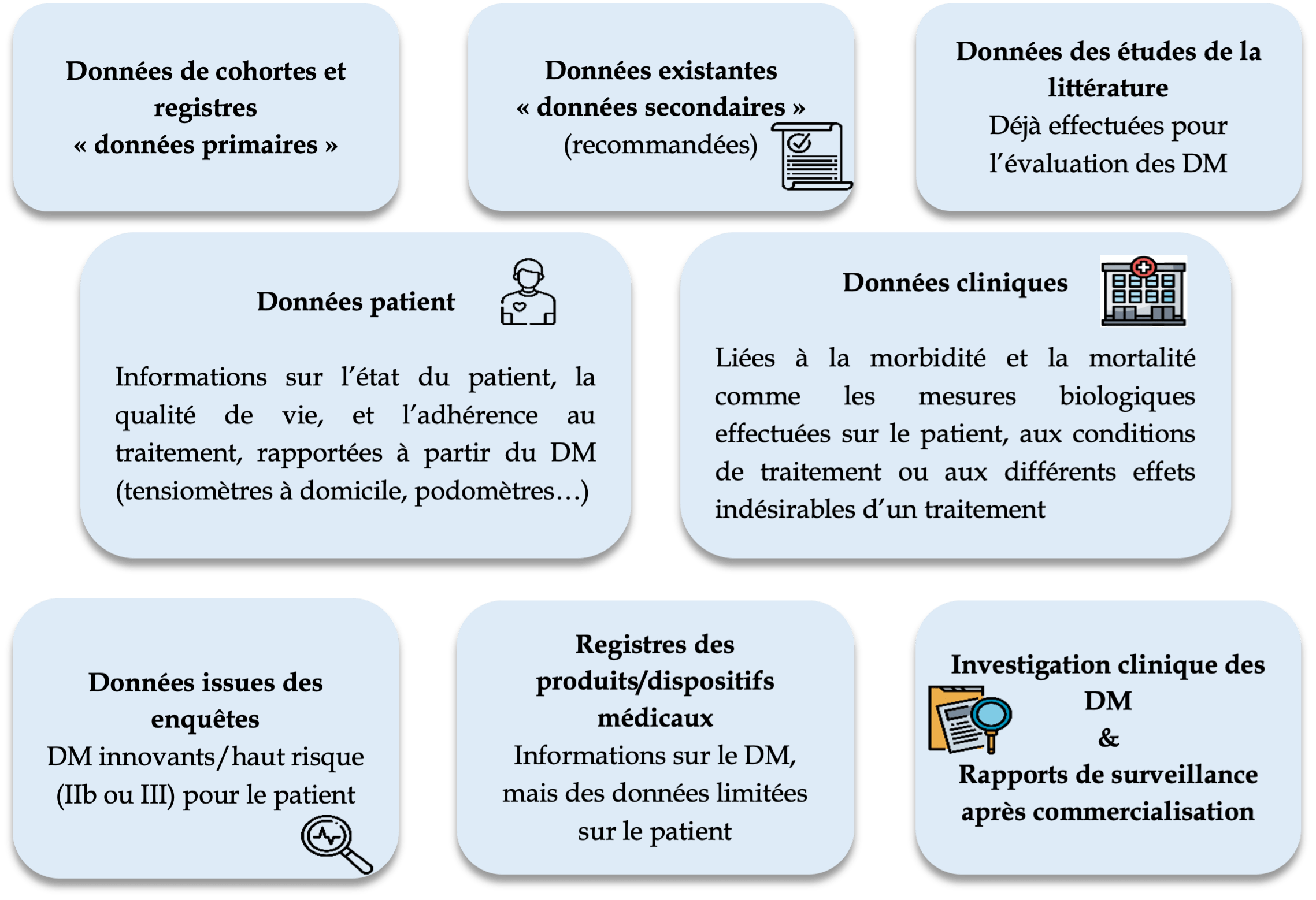
Les données en vie réelle concernent également toutes les données épidémiologiques sur les pathologies, leurs traitements en passant par l’historique de la maladie, l’efficience et l’efficacité thérapeutique du traitement. Ces données peuvent provenir de plusieurs sources différentes.
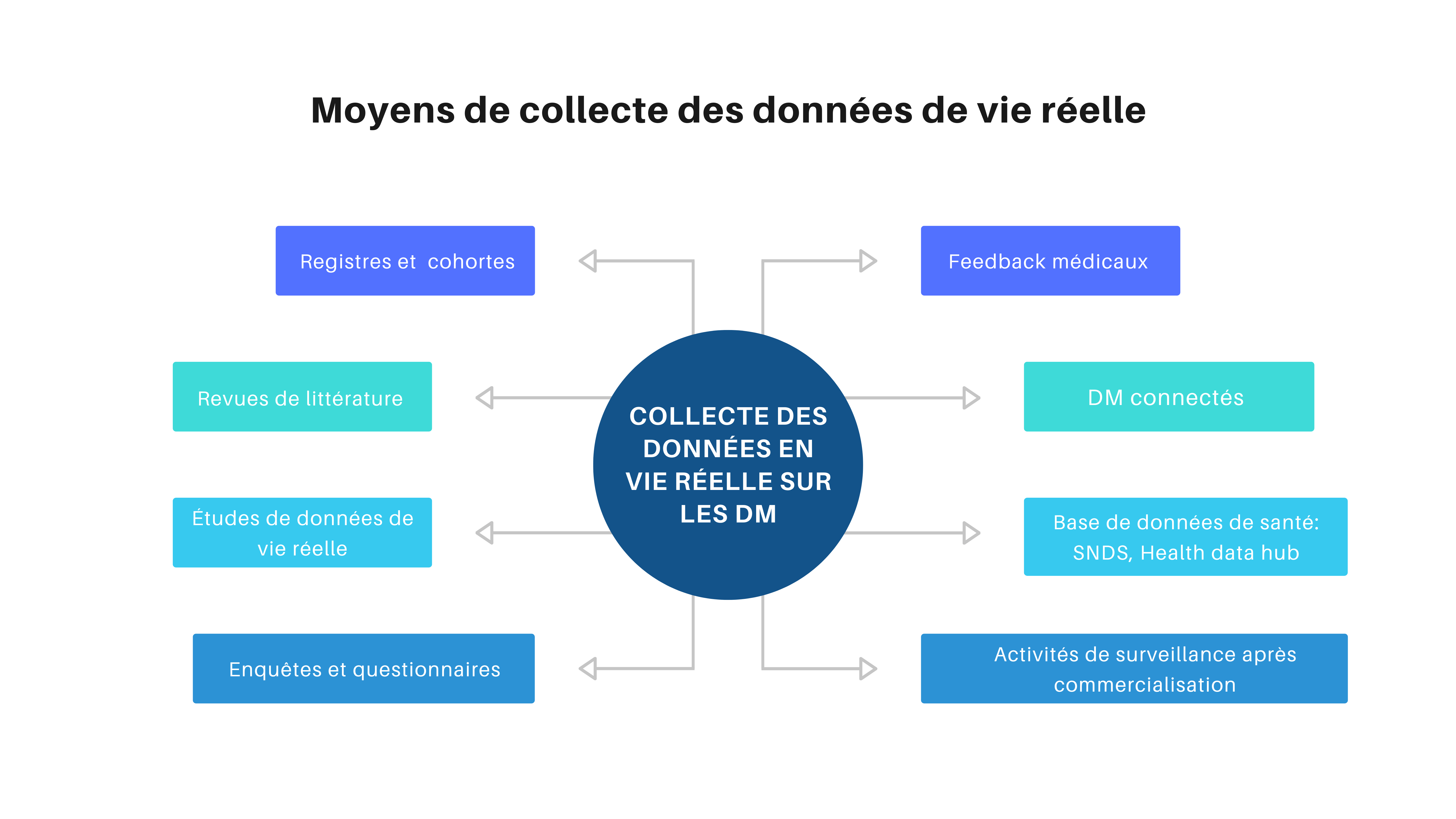
C) Collecte des données de vie réelle
Les données de vie réelle des DM peuvent entre autres découler de nombreuses sources comme présenté en Figure 3 :
Figure 3 : Les moyens de collecte des données de vie réelle [source : Auteur]
1) Les questionnaires
Les questionnaires (suivis d’une documentation sur la méthode de notation, d’analyse et d’interprétation des données collectées) peuvent servir de moyen de collecte pour les données de vie réelle. Ils offrent la possibilité de pouvoir mesurer la qualité de vie, les symptômes ressenties et la capacité fonctionnelle (déplacement…) du patient lié au traitement.
Il existe entre autres les questionnaires PROMs (Patient-Reported Outcome Measures) qui mesurent le résultat des soins perçu par le patient sans interprétation des réponses par un autre individu. Ces mesures peuvent se faire soit en auto-administration, c’est-à-dire que le patient répond seul au questionnaire, soit lors d’entretiens durant lesquels le patient est questionné par un professionnel qui saisit ses réponses [12].
De plus, ces mesures peuvent également se faire en collectant les retours des usagers comme des plaintes sur le traitement, des commentaires sur les réseaux sociaux ou grâce à la méthode du patient traceur. Cette méthode consiste à analyser le parcours pluriprofessionnel et pluridisciplinaire d’un patient en le comparant à des référentiels (réglementation, bonnes pratiques). Elle croise la perception du patient avec le point de vue des professionnels pour en tirer des conclusions sur les organisations, les traitements…
2) Les dispositifs médicaux eux-mêmes (cas des dm connectés)
Les données de vie réelle peuvent aussi être recueillies par des dispositifs médicaux connectés (saturomètre, pacemaker, tensiomètre…) qui peuvent contenir des logiciels classés comme DM. Cette catégorie de dispositifs est en plein essor aujourd’hui et permet aux fabricants de collecter ce type de données afin de pouvoir évaluer les performances cliniques de leurs dispositifs.
3) Les bases de données
Les données de vie réelle peuvent être collectées via le Health Data Hub dont le but est de faciliter l’accès aux données de santé respectueuses de la vie privée et qui pourraient être utilisées pour des recherches d’intérêt public par des acteurs privés ou publics. Cette base de données regroupe entre autres le Système National des Données de Santé (SNDS), le Système National d’Information Inter-Régimes de l’Assurance Maladie (SNIIRAM) ou encore le Programme de Médicalisation des Systèmes d’Information (PMSI) [13].
Le système national de données de santé [1]
Si un fabricant souhaite évaluer son dispositif uniquement sur la base des données du SNDS, cela sera difficile car le dispositif ne sera jamais étiqueté clairement, sauf s’il est le seul dispositif du marché à générer un type d’acte qui lui est spécifique. En revanche, cette base de données révèle tout son intérêt pour l’évaluation des dispositifs médicaux lorsqu’elle est utilisée de manière indirecte.
Dans le cas où le fabricant possèderait des données permettant d’identifier directement les individus, il pourra faire une demande auprès de la Commission Nationale de l’Informatique et des Libertés (CNIL) pour chaîner ses données à celles du SNDS. L’objectif de son étude doit être clair, il ne doit pas faire de la promotion de son dispositif mais bien de l’évaluation. Cette demande doit être déposée sur le Health Data Hub et sera soumise au Comité Éthique et Scientifique pour les Recherches, les Études et les Évaluations dans le domaine de la Santé (CESREES). Si l’avis de ce dernier est favorable, le projet sera transmis à la CNIL qui autorise ou non le traitement.
Du fait de la contrainte réglementaire, il est rare que les fabricants hébergent des données directement identifiantes telles que le numéro de sécurité sociale, les noms ou encore prénoms des individus. Toutefois, ils peuvent posséder des données indirectement identifiantes telles que la date de naissance, la date de décès, le lieu de résidence, la date ainsi que le lieu de soins etc. C’est à partir de ces variables communes que les deux bases peuvent être chaînées indirectement entre elles.
Ces données peuvent être extrêmement intéressantes pour évaluer un DM, car elles permettent d’avoir accès à toutes les dates et raisons d’hospitalisation, les recours aux urgences, les prises de médicaments (anti-douleurs etc.), les fréquences de consultations (même si la raison de la consultation reste inconnue) et les types d’examens. Il sera ainsi possible d’identifier quelques évènements pouvant être qualifiés « d’indésirables » ou « d’effets secondaires » surtout s’ils se répètent pour plusieurs patients.
[1] Réflexion menée par Claire Imbaud, ingénieur de recherche.
4) Les études de données de vie réelle
Il existe plusieurs types d’études et avant de déterminer les types d’études à utiliser, il est nécessaire de les justifier en fonction des thèmes de recherche identifiés, et de leur capacité à répondre de manière appropriée à la demande de données formulée par la Haute Autorité de Santé (HAS).
- Études observationnelles descriptives et non comparatives : elles permettent de documenter l’utilisation du DM en pratique dans la vie réelle, en décrivant les modalités d’utilisation du DM en conditions réelles (caractéristiques des patients et des prescripteurs, stratégies thérapeutiques, etc.), l’évolution clinique des patients ayant utilisé le DM au cours du temps, la qualité de vie ressentie par le patient ou la sécurité d’utilisation du DM [9],
- Études comparatives : elles permettent d’analyser l’efficacité, la sécurité, l’efficience du DM en le comparant avec un autre DM ou une autre modalité de traitement (modalité IRM vs Scanner). Ces types d’études peuvent également être des études en vie réelle comparatives, des essais pragmatiques, ainsi que des comparaisons avant/après pour décrire l’impact d’un changement de prise en charge [9],
- Études pour l’évaluation des dispositifs médicaux (non spécifiquement des études observationnelles) : recommandations pour l’évaluation de l’efficacité clinique comparative des dispositifs médicaux [14],
- Études de suivi post-mise sur le marché (études observationnelles prospectives ou registres) : elles permettent de recueillir des données complémentaires au cours de l’utilisation du DM en situation des soins quotidiens [14].
La HAS recommande d’enregistrer l’étude dans des bases de données publiques, comme sur le portail de l’Organisation Mondiale de la Santé (OMS), et suggère également que les résultats soient publiés dans une revue scientifique, et que les fabricants de DM collaborent avec le Health Data Hub afin de stocker les données d’études réelles pour une utilisation ultérieure [9].
Les données produites par ce genre d’études ne visent pas à remplacer les essais randomisés contrôlés mais interviennent davantage comme complément de preuve. Pour mieux illustrer la complémentarité de ces deux types d’études, un tableau récapitulatif des spécificités des deux types d’études est présent en Tableau 1. Ces études vont venir étoffer les connaissances sur le contexte de soins apportés aux patients, de leur maladie ainsi que sur l’efficacité et l’efficience du DM.
Les données de vie réelle vont davantage refléter la réalité d’utilisation du DM en s’affranchissant par exemple de la surveillance accrue des essais cliniques, ou encore en émettant des données d’efficacité sur des personnes non incluses dans les essais cliniques. Ainsi ces données pourront nous permettre de vérifier dans quelles conditions les résultats de l’essai clinique sont vérifiés.
Les ERC en double insu ont longtemps été considérés comme l’une des seules sources fiables en tant que preuve d’efficacité et de sécurité des DM. Les études observationnelles quant à elles ont longuement été délaissées en raison de leur apport de preuves jugé trop faible par la communauté scientifique. En 2013, le département de la santé et des services sociaux des États-Unis a publié un guide démontrant l’importance des données de vie réelle en abordant plusieurs protocoles suivant la situation du DM/produit de santé [15]. En 2014, COCHRANE (organisation) a également menée une étude visant à comparer les résultats de ces deux types d’essais et a conclu à une absence de différence renforçant ainsi l’utilisation des données de vie réelle dans les dossiers d’évaluation [12].
Tableau 1 : Récapitulatif des spécificités des études de vie réelle et des essais randomisés contrôlés [source : Auteur]
| Étude en vie réelle | Essai randomisé contrôlé | |
| Type d’étude | Observationnelle Non interventionnelle | Expérimentale Interventionnelle |
| Objectif principal | Efficience | Efficacité, sécurité, qualité |
| Perspective | Le malade est au centre de l’étude | La maladie est au centre de l’étude |
| Population observées | Large et non restreinte | Groupe homogène et restreint |
| Suivi | Non | Important |
| Durée de suivi | Moyen – Long terme | Court terme |
| Quand | Pendant le développement et toute la durée d’utilisation du DM | Uniquement durant le développement du DM |
| Comparateurs | Pas de comparateur défini : alternatives thérapeutiques prescrites dans la pratique courante | Placebo ou gold standard |
| Randomisation | Non | Oui |
| Coût | € | €€€ |
| Délai de mise en place | + | +++ |
| Validité de l’étude | Méthodologie contestée mais validité externe importante | Validité interne importante |
D) Exemples d'utilisation des données de vie réelle
Ces données possèdent une utilisation vaste et mondiale dont voici quelques exemples :
Exemple 1 : la montre connectée dans le suivi de la maladie de Huntington [16]
Intel et Teva (entreprise pharmaceutique) collaborent pour développer une montre connectée qui sera liée à une application permettant le suivi en temps réel des patients atteints de la maladie de Huntington. Le stockage et l’interprétation des données collectées sera fait sur la plateforme Cloud développée par Intel.
Le projet a pour but de générer des données de vie réelle objectives et continues concernant la progression de la maladie, ses effets et l’impact des traitements sur la qualité de vie des patients.
Exemple 2 : le projet CancerLinQ aux Etats-Unis [10]
L’American Society of Clinical Oncology (ASCO) a lancé en 2016 une plateforme numérique de big data, reliant et analysant les dossiers médicaux informatisés d’hôpitaux volontaires (environ 90 hôpitaux y étaient abonnés en 2016). Cette plateforme a pour but de créer un système apprenant et rapide qui analyse en temps réel les données observationnelles d’un nombre conséquent de patients atteints de cancer afin de fournir une aide clinique au praticien, faire progresser les connaissances et améliorer les soins.
Exemple 3 : les registres italiens et les médicaments coûteux [10]
L’agence italienne du médicament, l’AIFA (Agenzia Italiana del Farmaco), a mis en place des registres collectant des données en vie réelle autour des médicaments au coût de traitement élevé, et qui ont une efficacité incertaine ou variable en fonction des patients.
Chaque patient bénéficiant de ce type de médicament est placé sous surveillance médicale de sorte que les données cliniques, démographiques ainsi que les informations relatives à la prescription soient enregistrées jusqu’à la fin du traitement. Ces enregistrements sont obligatoires et conditionnent le remboursement, en effet l’objectif de ces données collectées est principalement de réduire l’impact budgétaire, l’incertitude sur l’efficacité et l’efficience afin d’en optimiser l’usage et améliorer la pertinence des prescriptions.
III) L'intérêt des données de vie réelle
A) Identifier la population cible et la population rejointe [9]
Les groupes de patients étudiés lors des essais cliniques (population cible) ne reflètent souvent pas la population qui va à la suite réellement bénéficier de ces soins (population rejointe), ainsi les informations collectées lors des essais comportent des incertitudes quant à l’utilisation du DM sur ces patients en termes de sécurité, d’efficacité et d’efficience.
Les essais randomisés ont généralement des critères d’inclusion assez stricts afin d’assurer une bonne reproductibilité des résultats. Ils n’incluent souvent pas les personnes à haut risque, présentant des comorbidités, les femmes enceintes et plus encore, omettant ainsi une partie de la population destinée aux soins évalués.
De plus, dans la pratique des soins courants, certaines prescriptions peuvent être exceptionnelles dû aux caractéristiques du patient en termes de contre-indication ou d’autres critères non pris en compte lors de l’étude clinique classique. Il est donc important de mener des études de vie réelle pour mieux caractériser la population rejointe.
B) Identifier la réalité des pratiques de soins courants [9]
Dans tout essai randomisé, l’ensemble des paramètres et facteurs pouvant influencer les résultats de l’étude sont très surveillés.
Parmi ces facteurs, on retrouve l’utilisation du DM. En effet, s’il n’est pas correctement utilisé, les résultats caractérisant l’efficacité de ce dernier peuvent alors être altérés. Ainsi, pour limiter toutes fausses conclusions, l’utilisation du DM évalué lors de l’essai clinique va être surveillée de près, les personnes encadrantes vont expliquer à plusieurs reprises comment le patient ou le personnel médical doit l’utiliser, à quelle fréquence etc.
Cette surveillance accrue à deux buts :
- S’assurer de la véracité des résultats de l’étude clinique,
- S’assurer de la bonne utilisation du DM pour qu’il soit le plus efficace possible.
Dans la pratique de soins courants, cette surveillance n’est malheureusement pas aussi soutenue. Ainsi, les personnes l’utilisant peuvent ne pas correctement l’utiliser ce qui peut potentiellement influencer son efficacité. Les données de vie réelle collectées quant à l’utilisation du DM et son efficacité en pratique de soins courants peuvent permettre de mettre en avant certains résultats non exposés lors des essais cliniques.
De plus, lors des essais, des mesures de surveillance et d’amélioration de l’observance thérapeutique peuvent être mises en place (prise de sang, rappels, visites programmées) tandis qu’en pratique de soin courante, les résultats sont fonctions du niveau d’observance habituel des patients qui va lui-même dépendre du patient ou de l’utilisation du DM comme sa pénibilité, l’efficacité ressentie, les effets indésirables etc.
L’utilisation du dispositif sera également différente entre une étude clinique classique et une étude observationnelle, que sa manipulation soit faite par un médecin ou un patient. Cette différence peut s’expliquer par l’impact du plateau technique ou encore par l’organisation de la prise en charge qui sont à prendre en compte lors de la transposabilité des résultats. Il en est de même pour les prescripteurs, les modalités de prescriptions ne sont pas toujours identiques lors d’une étude clinique et lors de la pratique courante ce qui peut être traduit par une nécessité non prise en compte lors de l’étude.
Enfin, le DM peut s’avérer être utilisé en parallèle à d’autres traitements en pratique de soins courants qui peuvent influencer l’efficacité du traitement aussi bien positivement que négativement et donc à terme impacter l’évaluation du DM par la HAS en ce qui concerne le rapport bénéfice / risque.
C) Identifier les effets à long terme du dispositif médical
Les études cliniques sont organisées sur un temps donné, les effets à long terme ou à très long terme sont donc rarement connus. Les études en vie réelle peuvent permettre d’étendre ces connaissances afin de valider ou non les hypothèses émises lors de la mise sur le marché du produit de santé et détecter des complications éventuelles.
L’objectif final sera de quantifier l’impact des différences observées en comparaison à l’hypothèse initiale en termes de sécurité, d’efficacité et d’efficience.
D) Évaluer la pertinence du dispositif sur l’amélioration de la qualité de vie des patients
1) Évaluer la pertinence [10]
« Un soin pertinent est le juste soin (actes, prescriptions, prestations), au bon patient, au bon moment, compte tenu des connaissances scientifiques actuelles (recommandations de la HAS, des sociétés savantes, etc.) » [17]. La pertinence d’un dispositif médical dépendra alors de l’ensemble de ces paramètres fixés par le fabricant et les autorités compétentes.
Cette pertinence n’est pas toujours évidente à mettre en avant lors des essais cliniques classiques où le critère de jugement principal se doit d’être objectif, reproductible, bien défini et mesurable de la même manière par tous les investigateurs de l’essai afin d’éviter tout biais. Ce critère est majoritairement observable, mais son observation (ou non) ne permet pas d’évaluer la pertinence de l’effet du DM sur les patients, ce qui peut justifier l’apport des données de vie réelle pour répondre à la problématique et ainsi réévaluer le DM. D’autant plus que l’ensemble des patients ne vont pas systématiquement avoir la même attente vis-à-vis du DM en ce qui concerne l’amélioration de leur qualité de vie.
2) Mesurer l’amélioration de la qualité de vie des patients
La qualité de vie contrairement à la durée de vie est une durée subjective, la création d’un outil permettant de mesurer cette variable a été réalisée par EuroQol (organisation). Il s’agit du questionnaire EQ-5D (annexe 1) qui a l’avantage d’être un outil générique utilisable sur plusieurs types de patient et différentes maladies.
Ce questionnaire peut être utilisé à plusieurs fins :
- Fournir un profil de santé du patient le jour où il remplit le questionnaire,
- Surveiller l’état de santé des groupes de patients à des moments particuliers (comme lors de l’orientation, l’admission, de la sortie et du suivi),
- Mesurer les changements d’un état de santé au fil du temps chez des patients individuels et dans les cohortes de patients : avant / après interventions ou traitements.
Utilisé dans 90 pays et disponible dans 200 langues, ce questionnaire évalue la mobilité, la capacité de prendre soin de soi, les activités quotidiennes, la douleur et l’inconfort ou encore la dépression et l’anxiété. Les patients vont remplir le questionnaire et estimer leur état de santé sur une échelle allant de 0 à 100 (0 étant le pire état de santé que le patient puisse imaginer et 100 le meilleur état de santé qu’il puisse imaginer). À partir de ces résultats, l’état de santé du patient va être converti en nombre qui pourra ensuite aider à prendre des décisions suivant l’objectif recherché.
Simple d’utilisation, il peut être utilisé en pratique de soins courants, lors d’essais cliniques ou encore lors d’un sondage concernant la santé. Lorsque les DM sont mis sur le marché, le score calculé par l’EQ-5D peut manquer de robustesse, les études de vie réelle vont s’avérer importantes dans ce cas. Elles vont venir compléter les connaissances déjà acquises sur les différentes maladies en estimant le score d’utilité de manière robuste par l’acquisition de la perception de la qualité de vie chez les patients associée à leur état de santé à l’aide du questionnaire auprès d’un échantillon représentatif de la population rejointe en France.
E) Contribuer à l’amélioration des essais randomisés contrôlés
Les données de vie réelle ne sont en aucun destinées à remplacer les essais classiques, la HAS a même émis un avis défavorable en ce qui concerne le remboursement d’un produit de santé lorsque les données de vie réelle sont utilisées à la place de l’ERC.
Récemment, certaines nouveautés en lien avec les données de vie réelle et les produits de santé présentées lors d’une proposition de loi concernant les médicaments et DM ont été adoptées par le Sénat [18]. L’encadrement du remboursement des médicaments innovants qui présentent une « absence de données cliniques suffisantes et pertinentes pour l’évaluation de l’amélioration du service médical rendu ; [...] une efficacité et une sécurité présumées [et qui] répond à un besoin thérapeutique majeur au regard des alternatives existantes [18] » prend désormais en compte les données de vie réelle en plus des données cliniques pour déterminer sa valeur thérapeutique relative. Ces données de vie réelle sont également intégrées dans l’évaluation de l’Amélioration du Service Médical Rendu (ASMR) des médicaments innovants. Désormais, le SNDS est mis à disposition pour permettre l’évaluation de l’efficacité en vie réelle des traitements aussi bien pour le titulaire de l’Autorisation de Mise sur le Marché (AMM) que pour l’exploitant de ce traitement.
L’accès à ces données de vie réelle via le SNDS a fait l’objet de la mise en place d’un régime particulier quant à l’évaluation des traitements en vie réelle dans le but de protéger les droits et libertés du patient. Ces données sont accessibles à toute personne (ou structure) publique ou privée, à but lucratif ou non, souhaitant les utiliser à des fins de recherche de santé publique après autorisation de la Commission Nationale de l’Informatique et des Libertés (CNIL) [18].
Dernièrement, la HAS a également créé une cellule sur les données de vie réelle dans le but de fournir un support méthodologique pour les industriels ainsi qu’une harmonisation des pratiques. Pour ce faire, la cellule a mis en place une feuille de route centrée autour de 4 axes qui sont « l’optimisation des procédures de suivi des demandes de données complémentaires en vie réelle, la standardisation de l’utilisation de ces données pour les (ré)évaluations des produits de santé, l’affirmation du rôle de la HAS dans la mise en place des registres en France et en Europe et enfin, améliorer la visibilité externe de la HAS sur les données en vie réelle » [19].
Mais malgré la mise en place de nouvelles lois dans ce domaine, la France a pris un réel retard dans l’utilisation de ces données impliquant des limites à leur utilisation.
Pour commencer, il existe un nombre trop faible d’experts pour assurer un suivi efficace et pertinent des organismes souhaitant s’approprier ces données dans le cadre d’un développement ou d’une demande de remboursement / réévaluation de leur DM. D’autant plus que ces experts doivent développer de nouvelles méthodologies pour utiliser ces données de manière efficiente : les data scientists doivent avoir des compétences biomédicales pour identifier et analyser les sources pertinentes tant qu’il y a des données de santé disponibles. De plus, une fois ces compétences acquises, il est important de prendre en compte d’éventuels biais pouvant être importés par l’Intelligence Artificielle (IA) impliquant des problèmes d’inclusion en se focalisant sur une population non suffisante car dans ce cas précis, les patients ne sont pas tirés au sort ce qui peut altérer les résultats.
Cette récolte de données se révèle être assez lourde, elle peut ainsi mettre en avant une fracture numérique entre les différentes parties prenantes. Selon la manière dont est menée le processus de récolte, les résultats des études peuvent être impactés par la qualité des données et le traitement de ces dernières par l’IA. Le problème de traitement des données est d’autant plus exacerbé par un manque de coopération entre les différents acteurs du domaine médical compliquant ainsi leur récolte et leur traitement efficace. Il n’existe par exemple pas de formats de données standards permettant un accès facilité à des données de meilleure qualité.
Enfin, les autorités de santé ont des attentes significatives en ce qui concerne l’utilisation de ces données en termes de pertinence et de preuve, de plus les fabricants ne doivent en aucun cas négliger l’importance du respect de la réglementation comme le Règlement Général sur la Protection des Données (RGPD) et les demandes d’accès aux différentes données qui peuvent nécessiter l’aval de l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) [18]. Ces données présentent aussi un intérêt pour les établissements de santé.
F) Cas des établissements de santé
1) Collecte des données de vie réelle
Les établissements de santé collectent des données de vie réelle pour :
- Évaluer leur parc biomédical,
- Anticiper sur les modalités liées à la réforme (prix, valeur, fiabilités, etc…),
- Anticiper les budgets lors du processus d’achat.
Évaluation du parc biomédical
Les données de vie réelle aident à mieux financer les décisions prises à la suite d’un diagnostic dans le parcours de soins du patient. La collecte des informations résultant de ces décisions permettra de recenser les dispositifs médicaux par efficacité, ordre d’utilité ou de priorité.
Anticipation sur les modalités liées à la réforme
Ces données sont également utiles afin de savoir si le dispositif médical est encore performant et sûr à la suite de nombreuses réutilisations. Si ce n’est pas le cas, ce dernier sera rebuté.
Anticipation des budgets d'achats
Les données de vie réelle sont aussi utiles quant au processus d’achat des dispositifs médicaux. La procédure d’achat d’un dispositif médical passe par une phase préparatoire avec la définition du besoin dans lequel sont précisés les attentes des utilisateurs et le type de dispositif. Durant cette phase, ces données vont permettre de mieux comprendre la place de ces dispositifs dans les soins de routine et ainsi de mieux anticiper leurs achats.
Bien qu’un grand nombre d’établissements de santé collectent des données de vie réelle, ils ne sont pas tous autorisés à les héberger.
2) Hébergement des données de vie réelle
Ces données peuvent être hébergées dans des Entrepôts de Données de Santé Hospitaliers (EDSH). La HAS en a recensé 22, dont 17 au sein d’un Centre Hospitalo-Universitaire (CHU), les 5 restants sont présents au sein d’autres types d’établissements de santé [20].
Les établissements de santé peuvent également héberger les données de vie réelle chez un hébergeur de données de santé agréé ou certifié dont les listes sont disponibles sur le site du ministère de la Santé et de la Prévention [21]. La certification est obtenue à la suite d’une évaluation de conformité vis-à-vis du référentiel de certification (notamment certification ISO 27 001 et ISO 20 000) par un organisme certificateur accrédité par le Comité Français d’Accréditation (COFRAC) ou un équivalent au niveau européen [22]. Contrairement aux hébergeurs agréés, les hébergeurs certifiés sont reconnus au niveau international. Le choix de l’hébergeur est libre, cependant, lors de son choix, l’établissement de santé doit prendre en compte les conditions de sécurité nécessaires au vu de la criticité des données en se référant aux exigences réglementaires.
Les données présentes dans ces EDSH peuvent également servir de base pour des études menées sur les DM. Pour qu’un tel projet soit mené, il doit au préalable être validé par un comité scientifique et éthique. La HAS a réalisé plusieurs projets expérimentaux afin d’éprouver le potentiel des EDSH dans ce cas. Ces derniers ont pu mettre en évidence leur utilité notamment dans le cadre de la contextualisation de l’utilisation des produits de santé et lors du développement d’indicateurs de qualité et de sécurité des soins. Ces entrepôts de données sont donc très prometteurs et pourraient servir à plus grande échelle [20].
IV) Cadre d'utilisation des données de vie réelle
Les données de vie réelle possèdent un cadre d’utilisation varié. Elles peuvent être utilisées lors des évaluations cliniques pour l'obtention du marquage CE, lors de la surveillance après commercialisation ou encore lors d’une demande de remboursement concernant les dispositifs médicaux.
A) Évaluation clinique et surveillance après commercialisation
Obligatoire pour toute classe de dispositifs médicaux, l’évaluation clinique repose sur des données cliniques permettant d’apporter des preuves cliniques quant à la sûreté et la performance du dispositif. Les données de vie réelle peuvent être utilisées dans ce cas pour renforcer les preuves cliniques avancées par les fabricants.
L’évaluation clinique doit aussi être documentée et tenue à jour en continu aussi bien avec des données favorables que défavorables. Afin de documenter l’évaluation clinique sur le long terme, le fabricant doit mettre en place un Suivi Clinique Après Commercialisation (SCAC) faisant partie intégrante du système de surveillance après commercialisation.
Le SCAC permet de collecter et d’évaluer les données cliniques provenant des dispositifs mis sur le marché et utilisés par l’Homme. Là aussi, les données de vie réelle peuvent être utilisées afin d’appuyer les points suivants :
- La confirmation de la sécurité et des performances du dispositif pendant toute sa durée de vie,
- L’identification des effets secondaires et des risques émergents inconnus jusqu’alors,
- S’assurer que les risques identifiés et que le rapport bénéfice/risque restent acceptables,
- L’identification des mésusages.
NB : les fabricants peuvent aussi utiliser les données de vie réelle dans le cadre de l’équivalence. Cette dernière permet l’utilisation de données cliniques issues d’un dispositif médical similaire existant afin d’éviter d’avoir recours aux essais cliniques. Toutefois, l’équivalence a fortement été restreinte dans le règlement, rendant son utilisation très hypothétique. En effet, désormais pour démontrer l’équivalence, les fabricants doivent convenir d’un contrat donnant accès à l’ensemble des données du produit du fabricant concepteur à fin qu’ils puissent s’appuyer sur les données cliniques du dispositif initial.
B) Remboursement du dispositif médical
Les données de vie réelle peuvent aussi être utilisées dans le cadre des demandes de remboursement des DM déposées par les fabricants.
1) Les modalités de remboursement [23]
Il existe différents modes de prise en charge des dispositifs médicaux, cela dépend entre-autres de leur usage et de leur lieu d’utilisation.
En ville, ils peuvent être remboursés de 2 manières différentes, via leur inscription sur la Liste des Produits et des Prestations Remboursables (LPPR) ou via leur inscription sur les nomenclatures des actes pris en charge par l’assurance maladie (NGAP, CCAM, NABM) lorsque leur utilisation est associée à la réalisation d’un acte professionnel.
En établissement de santé, la forfaitisation des soins permet d’intégrer les dépenses de la plupart des dispositifs dans les prestations d’hospitalisation à la suite de leur inscription sur la liste intra-GHS. Certains dispositifs coûteux, peuvent être inscrits sur la LPPR en plus de leur prise en charge dans les prestations d’hospitalisation.
Ces modalités de prise en charge des dispositifs médicaux font intervenir un acteur commun ; la Commission Nationale d’Évaluation des Dispositifs Médicaux et des Technologies de Santé (CNEDiMTS).
2) La CNEDiMTS [23]
La CNEDiMTS est une commission spécialisée de la HAS chargée d’émettre des avis relatifs au remboursement des dispositifs et des actes médicaux. Pour cela, elle se base sur le Service Attendu (SA) lors d’une première demande et sur le Service Rendu (SR) lors d’une demande de renouvellement. Les demandes peuvent être déposées par un fabricant (LPPR, intra-GHS) ou par les Conseils Nationaux Professionnels (CNP).
Le SA/SR est apprécié selon 2 critères, l’intérêt du produit/acte à l’échelle individuelle (effet thérapeutique, balance bénéfice/risque, comparaison avec ce qui est déjà existant) et l’intérêt de santé publique à l’échelle collective (estimation de la population cible, épidémiologie de la maladie visé par le dispositif/acte, impact du dispositif/acte sur la santé publique). Le service peut être jugé suffisant et dans ce cas, la commission émettra un avis favorable quant au remboursement du dispositif/acte. À l’inverse, elle émettra un avis défavorable si le service est jugé insuffisant.
Lorsque le service est suffisant, la commission apprécie l’Amélioration du Service Attendu (ASA) lors d’une première demande et l’Amélioration du Service Rendu (ASR) lors d’une demande de renouvellement. L’appréciation de l’ASA/ASR mesure ce que le dispositif/acte apporte de plus par rapport au traitement/acte de référence s’il existe. Elle se base sur les résultats des études cliniques comparatives et randomisées. À l’issu de l’appréciation, le dispositif/acte sera classé dans l’une des 5 catégories d’amélioration du service suivantes :
- Majeure (I),
- Importante (II),
- Modérée (III),
- Mineure (IV),
- Absente (V).
Si l’organisme (fabricant, CNP) est en désaccord avec les avis de la commission, il peut demander à être auditionné et devra alors argumenter sur les raisons de son désaccord ce qui peut s’avérer payant. En effet, en 2021, 34 % des avis ont été modifiés après une audition [24].
3) La tarification [23]
Si le SA est suffisant, la tarification du dispositif/acte peut avoir lieu. Pour les dispositifs inscrits sur la liste intra-GHS, les prix sont directement négociés auprès des établissements de santé. Pour les dispositifs inscrits sur la LPPR, les prix sont négociés entre le Comité des Produits de santé (CEPS) et les fabricants.
La tarification des actes est négociée entre les professionnels de santé et l’Union Nationale des Caisses de l’Assurance Maladie (UNCAM). Les établissements de santé, le CEPS et l’UNCAM utilisent entre-autres l’avis émis par la CNEDiMTS pour négocier le prix de remboursement du dispositif/acte.
4) L’approbation
Enfin, la décision finale relative à l’inscription sur la liste intra-GHS, la LPPR ou encore sur les nomenclatures des actes pris en charge par l’assurance maladie revient au ministre de la Santé. C’est cette validation d’inscription par le ministre qui va permettre le remboursement des dispositifs selon les différentes modalités de prise en charge. Pour mieux visualiser ce processus complexe, un schéma récapitulatif est présent en Figure 4.
Figure 4 : Schéma simplifié des processus de remboursement des DM [source : Auteur]
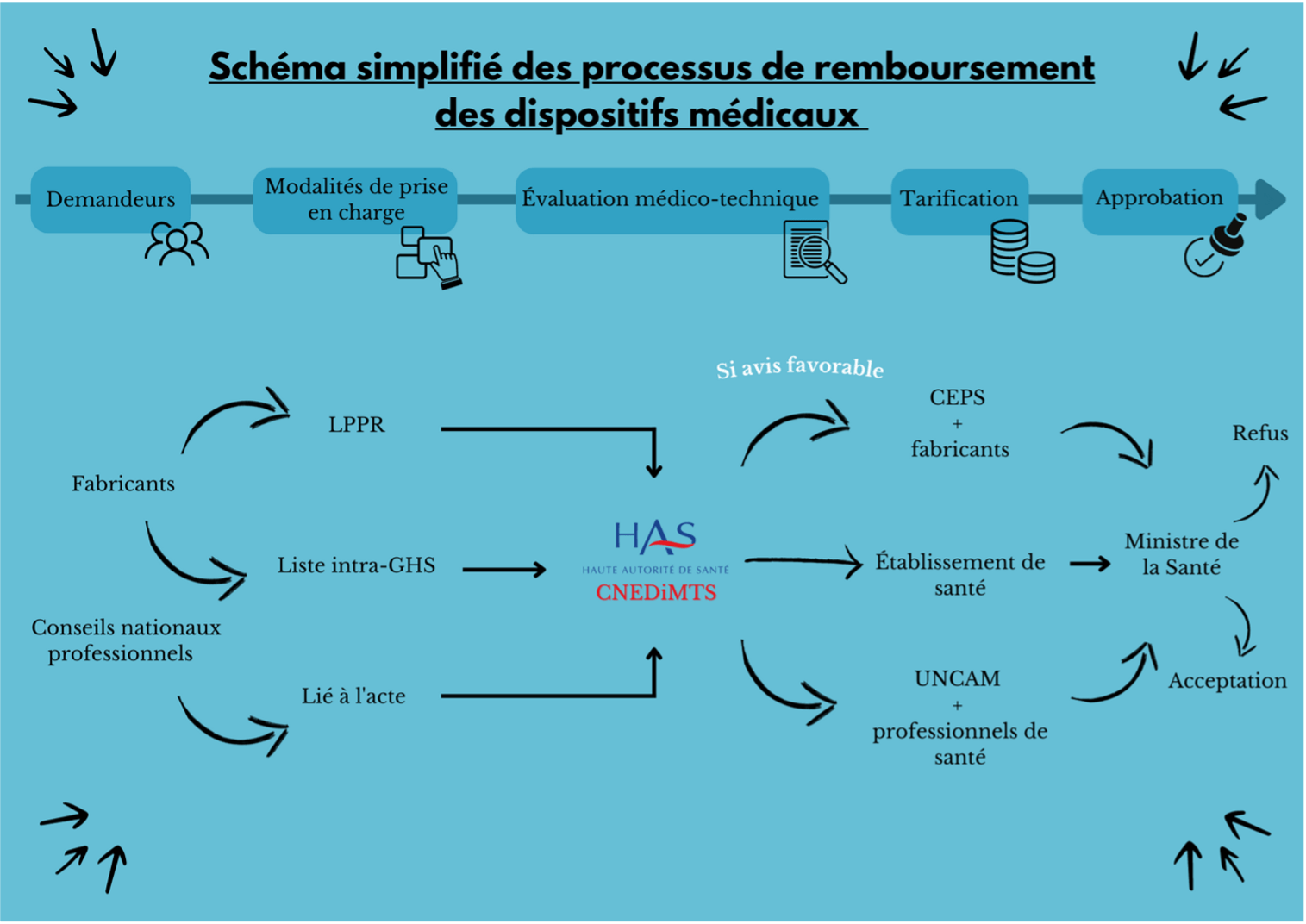
5) Demande d’études post-inscription
Il arrive que la CNEDiMTS demande des études post-inscription (EPI) après qu’elle ait émis un avis favorable quant au remboursement d’un dispositif médical.
Depuis 2019, le nombre d’études demandées est stable avec une vingtaine d’études demandées par an comme l’illustre le Tableau 2. Ce bilan a été réalisé à l’aide des rapports d’activité de la HAS et de la CNEDiMTS.
Tableau 2 Bilan des études post-inscription de 2014 à 2021 [source : Auteur]
| Année | Nombre de dossiers ayant reçu un avis favorable de la CNEDiMTS (1ère demande ou demande de renouvellement) | Nombre d’études post-inscription demandées | Pourcentage des études demandées |
| 2014 | 170 | 11 | 6% |
| 2015 | 137 | 8 | 6% |
| 2016 | 112 | 13 | 12% |
| 2017 | 238 | 11 | 5% |
| 2018 | 160 | 6 | 4% |
| 2019 | 164 | 21 | 13% |
| 2020 | 199 | 19 | 10% |
| 2021 | 181 | 18 | 10% |
* Bilan réalisé à l’aide des rapports d’activité de la HAS et de la CNEDiMTS.
Ces études font appel à des données en vie réelle et peuvent aussi bien être demandées lors d’une première demande de remboursement que lors d’une demande de renouvellement de remboursement. Les résultats des EPI doivent être présentés lors de la demande de renouvellement et serviront à l’élaboration de l’avis de cette dernière. Il est important de noter que la réalisation des EPI n’est pas obligatoire, cependant, la convention signée entre l’entreprise et le CEPS prévoit une diminution de tarif de remboursement en cas de non-réalisation de l’étude [25].
Les données de vie réelle pour négocier un meilleur prix : l'exemple de Jevtana® [10]
Jevtana® est un anticancéreux (médicament) ayant obtenu un avis favorable de la Commission de Transparence (CT) en 2011 pour son inscription sur la liste des médicaments agréés aux collectivités grâce à un Service Médical Rendu (SMR) important et une Amélioration du Service Médical Rendu (ASMR) mineure. Une année plus tard, le Zytiga®, un anticancéreux aux prescriptions similaires s’est vu octroyé un SMR important et une ASMR modérée ce qui lui a conféré un prix de remboursement plus élevé.
Le laboratoire produisant Jevtana® a alors demandé une réévaluation de l’ASMR de la spécialité. Pour ce faire, la demande était accompagnée de données de vie réelle comme des résultats d’analyse, des données de tolérance provenant des autorisations temporaires d’utilisations nominatives et de cohorte accordées en France, des données de deux études observationnelles (provenant de la littérature) sur le Zytiga® pour justifier leur demande.
Après évaluation de la demande, la CT a déterminé que l’ASMR était modérée et non mineure comme évalué en 2011 ce qui a permis de fixer un prix de remboursement plus élevé pour la spécialité.
Ces données possèdent un vaste cadre d’utilisation dont toutes les possibilités n’ont pas encore été explorées. De plus, elles possèdent un cadre réglementaire contraignant.
V) Réglementation autour des données de vie réelle
Les données de vie réelle, également identifiées comme des données à caractère personnel doivent être protégées avant de pouvoir être utilisées. Cette protection concerne tous les individus de la société et ce quel que soit leur âge. De là, intervient la réglementation des données, ne permettant leur utilisation qu’après avoir respecté la vie privée des individus, générateurs de ces données de vie réelle [26].
A) À qui s’applique le Règlement Général sur la Protection des Données (RGPD)
Ce règlement s’applique à tous les organismes établis ou non dans l'Union européenne et traitant des données personnelles d’individus se trouvant dans celle-ci. Il s’applique aussi bien au responsable de traitement qui détermine les finalités et les moyens du traitement qu’au sous-traitant qui traite des données pour le compte d’un responsable de traitement.
Le traitement de ces données peut être défini comme « toute opération ou tout ensemble d’opérations effectuées ou non à l’aide de procédés automatisés et appliqués à des données ou à des ensembles de données à caractère personnel » [3].
Les opérations suivantes sont considérées comme traitement de données :
- La collecte,
- L’enregistrement,
- La structuration,
- La conservation,
- La transmission,
- La modification,
- L’extraction,
- La communication,
- La mise à disposition.
NB : Cette réglementation ne concerne pas seulement les données informatisées, les fichiers papiers doivent être protégés de la même manière.
La première chose qu’un responsable de traitement doit faire avant de mettre en place un traitement de données de vie réelle est une analyse d’impact relative à la protection des données (AIPD). Ces données sont des données dites « sensibles » et bénéficient d’une protection renforcée. Par principe, le traitement de ces données est interdit. Toutefois, ces données peuvent être traitées si les personnes concernées ont donné leur consentement explicite (d’autres exceptions sont listées dans l’article 9.2 du RGPD).
Le consentement est défini comme toute manifestation de volonté libre (ni contrainte ni influencée), spécifique (pour un seul traitement pour une finalité donnée), éclairée (informations distinctes et non noyées parmi d’autres) et univoque (sans ambiguïté par une déclaration ou un acte positif de la personne) par laquelle la personne concernée accepte, par une déclaration ou par un acte positif clair, que des données à caractère personnel la concernant fassent l’objet d’un traitement [3]. Il est important de noter qu’une personne peut à tout moment retirer son consentement et ainsi mettre fin au traitement de ses données.
B) L’analyse d’impact relative à la protection des données (AIPD)
Cette analyse est un processus qui s’intéresse aux risques sur la vie privée et aux impacts potentiels qu’ils peuvent avoir sur les personnes concernées. Un accès illégitime, une modification ou encore une disparition des données de vie réelle peuvent être des exemples de risques sur la vie privée. L’AIPD a pour but de décrire un traitement de données de façon détaillé, d’évaluer sa conformité au RGPD, d’identifier les risques que ce traitement peut engendrer pour les personnes physiques concernées et de réduire ces risques à un niveau acceptable en mettant en place des mesures appropriées. Une fois cette analyse menée, le responsable de traitement peut mettre en place un traitement de données qui respecte les principes du RGPD.
C) Les principes de la protection des données
1) La licéité du traitement
Un traitement des données de vie réelle n’est licite que s’il est nécessaire à l’une des conditions ci-dessous :
- La sauvegarde d’intérêts vitaux,
- L’exécution d’une mission d’intérêt publique,
- Intérêts légitimes du responsable de traitement ou,
- Si les personnes concernées par le traitement des données ont donné leur consentement,
Le responsable de traitement doit choisir la condition la plus pertinente, et en plus de devoir respecter au moins l’une de ces conditions, le traitement doit avoir une finalité.
2) La finalité du traitement
Un traitement de données de vie réelle ne peut exister que s’il possède un objectif, aussi appelé finalité. Cette finalité doit être définie, justifiée et explicite. Cette dernière ne doit pas être détournée, l’utilisation des données ne peut être utilisée pour une finalité autre que celle prévue à l’origine. Si le responsable de traitement souhaite changer sa finalité, il doit en informer les personnes concernées qui maintiendront ou non leur consentement. Dès la définition de la finalité, le responsable de traitement doit prévoir une récolte de données la plus minimale possible.
3) La minimisation des données
La récolte des données de vie réelle doit être la plus minimale possible. C’est-à-dire que le responsable de traitement ne doit récolter que les données de vie réelle utiles et pertinentes qui permettent de répondre à la finalité du traitement. Il doit donc avoir une vraie réflexion et se poser les bonnes questions. Il peut par exemple se demander quelles données sont indispensables pour atteindre la finalité et distinguer ainsi, les données obligatoires des données facultatives. En plus, de la minimisation des données, la durée de conservation de ces dernières est aussi limitée.
4) La conservation limitée des données
Lorsque la finalité du traitement des données est atteinte, les données de vie réelle doivent être effacées, ou être anonymisées afin d’éviter toute réidentification des individus, ou encore être archivées. Le responsable de traitement des données ne doit pas garder des données sous prétexte qu’elles pourraient servir ultérieurement et doit définir une durée de conservation des données pour chaque traitement. Dans certains cas, la loi française prévoit une durée de conservation. À cette conservation limitée des données, s’ajoute l’obligation de sécurité.
5) L’obligation de sécurité
La sécurité des données de vie réelle doit être assurée par le responsable de traitement. Ce dernier est libre quant à la méthode de sécurisation choisie. Il peut par exemple opter pour la pseudonymisation ou le chiffrement des données. Les données ne doivent être accessibles qu’aux personnes autorisées (confidentialité), ne doivent pas être modifiées (intégrité) et doivent être disponibles en continu par les personnes autorisées (disponibilité). En plus de devoir être sécurisées, les données doivent aussi répondre au principe de transparence.
6) La transparence
Ce principe repose sur le fait que les personnes concernées par le traitement des données de vie réelle doivent être informées de l’utilisation des données les concernant et de la manière d’exercer leurs droits.
Cette obligation est du ressort du responsable de traitement et ce, que les données de vie réelle aient été collectées de manière directe ou indirecte. La collecte directe se fait directement auprès des individus, par exemple un dispositif médical qui permet de collecter des données de vie réelle. La collecte indirecte quant à elle, ne se fait pas directement auprès de l’individu mais sur une source de données comme le SNDS par exemple. Lorsque possible, les individus doivent être informés en cas de nouvelle finalité du traitement.
Dans tous les cas, le responsable de traitement devra fournir à l’individu :
- Son identité et ses coordonnées,
- La finalité et la base juridique du traitement,
- Les conséquences d’une fourniture de données,
- Les destinataires des données,
- La durée de conservation,
- Ses droits sur les données traitées (droit de l’individu),
- Son droit d’introduire une réclamation (en France, la CNIL),
- Les coordonnées du délégué à la protection des données (DPO),
- L’existence du droit de retirer son consentement (applicable si un consentement a eu lieu),
- L’existence d’un transfert hors de l'Union Européenne.
NB : Le RGPD autorise le transfert des données hors de l’Union Européenne seulement si un niveau de protection équivalent peut être assuré (les données anonymisées ne sont pas concernées). La commission européenne publie la liste des pays dits « adéquats » vers lesquels les données peuvent être transférées sans autorisation spécifique. Pour les autres pays, le transfert ne sera possible que sous certaines conditions (chapitre 5 du RGPD).
En cas de collecte indirecte, le responsable de traitement doit en plus fournir à l’individu, les catégories de données traitées ainsi que la source des données. S’il parvient à démontrer que les informations étaient déjà en possession de l’individu, il n’est alors pas tenu de fournir les informations citées ci-dessus.
Cette obligation de transparence est étroitement liée au droit des personnes qui s’est vu être renforcé par le RGPD.
7) Le droit des personnes
Les individus bénéficient de droits qui leur permettent de garder une certaine maîtrise des données de vie réelle qu’ils ont générées.
Le droit d'accès
Tout individu peut demander à un responsable de traitement si des données de vie réelle le concernant sont traitées par ce dernier. Si tel est le cas, le responsable de traitement est tenu de fournir à l’individu une copie des données le concernant et les mêmes informations qu’au point sur la transparence des données.
Le droit de rectification
Ce droit permet à l’individu de corriger les données inexactes ou de compléter des données.
Le droit d'opposition
Les individus peuvent s’opposer à tout moment au traitement des données de vie réelle qu’ils ont générées. Ils doivent donc être en mesure de retirer leur consentement à tout moment.
Le droit à la limitation
Lorsqu’un individu exerce son droit de rectification ou d’opposition, le responsable de traitement possède 1 mois pour traiter la demande. Pendant ce temps, l’individu peut exercer son droit à la limitation pour geler l’utilisation de ses données. Le responsable de traitement ne pourra alors plus utiliser ces dernières mais devra les conserver. Il devra aussi informer l’individu concerné avant la levée de la limitation.
Le droit à l'effacement
Un individu est en droit de demander l’effacement de ses données lorsque la finalité du traitement est atteinte, s’il retire son consentement ou encore si le traitement est illicite.
NB : Lorsqu’un individu exerce son droit de rectification, d’effacement ou de limitation, le responsable de traitement, lorsque possible et si cela ne demande pas d’efforts disproportionnés, doit informer tous les organismes à qui il a transmis les données personnelles visées par l'exercice de ces droits.
Le droit à la portabilité
Lorsque c’est techniquement possible, un individu peut demander au responsable de traitement de transférer les données de vie réelle qu’il a généré grâce à son activité à un autre responsable de traitement.
Le RGPD a aussi renforcé les responsabilités des responsables de traitement et des sous-traitants. En plus de devoir respecter les grands principes du règlement, les responsables de traitement doivent aussi être en mesure de démontrer leur conformité à tout moment.
D) Les responsabilités des acteurs
Les responsables de traitement doivent désigner un délégué à la protection des données (DPO) et tenir un registre. Si les acteurs ne sont pas conformes au RGPD, ces derniers s’exposent à des sanctions.
1) Le délégué à la protection des données (DPO)
La désignation d’un DPO est obligatoire pour les organismes aussi bien privés que publics traitant des données de vie réelle à grande échelle. Aucun seuil n’a été défini par les autorités de contrôle pour la notion de grande échelle. Ce sera apprécié au cas par cas avec notamment le volume des personnes concernées, la durée du traitement, le nombre de données traitées ou encore l’étendue géographique.
Au sein de l’organisme, le DPO assure les missions suivantes :
- Informer et conseiller l’organisme quant à ses obligations concernant la protection des données,
- Contrôler la conformité en s’assurant que les principes généraux du RGPD soient respectés,
- Être l’interface entre l’organisme, l’autorité de contrôle et les personnes concernées en répondant aux sollicitations de chacun de ces acteurs.
L’organisme est libre de choisir son DPO (interne, externe, personne physique ou morale) mais ce dernier doit avoir les connaissances et aptitudes requises pour mener à bien ses activités, être exempt de conflits d’intérêts (éviter de désigner les cadres occupant des postes de direction), bénéficier des moyens suffisants (temps, budget, accès aux bases de données, etc.) et avoir la capacité d’agir de façon indépendante, c’est-à-dire qu’il ne doit pas recevoir d’instructions de la part du responsable de traitement sur la manière dont il doit mener ses activités.
2) Le registre
Tous les organismes traitant des données de vie réelle sont dans l’obligation de tenir un registre sous forme écrite (format papier ou électronique). Cette mission peut être confiée au DPO ou à une autre personne en interne.
Un modèle de registre ainsi qu’une liste détaillée des éléments que le registre doit contenir sont disponibles sur le site de la CNIL, ce dernier doit entre-autres comprendre :
- Les parties prenantes intervenant dans le traitement des données,
- Les catégories de données traitées,
- Le but de ces données,
- Qui peut accéder à ces données et à qui elles sont communiquées,
- Leur durée de conservation ou encore,
- Leur méthode de sécurisation.
Ce registre permet d’avoir une vue d’ensemble de ce qui est fait des données traitées et doit être mis à jour régulièrement en fonction des changements des traitements et doit pouvoir être communiqué à la CNIL si celle-ci le demande.
3) Les sanctions
Le RGPD a renforcé les pouvoirs de sanctions des CNIL européennes et il est toujours intéressant de savoir ce à quoi est exposé l’organisme en cas de non-conformité au règlement lorsque des données de vie réelle sont traitées.
La CNIL peut décider d’adopter l’une des sanctions suivantes :
- Un rappel à l’ordre,
- Une injonction de mettre le traitement en conformité,
- Une limitation temporaire ou définitive du traitement,
- Une suspension des flux de données adressés à un destinataire situé dans un pays tiers,
- Un ordre de satisfaire aux demandes d’exercice des droits des personnes,
- Un retrait d’une certification,
- Une amende administrative en fonction de la gravité.
NB : Les amendes administratives peuvent atteindre 20 millions d’€ ou 4% du chiffre d’affaires mondial annuel, en sachant que c’est le montant le plus élevé qui sera retenu comme montant maximum. Par exemple, si 4% du chiffre d’affaires mondial annuel d’une entreprise représentent 200 millions d’€, c’est ce montant qui sera retenu comme montant maximum et non 20 millions d’€. Cela marche dans les 2 sens, si ces 4% représentent 300 000 €, alors ce sera le montant de 20 millions d’€ qui sera retenu comme montant maximum.
VI) Présentation de l'outil
L’émergence des données de vie réelle constitue un nouvel enjeu pour les fabricants de dispositifs médicaux. Il est alors essentiel de créer une cartographie énonçant les différents aspects relatifs à ces données afin de réaliser l’importance de leur utilisation et démontrer le respect des exigences du règlement (UE) 2017/745.
Cette cartographie interactive apporte un certain nombre d’éléments sur ces données, de l’explication du contexte à leur collecte, en passant par une description de leur utilisation dans les phases du cycle de vie du dispositif où elles s’avèrent être utiles (Figure 5). Un développement des problématiques auxquelles elles peuvent apporter une réponse est également présent.
Son principal objectif est de soutenir et accompagner les fabricants ainsi que l’ensemble des parties impliquées dans la conception d’un DM jusqu’à sa commercialisation. Les utilisateurs pourront ainsi consulter les différents items au gré de leurs envies et recueillir les informations dont ils ont besoin.
Figure 5 : aperçu de la cartographie interactive [source : Auteur]
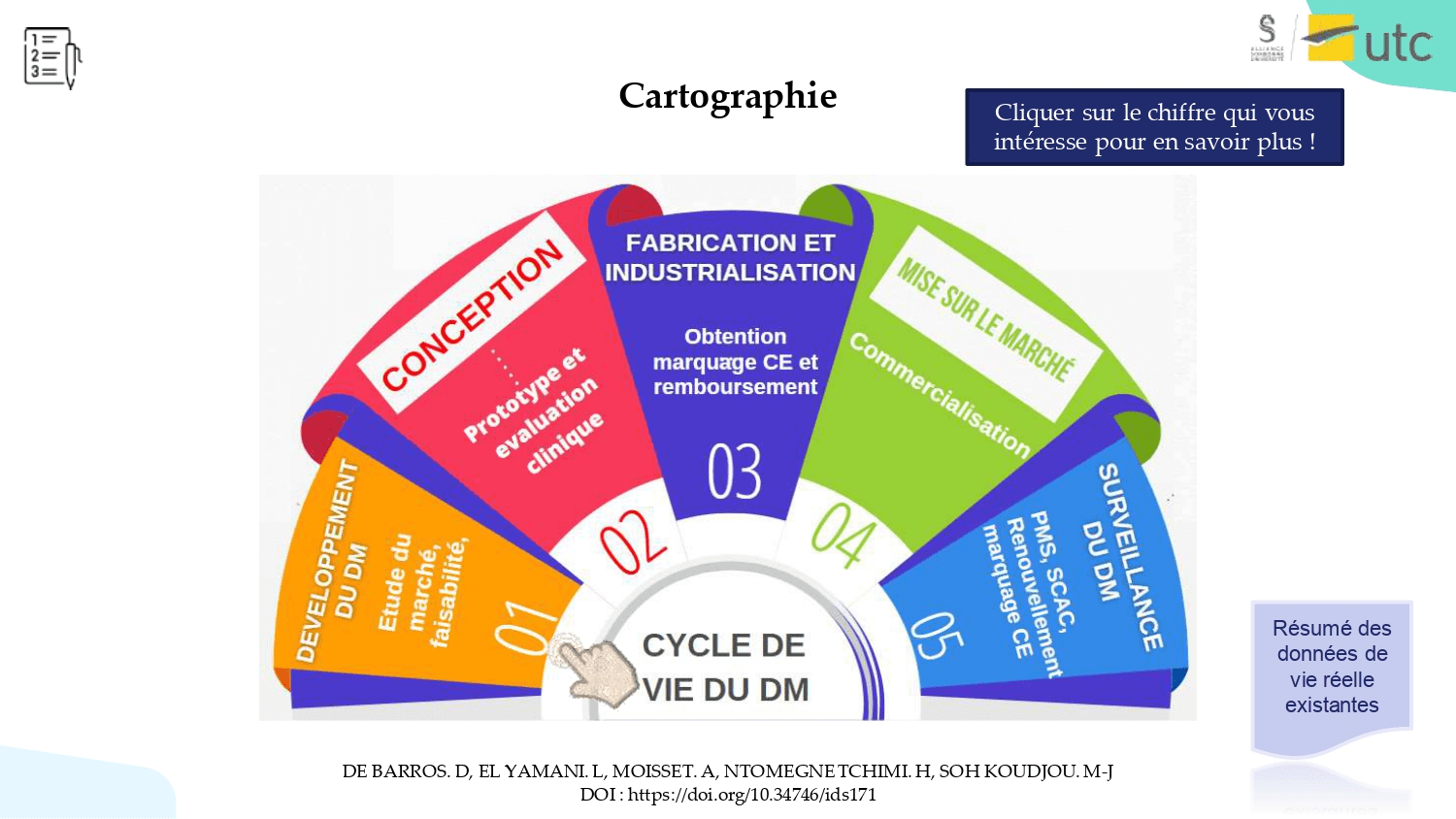
Afin de parfaire l’utilité de cet outil, une liste de guides et de référentiels portant sur les données de vie réelle en français et en anglais est mise à disposition dans l'onglet "téléchargements" afin d'approfondir les connaissances sur le sujet. Cette liste comporte diverses informations comme le titre du guide, son résumé, sa date de publication et le sujet traité.
VII) Conclusion
L’évaluation de la performance, de la sécurité, et de la pertinence des dispositifs médicaux est une exigence du règlement (UE) 2017/745. Cette évaluation permet l’entrée sur le marché, clarifie les décisions de remboursements du DM, et augmente continuellement le niveau de preuves quant à la sûreté et l’efficacité de ces derniers en s’appuyant principalement sur les essais cliniques.
Cependant, le développement technologique qui intègre l’intelligence artificielle a créé une incertitude concernant la mise en œuvre de ces essais dans les pratiques existantes et a renforcé les attentes des études de vie réelle. De ce fait, les données de vie réelle sont devenues un sujet de plus en plus important pour les fabricants. Elles peuvent être utilisées pour de nombreux objectifs différents tout au long du cycle de vie du dispositif médical, de son développement à sa commercialisation. Le point de départ de ce projet est né d'une problématique générale statuant sur le manque de visibilité de ces données, ce qui réduit leur transparence et rend l'évaluation des dispositifs médicaux difficile.
Pour cette raison, il semblait nécessaire de proposer aux professionnels de santé, fabricants, et parties prenantes, une cartographie interactive comme outil informationnel destiné à être utile, utilisable et utilisé, et qui aiderait à la compréhension de l'évaluation des dispositifs médicaux en vie réelle. Cet outil a été élaboré méticuleusement, en apportant les informations pertinentes, et en montrant la place des données de vie réelle dans chaque étape cruciale du cycle de vie du DM. Pour approfondir les connaissances sur le sujet, une liste de guides a été mise à disposition, et grâce à elle, les utilisateurs pourront rapidement s'approprier les informations.
S'appuyer sur notre outil permettra aux utilisateurs de mesurer la sécurité et la performance de leurs dispositifs médicaux, en vue de garantir au patient une expérience de soin optimale.
Notre cartographie vise à faire gagner du temps aux fabricants, en fournissant des informations précises, faciles d’accès et fiables. Comme tout outil, elle est soumise au principe de l’amélioration continue. De ce fait, elle devra suivre l’évolution du milieu réglementaire relatif aux données de vie réelle et en vue de s’y conformer, il est évident que notre outil devra être régulièrement évalué et mis à jour, en prenant en considération tous les détails, retours et informations qui seront remontés par les utilisateurs finaux.
Références bibliographiques
Annexes
Annexe 1 : questionnaire EQ-5D [27]


