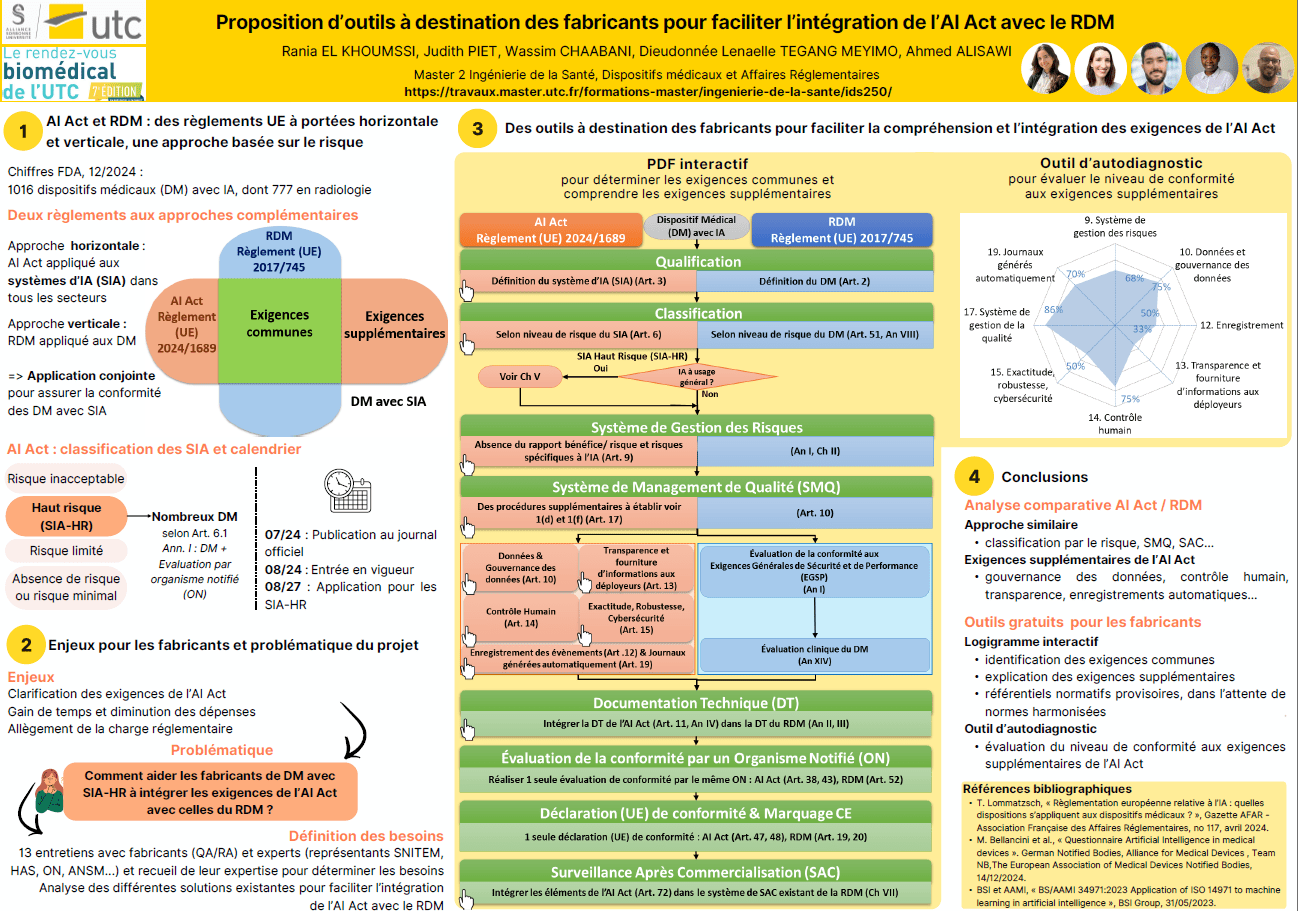
IDS250 – Proposition d’outils à destination des fabricants pour faciliter l’intégration de l’AI Act avec le RDM
DOI mémoire
https://doi.org/10.34746/ids250Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- Judith PIET : judith.piet@live.fr
- Wassim CHAABANI : wassim.chaabani12@gmail.com
- Rania EL KHOUMSSI : raniaelkhoumssi20@gmail.com
- Lenaelle Dieudonnée TEGANG MEYIMO : teganglenaelle@gmail.com
- Ahmed ALISAWI : a.issawwi@gmail.com
Citation
A rappeler pour tout usage : Judith PIET, Wassim CHAABANI, Rania EL KHOUMSSI, Lenaelle Dieudonnée TEGANG MEYIMO, Ahmed ALISAWI, « Proposition d’outils à destination des fabricants pour faciliter l’intégration de l’AI Act avec le RDM », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, réf n° IDS250, https://travaux.master.utc.fr/, DOI : https://doi.org/10.34746/ids250), janvier 2025, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids250/
Résumé
Le Règlement (UE) 2024/1689 établissant des règles harmonisées concernant l'intelligence artificielle (IA), aussi appelé "AI Act", a été publié au Journal officiel de l'Union européenne le 12 juillet 2024. Ce règlement a une portée horizontale et concerne tous les secteurs utilisant l'IA, notamment les dispositifs médicaux (DM). La plupart des DM utilisant de l'IA seront classés comme systèmes d'IA à haut risque (SIA-HR) selon l'AI Act, qui impose des exigences règlementaires pour les différentes parties prenantes lors du cycle de vie des systèmes.
Les fabricants de DM avec SIA-HR devront se conformer à la fois au règlement (UE) 2017/745 relatif aux DM (RDM) et à l'AI Act, ce qui représente un défi d'intégration réglementaire. Ce mémoire vise à répondre à la problématique suivante : Comment aider les fabricants à intégrer les exigences de l’AI Act avec celles du RDM ?
Pour répondre à cette problématique, nous avons d’abord réalisé une lecture croisée des deux règlements, dressé un état des lieux des guides existants, et mené des entretiens avec diverses parties prenantes du cycle de vie des DM avec SIA-HR. Nous avons produit deux outils à destination des équipes Assurance Qualité et Affaires Réglementaires (AQ/AR) chez les fabricants de DM avec SIA-HR, afin de comprendre exigences supplémentaires de l’AI Act par rapport au RDM, et d’évaluer la maturité du fabricant concernant ces exigences relatives à l’IA (PDF interactif et outil diagnostic).
Abstract
Regulation (EU) 2024/1689 laying down harmonized rules on artificial intelligence (AI), also known as the “AI Act”, was published in the Official Journal of the European Union on July 12, 2024. This regulation has a horizontal scope and concerns all sectors using AI, including medical devices (MDs). Most MDs using AI will be classified as high-risk AI systems (HR-AIS) under the AI Act, which imposes regulatory requirements for the various stakeholders during the systems' lifecycle.
Manufacturers of MDs with HR-AIS will have to comply with both Regulation (EU) 2017/745 relative to MDs (RDM) and the AI Act, representing a complex regulatory integration challenge. Our thesis aims to answer the following problem : How can we help manufacturers integrate AI Act and MDR requirements ?
To address this issue, we first cross-read the two regulations, carried out a review of existing guides and conducted interviews with various stakeholders in the lifecycle of MDs with SIA-HR. We have created two tools for Quality Assurance and Regulatory Affairs (QA/RA) teams at manufacturers of MDs with SIA-HR, in order to understand the additional requirements of the AI Act compared to the MDR, and to assess the manufacturer's maturity regarding these AI-related requirements (interactive PDF and diagnostic tool).
Téléchargements

Mémoire d’Intelligence Méthodologique
Poster
Introduction
Le règlement (UE) 2024/1689 du 13 juin 2024 établissant des règles harmonisées concernant l’intelligence artificielle (IA), également appelé AI Act, est un règlement à portée horizontale, qui concerne tous les secteurs utilisant des systèmes d’IA (SIA) [1]. Cette législation concerne donc également les fabricants de dispositifs médicaux (DM) utilisant l’IA, qui devront se conformer non seulement au règlement (UE) 2017/745 relatifs aux DM (RDM) [2], mais également à l’AI Act.
La grande majorité des DM avec IA seront considérés systèmes d’IA à haut-risque (SIA-HR) selon l’AI Act, qui liste une série d’exigences associées à cette classe. Les fabricants de DM font déjà l’objet d’une charge réglementaire importante relative à l’application du RDM, et un enjeu important de l’application de l’AI Act est d’éviter une double charge réglementaire. Notre mémoire vise à répondre à la problématique suivante : Comment aider les fabricants de DM avec SIA-HR à intégrer les exigences de l’AI Act avec celles du RDM ?
Dans le cadre de notre travail qui porte sur l’intégration de l’AI Act et du RDM, nous présenterons notre analyse des écarts entre ces deux règlements afin de faire ressortir et d’interpréter les exigences de l’AI Act déjà couvertes par le RDM, ainsi que les exigences supplémentaires (objectif 1). Ensuite, nous ferons l’état des lieux des guides à destination des fabricants de DM avec IA, et nous étudierons les besoins des fabricants sur la base d’entretiens, afin de proposer des outils d’intégration des deux règlements à destination des fabricants. Ces outils auront pour but de de faciliter la charge de travail des responsables d’affaires réglementaires pour la mise sur le marché des DM avec IA (objectif 2).
Chapitre I - AI Act et RDM, des règlements à portée horizontale et verticale, une approche basée sur le risque
1 L'AI Act : un règlement à portée horizontale concernant tous les secteurs utilisant l'IA
1.1 Définition et types d'IA
L’AI Act définit l'IA comme suit : « Un système basé sur une machine qui est conçu pour fonctionner avec différents niveaux d'autonomie et qui peut faire preuve d'adaptabilité après son déploiement, et qui, pour des objectifs explicites ou implicites, déduit, à partir des données qu'il reçoit, comment générer des résultats tels que des prédictions, du contenu, des recommandations ou des décisions qui peuvent influencer des environnements physiques ou virtuels » (article 3).
L’IA utilise des règles et théories issus des mathématiques, informatiques et statistiques, afin de simuler des fonctions cognitives telle que l’apprentissage, la mémorisation et la prise de décision. Elle englobe : l’apprentissage automatique (Machine Learning) et l’apprentissage profond (Deep Learning), qui est une sous branche de l’apprentissage automatique [3]. La Figure 1 décrit les différentes approches de l’IA, ses méthodes d’apprentissage et quelques types utilisés :
Figure 1 : Représentation des différentes approches, méthodes d'apprentissage et types d'IA. Source : Auteurs, inspiré de [4],[5],[6]
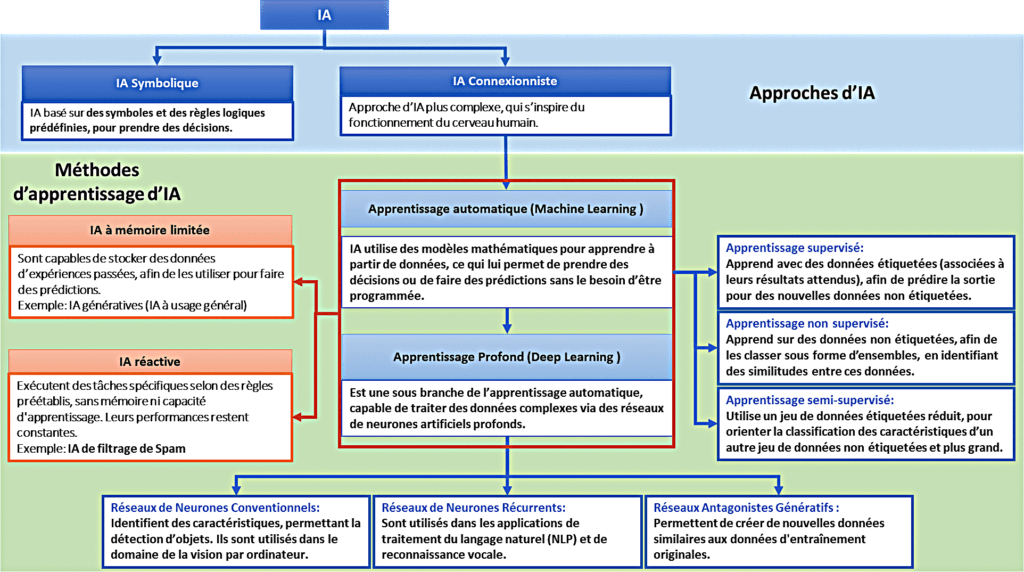
Les IA génératives ou à usage général sont un exemple d’IA à mémoire limitée. Elles sont capables de générer du contenu (images, vidéos, textes) et même d’exécuter d’autres tâches, comme répondre aux questions, ou traduction. Un exemple connu de ces modèles d’IA à usage général est ChatGPT.
1.2 Règlement européen sur l'IA
Ces dernières années, le domaine de l’IA connaît des progrès spectaculaires. Ces progrès offrent de larges opportunités pour l'innovation technologique et l'automatisation dans le cadre professionnel. Cependant, malgré sa valeur ajoutée indéniable et son énorme potentiel, l’IA pose des problèmes qui soulèvent des questions et des défis éthiques, juridiques et politiques. Face à ce constat, la Commission européenne a publié le 21 avril 2021 la proposition de règlement sur l’IA [7]. L’AI Act est finalement adopté le 13 mars 2024 au Parlement Européen [8]. Il est publié au Journal Officiel de l’Union Européenne le 12 juillet 2024.
Le règlement vise à harmoniser les règles de développement, de commercialisation et d'utilisation des systèmes d’IA (SIA) à travers l'Union Européenne (UE). Son objectif est également de promouvoir une IA sûre et fiable, axée sur l'humain, en établissant des garde-fous pour protéger la santé, la sécurité et les droits fondamentaux des citoyens européens (article 1). Ce règlement a une portée horizontale, ce qui signifie qu’il s’applique à tous secteurs utilisant l’IA (jouets, aéronautique, automobile, banque, assurance, ressources humaines etc.).
1.3 Classification des systèmes d'intelligence artificielle à haut risque
L’approche de l’AI Act est basée sur le risque, et les SIA sont classifiés en quatre niveaux de risques, chacun ayant ses propres exigences [9] :
- Risque inacceptable : les SIA qui sont interdits par la loi tels que la manipulation comportementale, l'exploitation des vulnérabilités des personnes et l’utilisation de la reconnaissance biométrique à distance en temps réel (article 5).
- Haut risque : dispositifs auxquels la majeure partie du texte se réfère. Cela concerne des produits tels que les machines, jouets, ascenseurs, de nombreux dispositifs médicaux, et les dispositifs médicaux de diagnostic in vitro qui sont réglementés (article 6).
- Risque limité : sont soumis à des obligations légères de transparence. Les développeurs et déployeurs doivent informer les utilisateurs finaux qu'ils interagissent avec une IA, comme dans le cas des chatbots (article 50).
- Risque minimal : l’AI Act ne spécifie pas d’exigences pour les SIA à risque minimal ou nul.
1.4 Exigences pour les fournisseurs de systèmes d'intelligence artificielle à haut risque
Les fournisseurs de SIA-HR doivent répondre aux exigences suivantes (articles 8 à 17) :
- Etablir un système de gestion des risques tout au long du cycle de vie du dispositif ;
- Mettre en place la gouvernance des données (données d’entraînement, de validation et de test des systèmes) ;
- Constituer la documentation technique (DT) pour démontrer la conformité du dispositif ;
- Concevoir le système de manière à enregistrer les événements importants ;
- S’assurer que le système est suffisamment transparent et que les informations pertinentes sont fournies, afin que les déployeurs puissent interpréter les sorties du système ;
- Permettre le contrôle humain du système via des interfaces homme-machine (IHM) ;
- Atteindre un niveau approprié d’exactitude, robustesse et cybersécurité, de façon constante ;
- Mettre en place un système de gestion de la qualité (SMQ) garantissant le respect du règlement.
1.5 Mise en application progressive
L’AI Act est entré en vigueur le 1er août 2024, avec une mise en application progressive (article 113) :
- 2 février 2025 : Interdiction des SIA présentant des risques inacceptables.
- 2 août 2025 : Application des règles pour les modèles d’IA à usage général et nomination des autorités compétentes dans les États membres.
- 2 août 2026 : Application des dispositions relatives aux SIA-HR définis dans l’annexe III.
- 2 août 2027 : Application des dispositions relatives aux SIA-HR définis dans l’annexe I (dont les DM).
1.6 Normes harmonisées
Ces étapes seront soutenues par des normes harmonisées, dont le respect vaudra présomption de conformité aux exigences du règlement. Le Comité Européen de Normalisation / Comité Européen de Normalisation en Electronique et en Electrotechnique (CEN/CENELEC) les rédige actuellement [10]. Les demandes de normalisation concernent les points ci-dessous [11] :
- Le système de gestion des risques pour les SIA ;
- La gouvernance et la qualité des ensembles de données utilisés pour construire les SIA ;
- La tenue de registres et les capacités d'enregistrement intégrées dans les SIA ;
- La transparence et l'information des utilisateurs des SIA ;
- La surveillance humaine des SIA ;
- Les spécifications de précision pour les SIA ;
- Les spécifications de robustesse des SIA ;
- Les spécifications en matière de cybersécurité pour les SIA ;
- Le système de gestion de la qualité pour les fournisseurs de SIA ;
- L'évaluation de la conformité des SIA.
2 Le RDM : un règlement à portée verticale concernant les DM
2.1 Objectifs principaux : sécurité, performance, transparence et traçabilité
Le RDM concerne la mise sur le marché, la mise à disposition et la mise en service des DM à usage humain et de leurs accessoires dans l’UE. Il vise à améliorer la sécurité et la performance des DM, assurer la protection des utilisateurs par le renforcement des exigences d’évaluation clinique et surveillance après commercialisation (SAC), améliorer la transparence tout en long du cycle de vie des DM et améliorer la traçabilité des produits sur le marché, et harmoniser les exigences afin de faciliter et d’assurer une application cohérente et uniforme entre les états membre de l’UE.
2.2 Champ d'application : types de dispositifs couverts
Le RDM établit un cadre réglementaire élargi et précis pour tous les DM mis sur le marché de l’UE.
Selon l’article 2, il définit un DM comme étant « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez, l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- Diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
- Diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
- Investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique.
- Communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus,
Et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens ».
Le champ d’application du règlement européen couvre :
- Les DM classiques tels que les instruments chirurgicaux, les implants, les logiciels médicaux et les équipements de diagnostic ;
- Les accessoires des DM, définis comme des articles destinés à être utilisés avec un DM pour permettre ou assister son utilisation ;
- Par rapport aux anciennes directives, il inclut les dispositifs sans finalité médicale listés en annexe XVI, comme les lentilles de contact colorées.
Cependant, les probiotiques sont exclus de la nouvelle législation.
2.3 Classes de risque et critères de classification pour les fabricants
Le RDM classe les DM en 4 classes, selon le niveau de risque lié à leur utilisation :
Tableau 1 : Classification des niveaux de risques et leur description
| Classe | Niveau de risque | Description |
| I | Faible | Contient 3 sous-classes : -Is : Dispositif stérile livré -Im : Dispositif avec fonction de mesure -Ir : Dispositif chirurgical réutilisable |
| IIa | Moyen | DM utilisés pour des applications temporaires ou de courte duré |
| IIb | Elevé | DM utilisés pour des traitements prolongés ou invasifs modéré |
| III | Haut risque | DM utilisés pour des interventions critiques ou invasives à haut risque |
La classification des risques est définie dans l'Annexe VIII du RDM et est basée sur les critères suivants : utilisation prévue, type d'appareil, exposition des patients, durée d'utilisation, caractère invasif, gravité du préjudice et niveau de risque. Le RDM contient 22 règles de classification. Ces règles ne sont pas seulement basées sur les risques associés au DM, mais aussi sur sa conception, sa méthode de fabrication, et d'autres facteurs tels que son mode d'utilisation et sa durée d'utilisation.
2.4 Résumé du processus de marquage CE selon le RDM
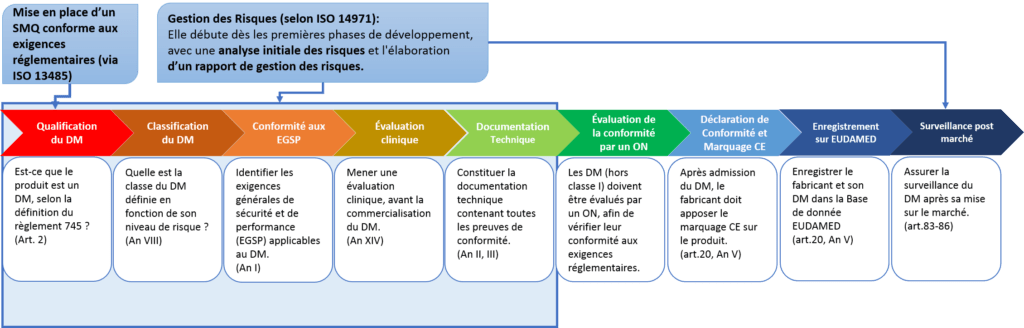
Un fabricant de DM désirant commercialiser son produit dans les marchés de l’UE doit suivre les étapes suivantes décrites dans la Figure 2 ci-dessous :
Figure 2 : Schéma représentant le processus du marquage CE d’un DM selon RDM. Source : Auteurs
Chapitre II - Intégration de l’AI Act et du RDM, enjeux réglementaires et analyse des écarts
1 Enjeux de l’AI Act pour les fabricants de DM
1. 1 Catégories de DM avec SIA proposés par les fabricants
L’IA en santé permet de résoudre des problèmes de diagnostic et de prise de décision pour des situations complexes, notamment dans le cas de patients affectés par plusieurs pathologies. En effet l’IA s’appuie sur des données d’entrées pour proposer un diagnostic ou un traitement dépendamment des situations, de manière patient-spécifique [12]. L’utilisation de l’IA en santé présente de nombreuses opportunités mais également des défis éthiques, de cybersécurité et d’algorithmes qui peuvent donner des faux résultats, ainsi que des risques liés à la confidentialité et à la sécurité des données en raison de la génération de grandes quantités de données [13], [14].
Le nombre de DM utilisant de l’IA est croissant. En Europe, l’estimation du nombre de DM utilisant l’IA est encore difficile. En effet, la base de données européenne sur les DM Eudamed ne permet pas encore de filtrer les DM selon le critère « IA », cependant plus de 2440 dispositifs de type « software » sont répertoriés dans la base de données [15]. Pour référence à un espace économique comparable, aux Etats-Unis, la base de données Food and Drug Administration (ou FDA) recensait 950 DM utilisant de l’IA approuvés depuis 1995, dont 721 dans le domaine de la radiologie. Sur ces 950 dispositifs, plus de la moitié ont été approuvés au cours des dernières années [16].
Les applications de l'IA dans le domaine médical se regroupent principalement en quatre grandes catégories :
- Gestion des maladies chroniques : Utilisation de l’IA pour surveiller en continu les patients via des capteurs intelligents. Exemple : Diabète, avec des dispositifs pour une gestion automatisée de l'insuline.
- Imagerie médicale : L'IA est intégrée aux dispositifs d'imagerie médicale pour améliorer la qualité des images tout en réduisant l'exposition aux radiations. Cette technologie optimise ainsi les résultats cliniques en offrant des images plus claires et plus précises.
- IA et IoT : L'intégration de l'IA avec l’IoT permet de surveiller de manière plus précise l'adhérence des patients aux protocoles de traitement. Cette approche aide à améliorer les résultats cliniques en offrant un suivi en temps réel et une meilleure gestion des soins.
- Interprétation des images radiologiques : L’IA améliore l'interprétation des images radiologiques en automatisant la détection de diverses anomalies, comme les fractures, la pneumonie ou les tumeurs.
Le machine learning est une méthode d'apprentissage utilisée dans les quatre domaines mentionnés ci-dessus. Cette technologie d'intelligence artificielle est employée pour analyser de grandes quantités de données, détecter des schémas complexes et automatiser certaines tâches d'interprétation et de diagnostic [17].
Selon l’AI Act (article 6.1), les DM utilisant un SIA et pour lesquels l’évaluation de la conformité est prévue par un ON dans le RDM sont classés à haut risque. Ainsi, théoriquement, des DM de classe I selon le RDM, qui n’ont pas besoin d’une évaluation par un ON et qui peuvent se contenter d’une auto-déclaration de conformité, ne seront pas classés à haut risque selon l’AI Act. Les DM pour lesquels une auto-déclaration de conformité n’est pas suffisante pour le RDM (classes Is : DM stérile, Im : avec fonction de mesure, Ir : chirurgical réutilisable, classes IIa, IIb et III) seront automatiquement classés comme SIA-HR.
1. 2 Problèmes éthiques et de sécurité liés à l’utilisation de l’IA dans les DM
L’AI Act vise entre autres à prévenir et atténuer les risques pour la sécurité dans le domaine de la santé. A cet effet, la désignation des autorités en charge de veiller à l'aspect réglementaire est en train d'être mise sur pied pour sécuriser les déployeurs (à savoir les utilisateurs) et les patients. L’utilisation de l’IA en santé doit en effet se faire de manière « sûre, fiable et éthique » (considérant 8), de façon à ne pas créer des effets néfastes ou induire de biais discriminatoires dans le traitement des patients. L’utilisation de l’IA dans les DM soulève plusieurs préoccupations éthiques majeures. Chaque type de préoccupation est associé à des défis éthiques spécifiques [18] (voir Tableau 2):
Tableau 2 : Préoccupations éthiques liés à l'IA dans les DM
| Préoccupation | Défis éthique | Explication |
| Preuves insuffisantes | Actions injustifiées | Lorsque les preuves soutenant l'efficacité de l'IA sont insuffisantes, des décisions médicales risquent d'être prises sans fondement solide, ce qui pourrait entraîner des actions injustifiées. |
| Preuves inexplicables | Opacité | Si les résultats de l'IA ne peuvent être expliqués de manière claire et transparente, cela engendre de l'opacité dans les processus décisionnels, réduisant la confiance et la responsabilité. |
| Preuves mal fondées | Biais | Des preuves fondées sur des données incorrectes ou mal interprétées peuvent entraîner des biais dans les décisions prises par l'IA, affectant ainsi l'équité et la fiabilité des résultats. |
| Résultats inéquitables | Discrimination | L’IA peut produire des résultats inéquitables, notamment des discriminations envers certains groupes de patients en raison de biais dans les données utilisées. |
| Effets transformateurs | Autonomie, vie privée informationnelle et vie privée de groupe. | L’IA peut transformer les pratiques médicales, ce qui soulève des questions concernant l’autonomie des patients. De plus, elle pose des défis pour la protection de la vie privée individuelle et de groupe. |
| Traçabilité | Responsabilité morale et distribuée, biais d’automatisation, sécurité, résilience et audit éthique | L'absence de mécanismes adéquats de traçabilité peut compliquer la désignation des responsabilités en cas d'erreur. Il en découle une responsabilité distribuée et des risques associés aux biais d'automatisation. Les systèmes d'IA doivent aussi garantir leur sécurité et leur résilience face aux cyberattaques, et des audits éthiques sont nécessaires pour assurer une utilisation responsable. |
Ces défis montrent l'importance de la régulation de l'IA pour garantir sa bonne utilisation dans les dispositifs médicaux afin de protéger les patients et renforcer la confiance des utilisateurs.
1. 3 Enjeux de surcharge réglementaire pour les fabricants et les ON
Selon le « Guide Bleu » de la Commission Européenne, plus d’un acte juridique de la législation d’harmonisation de l’UE peut s’appliquer aux dispositifs [19]. Dans le cas des DM utilisant l’IA, certaines exigences de l’AI Act peuvent compléter le RDM. Afin d’éviter une surcharge réglementaire pour les fabricants ainsi que pour les ON, déjà engorgés par la certification RDM, une certaine souplesse concernant l’application de la conformité des exigences est recommandée dans l’AI Act. Par exemple, le fournisseur pourrait décider d’intégrer une partie de la DT relative à l’AI Act dans la DT requises en vertu du RDM (article 8.2 de l’AI Act).
De plus, l’interprétation pratique de l’AI Act n’est pas encore claire pour les acteurs du système de santé, y compris les fabricants de DM [20], [21]. Selon l’AI Act, les fournisseurs de SIA devraient pouvoir démontrer la conformité avec les exigences de l’AI Act grâce au respect de normes harmonisées, et en leur absence, au moyen de spécifications communes établies par actes d’exécution (articles 40 et 41 de l’AI Act). Dans l’attente de ces normes harmonisées et actes d’exécution, les meilleures pratiques réglementaires restent à identifier [22].
Les fabricants de DM se trouvent confrontés à une complexité accrue des exigences réglementaires, notamment en raison de l’interaction entre le RDM et l’AI Act. Cette situation génère des coûts supplémentaires et nécessite des investissements conséquents pour garantir la conformité avec les deux régulations. Parallèlement, les ON doivent faire face à une forte demande en compétences techniques spécifiques, en raison du manque de personnel qualifié et des délais de certification souvent prolongés. L’application conjointe de ces législations peut donc entraîner une surcharge de travail, tant pour les fabricants que pour les ON, rendant d'autant plus crucial l’adoption de mesures visant à réduire cette complexité.
2 Analyse des écarts entre les deux règlements
2.1 Comparaison des sommaires
Une lecture des sommaires des deux règlements met en évidence des similarités importantes. Par exemple, les deux règlements comportent tous deux des chapitres sur les bases de données, le SAC, et les sanctions en cas de non-respect des exigences ; ainsi que des annexes sur la DT, les informations à fournir lors de l’enregistrement, et sur l’évaluation de la conformité sur la base d'un SMQ et de l'évaluation de la DT (voir les sommaires en Appendice 1). Dans notre lecture de l’AI Act, nous avons porté une attention particulière au chapitre III, qui détaille les exigences pour les SIA-HR.
2.2 Ecarts entre les définitions
Nous avons effectué une lecture croisée des définitions dans les deux règlements. Dans le Tableau 3, nous avons reporté les écarts les plus notables, ainsi que leur analyse. Il est important de noter que la notion d’incident grave dans l’AI Act ne se limite pas à la santé humaine, mais intègre également les impacts sociétaux et environnementaux du SIA. Nous avons également relevé de manière exhaustive les différences de définitions dans l’Appendice 2.
Tableau 3 : Comparaison des définitions entre l'AI Act et le RDM
| Définition | Idée générale selon l’AI Act | Idée générale selon le RDM | Ecart |
| Opérateur économique | Ajoute la notion de déployeur (à savoir l’utilisateur) | Fabricant, importateur, mandataire, distributeur, etc | Prend en compte les spécificités des systèmes évolutifs |
| Incident grave | Ajoute les risques aux infrastructures critiques, droits fondamentaux, et environnement | Risque pour la santé humaine | S'étend au-delà de la santé humaine pour intégrer les impacts sociétaux et environnementaux |
| SAC | Surveillance continue des SIA, y compris les biais algorithmiques | Collecte proactive de données sur les dispositifs | Anticipe les risques dynamiques propres aux SIA |
| Notice d'utilisation | Indications spécifiques pour le déployeur, y compris le contexte d’utilisation d’un SIA. | Instructions fournies par le fabricant pour la bonne utilisation d’un dispositif | Met l'accent sur les particularités techniques et contextuelles des SIA, incluant les biais et limitations possibles. |
2.3 Comparaison des exigences des deux règlements
Notre analyse comparative entre le RDM et l’AI Act révèle que ce dernier a largement conservé les approches du RDM en ce qui concerne l’approche méthodologique (classification selon niveau de risques et évaluation de la conformité par ON). Bien que l'AI Act s'applique à toutes les industries utilisant l'IA, les fabricants de DM seront les moins impactés, car ils sont déjà familiers avec des concepts nouveaux pour d'autres secteurs tels que les fabricants de jouets (comme la surveillance post-commercialisation).
Figure 3 : Les DM avec SIA-HR doivent répondre aux exigences règlementaires de l'AI Act et du RDM
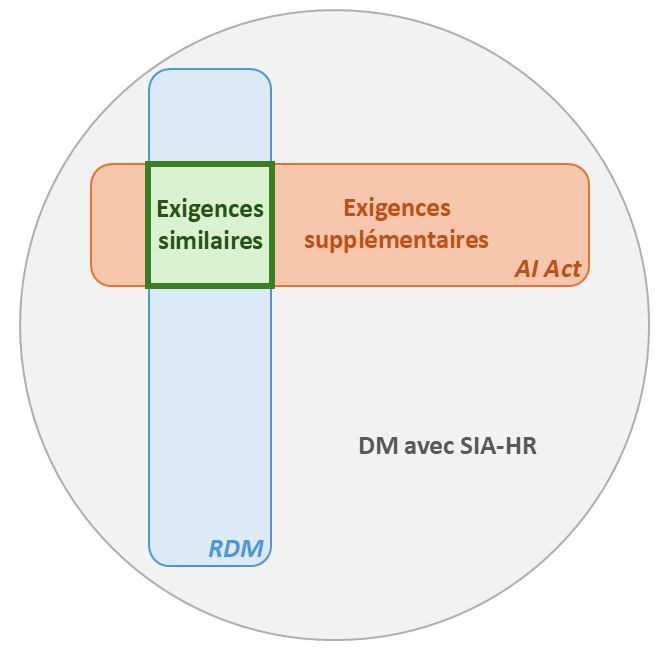
Les deux règlements ont des exigences similaires, comme :
- Le système de gestion des risques : qui est exigé par les deux réglementations, mais l'AI Act (Art.9) se distingue en mettant l'accent sur les risques portant atteinte aux droits fondamentaux. Il introduit également un nouveau principe stipulant que les risques résiduels et les risques résiduels globaux doivent être jugés acceptables, contrairement au RDM qui se concentre sur le rapport bénéfice-risque. Bien que l'AI Act n'évoque pas explicitement cette notion, les fabricants doivent néanmoins la prendre en compte lors du processus de marquage CE de leur SIA-HR.
- Le SMQ : le SMQ de l'AI Act (Art.17) partage la plupart des exigences du RDM, mais demande l’établissement de nouvelles procédures, qui concernent ; l’examen, validation et les tests avant et pendant le développement des SIA-HR (Art.17.1.d), ainsi que toutes les opérations qui traitent la gestion des données (acquisition, collecte, analyse, étiquetage, stockage, filtration, exploration, agrégation, conservation) (Art.17.1.f).
- La Documentation Technique (DT) : la DT de l’AI Act (Art.11, An IV), y compris les procédures et les documents nécessaires, peuvent être intégrée dans celle du RDM.
- L’évaluation de la conformité : le fabricant suit la procédure établie par le RDM. L'évaluation porte sur les deux textes, réalisée par le même ON, à condition que celui-ci soit habilité à auditer selon l'AI Act.
- La déclaration UE de conformité et le marquage CE : le fabricant peut établir une déclaration unique mentionnant à la fois l'AI Act et le RDM (Art.47). Un seul marquage CE peut être apposé, attestant du respect des exigences des deux réglementations (Art.48).
- Le SAC : les deux règlements exigent un plan et un système SAC. Le fabricant a la possibilité d’intégrer les éléments du SAC de son SIA-HR dans celui du RDM.
- Notification d'incidents graves : l’AI Act implique le même principe de signalement aux autorités compétentes locales, mais se différencie du RDM dans la définition de l’incident grave (voir tableau 4).
Notre étude a montré que l’AI Act contient des exigences supplémentaires et spécifiques aux SIA-HR, notamment :
- Les données et la gouvernance des données : L’AI Act impose aux fabricants d’avoir trois jeux de données différents (entrainement, validation, tests). Ces jeux de données doivent respecter des pratiques de gestion mentionnées dans Art.10.2. Ils doivent être pertinents et suffisamment représentatifs de leur environnement d’utilisation prévue, en tenant compte du cadre géographique, comportemental, contextuel, ethnique ou fonctionnel prévu.
- La transparence et fourniture d’informations aux déployeurs : les deux règlements exigent une notice d’utilisation, mais l’AI Act détaille son contenu (Art.13). La notice doit informer le déployeur ; des niveaux de précision du SIA, y compris les métriques, des mesures adoptées pour assurer robustesse et la cybersécurité su SIA et le contrôle humain, des outils d’interprétation des résultats, et d’utilisation correcte du SIA.
- Le contrôle humain : l’AI Act exige que les SIA-HR doivent avoir une IHM interprétable et contrôlable par les humains (Art.14). Les fabricants doivent aussi implémenter des mesures, afin que les déployeurs puissent comprendre les caractéristiques et fonctionnalités des SIA-HR, détecter et résoudre des problèmes, interpréter les résultats et avoir la possibilité d’utiliser l’IA, ignorer ces résultats ou même l’arrêter.
- L’exactitude, la robustesse, la cybersécurité : L’AI Act exige (Art.15) que les SIA-HR soient précis, performants, résilients face aux dangers et protégés contre les cyberattaques. Le fabricant doit, comme mentionné ci-dessus, indiquer dans sa notice d’utilisation les niveaux et indicateurs d’exactitude, accompagnés de méthodes de mesures de performance utilisées, les métriques. Il doit aussi intégrer des mesures techniques ou organisationnel pour assurer la robustesse de son SIA-HR, et solutions techniques capables de prévenir, détecter et maitriser des cyberattaques qui visent les jeux de données, les composants pré-entrainées, les éléments d’entrées et la confidentialité.
- L’enregistrement des évènements et les journaux générés automatiquement : l’AI Act exige que SIA-HR soient capables d’enregistrer automatiquement des évènements (Logs), afin de garantir la traçabilité du fonctionnement du SIA, ce qui permet d’identifier les situations à risques et faciliter le contrôle humain et les analyses de post-commercialisation (Art.12). L’article 19 évoque l’obligation de conserver ces journaux pour une période adaptée à la destination du SIA-HR, d’au moins 6 mois.
L’AI Act consacre un chapitre aux IA à usage général (Chapitre V), avec des exigences particulièrement strictes pour les SIA à risque systémique (définition 65 de l’AI Act : « un risque spécifique aux capacités à fort impact des modèles d’IA à usage général, ayant une incidence significative sur le marché de l’Union en raison de leur portée ou d’effets négatifs réels ou raisonnablement prévisibles sur la santé publique, la sûreté, la sécurité publique, les droits fondamentaux ou la société dans son ensemble, pouvant être propagé à grande échelle tout au long de la chaîne de valeur »). Notre mémoire ne s’est pas étendu sur ces exigences.
2.4 Analyse des référentiels normatifs disponibles pour les exigences supplémentaires de l’AI Act, dans l'attente de normes harmonisées
Dans l’attente de normes harmonisées et codes de bonne pratiques, de nombreux référentiels peuvent être envisagés pour aborder les exigences supplémentaires de l’AI Act par rapport au RDM. Nous les avons répertoriés et analysés afin d’évaluer leur pertinence (Tableau 4).
Tableau 4 : Propositions de référentiels normatifs dans l'attente de normes harmonisées
| Exigence supplémentaire | Référentiel dans l'attente de norme harmonisée | Spécifique | Commentaires | |
| DM | IA | |||
| Gestion des risques liés à l'IA (art. 9) | BS/AAMI 34971:2023 : application de l’ISO 14971 pour les SIA d’apprentissage automatique [23] | ✓ | ✓ | S'applique aux DM basés sur l'apprentissage automatique, tout du long du cycle de vie du DM, et est destinée à être utilisé conjointement avec la norme EN ISO 14971. |
| NF EN ISO/IEC 23894:2024 : Technologies de l'information - IA - Recommandations relatives au management du risque [24] | ✓ | |||
| ISO/IEC TR 24027:2021 Technologie de l'information — IA — Biais dans les SIA et dans la prise de décision assistée par IA [25] | ✓ | Biais liés aux SIA, en particulier en ce qui concerne la prise de décision assistée par l'IA. Les techniques de mesure et les méthodes d'évaluation des biais sont décrites. Toutes les phases du cycle de vie des SIA sont concernées. | ||
| NF EN 62304:2006 + NF EN 62304/A1:2018 Logiciels de DM — Processus du cycle de vie du logiciel [26] | ✓ | N'est pas exclusivement dédiée à la gestion des risques, mais l'aborde. | ||
| NIST AI Risk Management Framework (AI RMF 1.0) [27] | ✓ | Référentiel du National Institute of Standards and Technology (Etats-Unis) pour aider les organisations à gérer les risques associés à la conception, au développement, à l'utilisation et à l'évaluation de SIA, tout au long du cycle de vie du produit. | ||
| Données et gouvernance des données (art. 10) | ISO/IEC 38505-1:2017 Technologies de l'information — Gouvernance des technologies de l'information — Gouvernance des données Partie 1 : Application de l'ISO/IEC 38500 à la gouvernance des données [28] | Adapte les principes de gouvernance de l'ISO/IEC 38500 à la gestion spécifique des données, et offre un cadre pour évaluer, diriger et surveiller l'utilisation des données. | ||
| Enregistrement (art. 12) | ISO/IEC AWI 24970 Artificial intelligence AI system logging [29] | ✓ | En cours de développement par le ISO/IEC JTC 1/SC 42 | |
| Transparence et fourniture d’informations aux déployeurs (art. 13) | ISO/IEC TS 27560:2023 - Technologies pour la protection de la vie privée - Structure de l'information d'enregistrement du consentement [30] | Norme conçue pour la gestion du consentement, mais qui pourrait être adaptée pour répondre aux exigences de transparence. Sa structure standardisée permet de documenter et communiquer les informations requises sur les SIA-HR. | ||
| Contrôle humain (art. 14) | En cours de développement : ISO/IEC AWI 42105 Information technology — Artificial intelligence — Guidance for human oversight of AI systems [31] | ✓ | Guide en cours de développement par le ISO/IEC JTC 1/SC 42 | |
| AFNOR SPEC 2213 - Garantie Humaine des systèmes fondés sur l’intelligence artificielle en santé [32] | ✓ | ✓ | Fournit des lignes directrices sur la méthodologie de mise en œuvre du processus de garantie humaine de l’IA en santé (DM et non-DM). | |
| Exactitude, robustesse et cybersécurité (art.15) | ISO/IEC TR 24029-1:2021 Artificial Intelligence (AI) — Assessment of the robustness of neural networks Part 1 : Overview [33] | ✓ | Aperçu des méthodes d'évaluation de la robustesse des réseaux neuronaux utilisés dans les SIA. Décrit les concepts clés, les défis et les approches pour tester la robustesse des réseaux neuronaux face à diverses perturbations et attaques. | |
| ISO/IEC TR 24028:2020 Technologies de l'information - Intelligence artificielle - Examen d'ensemble de la fiabilité en matière d'intelligence artificielle [34] | ✓ | Aperçu complet des facteurs influençant la fiabilité des SIA, y compris l'exactitude, la robustesse et la cybersécurité. | ||
| FDA guidance : Cybersecurity in Medical Devices [35] | ✓ | ✓ | Aspects de cybersécurité, en particulier en ce qui concerne l'évaluation des risques, la robustesse des algorithmes et la gestion du cycle de vie des DM. | |
| Système de gestion de la qualité : 1.d) Des procédures d’examen et 1.f) Les systèmes et procédures de gestion des données (art. 17) | ISO/IEC 42001:2023 Technologies de l'information - Intelligence artificielle - Système de management [36] | ✓ | Attention : cette norme généraliste ne sera pas retenue comme norme harmonisée pour l'AI Act. | |
| ISO/IEC 5259-1:2024 Intelligence artificielle — Qualité des données pour les analyses de données et l’apprentissage automatique Partie 1 : Vue d'ensemble, terminologie et exemples [37] | ✓ | Evaluer et améliorer la qualité des données utilisées dans l'analyse de données et l'apprentissage automatique. Peut aider les fabricants à démontrer que leurs ensembles de données sont pertinents, représentatifs et exempts de biais. | ||
Chapitre III - Proposition d’outils d’intégration à destination des fabricants
1 État des lieux des solutions existantes d’intégration de l’AI Act avec le RDM
Depuis la préparation de l'AI Act, diverses initiatives et solutions ont été proposées pour mieux comprendre son contenu et évaluer son degré de convergence avec le RDM. Le Tableau 5 ci-dessous présente une synthèse et une analyse des solutions et guides publiés à ce jour :
Tableau 5 : État des lieux des guides existants
| Types d'auteurs | Auteurs | Nom du guide | Date de publication | Contenu du guide | Forme |
| Organismes Notifiés | GMED | « AI ACT » : Quelles exigences et délais pour les dispositifs médicaux [38] | Novembre 2024 | Guide très court à destination des fabricants de DM. Méthode basée sur le résumé de l’AI Act avec classification des SIA-HR, évoque les dates butoirs, un rappel des exigences communes et une brève explication pour les exigences supplémentaires. | Pdf d’une newsletter |
| IG-NB Alliance allemande des ON | Questionnaire « Intelligence Artificielle (IA ) dans les dispositifs médicaux » [39] | 15 décembre 2023 | Guide détaillé à destination des ON, fabricants et tiers intéressés. Il est basé sur la Guideline for AI for Medical Devices qui évoque des sujets tels que la documentation et les exigences logicielles. Des réponses font référence aux normes et réglementations. | Guide basé sur des questionnaires | |
| Fabricants | QUICKBIRD MEDICAL | Loi sur l’IA : Guide pour les fabricants de dispositifs médicaux conformément au MDR (2024) [40] | 11 septembre 2024 | Guide à destination des fabricants de DM. Il évoque les exigences couvertes par le RDM et les exigences supplémentaires en détails avec ce que les fabricants doivent faire. | Pdf contenant des schémas et des tableaux |
| Consultants | MRM | Règlement (UE) 2024/1689, dit AI Act et le RDM/IVDR [41] | Guide peu détaillé qui aborde les aspects généraux de l’AI Act. Il donne aussi les exigences que le fabricant doit respecter en plus des exigences du RDM. | PDF sous forme de points clés | |
| Article scientifique | Mateo Aboy Timo Minssen Effy Vayena | La loi européenne sur l’IA : Implications pour les produits médicaux numériques réglementés [42] | 6 septembre 2024 | Guide assez détaillé permettant la navigation dans la législation européenne sur l’AI Act avec des grands thèmes, qui évoque les exigences appliquées aux SIA-HR, leurs impacts sur les DM et les déployeurs. | Article basé sur les questions générales sur AI Act |
| Organisme indépendant à but non lucratif | EU Artificial Intelligence Act | Vérification de la conformité à la loi européenne sur l’IA [43] | Guide à destination de plusieurs organismes avec des questions et des réponses au choix ainsi que les références des articles traitant ces questions. | Site web interactif sous forme de questionnaire à choix multiples |
D’après l’état des lieux effectué, nous avons trouvé que la plupart des guides actuels sont soit très détaillés et complexes, soit assez superficiels, et donc peuvent engendrer des ambiguïtés pour le fabricant. Donc, nous proposons des outils intermédiaires et interactifs permettant de faciliter aux fabricants l’intégration des exigences de l’AI Act dans le processus de marquage CE selon RDM.
2 Conception d’outils à destination des fabricants de DM avec SIA
2.1 Entretiens avec les parties prenantes pour recueillir les besoins
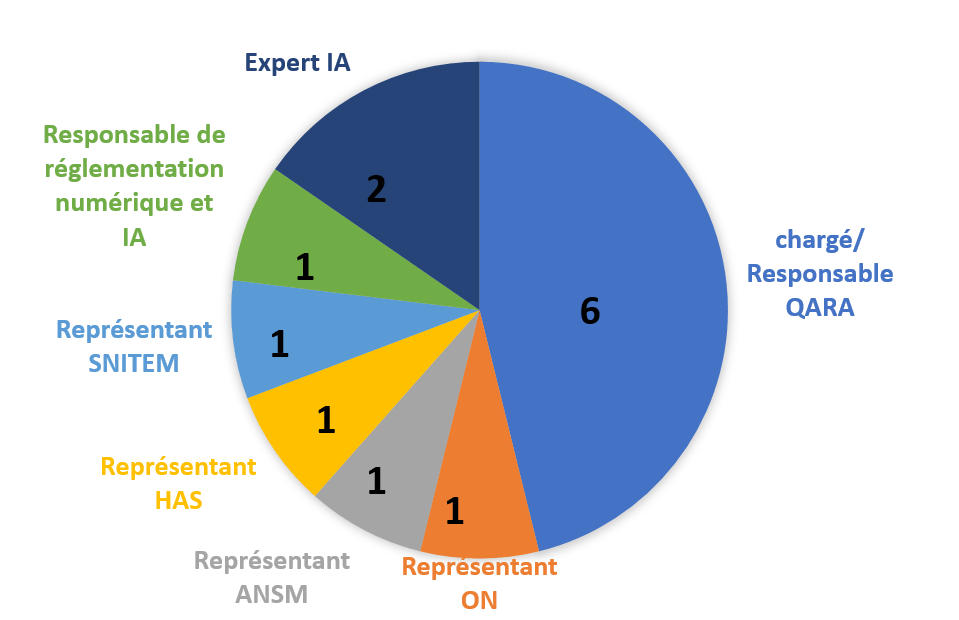
Pour établir les besoins de nos livrables, nous avons conduit des entretiens avec 13 experts occupant divers postes liés aux DM allant de chargés et responsables AQ/AR travaillant pour des fabricants de DM, ainsi que des représentants d'un organisme notifié, d'une autorité compétente et de syndicats (au niveau français et européen). La Figure 4 représente la répartition des entretiens faits :
Figure 4 : Répartition des postes des experts interviewés et de leur nombre. Source : Auteurs.
Les experts ont soulevé plusieurs points importants concernant les obstacles à surmonter, leurs inquiétudes, leurs craintes et leurs besoins pour se conformer à l’AI Act. Ils ont également partagé leurs préférences quant au guide, son contenu souhaité et sa forme désirée. Le tableau suivant représente les idées les plus fréquemment exprimées par eux (voir Tableau 6 ci-dessous) :
Tableau 6 : Synthèse des avis d'experts concernant l’AI Act
| Défis et inquiétudes | Besoins et attentes | Préférences du guide |
| - La disponibilité des ONs compétents du côté technique (IA) que réglementaire - Doutes sur la préparation de l’ON aux temps prévus - Déroulement des audits des deux règlements (simultanés ou séparées) - Risque de retard de la commercialisation des produits - Crainte de l’impact de l’AI Act sur l’innovation dans le marché européen - Ambiguïtés sur l’identification et l’application des normes harmonisées - Inquiétudes sur les nouvelles exigences et risques de l’AI Act (risque de biais, cybersécurité) | -Actions nécessaires à faire pour se conformer au règlement -Des conseils pour intégrer les exigences de l’IA Act au processus de marquage CE selon le RDM -Besoin d'un soutien des organismes européens de normalisation et des ON par normes ou guides -Cherchent des mesures légitimes et acceptées par les ON autres que NH pour répondre aux nouvelles exigences -Extension des délais -Des clarifications sont demandées sur des aspects spécifiques (mise en place des indicateurs de performance, l'évaluation des performances et la définition des changements majeurs d'algorithmes) -Avoir les moyens nécessaires pour y répondre (coût humain et économique) | - Analyse d’impact - Analyse d’écarts - Checklist - Logigramme - Illustrations claires -Interprétation compréhensible et non ambiguë - Identifier les similitudes et différences entre les deux règlements |
Pour notre outil, nous avons privilégié les avis les plus couramment exprimés par les experts. Tous s'accordent sur la nécessité d'intégrer les nouvelles exigences de l'AI Act dans le processus de marquage CE selon le RDM. Ils préconisent également de proposer des solutions pour se conformer à ces exigences, et de préciser certaines notions techniques spécifiques aux SIA. Concernant la forme, les experts préfèrent un logigramme, illustrant les tâches à réaliser et mettant en évidence les similitudes et différences entre les deux règlements, afin d'éviter une surcharge réglementaire. Ils soulignent également l'importance d'une check-list pour identifier les éléments essentiels à fournir et faciliter leur validation.
2.2 Guide PDF interactif
Cet outil est destiné aux fabricants de DM qui souhaitent comprendre les exigences supplémentaires de l’AI Act par rapport au RDM pour les DM avec IA. Entre le jalon 2 et le jalon 3, nous avons obtenu des retours d’experts à l’aide d’un questionnaire de satisfaction et d’entretiens pour affiner notre outil (voir Appendice 3).
L’outil est un PDF interactif, facile d’utilisation. La première page est un logigramme qui représente les étapes du marquage CE selon l’AI Act et le RDM, et qui met en exergue les exigences supplémentaires. L’utilisateur peut cliquer sur les exigences supplémentaires, et est amené vers des diapositives qui contiennent une interprétation de l’exigence, des exemples concrets et des référentiels normatifs dans l’attente de normes harmonisées.
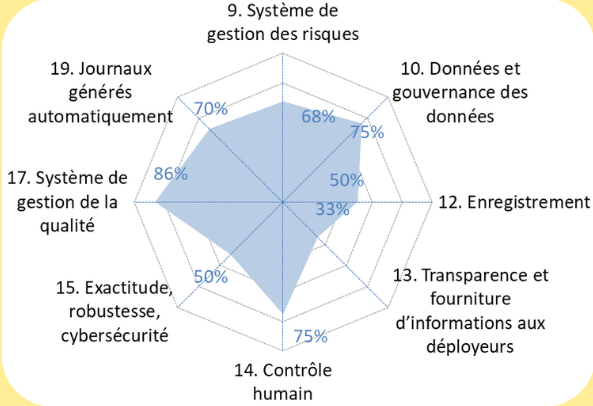
2.4 Outil d'autodiagnostic
Cet outil est destiné aux fabricants de DM qui connaissent déjà le RDM, et qui souhaitent commercialiser un DM avec SIA. L’outil d’autodiagnostic leur permet d’évaluer la maturité de leur produit et de leur SMQ, et de communiquer leurs résultats de manière claire et synthétique. Cet outil se présente sous forme de fichier Excel avec :
- Un premier onglet « mode d’emploi » ;
- Un deuxième onglet « exigences AI Act pour DM », où les exigences de l’AI Act pour le fabricant, qui s’appliquent aux fabricants de DM, sont listées. Le fabricant peut répondre à chaque sous-article du règlement avec un certain niveau de véracité, et indiquer en commentaire les modes de preuve existants.
- Un troisième onglet « résultats et actions », qui résume le niveau de conformité pour chaque article et sous-article à l’aide de graphiques radars et de tableaux de taux de conformité générés automatiquement.
Nos livrables abordent brièvement la nécessité de se référer au chapitre 5 de l'AI Act, pour les IA à usage général, sans en détailler les exigences. Le périmètre de nos outils concerne les IA non à usage général, estimant que cela concerne peu de DM.
Conclusion
Notre étude a mis en évidence les défis auxquels sont confrontés les fabricants de DM intégrant des SIA-HR face à l’intégration des exigences de l’AI Act avec celles du RDM. Nous avons identifié des exigences similaires entre ces deux règlements, par exemple en matière de SMQ et de gestion des risques, avec certaines particularités spécifiques à l’IA. L'AI Act introduit également de nouvelle exigences, telles que l'enregistrement automatique des journaux et le contrôle humain, qui nécessitent une attention particulière. La preuve de conformité à ces exigences pourra être incorporée dans la DT relative au RDM.
Les entretiens menés avec les parties prenantes ont révélé différents degrés de préparation à l’application de l’AI Act. En particulier, les fabricants expriment un besoin pressant de clarification de l'interprétation des exigences et d’établissement de référentiels normatifs harmonisés, tant pour la certification de leur société (par exemple pour le SMQ de l’IA) que pour la démonstration de conformité dans la DT. Les fabricants ont également partagé des inquiétudes concernant le manque potentiel d'ON qualifiés pour évaluer les aspects liés à l'IA, et le coût financier de la mise en conformité à l’AI Act.
Pour permettre aux fabricants de mieux comprendre l’impact de l’AI Act sur leur organisation et sur les dossiers techniques, dans l’attente de normes harmonisées ou spécification communes par la CE, nous avons développé un premier livrable sous forme de guide d'intégration interactif, qui explicite les exigences supplémentaires et propose des référentiels normatifs. Nous avons également conçu un deuxième livrable sous forme d’outil d’autodiagnostic permettant aux fabricants de DM avec SIA d’évaluer la maturité de leur conformité aux exigences supplémentaires imposées par l’AI Act.
Liste des abréviations
AI Act : Règlement (UE) 2024/1689 du 13 juin 2024 établissant des règles harmonisées concernant l’intelligence artificielle
AQ/AR : Assurance Qualité et Affaires Réglementaires
CEN/CENELEC : Comité Européen de Normalisation / Comité Européen de Normalisation en Electronique et en Electrotechnique
DM : Dispositif médical
DM/DIV : Dispositif médical de diagnostic in vitro
DT : Documentation Technique
EHDS : Espace européen des données de santé
EGSP : Exigences générales de sécurité et de performance (General safety and performance requirements)
FDA : Food Drug Administration
GEHN : Groupe d'Experts de Haut Niveau
IA : Intelligence Artificielle
IHM : Interface Homme-Machine
IoT : Internet des objets (Internet of things)
RDM : Règlement (UE) 2017/745 du 5 avril 2017 relatif aux dispositifs médicaux
ML : Machine learning
ON : Organisme Notifié
PSUR : Rapport périodique actualisé de sécurité
RGPD : Règlement Géneral sur la protection des données
SAC : Surveillance Après Commercialisation
SIA : Système d’intelligence artificielle
SIA-HR : Système d’intelligence artificielle à haut risque
SMQ : Système de management de la qualité
UE : Union Européenne
Références bibliographiques
[1] « Règlement (UE) 2024/1689 du Parlement européen et du Conseil du 13 juin 2024 établissant des règles harmonisées concernant l’intelligence artificielle et modifiant les règlements (CE) n° 300/2008, (UE) n° 167/2013, (UE) n° 168/2013, (UE) 2018/858, (UE) 2018/1139 et (UE) 2019/2144 et les directives 2014/90/UE, (UE) 2016/797 et (UE) 2020/1828 (règlement sur l’intelligence artificielle) (Texte présentant de l’intérêt pour l’EEE) », juill. 2024. Disponible sur : https://eur-lex.europa.eu/eli/reg/2024/1689/oj
[2] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra
[3] Inserm, « Intelligence artificielle et santé », 25 janvier 2024. Disponible sur : https://www.inserm.fr/dossier/intelligence-artificielle-et-sante/
[4] IBM, « Qu’est-ce que le machine learning (ML) ? | IBM », ibm.com. Consulté le : 15 décembre 2024. Disponible sur : https://www.ibm.com/fr-fr/topics/machine-learning
[5] J. Holdsworth et M. Scapicchio, « What Is Deep Learning ? », 17 juin 2024. Disponible sur : https://www.ibm.com/topics/deep-learning
[6] IBM Data and AI Team, « Understanding the different types of artificial intelligence », 12 octobre 2023. Disponible sur : https://www.ibm.com/think/topics/artificial-intelligence-types
[7] European Commission, « Proposal for a regulation of the European parliament and of the council laying down harmonised rules on artificial intelligence (artificial intelligence act) and amending certain union legislative acts », 21 avril 2021. Disponible sur : https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52021PC0206
[8] Parlement Européen, « Textes adoptés le mercredi 13 mars 2024 à Strasbourg », www.europarl.europa.eu. Consulté le : 24 septembre 2024. Disponible sur : https://www.europarl.europa.eu/doceo/document/TA-9-2024-03-13-TOC_FR.html
[9] CNIL (Commission nationale de l’informatique et des libertés), « Entrée en vigueur du règlement européen sur l’IA : les premières questions-réponses de la CNIL », 12 juillet 2024. Disponible sur : https://www.cnil.fr/fr/entree-en-vigueur-du-reglement-europeen-sur-lia-les-premieres-questions-reponses-de-la-cnil
[10] V. Desbordes, « Grand Défi IA : la saison 2 a débuté ce jeudi », 22 août 2024. Disponible sur : https://www.afnor.org/actualites/grand-defi-ia-bonnes-pratiques/
[11] P. Bezombes, « JTC 21 : Activities in the context of the AI Act », 29 avril 2024. Disponible sur : https://www.eurocontrol.int/sites/default/files/2024-04/20240429-flyai-forum-session-2-bezombes.pdf
[12] S. S. Gadde et V. D. R. Kalli, « Applications of Artificial Intelligence in Medical Devices and Healthcare », www.ijcstjournal.org, vol. 1, p. 182‑188, mai 2020, doi : https://doi.org/10.3233/SHTI240472
[13] E. Gómez-González et E. Gómez, « Artificial intelligence for healthcare and well-being during exceptional times », Joint Research Centre, Commission Européenne, JRC134715, nov. 2023. Disponible sur : https://ai-watch.ec.europa.eu/publications/artificial-intelligence-healthcare-and-well-being-during-exceptional-times_en
[14] E. Biasin, E. Kamenjašević, et K. R. Ludvigsen, « Chapter 4 : Cybersecurity of AI medical devices : risks, legislation, and challenges », in Research Handbook on Health, AI and the Law, in Law 2024. , Elgaronline, 2024. Disponible sur : https://www.elgaronline.com/edcollchap-oa/book/9781802205657/book-part-9781802205657-10.xml
[15] Commission Européenne, « EUDAMED - Base de données européenne sur les dispositifs médicaux », Site de la Commission Européenne. Consulté le : 5 janvier 2025. Disponible sur : https://ec.europa.eu/tools/eudamed/#/screen/search-device
[16] Center for Devices and Radiological Health, « Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices », fda.gov. Consulté le : 5 janvier 2025. Disponible sur : https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-aiml-enabled-medical-devices
[17] Inserm, « Intelligence artificielle et santé : Des algorithmes au service de la médecine », 25 janvier 2024. Disponible sur : https://www.inserm.fr/dossier/intelligence-artificielle-et-sante/
[18] B. Mittelstadt, « L’impact de l’intelligence artificielle sur les relations medecin-patient », Conseil de l’Europe, Rapport commandé par le Comité directeur pour les droits de l’Homme dans les domaines de la biomédecine et de la santé, déc. 2021. Disponible sur : https://rm.coe.int/inf-2022-5-report-impact-of-ai-on-doctor-patient-relations-f/1680a6885a
[19] Commission Européenne, « Le « Guide bleu » relatif à la mise en œuvre de la réglementation de l’UE sur les produits 2022 (Texte présentant de l’intérêt pour l’EEE) 2022/C 247/01 », Journal officiel de l’Union européenne, juin 2022. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/ALL/?uri=CELEX%3A52022XC0629%2804%29
[20] H. van Kolfschooten et J. van Oirschot, « The EU Artificial Intelligence Act (2024): Implications for healthcare », Health Policy, vol. 149, p. 105152, nov. 2024, doi : https://doi.org/10.1016/j.healthpol.2024.105152.
[21] S. Gilbert, « The EU passes the AI Act and its implications for digital medicine are unclear », Npj Digit. Med., vol. 7, no 1, p. 1‑3, mai 2024, doi : https://doi.org/10.1038/s41746-024-01116-6.
[22] J. Schmidt et al., « Mapping the regulatory landscape for artificial intelligence in health within the European Union », Npj Digit. Med., vol. 7, no 1, p. 1‑9, août 2024, doi : https://doi.org/10.1038/s41746-024-01221-6.
[23] BSI et AAMI, « BS/AAMI 34971:2023 », BSI Group, 31 mai 2023. Disponible sur : https://knowledge.bsigroup.com/products/application-of-iso-14971-to-machine-learning-in-artificial-intelligence-guide?version=standard
[24] AFNOR CN IA, « NF EN ISO/IEC 23894 Technologies de l’information - Intelligence artificielle - Recommandations relatives au management du risque », Ed. Afnor, Paris, www.afnor.org, 21 février 2024.
[25] ISO/IEC/JTC 1, « ISO/IEC TR 24027:2021 Technologie de l’information - Intelligence artificielle (IA) - Tendance dans les systèmes de l’IA et dans la prise de décision assistée par l’IA », Ed. Afnor, Paris, www.afnor.org, 1 novembre 2021.
[26] TC 62/SC 62A, « IEC 62304:2006 Logiciels de dispositifs médicaux - Processus du cycle de vie du logiciel », Ed. Afnor, Paris, www.afnor.org, 1 mai 2006.
[27] « Artificial Intelligence Risk Management Framework (AI RMF 1.0) », National Institute of Standards and Technology (U.S.), Gaithersburg, MD, NIST AI 100-1, janv. 2023. Disponible sur : http://nvlpubs.nist.gov/nistpubs/ai/NIST.AI.100-1.pdf
[28] ISO/IEC/JTC 1, « ISO/IEC 38505-1:2017 Technologies de l’information - Gouvernance des technologies de l’information - Gouvernance des données - Partie 1 : Application de l’ISO/IEC 38500 à la gouvernance des données », Ed. Afnor, Paris, www.afnor.org, 1 avril 2017.
[29] ISO/IEC JTC 1/SC 42, « Projet : ISO/IEC AWI 24970 Intelligence artificielle — Journalisation des systèmes d’IA », Ed. ISO - Organisation Internationale de Normalisation www.iso.org. Consulté le : 12 novembre 2024. Disponible sur : https://www.iso.org/fr/standard/88723.html
[30] ISO/IEC JTC 1/SC 27, « ISO/IEC TS 27560:2023 Technologies pour la protection de la vie privée - Structure de l’information d’enregistrement du consentement », Ed. Afnor, Paris, www.afnor.org, 8 août 2023.
[31] ISO/IEC JTC 1/SC 42, « Projet : ISO/IEC AWI 42105 Guidance for human oversight of AI systems », Ed. ISO - Organisation Internationale de Normalisation www.iso.org. Consulté le : 12 novembre 2024. Disponible sur : https://www.iso.org/fr/standard/86902.html
[32] AFNOR SPEC ETHIC IA, « AFNOR SPEC 2213 Garantie Humaine des systèmes fondés sur l’intelligence artificielle en santé », Ed. Afnor, Paris, www.afnor.org, 29 mai 2024.
[33] ISO/IEC/JTC 1, « ISO/IEC TR 24029-1:2021 Artificial Intelligence (AI) - Assessment of the robustness of neural networks - Part 1 : Overview », Ed. Afnor, Paris, www.afnor.org, 10 mars 2021.
[34] ISO/IEC/JTC 1, « ISO/IEC TR 24028:2020 Technologies de l’information - Intelligence artificielle - Examen d’ensemble de la fiabilité en matière d’intelligence artificielle », Ed. Afnor, Paris, www.afnor.org, 1 mai 2020.
[35] Center for Devices and Radiological Health, Guidance : Cybersecurity in Medical Devices : Quality System Considerations and Content of Premarket Submissions, FDA-2021-D-1158, 26 septembre 2023. Disponible sur : https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cybersecurity-medical-devices-quality-system-considerations-and-content-premarket-submissions
[36] ISO/IEC JTC 1/SC 42, « ISO/IEC 42001:2023 Technologies de l’information - Intelligence artificielle - Système de management », Ed. Afnor, Paris, www.afnor.org, 18 décembre 2023.
[37] ISO/IEC JTC 1/SC 42, « ISO/IEC 5259-1:2024 Intelligence artificielle - Qualité des données pour les analyses de données et l’apprentissage automatique - Partie 1 : Vue d’ensemble, terminologie et exemples », Ed. Afnor, Paris, www.afnor.org, 2 juillet 2024.
[38] GMED groupe LNE, « « AI ACT » : quelles exigences et échéances pour les dispositifs médicaux ? », 7 novembre 2024. Disponible sur : https://lne-gmed.com/fr/newsletters-fr/exigences-ai-act
[39] M. Bellancini et al., « Questionnaire Artificial Intelligence in medical devices ». German Notified Bodies, Alliance for Medical Devices , Team NB,The European Association of Medical Devices Notified Bodies, 14 novembre 2024. Disponible sur : https://www.team-nb.org/wp-content/uploads/2024/11/Team-NB-PositionPaper-AI-in-MD-Questionnaire-V1-20241125.pdf
[40] A. Fenkiw, « AI Act : Guide for medical device manufacturers according to MDR (2024) », 11 septembre 2024. Disponible sur : https://quickbirdmedical.com/en/ai-act-medical-device/
[41] QNET, « (EU) Regulation 2024/1689, the so-called “AI Act” (AIA), and the MDR/IVDR », MRM Medical Risk Management. Consulté le : 10 novembre 2024. Disponible sur : https://www.medical-risk.com/item/90-eu-regulation-2024-1689-the-so-called-ai-act-aia-and-the-mdr-ivdr
[42] M. Aboy, T. Minssen, et E. Vayena, « Navigating the EU AI Act : implications for regulated digital medical products », Npj Digit. Med., vol. 7, no 1, p. 1‑6, sept. 2024, doi : https://doi.org/10.1038/s41746-024-01232-3.
[43] Future of Life Institute, « EU AI Act Compliance Checker | Loi européenne sur l’intelligence artificielle », artificialintelligenceact.eu. Consulté le : 9 novembre 2024. Disponible sur : https://artificialintelligenceact.eu/fr/evaluation/verificateur-de-conformite-a-l-acte-de-l-ai-de-l-ue/
Appendices
Appendice 1 Sommaires de l’AI Act et du RDM
| AI Act | RDM |
| Nombre de considérants | |
| 180 | 101 |
| Chapitres | |
| I Dispositions générales II Pratiques interdites en matière d’IA III Systèmes d’IA à haut risque (SIA-HR) IV Obligations de transparence pour les fournisseurs et les déployeurs de certains SIA V Modèles d'IA à usage général VI Mesures de soutien à l'innovation VII Gouvernance VIII Base de données de l'UE sur les SIA-HR IX Suivi post-commercialisation, partage d'informations et surveillance du marché X Codes de conduite et lignes directrices XI Délégation de pouvoir et procédure de comité XII Sanctions XIII Dispositions finales | I Champ d’application et définitions II Mise à disposition sur le marché et mise en service des dispositifs, obligations des opérateurs économiques, retraitement, marquage CE et libre circulation III Identification et traçabilité des dispositifs, enregistrement des dispositifs et des opérateurs économiques, résumé des caractéristiques de sécurité et des performances cliniques et base de données européenne sur les DM IV ON V Classification et évaluation de la conformité VI Evaluation clinique et investigations cliniques Ch VII Surveillance après commercialisation, vigilance et surveillance du marché VIII Coopération entre les états membres, le groupe de coordination en matière de DM, les laboratoires spécialisés, les groupes d’experts et les registres de dispositifs IX Confidentialité, protection des données, financement et sanctions X Dispositions finales |
| Annexes | |
| I Liste de la législation d'harmonisation de l'Union II Liste des infractions pénales visées à l’art. 5, § 1, premier alinéa, point h) iii) III Systèmes d’IA à haut risque visés à l’art. 6, § 2 IV DT visée à l’art. 11, § 1 V Déclaration de conformité de l'UE VI Procédure d'évaluation de la conformité basée sur le contrôle interne VII Conformité sur la base d'une évaluation du système de gestion de la qualité et d'une évaluation de la DT VIII Informations à fournir lors de l'enregistrement des systèmes d'IA à haut risque conformément à l'art. 49 IX Informations à fournir lors de l'enregistrement des systèmes d'IA à haut risque énumérés à l'An III en ce qui concerne les essais en conditions réelles conformément à l'art. 60 X Actes législatifs de l'Union sur les systèmes d'information à grande échelle dans le domaine de la liberté, de la sécurité et de la justice XI DT visée à l'art. 53, § 1, point a) - DT destinée aux fournisseurs de modèles d'IA à usage général XII Informations relatives à la transparence visées à l'art. 53, § 1, point b) - DT à l'intention des fournisseurs de modèles d'IA à usage général aux fournisseurs en aval qui intègrent le modèle dans leur système d'IA XIII Critères de désignation des modèles d'IA à usage général présentant un risque systémique visés à l'art. 51 | I Exigences générales en matière de sécurité et de performances II DT III DT relative à la surveillance après commercialisation IV Déclaration de conformité UE V Marquage de conformité CE VI Informations à fournir lors de l'enregistrement des dispositifs et des opérateurs économiques conformément à l'art. 29, § 4, et à l'art. 31 ; principaux éléments de données à fournir à la base de données IUD avec l'IUD-ID conformément aux art.s 8 et 29 et système IUD VII Exigences auxquelles doivent satisfaire les organismes notifiés VIII Règles de classification IX Évaluation de la conformité sur la base d'un système de gestion de la qualité et de l'évaluation de la DT X Évaluation de la conformité sur la base de l'examen de type XI Évaluation de la conformité sur la base de la vérification de la conformité du produit XII Certificats délivrés par un organisme notifié XIII Procédure pour les dispositifs sur mesure XIV Évaluation clinique et suivi clinique après commercialisation XV Investigations cliniques XVI Liste des groupes de produits n'ayant pas de destination médicale prévue visés à l'art. 1er, § 2 XVII Tableau de correspondance |
Appendice 2 Ecarts des définitions dans les deux règlements

Appendice 3 Questionnaire pour l'amélioration du logigramme