
IDS301 – Analyse des méthodes de gestion des risques des fabricants de DM et proposition d’un outil d’aide à l’application du guide FD CEN ISO/TR 24971
DOI mémoire
DOI : https://doi.org/10.34746/ids301Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteures




Contacts
- KENGNIE Diane Price Aout : dianekengnie05@gmail.com
- KADIRI Chourouk : kadirichorouk@gmail.com
- OUHAB Sabrina : sabrinaouhab2002@icloud.com
- BENCHAOUCH Hafsa : benchaouchhafa@gmail.com
Citation
A rappeler pour tout usage : D. KENGNIE, C. KADIRI, S. OUHAB, H. BENCHAOUCH « Analyse des méthodes de gestion des risques des fabricants de DM et proposition d’un outil d’aide à l’application du guide FD CEN ISO/TR 24971 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, janvier 2026, https://travaux.master.utc.fr/, réf n° IDS301, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids301/, https://doi.org/10.34746/ids301
Résumé
La gestion des risques est un élément central de la sécurité et de la conformité réglementaire des dispositifs médicaux. Elle est encadrée par la norme NF EN ISO 14971:2019 et complétée par le guide d’application FD CEN ISO/TR 24971, qui vise à faciliter sa mise en œuvre opérationnelle.
Ce projet a pour objectif d’analyser les pratiques réelles de gestion des risques chez les fabricants de dispositifs médicaux et d’identifier les limites rencontrées dans l’application des exigences normatives. Une enquête terrain, réalisée sous forme de questionnaire auprès de fabricants de tailles variées, a permis d’évaluer le niveau de maîtrise de la norme, les méthodes d’analyse des risques utilisées ainsi que l’usage du guide FD CEN ISO/TR 24971.
Les résultats montrent une application globalement satisfaisante de la norme ISO 14971, mais une utilisation encore partielle de son guide d’application. Les méthodes les plus employées restent l’AMDEC, les matrices de criticité et les check-lists internes, majoritairement basées sur des approches qualitatives. Les répondants expriment un besoin d’outils pédagogiques et opérationnels pour harmoniser les pratiques et améliorer la compréhension des exigences réglementaires.
Sur la base de ces constats, ce travail propose le développement d’un outil interactif d’aide à l’application du guide FD CEN ISO/TR 24971, destiné à accompagner les fabricants dans la structuration et l’amélioration de leur processus de gestion des risques.
Abstract
Risk management is a central element of medical device safety and regulatory compliance. It is governed by standard NF EN ISO 14971:2019 and supplemented by application guide FD CEN ISO/TR 24971, which aims to facilitate its operational implementation.
The aim of this project is to analyse the actual risk management practices of medical device manufacturers and to identify the limitations encountered in the application of the standard requirements. A field survey, conducted in the form of a questionnaire among manufacturers of various sizes, assessed the level of mastery of the standard, the risk analysis methods used and the use of the FD CEN ISO/TR 24971 guide.
The results show that the ISO 14971 standard is generally applied satisfactorily, but that its application guide is still only used to a limited extent. The most commonly used methods remain FMEA, criticality matrices and internal checklists, which are mainly based on qualitative approaches. Respondents expressed a need for educational and operational tools to harmonise practices and improve understanding of regulatory requirements.
Based on these findings, this work proposes the development of an interactive tool to assist in the application of the FD CEN ISO/TR 24971 guide, designed to support manufacturers in structuring and improving their risk management processes.
Téléchargements

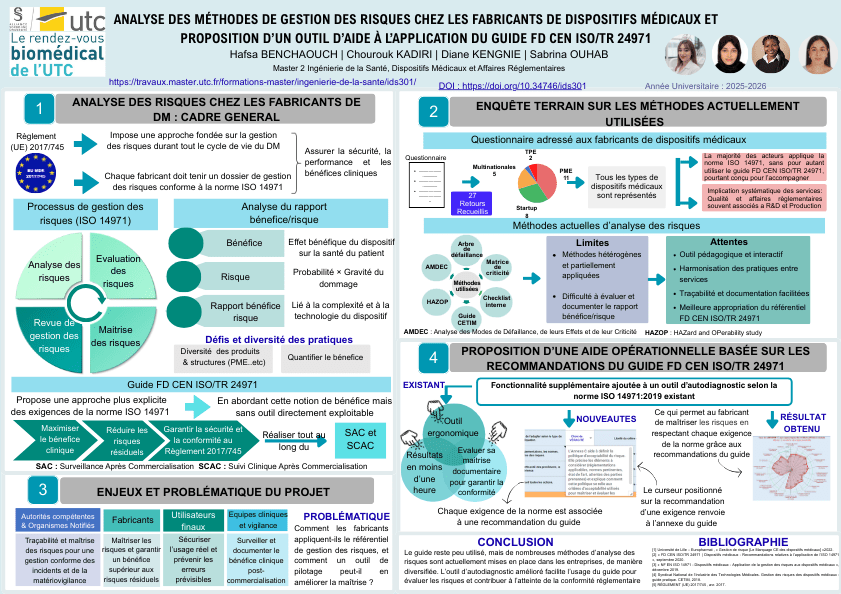
Selon le référentiel ISO 14971 et son guide d’application FD CEN ISO/TR 24971.
Mémoire Complet
Analyse des méthodes de gestion des risques des fabricants de DM et proposition d’un outil d’aide à l’application du guide FD CEN ISO/TR 24971
Introduction
Dans le domaine des dispositifs médicaux (DM), le risque est un élément important pour garantir la sécurité des patients et la conformité réglementaire. Ce risque combine la probabilité de survenue d’un dommage et la gravité de ce dommage ; La gestion des risques, correspond à l’application systématique de politiques, de procédures et de pratiques visant à analyser, évaluer, contrôler et maîtriser les risques [1]. C’est dans ce cadre que s’inscrit la norme NF EN ISO 14971, référence internationale pour la gestion des risques appliquée aux dispositifs médicaux. Cette norme établit un processus permettant aux fabricants l'identification jusqu’à la maîtrise des risques associés à leurs dispositifs et à leurs accessoires tout au long du cycle de vie. Cette norme décrivant principalement les exigences à respecter est en association avec le guide FD CEN ISO/TR 24971 qui fournit des recommandations pratiques et des approches que les fabricants peuvent exploiter pour développer, mettre en œuvre et maintenir un processus de gestion des risques. Ce guide constitue ainsi une aide précieuse pour l’application concrète et opérationnelle de la gestion des risques dans le domaine des dispositifs médicaux [2,3].
Le présent document a pour ambition de recenser les méthodes d’analyse des risques applicables tout au long du cycle de vie d’un dispositif médical, de clarifier la notion de bénéfice/risque, souvent encore mal appréhendée par les professionnels dans la démarche de maîtrise des risques, et de concevoir un outil attractif, ergonomique et communicatif inspiré du guide FD CEN ISO/TR 24971. Cet outil vise à accompagner les fabricants et les néophytes dans la mise en œuvre efficace de la gestion des risques au sein de leur entreprise.
Ce mémoire sera organisé en trois grandes parties principales : la première partie sur La gestion des risques des dispositifs médicaux, la deuxième sur la méthodologie de la gestion des risques et le rapport bénéfice/risque puis la troisième sur la proposition d’un outil d’aide à l’application du guide FD CEN ISO/TR 24971.
Chapitre 1 - Cadre général, enjeux et objectifs de l’analyse des risques chez les fabricants de dispositifs médicaux
1 - Contexte, enjeux, problématique, objectif sur l’analyse des risques des dispositifs médicaux
a) Contexte général et normatif
Le terme « risque » occupe une place centrale dans le Règlement (UE) 2017/745 (MDR), reflétant la volonté d’intégrer la maîtrise des risques comme principe structurant la conformité réglementaire. Ce Règlement, pleinement applicable depuis mai 2021, impose aux fabricants une approche fondée sur le risque tout au long du cycle de vie des dispositifs médicaux [4] : de la conception à la mise en rebut ou réforme, passant par la surveillance après commercialisation du DM. Chaque fabricant doit ainsi établir et maintenir un dossier de gestion des risques documenté, conforme à la norme NF EN ISO 14971 et à son guide d’application (FD CEN ISO/TR 24971) [2].
La gestion des risques, telle que définie par la norme ISO 14971, constitue un processus structuré et continu qui comprend plusieurs étapes successives [3]:
- L’identification des risques, qui consiste à recenser l’ensemble des phénomènes dangereux et des situations dangereuses pouvant conduire à un dommage pour le patient ou l’utilisateur [17];
- L’évaluation des risques, au cours de laquelle chaque risque est analysé en fonction de sa probabilité de survenue et de la gravité potentielle de ses conséquences, afin d’en déterminer la criticité et l’acceptabilité au regard du rapport bénéfice/risque ;
- La maîtrise des risques, qui vise à éliminer ou à réduire les risques jugés inacceptables, notamment par des modifications de conception, l’ajout de dispositifs de sécurité ou la communication d’avertissements clairs à l’utilisateur ;
- L’évaluation des risques résiduels, destinée à vérifier que les mesures de maîtrise ont effectivement réduit le niveau de risque et que les risques restants demeurent acceptables ; Enfin, la norme prévoit une revue finale, permettant de s’assurer que les mesures correctives n’ont pas introduit de nouveaux risques ou modifier ceux déjà évalués [1].
Cependant, malgré ce cadre normatif précis et les exigences du MDR, les fabricants rencontrent des difficultés à appliquer de manière homogène et efficace ce processus tout au long du cycle de vie du dispositif. La diversité des produits (dispositifs implantables, logiciels médicaux, instruments chirurgicaux, etc.) et la variété des structures industrielles (PME, start-up, grands groupes) engendrent une hétérogénéité des pratiques [5]. L’objectif est de prévenir tout dommage potentiel pour le patient ou l’utilisateur, tout en garantissant que le dispositif conserve son efficacité et son bénéfice clinique [4]. Le règlement met un accent particulier sur la balance bénéfice/risque, exigeant des fabricants qu’ils démontrent que les bénéfices cliniques d’un dispositif surpassent ses risques résiduels [4]. La mesure du bénéfice reste un défi méthodologique : comment quantifier l’amélioration de la santé ou le service rendu au patient ? Le guide ISO/TR 24971 et les documents de référence internationaux (MDCG, IMDRF) apportent des pistes d’interprétation, mais la quantification du ratio bénéfice risque demeure complexe.
Historiquement, les méthodes d’analyse des risques appliquées aux dispositifs médicaux (telles que l’AMDEC - Analyse des Modes de Défaillance, de leurs Effets et de leur Criticité) proviennent du secteur industriel [6]. Leur adaptation au domaine médical s’avère nécessaire, car les dispositifs médicaux présentent des risques spécifiques d’ordre clinique, en plus des risques techniques ou organisationnels. Selon le type de produit, la nature des risques varie : un dispositif stérile doit maîtriser les risques de contamination, tandis qu’un dispositif numérique (SaMD) doit anticiper les risques de défaillance logicielle ou de mauvaise appropriation des fonctionnalités [4]. Cette diversité des produits s’accompagne d’une hétérogénéité des pratiques de gestion du risque entre les structures, qui doivent pourtant toutes répondre à la même exigence réglementaire. Certaines entreprises disposent d’équipes dédiées à la gestion du risque, d’autres confient cette responsabilité au chargé d’affaires réglementaires. Dans tous les cas, la démarche doit s’inscrire dans une analyse continue, intégrant les données de la vie réelle, les retours d’incidents et les informations issues de la vigilance et de la surveillance post-commercialisation. Ces informations peuvent être synthétisées sous forme de tableaux, illustrant par exemple le nombre de signalements, leur origine et la gravité des incidents rapportés
Ces données illustrent la diversité des incidents signalés et soulignent l’importance d’une collecte systématique et structurée pour alimenter le processus de gestion des risques et la surveillance post-commercialisation.
Le FD CEN ISO/TR 24971 permet de clarifier la distinction entre danger (source potentielle de dommage) et risque (probabilité et gravité d’un dommage) et de structurer la démarche de maîtrise des risques en intégrant la notion de bénéfice clinique [2]. Cette approche est essentielle pour garantir que les avantages pour le patient compensent les risques résiduels associés à l’utilisation du dispositif. Pour renforcer cette démarche, la norme expérimentale française XP S 99-223 fournit un cadre méthodologique complémentaire, détaillant comment documenter et évaluer le rapport bénéfice/risque. Elle s’appuie sur les activités de gestion des risques, Bien qu’elle ne fixe pas de seuil de risque acceptable, elle offre des recommandations concrètes pour guider les fabricants dans leurs décisions [7].
b) Enjeux sur la gestion des risques selon les acteurs
Les enjeux de la gestion des risques des dispositifs médicaux concernent principalement la sécurité des patients, la conformité réglementaire et la compétitivité, la pérennité et la réussite des organisations. Plus précisément, selon les différents acteurs notamment les autorités compétentes, les organismes notifiés (ON) et les fabricants la gestion des risques constitue un élément central auquel chacun contribue afin de garantir un haut niveau de sécurité pour les patients. Le tableau ci-après présente ces enjeux selon chaque acteur.
Tableau 1 : les enjeux de la gestion des risques en fonction des acteurs [source : auteures]
| Acteurs | Enjeux de la gestion des risques |
| Autorités compétente et Organismes notifiés | Harmoniser la compréhension de l’évaluation des risques, exigé une documentation rigoureuse de la maîtrise des risques basée sur des méthodes solides, évaluation le rapport bénéfice/risque afin d'assurer une sécurité des patients uniforme [3,4]. |
| Fabricants (ingénieurs qualité, R&D, affaires réglementaires) | Les enjeux présentent des aspects à la fois techniques et opérationnels notamment adapter les méthodes industrielles aux exigences spécifiques des dispositifs médicaux, définir de manière claire les concepts de danger et de risque tout au long du cycle de vie du produit qu'il s'agisse de risques financiers, humains, techniques ou externes, démontrer que le bénéfice clinique l'emporte sur les risques résiduels [3,8]. |
| Patients, utilisateurs finaux et professionnels de santé | Les enjeux se concentrent sur les conditions réelles d'utilisation des dispositifs. Même en cas de conformité technique, une utilisation incorrecte, une interface peu intuitive ou un environnement inapproprié peuvent entraîner de nouveaux risques. Il est donc primordial d'anticiper les erreurs d'utilisation prévisibles, de prendre en compte les caractéristiques de la population cible, et d'intégrer des mesures de sécurité d'utilisation telles que l'ergonomie, la signalétique, les alarmes et les verrouillages [3]. |
| Équipes cliniques et de vigilance | Les équipes cliniques et de vigilance jouent un rôle crucial dans la collecte, l'analyse et l'exploitation des données cliniques et de surveillance post-commercialisation. Leur objectif est de quantifier et documenter le bénéfice clinique, de surveiller en continu les risques et de détecter précocement les signaux, tout en ajustant les mesures de maîtrise des risques en fonction des données réelles du terrain [3,4]. |
c) Cadrage du projet
Les pratiques des fabricants dans la gestion des risques demeurent très diversifiées en fonction de la nature des dispositifs et de la taille des entreprises. Cette diversité soulève des questionnements quant à la mise en œuvre concrète des procédures de gestion des risques tout au long du cycle de vie du dispositif par les acteurs du secteur, ainsi que sur l’intégration de la dimension du bénéfice clinique dans cette approche. L’ensemble de ces éléments conduit à la formulation de la problématique suivante ::
“ En se basant sur la norme ISO 14971, en complément du guide FD CEN ISO/TR 24971, comment aider les fabricants de dispositifs médicaux à mieux piloter la gestion des risques tout en intégrant le bénéfice clinique ? ”
d) Objectif du projet
L’objectif principal de ce projet est de dresser un état des lieux des pratiques méthodologiques de gestion des risques chez les fabricants de dispositifs médicaux, en s’appuyant sur les exigences de la norme ISO 14971 et sur son guide d’application FD CEN ISO/TR 24971.
Le projet vise à identifier les méthodes, outils et démarches réellement utilisés sur le terrain, à repérer les difficultés rencontrées par les entreprises dans l’évaluation et la mise à jour continue du rapport bénéfice/risque, et à mettre en évidence les écarts entre la théorie normative et la pratique industrielle.
Enfin, le travail aboutira à la conception d’un outil ergonomique et pédagogique permettant :
- Faciliter l’appropriation des exigences réglementaires par les différents acteurs,
- Améliorer la compréhension du lien entre maîtrise du risque et bénéfice clinique dans la conformité globale des dispositifs médicaux,
- Favoriser le choix de la méthode de gestion des risques la plus adaptée selon le type de dispositif ou la maturité de l’entreprise.
2 - Analyse du guide FD CEN ISO/TR 24971
Le FD CEN ISO/TR 24971:2020 est un rapport technique publié par l’Organisation internationale de normalisation (ISO) et adopté par le Comité Européen de Normalisation (CEN). Il a été élaboré pour compléter la norme ISO 14971:2019, intitulée « Dispositifs médicaux Application de la gestion des risques aux dispositifs médicaux ». Alors que la norme ISO 14971 énonce les exigences fondamentales à respecter pour la gestion des risques, le guide 24971 fournit des recommandations pratiques, des interprétations détaillées et des exemples concrets pour aider les fabricants à appliquer efficacement ces exigences dans la pratique industrielle. En d’autres termes, la norme ISO 14971 définit ce qu’il faut faire, tandis que le FD CEN ISO/TR 24971 explique comment le faire concrètement.
Ce guide vise principalement à clarifier les concepts de la norme ISO 14971, à faciliter sa mise en œuvre pratique à toutes les étapes du cycle de vie d’un dispositif médical, à harmoniser les pratiques entre les fabricants et les organismes de contrôle, et à renforcer la conformité réglementaire aux exigences européennes et internationales. Il explicite chaque article de la norme et fournit des conseils méthodologiques, des exemples d’application, des liens avec d’autres normes connexes telles que l’ISO 13485 ou l’IEC 62366, ainsi que des recommandations adaptées selon le type de dispositif médical.
Le guide FD CEN ISO/TR 24971 reprend la structure générale de la norme ISO 14971 mais l’enrichit de précisions pratiques. Chaque chapitre du guide renvoie à un article correspondant de la norme ISO 14971 et en détaille la mise en œuvre concrète.
Dans le cadre de ce projet, le guide FD CEN ISO/TR 24971 joue un rôle essentiel car il fournit la base de connaissance normative sur laquelle repose l’outil à concevoir. L’outil doit non seulement respecter la structure imposée par la norme ISO 14971, mais également intégrer les recommandations pratiques du guide.
Chapitre 2 - Méthodologie, état de l’art de la gestion des risques et concept du rapport bénéfice risque chez les fabricants de dispositifs médicaux
1 - Réalisation d’un questionnaire à l’attention des fabricants de DM
a) Présentation du questionnaire
Afin de comprendre les pratiques réelles de gestion des risques au sein des entreprises du secteur, une enquête en ligne a été réalisée sous la forme d’un questionnaire Google Forms(Figure 1).
Ce sondage a été diffusé auprès de plusieurs fabricants de dispositifs médicaux. L’objectif est d’évaluer la mise en œuvre concrète de la norme NF EN ISO 14971 :2019 et de son guide FD CEN ISO/TR 24971 :2020, d’identifier les méthodes d’analyse utilisées, les difficultés rencontrées, ainsi que les attentes des acteurs en matière d’outils pédagogiques ou opérationnels pour la maîtrise des risques.
Figure 1 : Page d'accueil du questionnaire [source : auteures]

Le questionnaire est structuré autour de quatre sections principales. La première, consacrée au profil de l’entreprise, vise à identifier son type, la portée géographique de ses activités, les types de dispositifs fabriqués ainsi que leur classe de risque. La deuxième section porte sur la connaissance et l’application des normes, et évalue le niveau de maîtrise et d’intégration de la norme ISO 14971 et de son guide FD CEN ISO/TR 24971, tout en identifiant les fonctions impliquées dans la gestion des risques et les besoins éventuels en formation complémentaire. La troisième section traite des méthodes et outils d’analyse des risques, en recensant les approches employées, le type d’approche adoptée, ainsi que les outils et sources de données utilisés pour l’évaluation du rapport bénéfice/risque. Enfin, la dernière section, dédiée aux perspectives et attentes, comprend des questions ouvertes permettant de recueillir les bonnes pratiques, les recommandations visant à améliorer la maîtrise des risques et les attentes relatives à la conception d’un outil pédagogique ou d’aide à l’application de la norme.
Cette structuration permet d’obtenir une vision complète et hiérarchisée du niveau de maturité des fabricants dans la gestion des risques, tout en facilitant l’analyse quantitative et qualitative des réponses recueillies.
b) Recueil des résultats
Le questionnaire a recueilli les réponses d’un échantillon varié de fabricants de dispositifs médicaux, représentant plusieurs catégories d’entreprises(Figure 2) : des multinationales, des PME, des start-up, des TPE et une ETI. La majorité d’entre elles opèrent en dehors de l’Union européenne, traduisant une ouverture internationale significative.
Figure 2 : Résultats du sondage - section profil des fabricants - type d'entreprise [source : auteures]
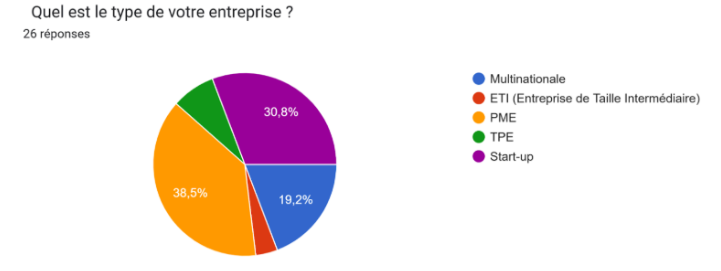
Les types de dispositifs concernés couvrent l’ensemble du spectre réglementaire, incluant les dispositifs médicaux (DM), les dispositifs médicaux implantables actifs (DMIA), les dispositifs médicaux de diagnostic in vitro (DMDIV) et les logiciels en tant que dispositifs médicaux (SaMD), avec des classes de risque allant de I à III. En termes d’organisation interne, la fonction Qualité et la fonction Affaires Réglementaires apparaissent systématiquement impliquées dans la gestion des risques, souvent en lien avec la R&D et la production, traduisant une approche pluridisciplinaire de plus en plus courante.
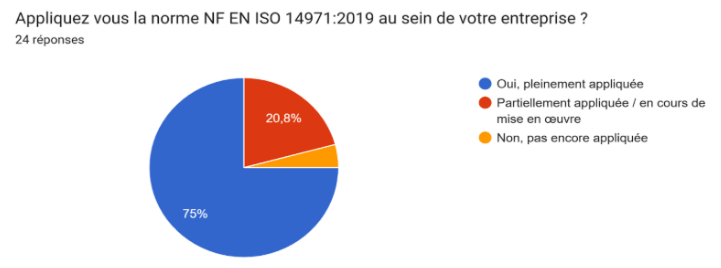
La majorité des répondants déclarent appliquer pleinement la norme ISO 14971(Figure 3), parfois en cours de déploiement dans les structures plus récentes (notamment les start-up). Le niveau de connaissance de la norme varie de moyen à expert (3 à 5 sur 5), confirmant une appropriation globalement satisfaisante du cadre réglementaire, bien que certaines entreprises, notamment les plus petites, présentent encore des lacunes ou une absence de formalisation complète.
Figure 3 : Niveau d’application de la norme ISO 14971 : 2019 [source : auteures]
En parallèle, près de la moitié (≈50 %) des participants indiquent utiliser le guide FD CEN ISO/TR 24971 associé à la norme, dont 29 % de manière partielle ou en cours de mise en œuvre. Ce résultat montre que si la norme ISO 14971 est bien intégrée, le recours systématique à son guide d’application reste encore inégal, bien qu’il constitue un appui essentiel pour structurer la démarche de gestion des risques.
Sur le plan méthodologique, les outils les plus utilisés pour l’analyse des risques sont l’AMDEC, la matrice de criticité et les check-lists internes. Plusieurs entreprises combinent différentes approches selon le type de produit ou la phase du cycle de vie. La majorité privilégie une évaluation qualitative (échelle gravité/probabilité, jugement d’expert), tandis que certaines structures plus matures ou disposant de données post-market intègrent des analyses quantitatives.
Les sources d’information les plus citées incluent les résultats de la surveillance post-commercialisation (PMS), les références normatives et réglementaires, ainsi que les données internes.
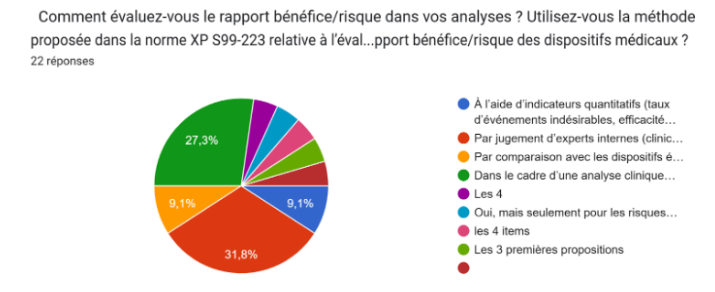
Concernant la gestion du rapport bénéfice/risque, la plupart des entreprises indiquent s’appuyer sur des données cliniques ou des jugements d’experts internes, en cohérence avec les exigences de la norme ISO 14971 et du règlement (UE) 2017/745. Quelques répondants soulignent la nécessité d’un outil facilitant la traçabilité entre les mesures de maîtrise des risques, les données d’entrée et les vérifications associées.
Figure 4 :Approches d’évaluation du rapport bénéfice/risque [source : auteures]
Les répondants soulignent le besoin d’outils pédagogiques et de guides pratiques pour harmoniser les pratiques et renforcer la compréhension des exigences réglementaires. Ils insistent aussi sur la nécessité d’une implication collective (Qualité, R&D, Production, Direction) dans la gestion des risques. Globalement, si la norme est bien appropriée, la maturité reste variable : les grandes entreprises disposent de processus structurés, tandis que les plus petites expriment un besoin d’accompagnement et d’outillage pour consolider leurs pratiques.
2 - Analyse du rapport bénéfice/risque chez les fabricants de dispositifs médicaux
Un bénéfice est un effet favorable d’un dispositif médical qui, lors de son utilisation prévue, produit une incidence positive pour le patient, de manière directe ou indirecte [9]. L’estimation de ce bénéfice repose sur la caractérisation de sa probabilité et de son importance.
La notion de balance bénéfice/risque a pris une importance croissante au fil des années, notamment à la suite de l’augmentation des incidents de matériovigilance, tels que les scandales des prothèses mammaires PIP ou des implants de contraception ESSURE [10]. En réponse à ces événements, le Règlement (UE) 2017/745 relatif aux dispositifs médicaux (RDM) impose désormais aux fabricants de démontrer un rapport bénéfice/risque favorable pour leurs dispositifs afin d’assurer la sécurité des patients [9].
Les fabricants reconnaissent de plus en plus l’importance de cette démarche, tout en soulignant sa complexité, car la balance bénéfice/risque varie selon la nature, la technologie et la finalité clinique de chaque dispositif [9]. Cette variabilité rend l’évaluation du rapport bénéfice/risque particulièrement délicate et représente un véritable défi pour l’obtention du marquage CE. D’ailleurs, une enquête menée auprès de plusieurs fabricants a montré qu’ils utilisent différentes approches et méthodes d’évaluation du rapport bénéfice/risque pour répondre à cette exigence réglementaire, notamment à partir de données qualitatives, telles que le jugement d’experts et/ou quantitative (par analyse clinique par exemple).
Dans ce contexte, la norme XP S 99-223, révisée en décembre 2023, s’inscrit dans la démarche d’évaluation du rapport bénéfice/risque. Elle précise les exigences à respecter et décrit de manière détaillée la méthode d’évaluation de ce rapport, afin de garantir l’efficacité et la sécurité pour les patients. Cette nouvelle version introduit plusieurs modifications, notamment la suppression de certaines exigences et la clarification selon laquelle il n’est pas nécessaire d’évaluer la balance bénéfice/risque lorsqu’un risque est déjà jugé acceptable après sa maîtrise [11].
Le rapport bénéfice/risque dépend donc à la fois de la complexité du dispositif et de la technologie mise en œuvre. Il est évalué sur la base des risques résiduels et globaux, qui doivent être acceptables au regard du bénéfice clinique attendu [9]. L’objectif principal est de réduire le niveau de risque tout en optimisant le niveau de bénéfice pour le patient.
Ce bénéfice peut être renforcé en améliorant les conditions d’utilisation, en augmentant les performances du produit, ou encore en renforçant la formation des utilisateurs ; et ce rapport bénéfice/risque ne se limite pas à la phase de conception ou de mise sur le marché : il fait l’objet d’un suivi continu après l’obtention du marquage CE, et doit être réévalué régulièrement à la lumière de toute nouvelle information pertinente recueillie lors de la surveillance post-commercialisation [9,10,11].
3 - Les solutions techniques existantes
a) Présentation des méthodes et outils actuellement utilisés dans l’industrie (AMDEC, APR, logiciels, matrices de risque, etc.)
Sur le terrain, les fabricants de DM déploient différentes méthodes d'analyse des risques, adaptées à la nature de leurs dispositifs, à leur niveau de criticité et à leur propre maturité organisationnelle.
a-1) L'AMDEC (Analyse des Modes de Défaillance, de leurs Effets et de leur Criticité)
Très répandue à la fois dans le secteur biomédical et industriel, l'AMDEC permet d'identifier systématiquement les défaillances potentielles d'un produit ou d'un processus. Elle évalue trois dimensions clés : la gravité des conséquences, la probabilité que la défaillance survienne, et la capacité à la détection à temps(Figure 5). La combinaison de ces critères aboutit au calcul de l'Indice de Priorité de Risque (IPR), ou Risk Priority Number (RPN), selon la formule :
RPN = Gravité × Probabilité d’Occurrence × Détection
Figure 5:Diagramme évaluation de la Criticité [18,19]
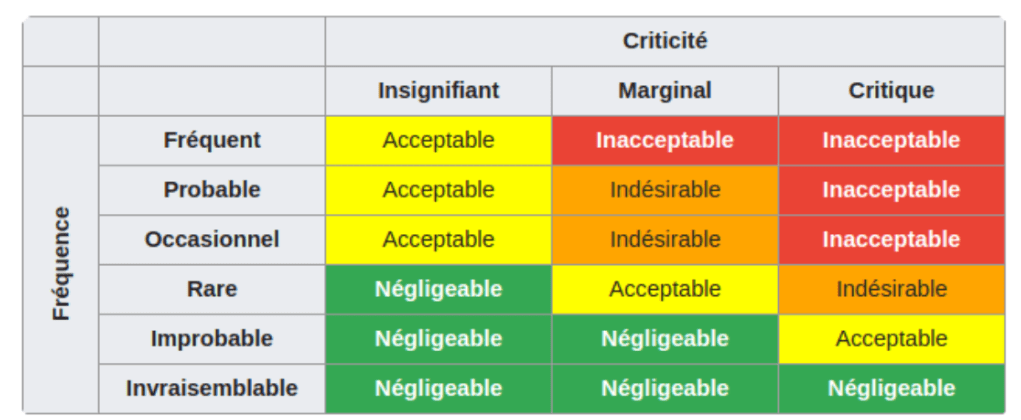
Cette approche s'avère particulièrement pertinente pour les dispositifs électromécaniques, les implants ou les équipements de diagnostic in vitro dans le secteur des DM, et s'inscrit pleinement dans la démarche préconisée par la norme ISO 14971 [9,12].
a-2) L'APR (Analyse Préliminaire des Risques)
L'Analyse Préliminaire des Risques intervient généralement en amont du développement, dès les premières réflexions sur la conception. Elle vise à repérer les dangers principaux associés à l'utilisation du dispositif, avant même que son design ne soit définitivement arrêté. Son principe est simple mais efficace : se demander « Que pourrait-il arriver de fâcheux ? » (Situation dangereuse) et « Quelles en seraient les répercussions ? » (Dommages). Elle s'avère précieuse lors des phases de concept et de faisabilité, notamment pour les dispositifs innovants ou les prototypes de recherche [2,9].
a-3) Le HAZOP (Hazard and Operability Study)
Initialement développée dans l'industrie chimique, la méthode HAZOP examine les écarts potentiels par rapport au fonctionnement normal d'un système complexe. Elle trouve naturellement sa place dans l'analyse des dispositifs médicaux impliquant des fluides, des réactifs ou des réactions chimiques ; pensons par exemple aux équipements de dialyse ou aux systèmes d'analyse in vitro. Le HAZOP s'appuie sur des mots-guides ("plus", "moins", "absence", "inversion", etc.) pour passer au crible chaque étape du processus étudié [13].
a-4) L'ETA et le FTA (Event Tree Analysis / Fault Tree Analysis)
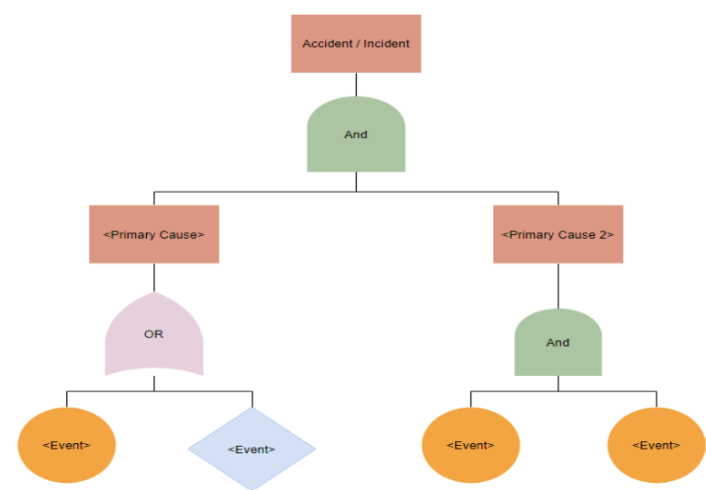
Ces deux méthodes graphiques aident à modéliser les enchaînements d'événements susceptibles de conduire à une situation dangereuse(Figure 6).
- Le FTA (Analyse par arbre de défaillance) part d'un événement indésirable comme un choc électrique pour remonter vers ses causes racines.
- L'ETA (Analyse par arbre d'événements) adopte la démarche inverse : elle prend pour point de départ un événement déclencheur pour explorer ses conséquences potentielles.
Le guide FD CEN ISO/TR 24971 recommande ces approches, notamment dans sa section 5.4.7, qui montre comment articuler les séquences d'événements pour estimer le risque global.
Figure 6 :Arbre de défaillance simplifié mettant en lumière la séquence Danger → Événement → Situation [20]
a-5) Les outils numériques et logiciels
Les fabricants s’appuient de plus en plus sur des solutions logicielles afin de garantir la traçabilité et la conformité de leur gestion des risques :
- ERM by Knowllence (Medical Device Suite) : logiciel certifié conforme à l’ISO 14971:2019, permettant de générer automatiquement un dossier de validation du module RM (module de Gestion des risques ou Risk Management ) 14971 [14].
- Ennov Risk Manager, Greenlight Guru, MasterControl, Q-Pulse : plateformes certifiées, intégrées au système de management qualité (ISO 13485).
- Excel ou Smartsheet : outils accessibles, souvent privilégiés par les PME pour élaborer leurs matrices de risques sur mesure.
- Logiciels spécialisés en cybersécurité (comme MedISA ou TIR57 Toolkit) : dédiés à l'évaluation des vulnérabilités de sécurité [2,15].
a-6) Les guides et ressources de référence
Pour accompagner la mise en œuvre des normes, plusieurs guides pratiques ont été conçus afin d'aider les fabricants à gérer efficacement les risques liés aux dispositifs médicaux. Le guide du SNITEM, intitulé “Gestion des risques des dispositifs médicaux” (CETIM, 2016), disponible en français et en anglais, présente une approche structurée en six étapes fondamentales, depuis la détection des dangers jusqu'au suivi post-commercialisation. Il offre également des exemples concrets d'application, montrant comment documenter et évaluer les risques à chaque phase. Ce guide accorde une attention particulière aux risques associés aux logiciels, en accord avec la norme NF EN 62304, et souligne l'importance des tests et de la validation pour les dispositifs intégrant des logiciels critiques. Parallèlement, le FD CEN ISO/TR 24971:2020 vient enrichir ces recommandations en partageant des bonnes pratiques issues du terrain, notamment pour identifier systématiquement les dangers (article 5.4), estimer et évaluer les risques (article 5.5), et définir des mesures de réduction adaptées. Ces ressources s'avèrent indispensables pour assurer la sécurité et la conformité réglementaire des dispositifs médicaux [2].
b) Analyse critique de leurs limites au regard du guide FD ISO/TR 24971
Bien que ces approches présentent des qualités indéniables, elles révèlent certaines lacunes importantes lorsqu'on les examine au regard des recommandations du guide FD CEN ISO/TR 24971:2020. Ce dernier propose en effet une interprétation plus nuancée et contextuelle de la norme ISO 14971.
- Approche centrée sur la défaillance et non sur la situation dangereuse :
Les méthodes traditionnelles, comme l'AMDEC, se concentrent principalement sur l'analyse des pannes techniques, mais ont tendance à négliger les aspects liés à l'utilisation réelle, à l'environnement et aux facteurs humains.
Pourtant, le guide FD CEN ISO/TR 24971 (article 5.4) insiste sur la nécessité d'intégrer les mauvais usages raisonnablement prévisibles et les caractéristiques de sécurité, même en l'absence de défaillance - pensons par exemple aux bistouris, aux lasers ou aux rayonnements.
- Une sous-estimation des risques systémiques et de cyber sécurité :
Les outils conventionnels ne prennent pas suffisamment en compte les vulnérabilités de sécurité, pourtant clairement identifiées dans le guide (article 5.4.6).
Celui-ci souligne que des incidents comme la perte de confidentialité, l'altération des données ou l'indisponibilité du système peuvent avoir des conséquences indirectes graves pour les patients.
- Une intégration insuffisante de l'équilibre bénéfice/risque :
La majorité des outils se contentent d'établir des scores de criticité sans véritablement intégrer la valeur clinique des bénéfices.
Le FD CEN ISO/TR 24971 introduit dès l'article 7.4 une approche plus équilibrée, fondée sur la justification du bénéfice clinique par rapport au risque résiduel.
- Un manque de pédagogie et d'accompagnement normatif :
Les solutions logicielles actuelles n'offrent pas d'explications contextuelles des exigences ISO 14971, ni d'aide à la décision concernant les critères d'acceptabilité.
À l'inverse, le FD ISO/TR 24971 propose des annexes pratiques (A, C, F, H) et des questions guides pour orienter l'analyse, comme par exemple : "Le dispositif pourrait-il délivrer plus d'énergie que prévu ?"
Tableau 2 : Tableau synthétique qui résume les méthodes existantes, leurs limites face au guide FD ISO/TR 24971 : 2020 et les améliorations proposées [source : Auteures]
| Méthode / Outil | Limites par rapport au FD ISO/TR 24971 | Améliorations proposées |
| AMDEC | Se concentre sur les défaillances techniques ; néglige les usages réels, facteurs humains et environnement | Ajouter identification systématique des situations dangereuses et des mauvais usages raisonnablement prévisibles ; intégrer les facteurs humains et environnementaux |
| APR | Principalement en amont, ne couvre pas les risques émergents ni la cyber sécurité | Compléter par l’évaluation des risques logiciels, cyber sécurité, interactions patient-dispositif et usages imprévus |
| HAZOP | Examen limité aux écarts de fonctionnement ; peu utilisé pour la dimension clinique et bénéfice/risque | Inclure l’évaluation des conséquences cliniques et l’intégration bénéfice/risque pour chaque scénario |
| FTA / ETA | Approche statique, focalisée sur les événements ; ne prend pas en compte la surveillance post-commercialisation | Ajouter suivi dynamique des incidents post-commercialisation et mise à jour automatique des matrices de risques |
| Outils logiciels (ERM, Ennov, Excel, etc.) | Traçabilité assurée mais peu pédagogique ; peu d’explications contextuelles sur les exigences normatives | Intégrer modules pédagogiques, liens normatifs automatiques, indicateurs visuels de maturité, alertes et suivi post-commercialisation |
| Guides et ressources (SNITEM, FD ISO/TR 24971) | Guide théorique : nécessite interprétation ; peu intégré aux outils industriels | Créé interface interactive reliant norme et guide, fiches dynamiques dangers/risques, critères d’acceptabilité, évaluation bénéfice/risque |
c) Enseignements tirés pour l’amélioration des pratiques et conception d’un outil adapté.
Lorsqu’on examine de manière critique les méthodes actuelles de gestion des risques telles que l’AMDEC, l’APR ou la FTA, on constate plusieurs limites persistantes : une approche trop compartimentée, une application irrégulière des exigences normatives et l’absence d’outils intégrés permettant d’établir un lien concret entre la norme ISO 14971:2019 et son guide d’application FD ISO/TR 24971:2020.
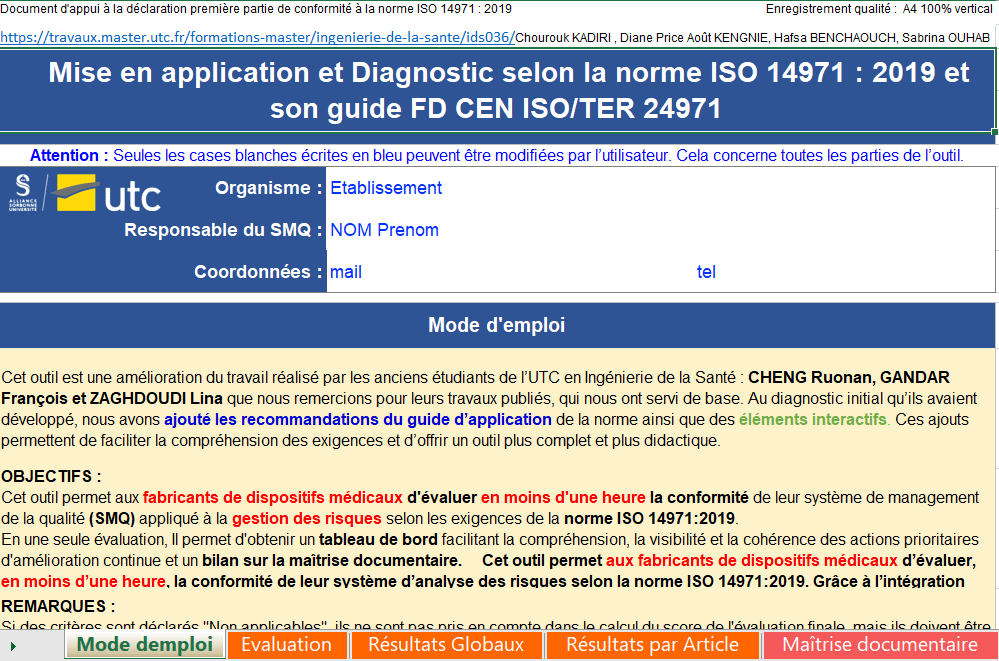
Pour remédier à cela, un outil déjà existant fera l’objet d’une amélioration en termes de positionnement et de pédagogie, afin d’intégrer des moyens d’accéder plus rapidement à des exemples concrets issus du guide.
Cet outil d’autodiagnostic, développé par d’anciens étudiants de l’UTC dans la mention IDS, a été conçu selon la norme ISO 14971:2019. Il permet d’évaluer le niveau de conformité atteint en matière de gestion des risques :
- Il facilite l’auto-évaluation progressive, l’identification des points faibles et le suivi de l’amélioration continue.
- Il contribue également à la dimension pédagogique, en permettant aux équipes qualité et réglementaires de visualiser rapidement le degré de conformité et la robustesse de leur démarche de processus de gestion des risques [2,16].
Chapitre 3 - Proposition d’une aide opérationnelle sur la mise en application du guide sur la gestion des dispositifs médicaux
1 - Lien entre la norme ISO 14971 et le guide FD CEN ISO/TR 24971
La norme ISO 14971 et le guide FD CEN ISO/TR 24971 sont des référentiels importants pour comprendre et appliquer la gestion des risques dans le domaine des dispositifs médicaux. Ces deux documents visent à établir des pratiques de gestion des risques afin de garantir la sécurité et l’efficacité des dispositifs médicaux.
L’ISO 14971 fournit un cadre général pour la gestion des risques, tandis que le FD CEN ISO/TR 24971 sert de guide pratique pour l’application de cette norme. Il aide à interpréter et à mettre en œuvre les exigences de l’ISO 14971 dans un contexte spécifique, en proposant des exemples d’application concrets. Ce guide, dont la structure suit celle de la norme, fournit des recommandations sur la planification, l’identification, l’évaluation et le contrôle des risques, rendant ainsi le processus plus accessible et applicable pour les fabricants.
Il est régulièrement mis à jour afin de refléter l’évolution des pratiques et des exigences réglementaires, garantissant ainsi que les fabricants restent conformes aux meilleures pratiques du secteur.
2 - Cartographie interactive de l’outil d’aide opérationnelle à l’application du guide FD 24971
Un outil Excel interactif, développé par d’anciens étudiants de l’UTC (CHENG Ruonan, GANDAR François et ZAGHDOUDI Lina) comme déjà évoqué plus haut, a été repris et amélioré afin de couvrir l’intégralité des recommandations du guide. Il a été conçu pour faciliter son appropriation et son application opérationnelle, afin de répondre à une inégalité de son application contrairement à la norme ISO 14971 qui lui est associée et donc sont complémentaire, constatée à travers les enquêtes. L’outil, disposant déjà d’une fonction de diagnostic de la conformité d’un dispositif médical par rapport au référentiel ISO 14971:2019, permet une navigation et une évaluation par chapitre pour chaque exigence de la norme, accompagnée des recommandations pour ces exigences selon le guide, afin de faciliter une meilleure compréhension et application des exigences de la norme(Figure 7).
Figure 7 : Représentation de la partie évaluation suivant les exigences avec des recommandations [source : Auteures]
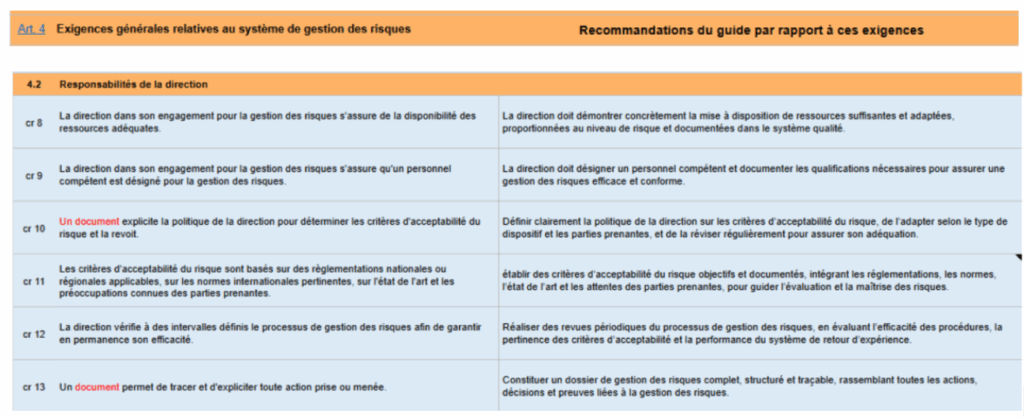
Cette organisation modulaire offre un parcours structuré, logique et progressif, garantissant que l’ensemble des exigences du guide est pris en compte. Elle permet à l’utilisateur de visualiser rapidement l’état d’avancement du processus de gestion des risques et de naviguer aisément entre les différentes étapes. Les exigences respectées ou non peuvent ainsi être identifiées de manière synthétique, facilitant la compréhension globale du guide et son utilisation en pratique.
Chaque onglet contient des check-lists et des critères d’évaluation(Figure 8) permettant d’indiquer si les exigences sont conformes, partiellement conformes ou non conformes. Ces éléments sont accompagnés de notes explicatives destinées à clarifier les attentes du guide et à aider l’utilisateur dans son interprétation. Une illustration de la structure de l’outil et de l’articulation entre les différents onglets est présentée en figure afin d’en faciliter la compréhension.
Figure 8 : Critères et échelle d’évaluation [source : Auteures]
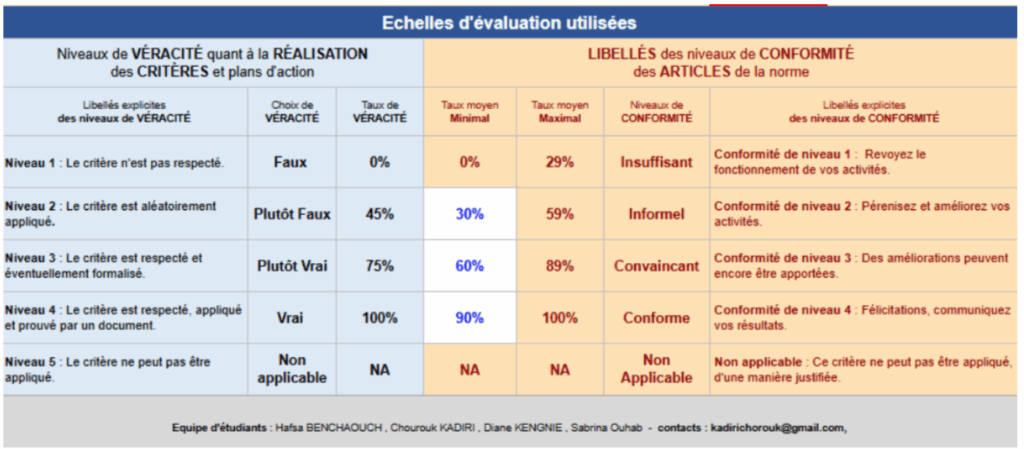
a) Méthode d’utilisation de l’outil
L’utilisation de l’outil repose sur une démarche simple et reproductible. L’utilisateur parcourt chaque exigence correspondant aux chapitres de la norme et recommandation du guide, en s’appuyant sur des check-lists afin de vérifier la conformité des pratiques mises en place. Les critères cochés permettent d’obtenir une vision globale des points conformes, des écarts identifiés et des actions d’amélioration potentielles. Cette méthode favorise une évaluation structurée, homogène et traçable.
Figure 9 : Les différents points de l’outil d’autodiagnostic basé sur le référentiel ISO 14971:2019 et son guide d’application [source : Auteures]

b) Conception et amélioration de l’outil
La conception de cet outil s’appuie sur les résultats de l’enquête terrain réalisée auprès de professionnels du domaine, qui ont mis en évidence des remarques sur l’application réduite du guide FD CEN ISO/TR 24971 face à la norme sur la gestion des risques. Afin de répondre à ces besoins, l’outil a été développé comme une amélioration d’un outil d'autodiagnostic existant, en intégrant une structuration plus claire, des critères d’évaluation plus détaillés et une approche plus intuitive pour les utilisateurs.
Figure 10 : page d’accueil de l’outil [source : Auteures]
Ainsi, cet outil constitue un support pratique et opérationnel pour les équipes qualité et affaires réglementaires, leur permettant d’évaluer la conformité des fabricants de manière rapide, fiable et reproductible, tout en facilitant l’appropriation du guide FD CEN ISO/TR 24971 dans un contexte professionnel. Des bulles contenant les résumés des annexes pour plus de détails ont été intégrées(Figure 11).
Figure 11 : Extrait de l’outil d’aide à l’application de l’ISO 14971 intégrant des bulles d’information explicatives [source : Auteures]

Les résultats sont d’abord visualisés sur un tableau de bord, qui renseigne sur le niveau de conformité et de véracité par rapport à la norme ISO 14971:2019.
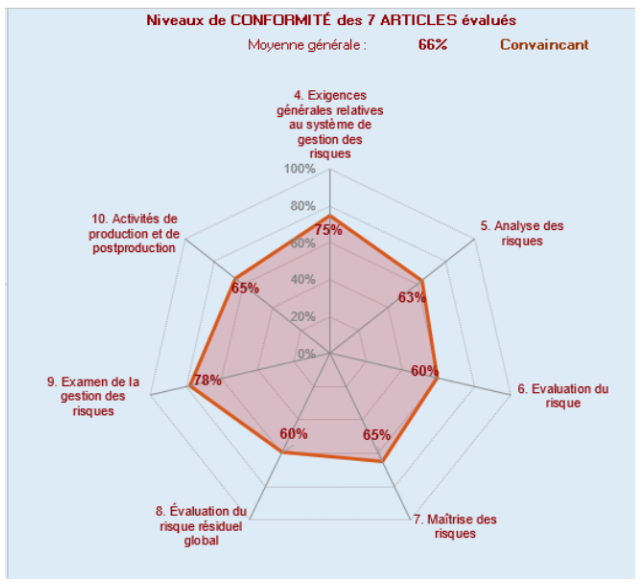
Un bilan global(Figure 12) est ensuite présenté sous la forme d’un graphe radar, montrant le niveau de conformité en pourcentage pour l’ensemble des sous-articles évalués de la norme, avec la possibilité d’ajouter des commentaires sur les résultats obtenus.
Figure 12 : Représentation du niveau global de conformité à la norme ISO 14971:2019 sous forme de graphe radar [source : Auteures]
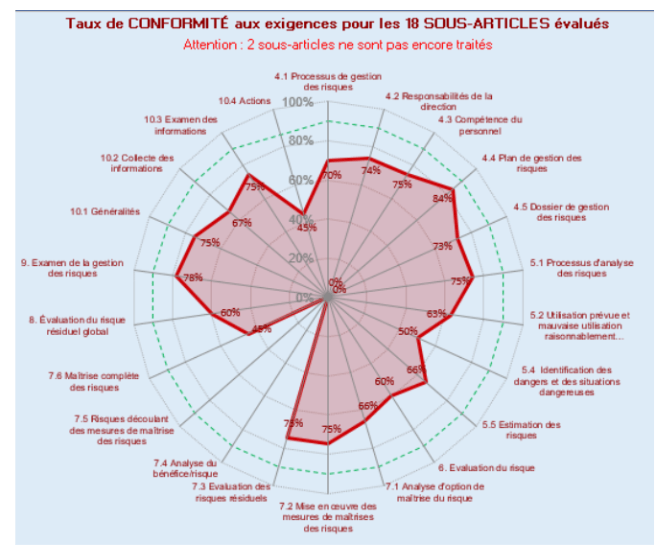
Enfin, les résultats détaillés par article (Figure 13) de l’autodiagnostic sont présentés dans l’onglet suivant, après les résultats globaux, afin de faciliter l’analyse du taux de conformité et la mise en place d’un système d’amélioration continue.
Figure 13 : Présentation détaillée des résultats de l’autodiagnostic par article de la norme ISO 14971:2019 [source : Auteures]
3 - Limites et perspectives de l’outil
Malgré sa pertinence, l’outil présente certaines limites. Il dépend fortement de la qualité et de la complétude des données saisies, et ne remplace pas l’expertise humaine pour la validation des mesures de maîtrise et des décisions critiques. Certaines analyses avancées peuvent nécessiter des logiciels spécialisés complémentaires.
En termes de perspectives, des améliorations pourraient inclure l’ajout de fonctionnalités automatisées pour la priorisation des risques et la génération d’alertes sur les risques critiques, le développement d’une version collaborative en ligne pour permettre un travail simultané des équipes multidisciplinaires.
Conclusion
Ce travail a permis de mettre en évidence les enjeux majeurs de la gestion des risques des dispositifs médicaux, dont l’objectif principal est de garantir un haut niveau de sécurité des patients tout en assurant la conformité réglementaire des produits tout au long de leur cycle de vie. Encadrée par la norme ISO 14971 et son guide d’application FD CEN ISO/TR 24971, cette démarche repose sur une identification systématique des pratiques existantes d’analyse des risques, une évaluation rigoureuse des risques et une démonstration documentée du rapport bénéfice/risque.
L’analyse des exigences normatives, complétée par une enquête terrain menée auprès de fabricants de dispositifs médicaux de tailles et de profils variés, a mis en évidence une appropriation globalement satisfaisante des principes de la gestion des risques, mais également une forte hétérogénéité des pratiques. Les méthodes mises en œuvre diffèrent selon la maturité organisationnelle, la nature des dispositifs développés et les ressources disponibles, allant de méthodes structurées telles que l’AMDEC ou l’arbre de défaillance à des approches plus simplifiées reposant sur des matrices de criticité ou des check-lists internes. Bien que reconnu comme pertinent, le guide FD CEN ISO/TR 24971 reste encore partiellement intégré dans les processus des entreprises.
Les résultats soulignent également des difficultés récurrentes liées à la complexité des méthodes, au manque d’outils pédagogiques harmonisés et à la coordination entre les différents acteurs impliqués (Qualité, R&D, Affaires Réglementaires, Production, clinique). Ces constats sont particulièrement marqués au sein des petites et moyennes entreprises, qui expriment un besoin accru d’accompagnement méthodologique et d’outillage opérationnel pour renforcer la maîtrise des risques.
Dans ce contexte, ce travail a conduit au développement d’un outil opérationnel visant à faciliter l’application du guide FD CEN ISO/TR 24971 dans la pratique industrielle. Conçu comme une amélioration d’un outil existant, il s’appuie à la fois sur une analyse approfondie du guide et sur les résultats de l’enquête terrain, afin de répondre aux difficultés identifiées en matière de compréhension, de structuration et de mise en œuvre des exigences. Sa structuration modulaire sous forme de fichier Excel permet une approche progressive et logique de la gestion des risques, tout en offrant une vision synthétique et traçable du niveau de conformité des fabricants.
Les check-lists, critères d’évaluation et notes explicatives constituent un véritable support d’aide à la décision pour les équipes qualité et affaires réglementaires, favorisant une évaluation homogène, reproductible et adaptée aux réalités industrielles. Ainsi, ce travail contribue à renforcer l’opérationnalisation du guide FD CEN ISO/TR 24971, à rapprocher les exigences normatives de la pratique professionnelle et à soutenir une approche cohérente et pluridisciplinaire de la gestion des risques des dispositifs médicaux.
Liste des acronymes
AMDEC : Analyse des Modes de Défaillance, de leurs Effets et de leur Criticité
APR : Analyse Préliminaire des Risques
DMDIV : Dispositif Médical de Diagnostic In Vitro
DM : Dispositif Médical
EN : Norme Européenne (European Norm)
ERM : Enterprise Risk Management (Gestion des risques d’entreprise)
ETA : Event Tree Analysis (Analyse en arbre d’événements)
FD : Fascicule de Documentation
FTA : Fault Tree Analysis (Analyse par arbre de défaillance)
HaZOP : Hazard and Operability Study (Étude des dangers et de l’opérabilité)
IPR : Indice de Priorité de Risque
ISO : International Organization for Standardization (Organisation internationale de normalisation)
MedISA : Medical Information Security Analysis (Analyse de la sécurité des systèmes d’information médicaux)
NF : Norme Française
ON : Organisme Notifié
PMS : Post-Market Surveillance (Surveillance après commercialisation)
PME : Petite et Moyenne Entreprise
R&D : Recherche et développement
RDM : Règlement relatif aux Dispositifs Médicaux
RPN : Risk Priority Number (Numéro de priorité du risque)
SaMD : Software as a Medical Device (Logiciel dispositif médical)
SNITEM : Syndicat National de l’Industrie des Technologies Médicales
TIR57 : Technical Information Report 57 (rapport technique numéro 57)
UE : Union Européenne