IDS036 - La gestion des risques liés aux dispositifs médicaux chez les fabricants selon la norme ISO 14971 : 2019
DOI mémoire
https://doi.org/10.34746/4eaz-7f06Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

CHENG Ruonan 
GANDAR François 
ZAGHDOUDI Lina
Contacts
Citation
A rappeler pour tout usage : R. CHENG, F. GANDAR, L. ZAGHDOUDI, « La gestion des risques liés aux disppositifs médicaux chez les fabricants selon la norme ISO 14971 : 2019 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids036 ; https://doi.org/10.34746/4eaz-7f06
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_02_idsap
Résumé
La gestion des risques chez les fabricants de dispositifs médicaux de la Communauté Economique Européenne peut s’appuyer sur la norme harmonisée ISO : 14971 dont la dernière version a été publiée en décembre 2019.
Les contraintes de délai vis-à-vis de l’application obligatoire en mai 2020 du nouveau règlement européen 2017/745 portant sur les dispositifs médicaux nécessite pour les fabricants de faire évoluer leurs systèmes de management des risques pour maintenir leur conformité aux exigences essentielles, et in fine leur accès au marché unique.
Le manque d’outils et méthodes gratuites adaptées spécifiquement à la gestion des risques chez les fabricants de dispositifs médicaux ne leur permet pas d’analyser rapidement et facilement leur niveau de conformité à ces nouvelles exigences.
Une aide opérationnelle composée de trois outils complémentaires a été réalisée dans le but de fournir un accompagnement aux fabricants dans cette transition de cadre réglementaire. Simple d’utilisation, au format facilement diffusable, elle se destine à permettre un suivi durable et personnalisable du système de management des risques lié aux dispositifs médicaux.
Elle permet une meilleure identification des changements apportées aux exigences essentielles, une compréhension approfondie de la structure de la norme et des éléments de preuve documentaire associés, ainsi que la réalisation d’un diagnostic dans le but de donner un aperçu du niveau de conformité de la gestion des risques chez le fabricant et d’en identifier les axes d’améliorations.
Gratuite et disponible sur internet, elle est adaptée à tout type de fabricant de dispositifs médicaux, et requiert seulement pour son fonctionnement les logiciels issus du pack Microsoft Office®.
Abstract
To comply with European regulations concerning risk management, medical devices manufacturers may use the standard ISO 14971, which was last published in December 2019.
As the new Regulation (EU) 2017/745 is set to be put in effect in May 2020, those companies need to adapt their management systems in order to maintain compliance with internal market policies.
The lack of free methods or tools specifically designed for medical devices risk management don’t allow manufacturers to quickly and easily diagnose their conformity levels to those regulations.
A toolbox has been created in order to give a hand companies throughout this regulatory transition. It is made of three different files that are easy to share or use, and is designed to help them build, improve, and follow their own custom risk management system.
It allows a better identification to changes brought by the regulatory transition, an improved understanding of the standard structure and its documentary requirements. The toolbox also furnishes a diagnostic tool to assess the manufacturer’s risk management system conformity, in order to point out improvement axis.
Online and free, it is made for every medical device manufacturer, regardless of its size. To run those tools on a computer it is only required to have the Microsoft Office™ pack installed.
Téléchargements




Mémoire complet :
La gestion des risques lié aux dispositifs médicaux chez les fabricants selon la norme ISO 14971 : 2019
Remerciements
Nous souhaiterions remercier en tout premier lieur nos suiveurs, G. Farges et PM. Felan, pour nous avoir accompagnés tout au long de ce projet et nous avoir permis, par leurs conseils et recommandations, de surmonter ses difficultés.
Nous voulons également remercier Mme König, pour son écoute, son expérience et son savoir dont elle a sur nous faire profiter.
Enfin, nous aimerions remercier l’ensemble de l’équipe pédagogique du Master Ingénierie de la Santé qui nous permis de réaliser ce projet au sein de l’Université de Technologie de Compiègne.
Introduction
La gestion des risques est effectuée tout au long du cycle de vie du dispositif médical. Elle comprend l’analyse, l’évaluation, le contrôle et la maîtrise des risques, qui se définissent par la conjugaison de l’occurrence et de la gravité d’un dommage.
Chez les fabricants c’est un processus répondant à des exigences réglementaires essentielles de performance et de sécurité dont la conformité est vérifiée en vue de l’obtention du marquage CE, et donc de l’accès au marché européen du dispositif médical.
L’ISO 14971, « Application de la gestion des risques aux fabricants de dispositifs médicaux » est le référentiel utilisé par les organismes notifiés pour l’évaluation de la conformité du produit : c’est une norme harmonisée, créée par l’Organisme International de Standardisation (ISO) sous le mandat du Parlement Européen.
L’évolution de la réglementation des dispositifs médicaux en Union Européenne, avec l’arrivée des règlements 2017/745 et 2017/746, a nécessité une révision systématique des normes harmonisées associées.
La norme ISO 14971 : 2019 a été publiée le 10 décembre 2019 mais n’est pas encore réémise par l’Agence Française de Normalisation en langue française.
Pour les fabricants, l’enjeu de cette adaptation aux exigences réside dans un possible refus d’un renouvellement du marquage CE de leur produit.
La question étant : Comment aider les fabricants de dispositifs médicaux à ajuster leur plan de gestion des risques en répondant aux exigences de la norme ISO 14971 : 2019 ?
Il s’agira ici, de détailler une aide opérationnelle aux fabricants, destinée à leur faciliter la compréhension de cette évolution normative, ainsi que l’adaptation aisée et rapide de leur processus de gestion des risques à cette transition réglementaire.
Cette aide comprend :
- Un livret de comparaison NF EN ISO 14971 : 2013 / ISO 14971 : 2019
- Une cartographie du projet de norme, détaillant les exigences et documents associés
- Un outil d’autodiagnostic, qui permet au fabricant de s’évaluer sur sa conformité, et d’en tirer des axes prioritaires d’améliorations ainsi que des actions à entreprendre.
I. La gestion des risques des dispositifs médicaux
A. Le secteur du dispositif médical en chiffres
a. Une marché croissant et nnovant
Le secteur des dispositifs médicaux en France représente 85 000 personnes. La dynamique du secteur montre une évolution des effectifs de 1,5 % et 3 % en moyenne par an depuis 2013 [1].
D’après le Syndicat National de l’Industrie des Technologies Médicales (SNITEM), il existe plus de 1300 entreprises en France dont 92% de TPE/PME. Le chiffre d’affaires des entreprises biomédicales sur le marché est de 28 milliards d’euros [1].
La répartition géographique des entreprises de dispositifs médicaux est assez inégale en France (présenté en Figure 1).
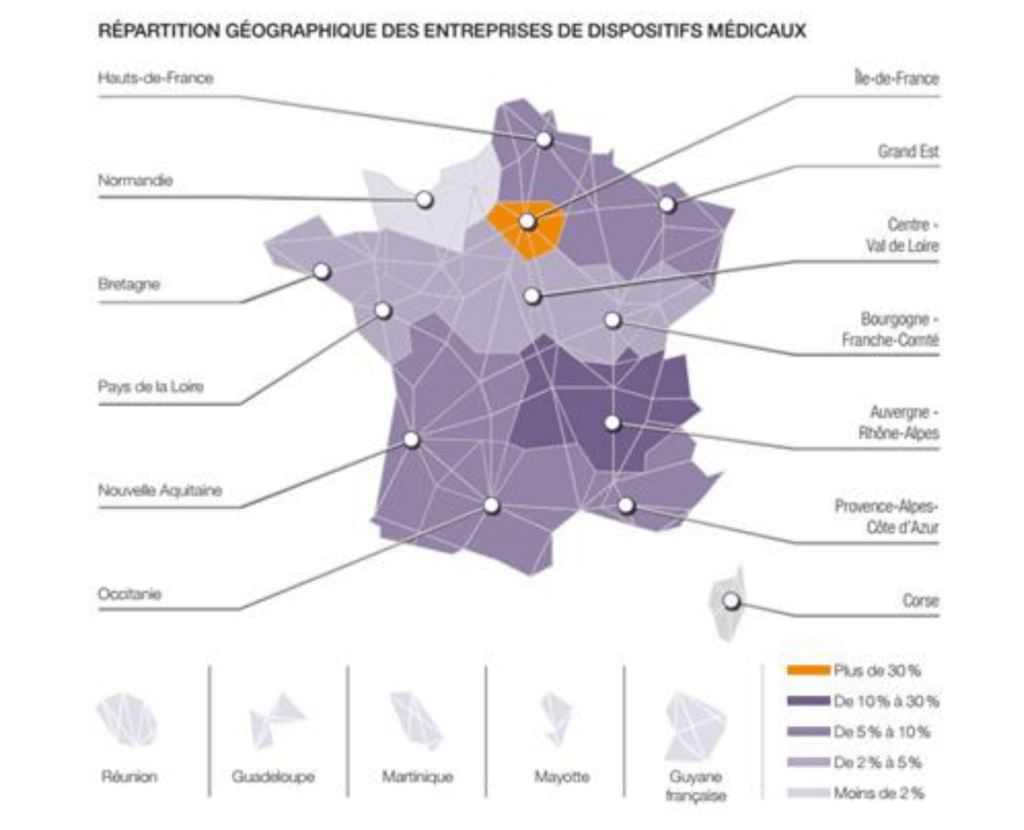
Les entreprises actives en recherche et développement (R&D) sont supérieures à 50%. Le nombre de brevets par an en Europe est de 12 000 soit un brevet toutes les 50 minutes [1]. L’ensemble des entreprises françaises et européennes des dispositifs médicaux conçoivent et commercialisent des équipements ayant l’obligation de posséder le marquage CE.
Alors que le secteur déploie de lourds capitaux pour ses activités de recherche et de développement, il doit également surveiller les produits et les risques inhérents à ceux-ci au vu des enjeux patients et utilisateurs : c’est pourquoi la matériovigilance a pour objectif d’éviter que ne se reproduisent des incidents et risques graves (article L.512-2).
b. Des scandales médiatisés
Au cours de l’année 2016, ce sont 15 961 effets indésirables de DM et DMDIV qui ont été déclarés en matériovigilance auprès de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) [2]. Ces signalements d’incidents liés à l’utilisation des dispositifs médicaux après leur mise sur le marché témoignent de la vigilance des institutions publiques vis-à-vis de l’enjeu de santé publique. En effet, le dysfonctionnement d’un appareil lors d’une opération ou d’un soin mettrait en danger la sécurité du patient.
Jeudi 4 avril 2019, l’ANSM a annoncé, l’interdiction des implants mammaires macro-texturés en silicone et des implants en polyuréthane parce que ces implants peuvent poser l’augmentation significative des cas de Lymphome Anaplasique à Grandes Cellules (LAGC) depuis 2011. Ceci quatre mois après la publication de l'enquête « Implant files » [3].
Ce n’est pas le seul scandale que « Implant files » relève. Publiée en novembre 2018, l’enquête, qui est réalisée par 59 médias de 36 pays relève de nombreux scandales des dispositifs médicaux. Elle a souligné que la France manque de contrôle sur des implants médicaux, ce qui entraîne de plus en plus d'accidents médicaux. En France, selon les chiffres de l’ANSM, qui tient un répertoire des signalements de matériovigilance, le nombre d'incidents liés à ces implants aurait doublé en dix ans, avec plus de 18 000 cas en 2017 et environ 158 000 incidents en dix ans, selon « le Monde » [4].
S’il est nécessaire pour le parlementaire de créer des textes destinés à protéger le citoyen, il convient également de les faire évoluer de pair avec les innovations techniques qui transforment peu à peu l’état de l’art et le cadre même des produits : ce faisant, les normes associées à la réglementation doivent alors être mises à niveau. C’est le cas pour l’ISO 14971 : “Application de la gestion des risques aux dispositif médicaux”.
B. Le context de la norme ISO 14971
Une norme est un ensemble de règles techniques et critères qui définissent un type d’objet, un produit ou un procédé : ces règles évoluent naturellement avec les évolutions technologiques ou organisationnelles. Si la NF EN 1441 : 1998, première norme relative aux risques liés aux dispositifs médicaux, ne portait que sur l’analyse des risques en donnant un standard de procédure, aujourd’hui sa descendante harmonisée englobe l’ensemble de la gestion des risques tout au long du cycle de vie du dispositif médical [5].
a. Les nouveaux règlements 2017/745 et 2017/746
L’entrée en vigueur printemps 2020 des règlements européens 2017/745 [6] et 2017/746 [6] relatifs aux dispositifs médicaux (DM) et aux dispositifs médicaux de diagnostic in vitro (DMDIV), remplaçants des directives 93/42/CEE [7] et 90/385/CEE [9, p. 385], apporte des modifications aux règles de classement des dispositifs médicaux, revoit et uniformise les modalités d’évaluation par les organismes notifiés, et impose donc aux fabricants de revoir l’ensemble de leurs produits vis-à-vis des exigences essentielles applicables.
Si le marquage CE peut encore être valable un certain temps, le renforcement de la réglementation ne garantit pas aux constructeurs qu’ils puissent le renouveler facilement, la situation réglementaire de leurs dispositifs pouvant avoir évoluée : ils doivent alors certes revoir l’ensemble de la documentation technique, mais également leurs systèmes de gestion de la qualité et des risques.
Au 28 janvier 2020, les organismes notifiés pour le règlement 2017/745 sont seulement neuf contre près de trente pour l’ancienne directive 93/42/CEE [5] . Cet écart s’explique parce que même ces organismes mettent un certain temps à obtenir les autorisations et accréditations nécessaires, mais également par le fait que certains d’entre eux ont décroché car ils ne peuvent gérer la hausse d’activité que ce passage d’un texte à l’autre requiert.
Actuellement, dans cette période de transition entre ces deux réglementations dans une course effrénée au marquage CE, alors que les rares organismes déjà notifiés croulent sous les demandes des fabricants, qui ont pour certains beaucoup de modifications à apporter pour espérer obtenir ce gage de qualité et fiabilité qui leur permet de vendre le produit marqué sur le marché européen [11].
b. Les normes harmonisé et la norme ISO 13485
Si la commission européenne oblige par la réglementation le fabricant à satisfaire des exigences essentielles pour obtenir le marquage CE par un organisme notifié, elle peut mandater des organismes de normalisation - l’ISO et le CEI selon le domaine d’expertise technique - pour établir des normes harmonisées. Le respect de ces textes référentiels par le constructeur confère une présomption de conformité aux exigences essentielles européennes.
Pour les dispositifs médicaux, la norme harmonisée “centrale” est l’ISO 13485, qui est relative au système de management de la qualité mis en place chez le fabricant [12]. C’est l’ISO 13485 qui est auditée dans le cadre de l’évaluation système demandée pour l’obtention du marquage CE. Elle est donc naturellement sujette à de nombreuses certifications chez les constructeurs de dispositifs médicaux pour montrer patte blanche à l’auditeur [13].
Le respect de l’ISO 13485 appelle celui de l’ISO 14971 ; en effet, elle incite le fabricant à se référer à la norme 14971 en ce qui concerne la gestion des risques. Le maillage entre ces deux normes explique un faible de taux de certification concernant l’ISO 14971, la certification ISO 13485 l’englobant.
c. La norme 14971
La norme ISO 14971 concerne “l’application de la gestion des risques aux dispositifs médicaux”, et s’adresse aux fabricants comme aux autres acteurs de cette industrie. Son champ d’application s’étends sur l’ensemble du cycle de vie du dispositif médical, de la conception (ou l’étude des dangers et risques associés doit dès lors être prise en compte) à la post-commercialisation (ou le fabricant suit la matériovigilance relative à son produit ainsi que les événements indésirables liés) jusqu’à la réforme du dispositif médical[14, p. 14971].
La norme ISO 14971 : 2019 apporte des changements à la dernière version en date de 2013 ; elle s’appuie sur les changements réglementaires entre les anciennes directives et les nouveaux règlements, dans la suite de la NF EN ISO 13485 : 2016, et se destine donc à être un outil de transition pour les fabricants lorsqu’elle sera finalisée [15].
La date de parution de la norme ISO 14971 est le 10 décembre 2019.
II. Etat de l’art de la gestion des risques chez les fabricants de dispositifs médicaux
A. Réalisation d'un sondage à l'attente des fabricants
Afin d’évaluer l’existant et de proposer des solutions opérationnelles, un questionnaire Google Form™ a été soumis à 496 fabricants par mail et partagé sur le réseau professionnel LinkedIn™.
a. Présentation de sondage
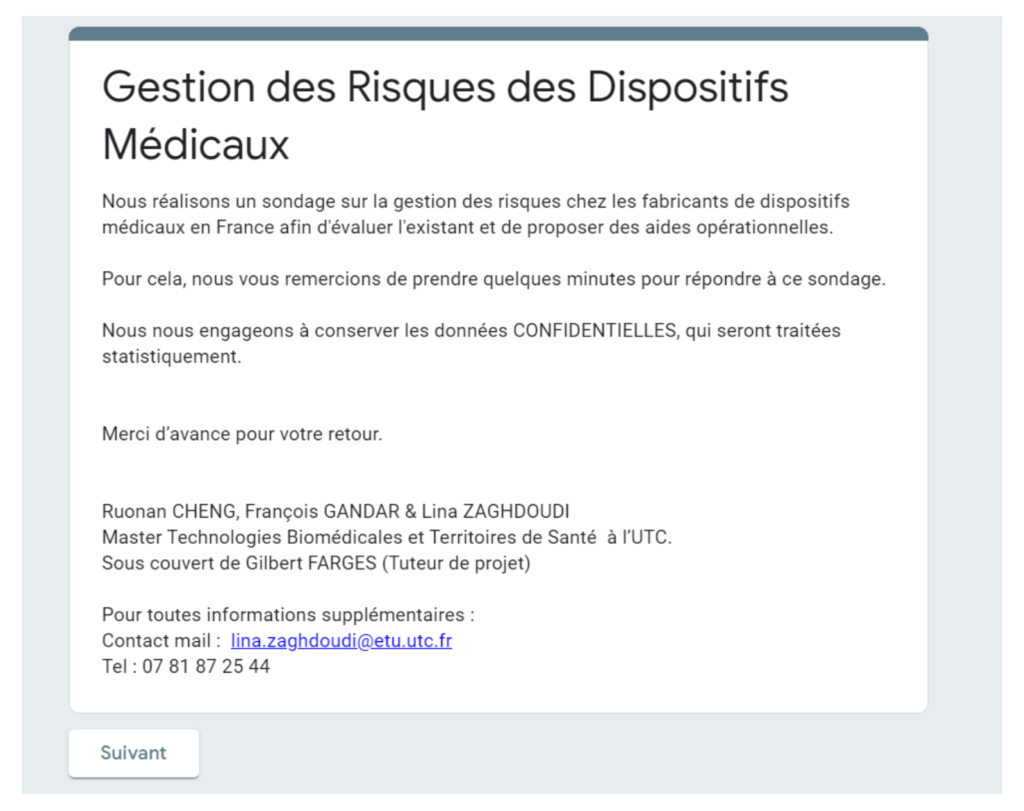
Ce questionnaire sur la “gestion des risques des dispositifs médicaux” est composé de plusieurs parties :
La première partie est une présentation de l’objectif présenté en Figure 2. ci-dessous :
Ensuite, en section 2, le Profil du fabricant y est renseigné en s’intéressant au type d’entreprise (multinationale, ETI, PME, TPE, start-up), si elle exporte au sein ou dehors de l’UE, sa catégorie de produits fabriqués (DM/DMIA/DMDIV ou autre), sa classe de risque du ou des produits (I, Is, Im, Ir, IIa, IIb, III) et enfin si elle dispose des fonctions d’Affaires Réglementaires, de responsable Qualité, de responsable Risque en interne ou externalisé.
Une fois informé sur le profil du fabricant, la section 3 s’intéresse à la Connaissance de la norme avec une échelle de degré de 0 à 5. Un 0 signifierait une non-connaissance de la norme, à l’inverse, un 5 représenterait une personne experte. Le fabricant est questionné sur l’intérêt par une formation ISO 14971, son exploitation au sein de son entreprise ou de ses sous-traitants, sa certification ISO 14971, et la connaissance et l’usage de l’outil de diagnostic version 2013 présenté dans la partie II.3.
Et enfin une dernière partie personnalisée sur l’Exploitation de la norme et de l’outil de diagnostic version 2013, le fabricant informe des types d’outils, méthodes ou approches utilisés en gestion des risques mais aussi en analyse des risques ainsi que d’autres questions plus techniques sur l’usage d’Excel ou encore la durée idéale pour effectuer un diagnostic avec l’outil.
b. Résultat du sondage
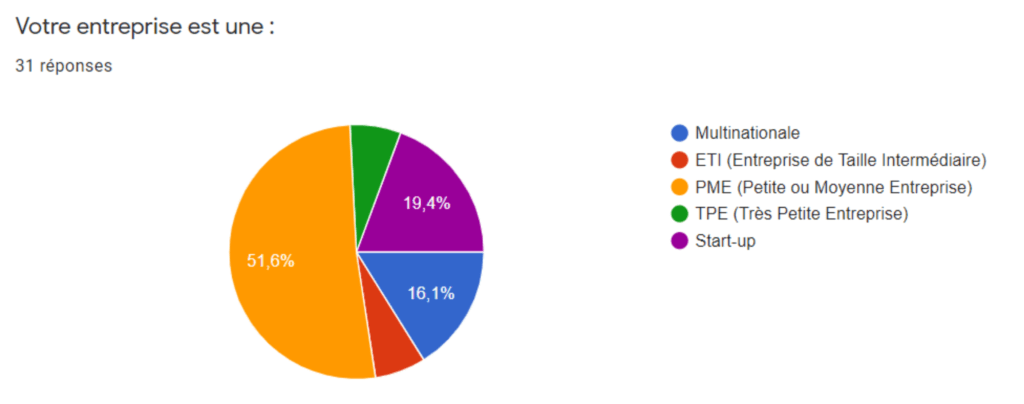
A l’issue de ce sondage, le taux de réponses est de 6,5% en une semaine. La grande majorité des entreprises étaient des PME, TPE et start-ups, ce qui est représentatif du secteur de l’industrie biomédicale (Figure 3).
82,1% de ces fabricants exportent au sein de l’UE et 60,2% de ces mêmes entreprises exportent en dehors de l’entreprise. Cet écart au niveau de l’exportation des DM au sein de l’UE, qui devrait être de 100%, dû au fait que nos entreprises soient Européennes, pourrait s’expliquer par le fait que certaines start-ups ne disposent pas du marquage CE ou bien qu’elle n’exporte par leur produit au sein de l’UE puisqu’elles les commercialisent uniquement en France.
Des informations complémentaires sur le profil des fabricants sont renseignés en Annexe I.
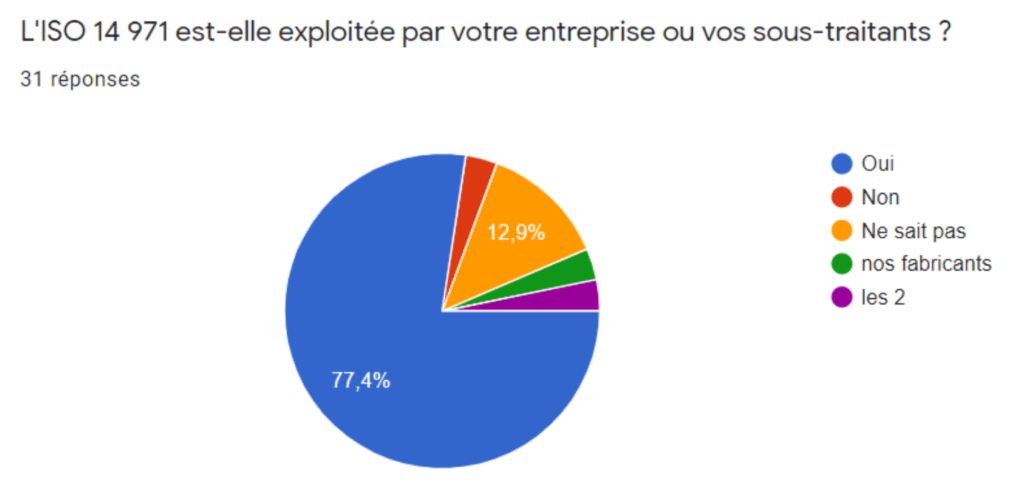
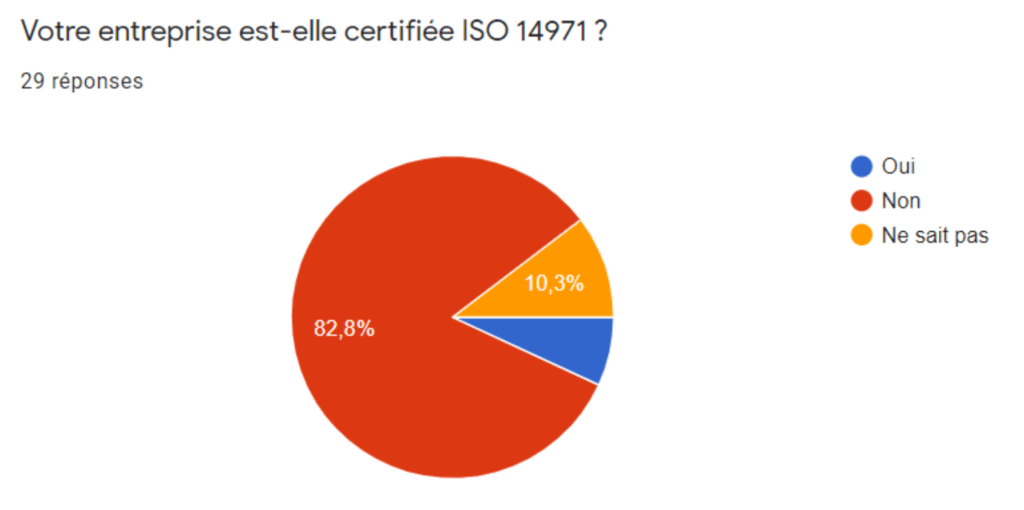
Sur 31 réponses, 77,5% de fabricants connaisse la norme dont 6,5% se considérant comme expert. De même, cette norme est très exploitée (Figure 4) mais peu d’entreprises sont certifiées ISO 14 971 (Figure 5). Ceci est dû au chainage de cette norme à la norme ISO 13485 qui est la norme harmonisée la plus utilisée. Cela s’explique par le fait que l’évaluation de conformité du système de management de la qualité pour obtenir le marquage CE se fait sur le référentiel ISO 13485 et non ISO 14971. Les fabricants ont plus d’intérêt à se faire certifié leur système de management de la qualité par la norme ISO 13485 qui vaut également présomption de conformité. Ce faible taux de certification peut favoriser l’usage d’un outil d’autodiagnostic et d’une auto-certification ISO 17050.
Ce sondage a permis aussi permis de lister les principaux types d’outils, méthodes ou approches utilisés pour la gestion des risques des dispositifs médicaux associée aux processus de l’entreprise : des outils Excel, la norme 14971, la norme ISO 13485, le guide du SNITEM, des outils propres à l’entreprise, la méthode Analyses des Modes de Défaillances de leurs Effets et leur Criticité, l’ERM by Knowllence.
Les types d’outils, méthodes ou approches utilisés pour la gestion et l’analyse des risques spécifiques aux dispositifs médicaux sont développés en Annexe IV.
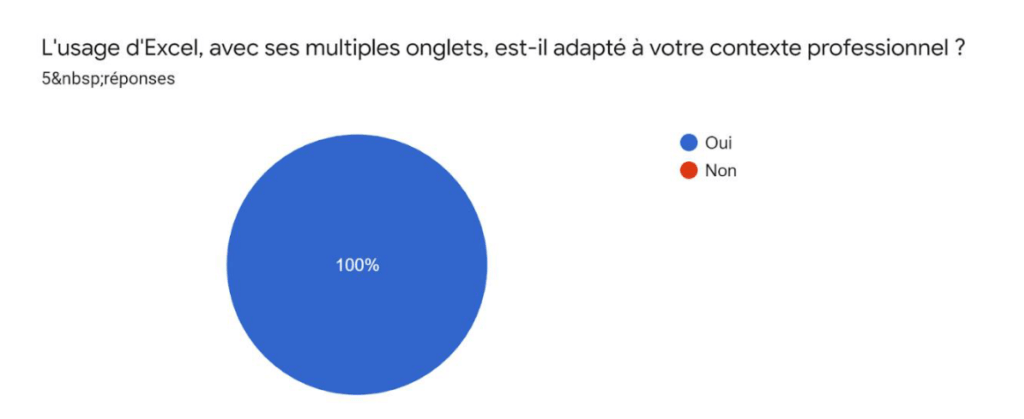
L’usage d’Excel est adapté au contexte professionnel du fabricant (Annexe V) avec une durée idéale d’utilisation minimale de 10 minutes. L’outil proposé en format Excel est donc adapté au public cible.
B. Les solutions techniques existants
a. Analyse des Modes de Défaillances, de leurs Effets et de leur Criticité (AMDEC)
Un cinquième des personnes ayant répondu à ce sondage utilise l’AMDEC pour réaliser une analyse des risques dans leur gesstion des risques. C’est l’Analyse des Modes de Défaillance, de leurs Effets et de leur Criticité.
C’est une méthode qui permet aux concepteurs de prévoir les défaillances potentielles et de prévenir leurs incidences en fabrication.
Utilisée à l’origine dans l’industrie aéronautique (NASA) puis généralisée à d’autres domaines, elle identifie la manière dont un produit ou un process peuvent défaillir et permet de prendre les dispositions pour prévenir les risques. Elle permet donc de rechercher des solutions.
Cette méthode a pour objectif de trouver des “remèdes” aux défaillances en procédant selon deux modes complémentaires, par composant ou bien par fonction.
b. Le guide « Gestion des risques des dispositifs médicaux » version 2016 du SNITEM
Ce guide a été élaboré par le Centre Technique des Industries Mécaniques (CETIM) à l’initiative du Syndicat National de l’Industrie des Technologies Médicales (SNITEM) en 2014 puis a été revu en 2016 [16].
La particularité de ce guide est qu’il est bilingue (français/anglais) et qu’il prend en compte les risques logiciels liés à l’utilisation des dispositifs médicaux.
Etant donné que tout dispositif médical mis sur le marché européen doit répondre à des exigences essentielles de sécurité et de performance prescrites par les anciennes directives européennes et transcrites en droit français dans le Code de la Santé Publique. Ce guide visait à aider les fabricants en leur fournissant une aide opérationnelle de référence pour la réalisation de leurs analyses de risques et pour la mise en œuvre de leur démarche de gestion des risques, basé sur les normes ISO 14971 : 2013 et NF EN 62304 (pour les risques logiciels).
Elle propose un processus se déroulant en six phases clés, des outils et des exemples de leur application. Elle délivre également des informations sur la norme EN 62304 dans le processus de gestion des risques de l’ISO 14971, avec des exemples concrets [16].
c. Logiciel de gestion du risque : ERM by Knowllence (Enterprise Risk Management)
Ce logiciel de gestion du risque “Medical Device Suite” proposé par Knowllence est disponible sur abonnement et propose un module logiciel dédié à l'analyse des risques au regard de la norme ISO 14971 : Dispositifs médicaux — Application de la gestion des risques aux dispositifs médicaux.
A l’issu de l’utilisation de ce logiciel, un dossier complet de validation est élaboré et est fourni à chaque sortie pour le module RM 14971 [17].
d. Autres outils utilisés
D’autres outils propres à chaque professionnel peuvent être utilisés pour la gestion ou l’analyse des risques. Comme exemple, des outils développés ont été développés sur Excel ou encore des réunions d’équipe sont organisées.
Il y a également :
- Analyse Préliminaire des Risques (APR)
- Analyse par Arbre de Pannes (AAP)
- Analyse des modes de défaillances et de leurs effets (AMDE)
- Étude des phénomènes dangereux et de faisabilité (HAZOP en anglais)
- Analyse des risques et des points critiques pour leur maîtrise (ARMPC)
Dans le cadre de travaux de master Ingénierie de la Santé à l’UTC, une grille d'autodiagnostic a également été mise en place par des étudiants en 2018 [18].
C. L’outil d’autodiagnostic pour la NF EN ISO 14971 : 2013
Cet outil d’autodiagnostic de la norme NF EN ISO 14971 : 2013, réalisé en 2018 par Elem AYNE, Valérian BAYEUX et Dylan WANNEPAIN [18], [19], se destine à l’attention des fabricants de dispositifs médicaux qui souhaitent évaluer leur conformité. Ayant pour objectif de les aider à améliorer leur plan de gestion des risques dans l’optique de garantir l’obtention du marquage CE pour leurs produits, l’outil risque de devenir désuète du fait de l’évolution de la norme en une nouvelle version.
Sous un format de classeur Microsoft™ Excel™, il s’articule autour de 6 feuilles comprenant :
- Un mode d’emploi, qui explicite la démarche ainsi que l’utilisation de l’outil ;
- Une évaluation, où l’utilisateur autodiagnostique la conformité de son organisme vis-à-vis des exigences essentielles relatives à la gestion des risques en 72 critères à infirmer/confirmer ;
- Des résultats globaux, qui permettent une visualisation globale des résultats pour chacun des articles en un graphique ;
- Des résultats par article, qui apportent des graphiques par article avec les résultats de chacun des critères associés ;
- Une maitrise documentaire, rendant compte de l’état de conformité du système documentaire lié à la gestion des risques ;
- Une auto déclaration selon l’ISO 17050 éditable rapidement pour communiquer sur la conformité à la norme NF EN ISO 14971 : 2013 à la suite d’une évaluation.
III. Proposition d’une aide opérationnelle sur la gestion des risques des dispositifs médicaux
A. Livret de comparaison des versions 2013 et 2019
Afin de percevoir les changements concrets du texte, il est nécessaire d’établir une comparaison des deux versions de norme visées : la NF EN NF ISO 14971 : 2013 ainsi que la ISO 14971 : 2019, afin de donner un aperçu facilement consultable à l’attention des acteurs concernés.
Ce livret met en exergue les différences entre les deux versions de normes par une police rouge de telle sorte qu’une entreprise puisse connaître visualiser rapidement les points clefs de cette évolution
Ce document s’adresse aux professionnels/fabricants souhaitant mettre à jour leur gestion des risques au sein de leur établissement. Cette comparaison leur faciliterait la compréhension du passage de l'ancienne norme EN NF ISO 14971 : 2013 à la nouvelle version norme ISO 14971 : 2019.
Pour une meilleure compréhension de l’articulation et de la structure de la version 2019 de la norme, une cartographie interactive a été réalisée.
B. Cartographie interactive de la norme ISO 14971 : 2019
Cet outil se destine à faciliter la compréhension de la norme, par la navigation de l’utilisateur à travers ses articles et exigences.
Le répertoriage des différentes exigences de la norme ISO 14971 : 2019 s’effectue par la réalisation d’une Analyse Normative Opérationnelle (ANO) du texte, qui ont ensuite été implémentées dans cette cartographie.
Le logiciel iDraw™ a été utilisé pour la réaliser de manière graphique. Les images obtenues ont alors été insérées dans un diaporama, dans lequel ont été définis des liens hypertextes. Exportée sous format PDF™, elle est compatible avec de nombreux médias, légère, et simple d’utilisation.
L’arborescence de la cartographie s’articule en 3 niveaux :
- Le premier niveau permet de voir une vue globale et détaillée de la norme ;
- Le second permet de voir les sous-articles dans chaque article, si celui-ci en contient ;
- La dernière partie permet de voir les critères associés à chaque sous-article.
Comment utiliser la cartographie ?
Pour naviguer au sein de la cartographie, il suffit de cliquer sur les zones d’intérêts spécifiées. La norme se divise en 7 articles principaux (de l’article 4 à l’article 10). Une vue globale introduit l’utilisateur dans la cartographie (Voir Figure 6).
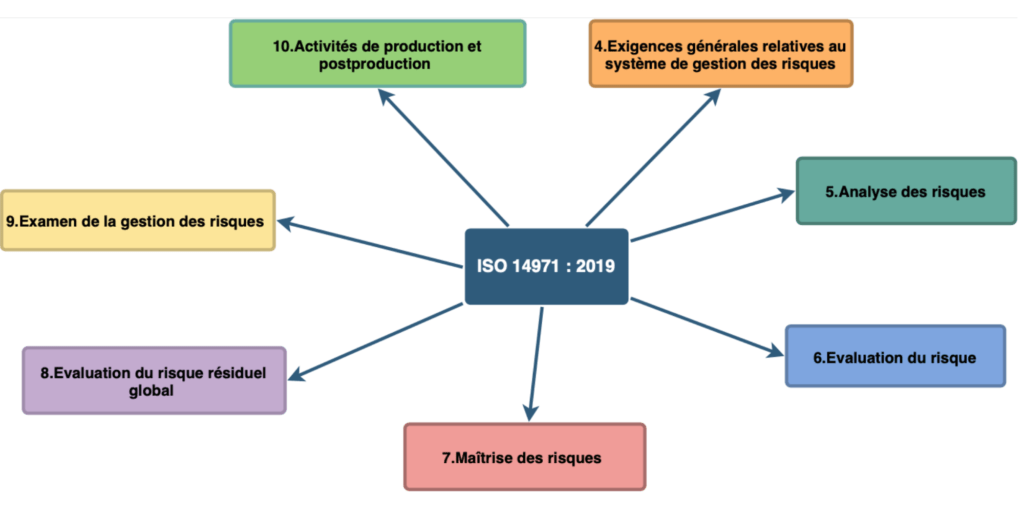
Pour naviguer parmi les articles, il suffit de cliquer sur l'un des articles pour obtenir plus d'informations. Les sous-articles sont ensuite déroulés (Voir Figure 7).
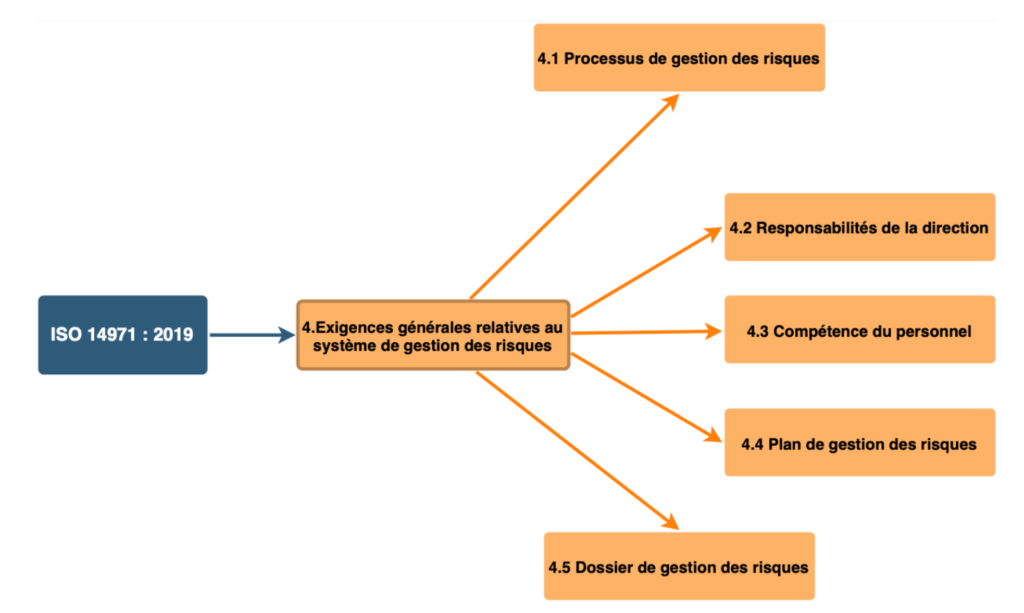
En cliquant ensuite sur un sous-article, les critères simplifiés associés sont affichés (Voir Figure 8). Aussi, les informations documentées associées aux exigences sont mentionnées par la présence d’un encadré rouge accompagné d’un logo.
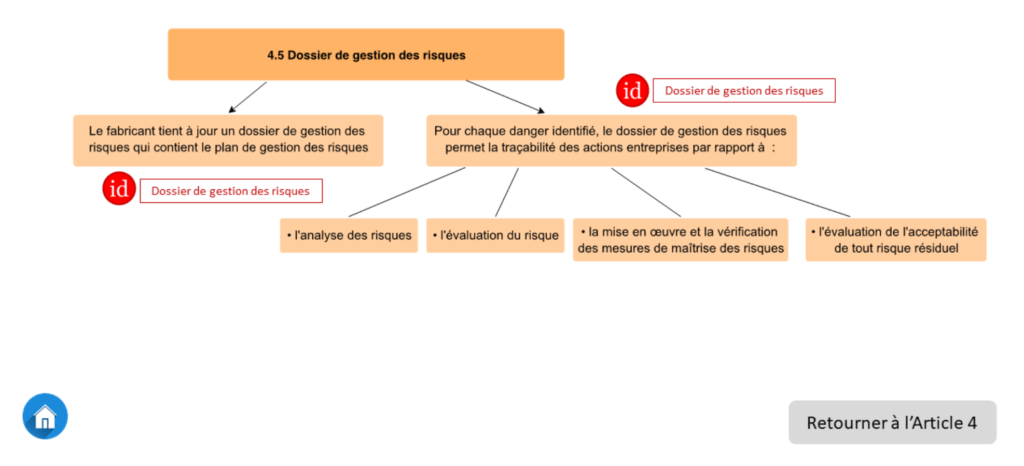
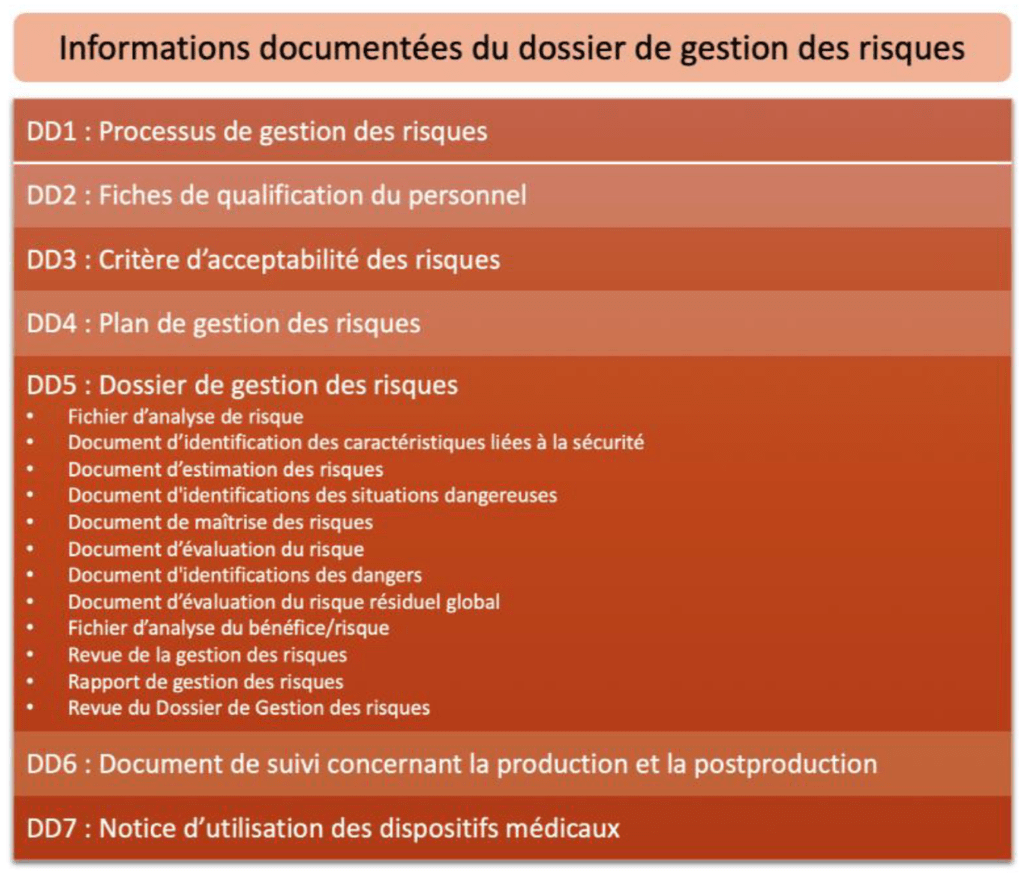
La dernière page de la cartographie liste l'ensemble de ces informations documentées, destinées à apporter une preuve de la conformité de l’organisme aux exigences réglementaires (voir Figure 9) :
C. Outil d’autodiagnostic ISO 14971 : 2019
L’outil d’autodiagnostic est l’élément central qui va permettre au fabricant d’évaluer sa conformité à la norme ISO 14971 : 2019 et ainsi définir les axes d’amélioration à prioriser. Reprenant le format Excel™ de l’outil de diagnostic à la version NF EN ISO 14971 : 2013, ce classeur s’oriente avec les mêmes onglets présentés en Figure 10 :
- Un mode d’emploi, qui donne le mode de fonctionnement et de paramétrage de l’outil ;
- Une évaluation, ou le fabricant évalue la conformité de son processus de gestion des risques vis-à-vis des nouvelles exigences essentielles sur 90 critères à infirmer/confirmer ;
- Des résultats globaux, qui donnent une vision graphique globale de la situation de conformité de l’organisme, et permet d’en tirer des axes d’améliorations, dont le suivi et possible ;
- Des résultats par article, qui donnent les résultats de chacun des critères par article visé ;
- Une maitrise documentaire, qui évalue la maturité du système documentaire de l’organisme lié à la gestion des risques ;
- Une page permettant d’éditer une auto déclaration selon l’ISO 17050 afin de communiquer efficacement sur son niveau de conformité à la norme ISO 14971 : 2019 à la suite d’une évaluation.

Dans le mode d’emploi en Figure 11, les seules cases modifiables par l’utilisateur ont un fond blanc et une police bleu vif, standard défini dans l’objectif de capter l’attention lors de l’usage.
Les onglets comportent des en-têtes permettant de figer les métadonnées relatives à la procédure d’évaluation : identité de l’utilisateur, de l’organisme ainsi que la date.
Il est possible de personnaliser les objectifs de taux de conformité dans l’onglet « Mode d’emploi » en fonction de l’objectif de conformité visé.

La personnalisation des seuils de conformité peut contribuer au développement progressif d’une culture qualité par l’amélioration continue des performances et des objectifs annuels de la structure utilisatrice de l’outil. Les autres seuils se recalculent afin de conserver une pertinence dans le diagnostic.
L’évaluation se déroule autour de 90 critères, qui sont des transcriptions des exigences essentielles formulées dans la norme. L’utilisation en synergie de la cartographie et du livret de comparaison des versions avec l’outil permettent à l’utilisateur de mieux comprendre le référentiel, son application au sein de l’entreprise et de s’informer sur ses évolutions lors du diagnostic.
L’évaluateur doit alors choisir entre les différents niveaux de véracité :
- Vrai (le critère est satisfait)
- Plutôt Vrai (le critère est satisfait, mais des améliorations sont possibles)
- Plutôt Faux (le critère n’est satisfait que partiellement)
- Faux (le critère n’est pas satisfait du tout)
- Non-Applicable (non comptabilisé mais à justifier)
Un code couleur reprend les niveaux de véracité : vert foncé pour « Vrai », vert clair pour « Plutôt Vrai », il tend à exprimer une non-conformité mineure lorsque le niveau de véracité est « Plutôt Faux » avec un formatage conditionnel orange, et à alerter l’utilisateur pour une non-conformité majeure « Faux » avec une coloration rouge.
Les mots en rouge dans les critères expriment un mode de preuve documentaire attendu et le signale donc à l’attention de l’évaluateur en ressortant du texte brut.
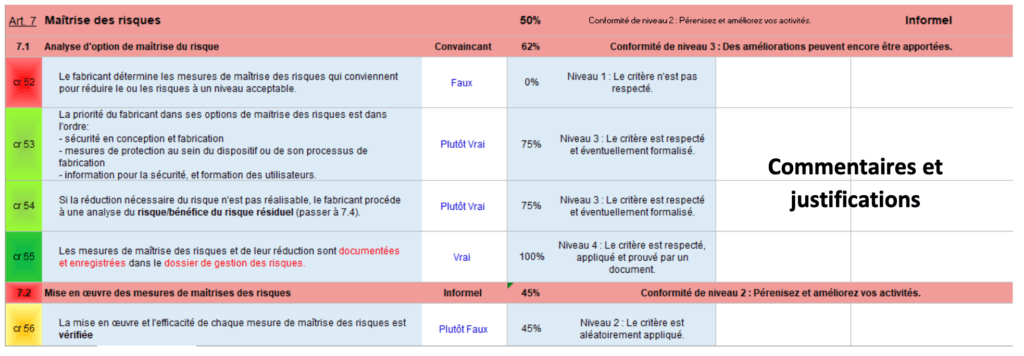
En Figure 12, le début de l’article 7 est présenté dans l’onglet évaluation.
Pour donner suite à l’évaluation, 2 onglets permettent d’obtenir une appréciation globale et détaillée des résultats. Des graphiques permettent d’illustrer la conformité des différents critères et de rendre l’information rapidement décryptable par un lecteur candide (Figure 13) ; il est possible d’utiliser les feuilles pour définir des plans d’actions par article en identifiant des axes d’améliorations à partir des articles et critères dont le niveau de véracité ne satisfait pas aux objectifs de conformité de l’organisme à la norme 14971 : 2019.

La feuille « Maitrise documentaire », en Figure 14 , s’attache à signaler à l’utilisateur quelles pièces documentaires lui manquent afin de satisfaire aux exigences essentielles en évaluant la véracité des critères liés à ceux-ci ; il donne une représentation graphique du niveau de maturité des documents visés. Il aide la structure utilisatrice de l’outil à évaluer la conformité de ses preuves documentaires et a fortiori à identifier les axes d’amélioration. Les indicateurs sont calculés par une moyenne en fonction des niveaux de conformité des critères dans lesquelles la preuve documentaire exigée par la norme est liée à une pièce documentaire.
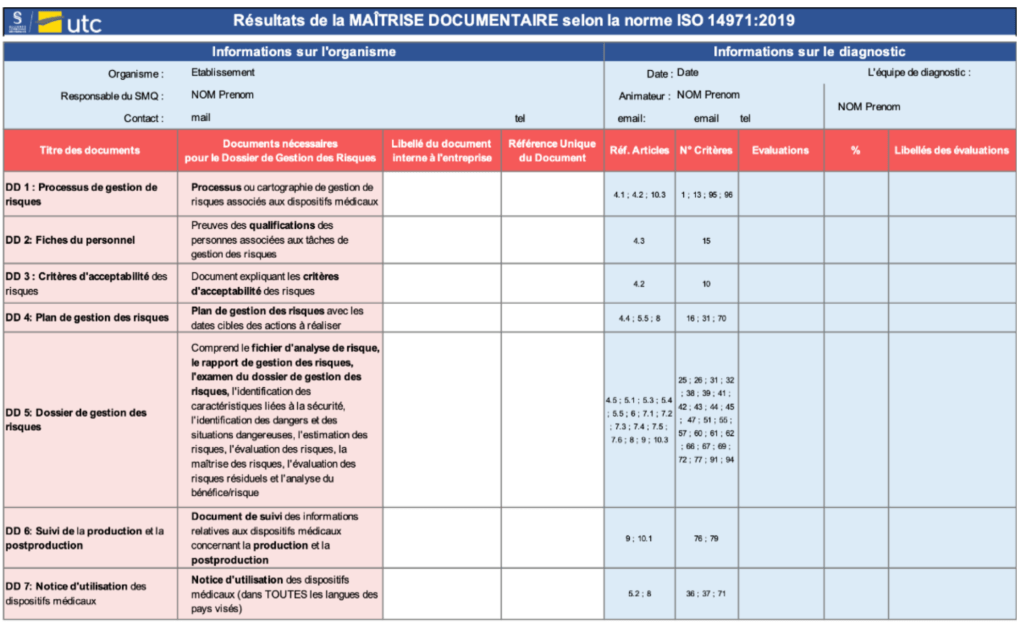
Enfin, le dernier onglet « Déclaration ISO 17050 » consiste en une pré-édition automatique d’une auto-déclaration selon l’ISO 17050 que remplit l’utilisateur pour communiquer sur ses résultats et sa conformité aux exigences réglementaires applicables. Cette déclaration ne fait pas office de preuve de conformité, mais peut faire présomption de conformité aux exigences réglementaires applicables si les résultats le permettent.
Conclusion
Les trois outils qui constituent cette aide opérationnelle sont destinés à l’utilisation des fabricants afin de faciliter leur transition de la version 2013 de l’ISO 14971 à la version 2019 en accord avec la nouvelle réglementation européenne relatives aux dispositifs médicaux.
Elle souhaite apporter une solution facile d’utilisation à cette démarche de transformation, afin de réduire le risque pour le fabricant de faillir à la satisfaction des exigences réglementaires sans la satisfaction desquelles il ne peut obtenir le marquage CE, et donc vendre ses dispositifs médicaux sur le marché.
A travers ce Mémoire d’Intelligence Méthodologique, il est possible pour les fabricants de se familiariser avec l’ensemble des exigences de la norme (cartographie), d’en percevoir les évolutions (livret de comparaison) et d’en réaliser l’évaluation point par point (outil de diagnostic).
La réalisation d’un sondage a permis d’identifier une population de professionnels favorables au test de cet ensemble d’outils, et témoigne de l’intérêt porté par les fabricants. Des améliorations supplémentaires, en sus du portage à la version française lors de sa publication par l’AFNOR, pourront donc être apportées.
Références bibliographiques
[1] Syndicat National de l’Industrie des Technologies Médicales, « SNITEM, “Le marché français des dispositifs médicaux” », 2018. [En ligne]. Disponible sur : https://www.snitem.fr/.
[2] « Synthèse d’activité 2016 ». Edition ANSM, www.ansm.sante.fr, 20-sept-2017.
[3] Emeline Cazi et Stéhane Horel, « Prothèses mammaires : la France interdit certains modèles liés à un cancer », https://www.lemonde.fr/societe/article/2019/04/03/protheses-mammaires-interdiction-historique-de-certains-modeles_5445336_3224.html, 03-avr-2019.
[4] Chloé Hecketsweiler et Stéphane Horel, « « Implant Files » : un scandale sanitaire mondial sur les implants médicaux », https://www.lemonde.fr/implant-files/article/2018/11/25/le-manque-de-controle-des-dispositifs-medicaux-met-en-peril-la-securite-de-millions-de-patients_5388424_5385406.html, 25-nov-2018.
[5] AFNOR, « Norme NF EN 1441 - Dispositifs médicaux - Analyse des risques ». Afnor Editions, www.afnor.org, avr-1998.
[6] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) ». Ed. JO L 117, http://data.europa.eu/eli/reg/2017/745/oj, 05-mai-2017.
[7] « Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission (Texte présentant de l’intérêt pour l’EEE. ) ». Ed. JO L 117, http://data.europa.eu/eli/reg/2017/746/oj, 05-mai-2017.
[8] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux ». Ed JO L 169, http://data.europa.eu/eli/dir/1993/42/oj, 12-juill-1993.
[9] « Directive 90/385/CEE du Conseil, du 20 juin 1990, concernant le rapprochement des législations des États membres relatives aux dispositifs médicaux implantables actifs ». Ed. JO L 189, http://data.europa.eu/eli/dir/1990/385/oj, 20-juill-1990.
[10] « EUROPA - European Commission - Growth - Regulatory policy - NANDO ». [En ligne]. Disponible sur : https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34. [Consulté le : 17-déc-2019].
[11] M. Durand et E. Seris, « Le marquage CE », IRBM, vol. 31, no 1, p. 30‑35, févr. 2010, doi : 10.1016/j.irbm.2009.11.005.
[12] « Norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires ». Editions Afnor, Paris, www.afnor.org, avr-2016.
[13] G. Promé, « Normes Harmonisées relatives aux Dispositifs Médicaux », Qualitiso, 02-mai-2014. [En ligne]. Disponible sur : https://www.qualitiso.com/normes-harmonisees-dispositifs-medicaux/. [Consulté le : 17-déc-2019].
[14] AFNOR, « Norme NF EN ISO 14971 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux ». Editions Afnor, Paris, www.afnor.org, 05-janv-2013.
[15] « Norme ISO 14971:2019 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux ». Editions ISO, Genève, www.iso.org, 10-déc-2019.
[16] CETIM et SNITEM, « Le guide « Gestion des risques des dispositifs médicaux » version 2016 disponible ». Edition CETIM, www.cetim.fr.
[17] « Les évolutions de l’ISO 14971 2019 et de Medical Device Suite », Ed. Knowllence Wwwknowllencecom, juin 2019.
[18] D. Wannepain, V. Bayeux, et E. Ayne, « La gestion des risques liés aux dispositifs médicaux selon la norme NF EN ISO 14971:2013 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositif Médical et Affaires Règlementaires (DMAR), Mémoire de projet, www.travaux.master.utc.fr, puis « IDS » réf n° IDS002, janv. 2019.
[19] E. Ayne, V. Bayeux, D. Wannepain, et G. Farges, « Accompagner les fabricants dans leur gestion des risques des dispositifs médicaux selon la norme NF EN ISO 14971:2013 », IRBM News, vol. 40, no 2, p. 69‑73, avr. 2019, doi : 10.1016/j.irbmnw.2019.02.004.
Annexes
Annexe I : Résultats du sondage à l’intention des fabricants - Section Profil du fabricant
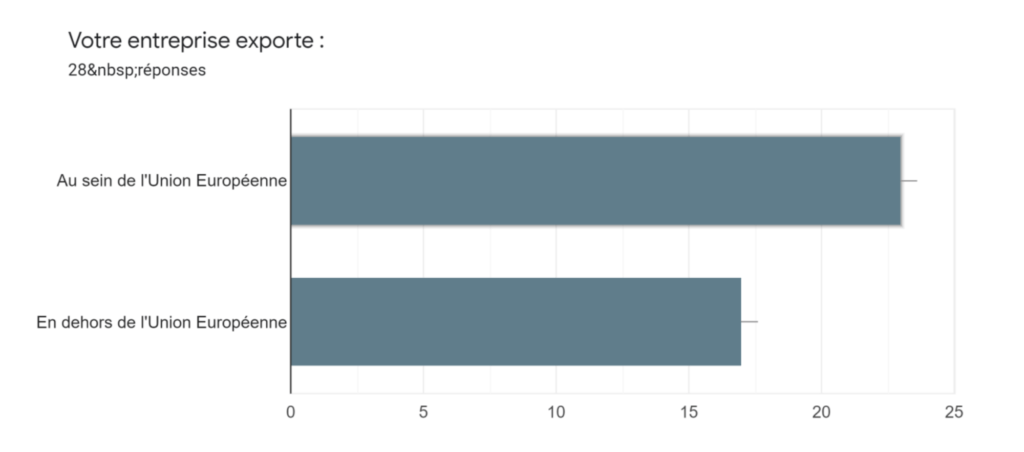
Annexe Ia : Exportation de l’entreprise au sein ou en dehors de l’UE - Source : auteurs
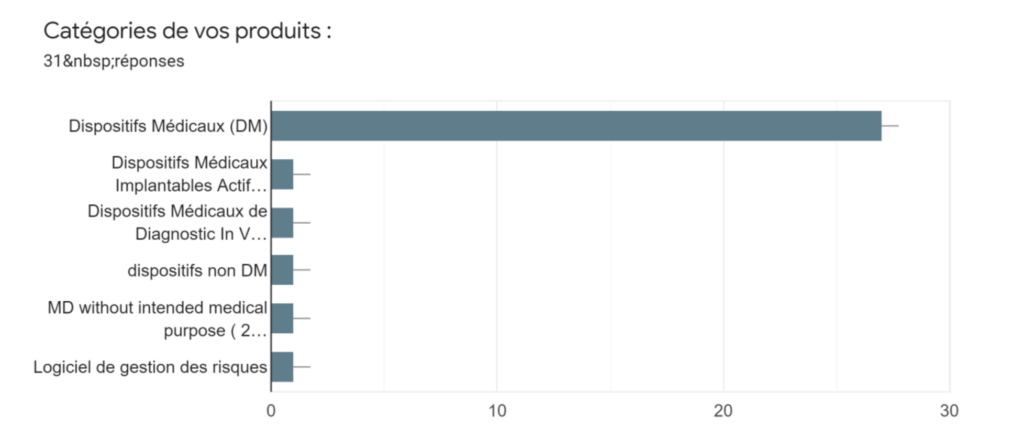
Annexe Ib : Catégories des produits des fabricants - Source : auteurs
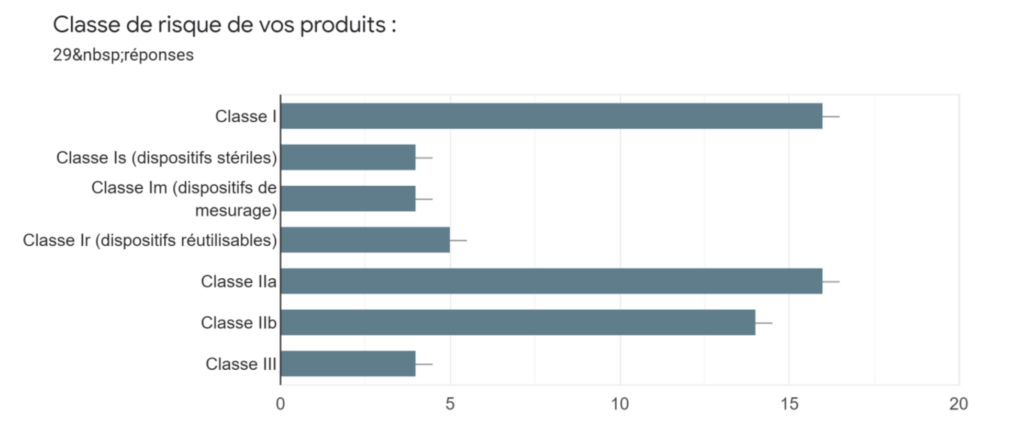
Annexe Ic : Classe de risque des DM - Source : auteurs
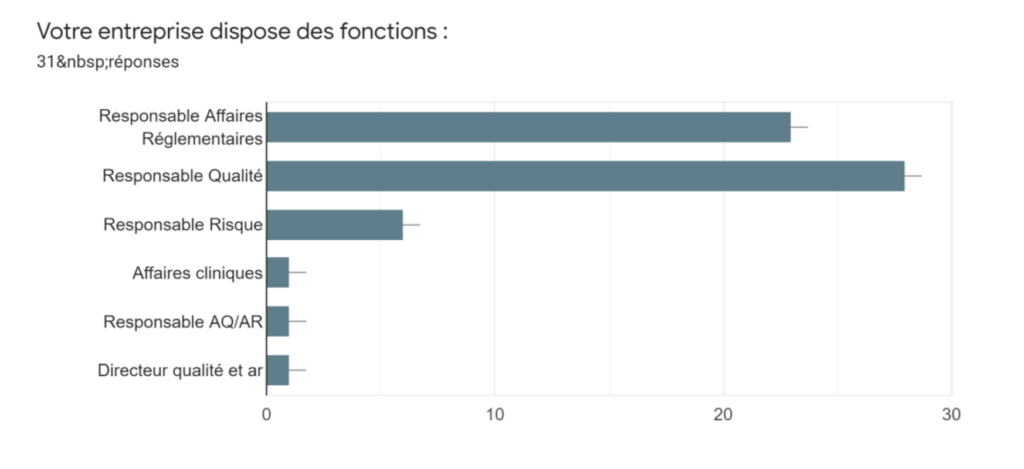
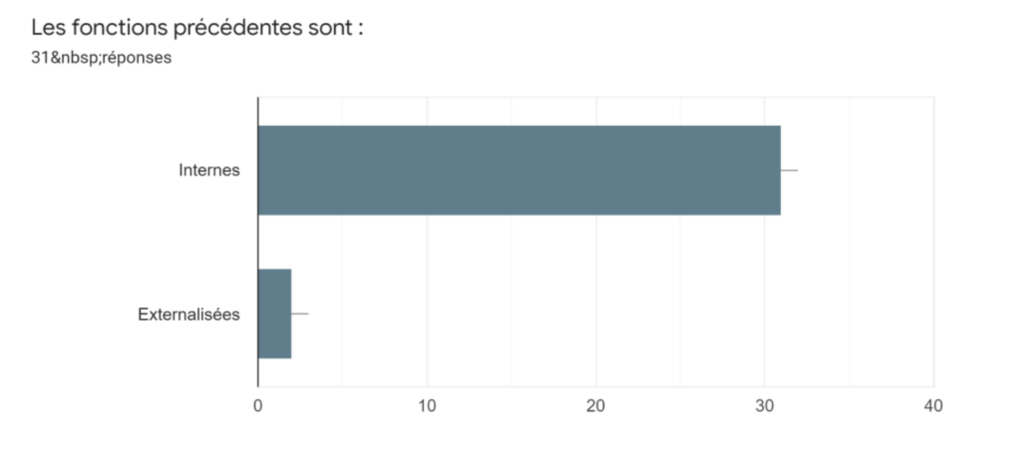

Annexe Id : Fonctions disponibles au sein de l’entreprise - Source : auteurs
Annexe II : Résultats du sondage à l’intention des fabricants - Section Connaissance de la norme
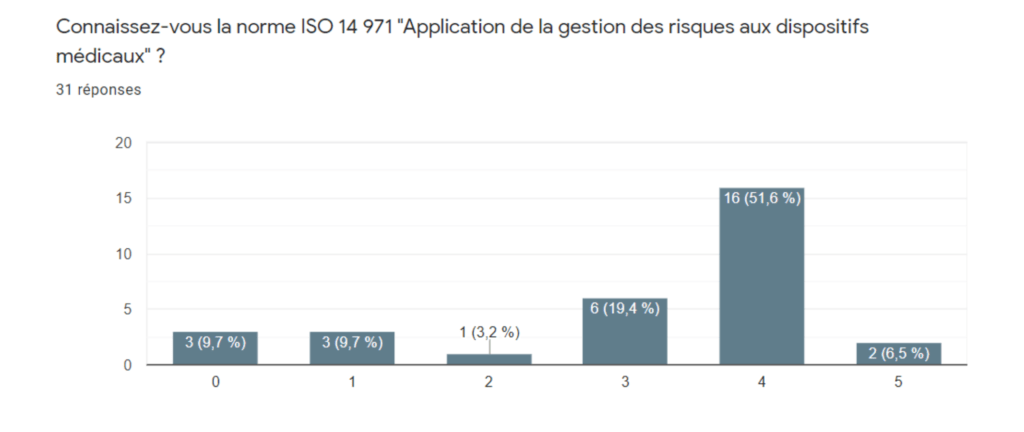
Annexe IIa - Connaissance de la norme ISO 14971 - Source : Auteurs
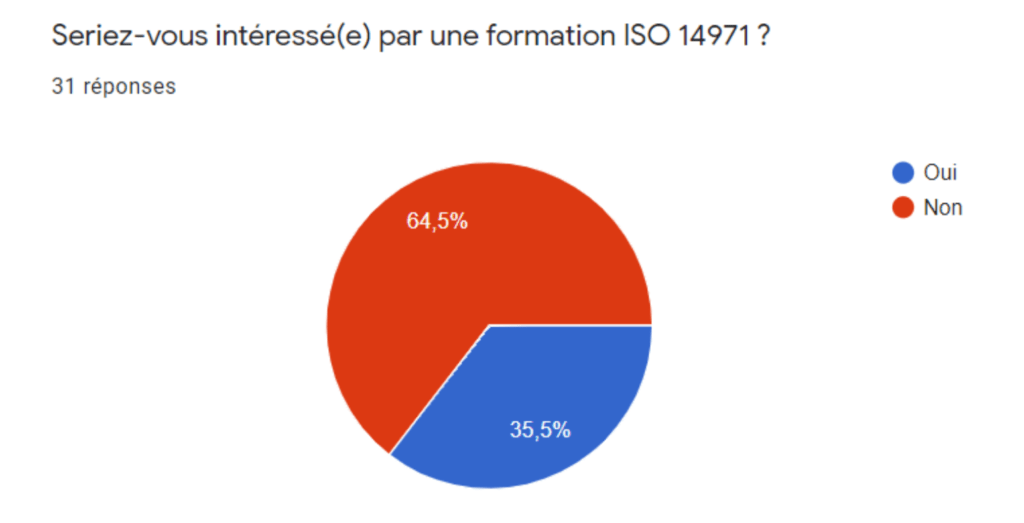
Annexe IIb - Intérêt pour une formation ISO 14971
Annexe III : Résultats du sondage à l’intention des fabricants - Section Utilisation de l’outil de diagnostic sur l’ISO 14971 version 2013
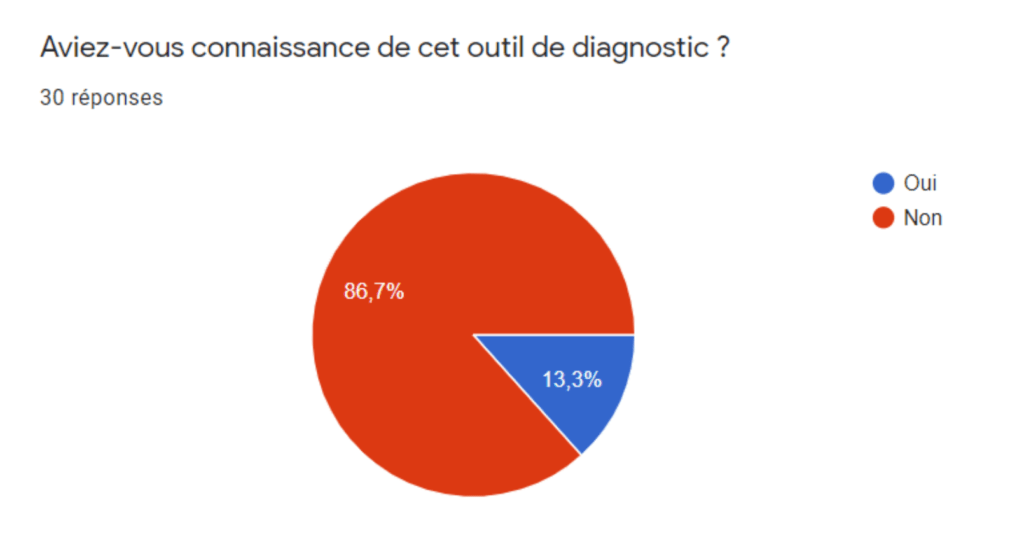
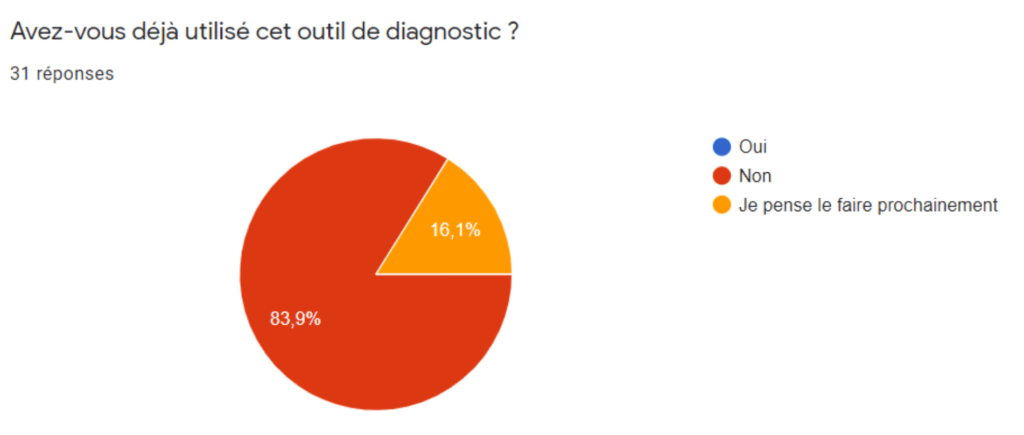
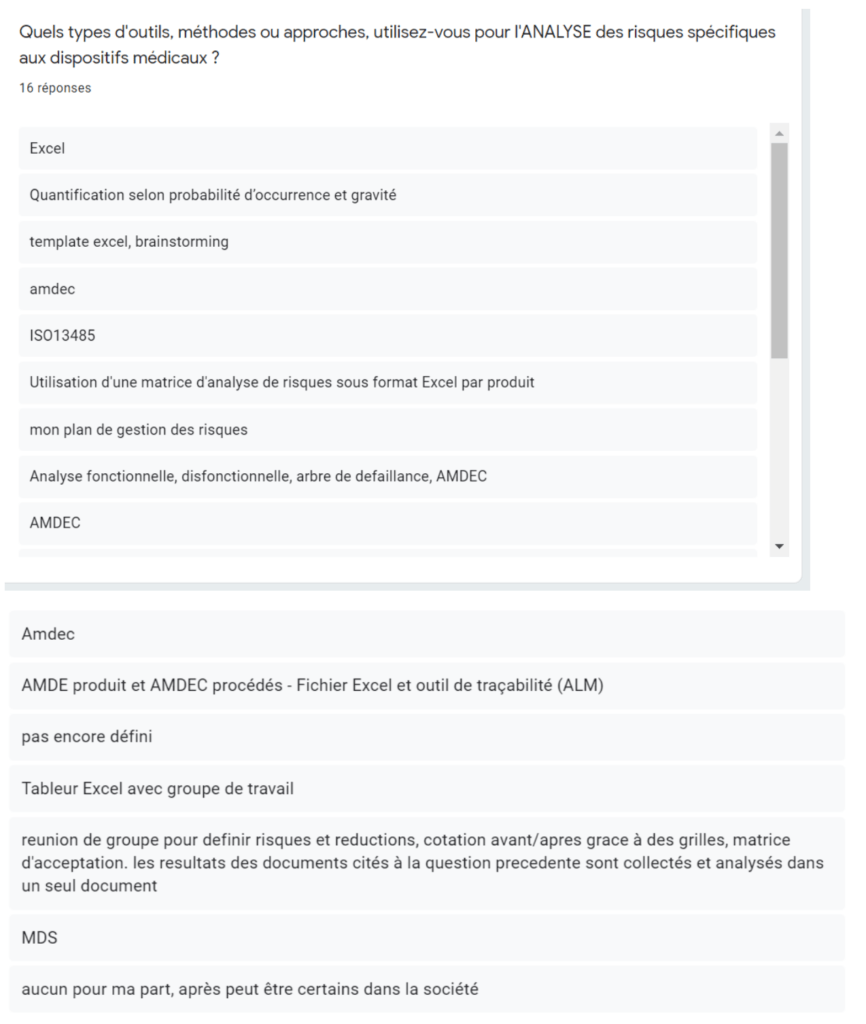
Annexe IV : Résultats du sondage à l’intention des fabricants - Section Utilisation de la norme

Annexe IVa - Types d’outils, méthodes ou approches utilisés pour la GESTION des risques par le fabricant
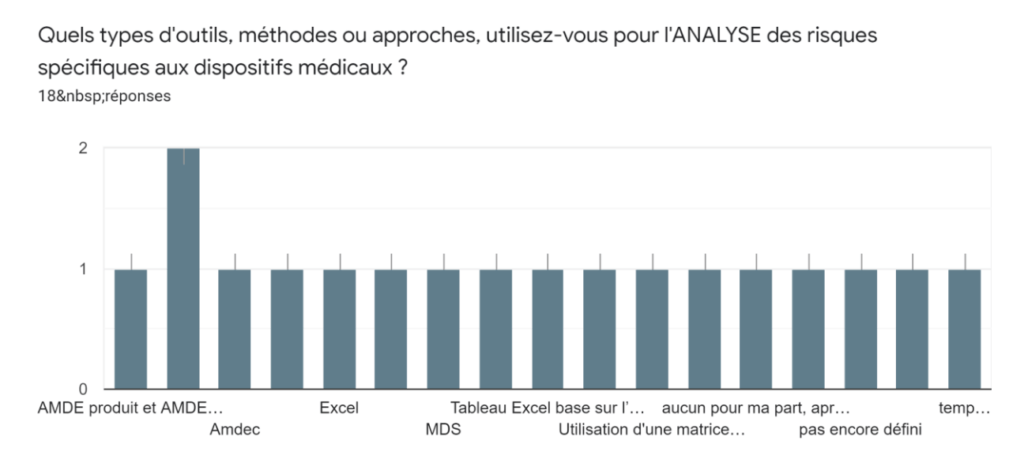
Annexe IVb - Types d’outils, méthodes ou approches utilisés pour l’ANALYSE des risques faite par le fabricant
Annexe V : Résultats du sondage – Usage d’Excel adapté au contexte professionnel