
IDS049 - Élaboration de la Documentation Technique selon le Règlement 2017/746 - Mise à jour et amélioration du Système de Management de la Qualité selon ISO 13485 : 2016
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Contact
Citation
A rappeler pour tout usage : CHEDJOU TAKAM Jean Ernest, « Élaboration de la Documentation Technique selon le Règlement 2017/746 & Mise à jour et Amélioration du Système de Management de la Qualité selon ISO 13485 : 2016 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs Médicaux et Affaires Réglementaires (DMAR), https://travaux.master.utc.fr/, puis "IDS", réf "IDS049", Septembre 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids049/
Résumé
Maîtriser les risques lors de la conception du dispositif, maintenir et améliorer les performances durant tout le cycle de vie du produit et garantir un niveau élevé de sécurité aux patients et utilisateurs sont les obligations des fabricants qui mettent les produits sur le marché européen. Les normes, guides, directives et règlements ont pour but de les aider dans cette tâche.
Depuis 1998, c’est la directive européenne IVDD (98/79/CEE) [1], qui définit les exigences pour mettre un dispositif médical de diagnostic in vitro (DMDIV) sur le marché. La société Bordier Affinity Products SA (BAP) commercialise des produits qui ne nécessitent pas l’évaluation de la conformité (documentation technique) par un organisme notifié pour obtenir le marquage CE. Une simple auto-déclaration UE de conformité et un certificat ISO 13485 sont suffisants pour commercialiser sur le marché de l’UE selon l’IVDD.
Avec la publication en 2017 du nouveau règlement européen IVDR (2017/746) [2], les produits de la société ont changé de classe de risque. Les exigences de cette classe sont renforcées par rapport à celle de l’IVDD. Pour obtenir le marquage CE à partir de 2022, la société BAP devra passer obligatoirement par un organisme notifié qui devra évaluer la documentation technique des produits et auditer le système de management de la qualité (SMQ).
Afin de mener à bien cette transition, l’entreprise Bordier Affinity Products SA a fait appel à un stagiaire formé dans le domaine des Affaires Règlementaires. Son rôle et le sujet de ce mémoire, ont été d’élaborer la documentation technique des produits BAP selon l’IVDR.
Mots clés : Gestion des risques, sécurité et performances des dispositifs, directive 98/79/CEE, règlement 2017/746, mise sur marché, conformité réglementaire, déclaration UE de conformité, certificat ISO 13485, documentation technique, système de management de la qualité.
Abstract
Controlling risks during device design, maintaining and improving performance throughout the product's life cycle and ensuring a high level of safety for patients and users are the obligations of manufacturers who place products on the European market. The standards, guides, directives and regulations are intended to help them in this task.
Since 1998, European Directive IVDD 98/79 / EEC [1] defines the requirements for placing an in vitro diagnostic medical device (IVDD) on the market. Bordier Affinity Products SA (BAP) markets products that do not require conformity assessment (technical documentation) by a notified body to obtain CE marking. A simple EU self-declaration of conformity and an ISO 13485 certificate are sufficient to market in the EU market according to IVDD.
With the publication in 2017 of the new European regulation IVDR 2017/746 [2], the company's products have changed risk class. The requirements of this class are reinforced compared to that of IVDD. To obtain the CE marking from 2022, the BAP Company will have to go through a Notified Body which will have to evaluate the technical documentation of products and audit the quality management system.
In order to successfully complete this transition, Bordier Affinity Products SA called on an intern trained in Regulatory Affairs. Its role, and the subject of this brief, was to develop the technical documentation of BAP products according to the IVDR.
Keywords : Risk management, device safety and performance, directive 98/79 / EEC, regulation 2017/746, placing on the market, regulatory compliance, EU declaration of conformity, ISO 13485 certificate, technical documentation, quality management system.
Téléchargements

Élaboration de la Documentation Technique selon le Règlement 2017/746 & Mise à jour et Amélioration du Système de Management de la Qualité selon ISO 13485 : 2016
Élaboration de la Documentation Technique selon le Règlement 2017/746 & Mise à jour et Amélioration du Système de Management de la Qualité selon ISO 13485 : 2016
Mémoire complet :
Élaboration de la Documentation Technique selon le Règlement 2017/746 ET Mise à jour et Amélioration du Système de Management de la Qualité selon ISO 13485 : 2016
Remerciements
L’aboutissement de ce travail n’aurait pas été possible sans l’aide et le soutien de plusieurs personnes, à qui du fond du cœur je témoigne toute ma reconnaissance.
Je remercie DIEU de m’avoir donné motivation, courage et santé durant tout mon parcours d’étude, qui été parsemé de nombreux embuches.
Je présente ma profonde gratitude à l’endroit de nos responsables de master Mme. Isabelle CLAUDE, M. Jean Mathieu PROT et M. Gilbert FARGES, ainsi qu’à notre secrétariat du master Mme. Françoise MERESSE et Morgane BOUFFLERS pour leur disponibilité, leurs conseils, leurs encadrements et leur soutien pendant toute cette période ;
Merci à Mme Virginie SALSAC, pour l’encadrement, la disponibilité, les précieux conseils, le soutien, les encouragements et le suivi durant ce stage, ce qui a contribué à alimenter ma réflexion au quotidien ;
Merci à M. Nicolas BEYLS, pour l’encadrement, la disponibilité, l’accueil chaleureux au sein de l’entreprise, ses précieux conseils, son soutien depuis le début du stage, sa clairvoyance, et son expérience qui m’ont été d’une grande aide pendant mes activités, et pour l’aboutissement de ce mémoire ;
Merci à M. Clément BORDIER, M. Florent GENOUX, et tout le personnel de BIOKEMA, pour l’accueil, le soutien, l’agréable interaction, la bienveillance qui m’ont permis de facilement m’intégrer au sein de l’équipe et de l’entreprise ;
Merci à tous le personnel enseignant de l’UTC (externe comme interne) qui ont bien voulu partager leurs connaissances et expérience avec nous, pour atteindre nos objectifs académiques ;
Merci à toute la Famille MAGNE et MAKOUGOUM pour la fraternité, soutien inconditionnel et leur amour à mon égard ;
Je terminerai en remerciant tous mes ami(e)s, camarades de promotion et aîné(e)s académiques pour les encouragements, le sourire, et le dévouement durant cette aventure.
Introduction
Afin de valider ma dernière année universitaire, j’ai effectué un stage de fin d’études de 6 mois dans les services Assurance Qualité et Affaires réglementaires de la société Bordier Affinity Products SA[3]. L’entreprise conçoit, fabrique et commercialise les kits pour le diagnostic des infections parasitaires et mycologiques depuis 1991.
Ces kits sont des dispositifs médicaux de diagnostic in vitro (DMDIV) qui permettent d’aider au diagnostic des maladies infectieuses négligées. La plupart de ces maladies sont transmissibles de l’animal (domestiques et/ou sauvages) à l’homme, majoritairement dans des zones tropicales.
Ces maladies touchant les pays pauvres, il y a très peu de concurrents identifiés dans ce secteur. Les produits BAP sont très appréciés par les utilisateurs dus à leur qualité, leur précision, leur sensibilité et leur spécificité. Chez BAP, la santé et sécurité des patients et utilisateurs est une priorité.
La santé et la sécurité des patients et des utilisateurs peut être assurée en respectant les règlementations, les normes internationales et en surveillant les évolutions réglementaires pendant tout le cycle de vie du produit depuis sa conception jusqu’à sa mise sur le marché. L’élaboration des dossiers techniques est la preuve du respect des exigences réglementaires et normatives pour garantir en permanence, des produits sûr et performants aux utilisateurs.
Ce mémoire présente les activités menées durant ces six mois de stage :
- Structure d’accueil,
- Différentes tâches réalisées
- Objectifs à atteindre pour garantir sécurité et performance des dispositifs développés
- Démarche utilisée, les aspects réglementaires pris en compte et les solutions apportées.
- Difficultés rencontrées, les perspectives envisagées.
I. Présentation de Bordier Affinity Products SA et de son environnement
I.1. Description
Bordier Affinity Products SA est une entreprise de biotechnologie qui effectue les activités de conception, fabrication et commercialisation de kits pour l’aide au diagnostic des maladies parasitaires et fongiques humaines (DMDIV). La société est composée du fondateur monsieur Mr Clément BORDIER, de deux employés à plein temps (NB et FG) qui assurent toutes les tâches de la société (achat, conception, production, vente, contrôle qualité, assurance qualité et affaires règlementaires) et de deux employés à temps partiel pour la production (voir annexe II.1). La société est organisée en différents services distincts que sont : Achat, Conception, Production, Vente, Affaires Règlementaires, Assurance Qualité… L’organigramme de la société BAP est très simple, Mr Clément Bordier (CB) et son fils Edgard Bordier (EB) assurent la direction, le reste des activités est assuré par les deux employés à temps plein.
I.2. Présentation
La société Bordier Affinity Products SA est basée à Crissier, proche de Lausanne en Suisse. Elle est locataire dans les locaux de la société BIOKEMA[4]. La société a été créée en 1991 par le professeur Clément BORDIER. A cette époque il était professeur de parasitologie et employé de BIOKEMA. Voulant développer sa propre activité, il a donc décidé de créer son entreprise, aux vues de ses compétences et de son réseau de connaissances. BIOKEMA a mis à disposition des locaux pour démarrer son entreprise. Mr Bordier a commencé avec 3 produits développés et commercialisés par lui-même et aujourd’hui on dénombre 14 produits vendus dans plusieurs pays dans le monde. Les clients sont les laboratoires de parasitologie & mycologie des hôpitaux, les laboratoires d’analyses médicales et les laboratoires de recherches.
Le kit est composé de tous les réactifs nécessaires à la réalisation du test en laboratoire (voir Figure 1 et 2). Tous les produits sont basés sur la technique Enzyme-Linked ImmunoSorbent Assay (ELISA) sauf un produit (#8100) basé sur la technique ImmunoFluorescence (IF) :
# 6100 Aspergillus fumigatus / # 9200 Toxocara canis / # 9300 Echinococcus multilocularis / # 9310 Echinococcus multilocularis / # 9350 Echinococcus granulosus / # 9400 Acanthocheilonema viteae / # 9450 Strongyloïdes ratti / # 9500 Leishmania infantum / # 9550 Entamoeba histolytica / # 9600 Schistosoma mansoni / # 9650 Fasciola hepatica / # 9700 Taenia solium / # 9750 Trichinella spiralis / # 8100 Anticorps monoclonaux anti-microsporidies


I.3. Environnement
Dans le but de garantir la qualité permanente des produits développés, l’entreprise Bordier Affinity Products SA est certifiée ISO 13485 [6] depuis 2016. BAP est une petite entreprise positionnée sur un marché de niche c’est-à-dire spécialisé sur des maladies négligées, ce qui explique que seuls 2 concurrents au monde développent de tels produits. Ces produits sont majoritairement des tests basés sur la technologie ELISA pour détecter des anticorps dans le sérum humain.
De l’antigène (protéine extraite de parasite) est fixé au fond de puits en plastique (voir figure 3). Les contrôles et les échantillons de patients sont déposés dans des puits distincts. Un lavage permet d'éliminer les anticorps non spécifiques. La présence d'anticorps spécifiques vis-à-vis des antigènes est détectée avec un conjugué protéine A - phosphatase alcaline. La protéine A du conjugué permet en effet de fixer les immunoglobulines. La phosphatase alcaline est une enzyme ayant la capacité de transformer un substrat incolore en un produit de dégradation jaune. Un deuxième lavage permet d'éliminer le conjugué non fixé aux anticorps. Le substrat (composant incolore) est déposé dans les puits. La réaction est enfin bloquée par un tampon qui inhibe l’enzyme du conjugué. L'intensité de coloration du substrat après dégradation par l’enzyme du conjugué, mesurée avec un lecteur de microplaque ou un automate ELISA (spectrophotomètre), permet la quantification des anticorps spécifiques.

Les principaux atouts de la société BAP sont :
- Le nombre personne réduit qui rend très efficace le suivie des clients et la réactivité par rapport à la production (facile de programmer des productions en fonction des commandes)
- Le temps de réponses aux clients est très court
- Les deux employés sont formés à toutes les tâches de l’entreprise (interchangeabilité)
- La personne qui traite les requêtes et les réclamations des clients est la même qui conçoit, fabrique, réalise le contrôle qualité… (donc les réponses fournies sont fiables)
- Le positionnement sur un marché niche, avec seulement deux concurrents identifiés.
Comme toute entreprise, BAP n’a pas que des atouts, elle a aussi des faiblesses qui sont :
- Localisation en suisse induisant des charges importantes ce qui entraine des produits couteux et des problèmes d’exportation vers le marché européen.
- Pas de service de marketing ou de représentant commercial pour aller rencontrer les clients et leur parler des produits développés ou prospecter des nouveaux produits…
I.4. Projet en cours de développement
Actuellement la société est en train de développer un 15ième produit (projet ASCARIS) et un contrat de sous-traitance est en cours d’étude avec une entreprise agroalimentaire qui veut se lancer dans le domaine médicale (conception, développement et commercialisation d’un kit de diagnostic du COVID 19 par PCR). Comme cette entreprise n’est pas encore certifié ISO 13485, ils souhaitent sous-traiter la production à Bordier.
II. Présentation et analyse des missions réalisées
II.1. Contexte et enjeux liés aux missions de stage
L'entreprise Bordier Affinity Products SA doit se conformer aux exigences de sécurité et performances énoncées dans le nouveau règlement européen d’ici Mai 2022, s’il veut conserver son marquage CE et continuer de commercialiser ses produits sur le marché européen [2]. Sur le plan international, plusieurs pays hors UE partagent de nombreuses exigences avec le règlement et requiert le marquage CE pour autoriser la commercialisation. BAP commercialise ses 14 produits (tous marqués CE selon la directive IVDD) dans environ 29 pays dans le monde (UE, Suisse, Iran, Corée, Australie, USA, Chili, Israël...).
Selon la directive [1], 80% des produits (classe autres tests) ne nécessitaient qu’une auto-déclaration de conformité pour l’obtention du marquage CE et seuls 20% des produits (liste A et liste B) nécessitaient un organisme notifié pour évaluer la conformité des produits. Dans le nouveau règlement IVDR [2], la classe A qui nécessite une auto-déclaration de conformité, regroupe de moins de 20% des produits et les autres dispositifs se répartissent entre les classes B, C et D, nécessite l’intervention d’un organisme notifié pour évaluer la conformité des produits développés. BAP se retrouve dans le cas du changement de classification.
Il y a moins d’ON dans le secteur des DMDIV par rapport au secteur des DM, notamment une vingtaine selon la directive IVDD contre une cinquantaine selon la directive 93/42/CEE enregistrés dans NANDO. Seulement 11 membres de teamNB ont manifesté l’intention de soumettre leur candidature pour le nouveau règlement 2017/746. Actuellement 16 ON sont déjà notifiés [6] selon le règlement 2017/745 contre seulement 4 ON déjà notifiés [7] selon le règlement IVDR. En somme, quatre fois plus de dispositifs nécessiteront l’évaluation de la conformité avec deux fois moins d’organismes notifiés et plusieurs exigences nouvelles et renforcées ainsi que des études cliniques à revoir.
II.2. Problématique et objectifs du stage
Ce travail a pour but de tenter de résoudre la problématique suivante : Quelle démarche entreprendre pour accompagner l’entreprise BAP dans sa phase de transition vers le nouveau règlement 2017/746 ? Pour répondre à cela, il va falloir mettre en place une démarche opérationnelle à suivre afin d’élaborer la documentation technique des produits de l’entreprise BAP conformément à ce nouveau règlement.
II.3. Moyens et méthodes mis en œuvre
II.3.1. Démarche opérationnelle vers la conformité réglementaire
Afin d’agir de façon efficace et efficiente dans la hiérarchisation et l’exécution des tâches pendant cette période de stage, une démarche d’amélioration continue dite opérationnelle a été élaborée. Il s’agit en effet du cycle CAPD (voir Figure 4) qui consiste à :
- Check : Vérifier
Dans un premier temps, il est question d’analyser le système de management de la qualité déjà existant dans l’entreprise BAP ainsi que le niveau de conformité aux exigences du règlement IVDR, afin d’identifier les axes d’amélioration pour mettre à jour le système de BAP selon la norme ISO 13485 [8] et de mettre en œuvre des plans d’action de mise en conformité au nouveau règlement IVDR. Cette étape permet de comprendre le fonctionnement des différents processus de l’entreprise et les interactions entre ces processus.
- Act : Agir
Suite à cette analyse, les procédures, processus et enregistrements nécessitant des améliorations et des mises à jour sont donc identifiés ainsi que les chapitres et annexes du règlement IVDR à prendre en compte dans l’élaboration des documentations techniques. Il s’agit notamment des procédures de veilles de règlementaires, procédures d’analyses d’écarts règlementaires, procédures de preuves de conformités, procédures liste des normes, règlements et guides applicables aux produits Bordier, procédures de gestion des risques, procédures d’aptitudes à l’utilisation et les notices. Parmi ces procédures, certaines existaient déjà dans l’entreprise mais nécessitaient des mises à jour et d’autres étaient inexistantes et nécessitaient d’être créées.
- Plan : Planifier
Un plan des tâches à réaliser a été élaboré afin de répartir les activités sur toute la durée du stage. Des plans d’action ont été élaborés pour la mise en conformité à la norme ISO 14971 : 2019 et aux Annexes 1, 2 et 3 de l’IVDR.
- Do : Faire
Lors de cette phase, il est question de réaliser toutes les activités qui ont été planifiées pour la mise à jour du système de management de qualité selon la norme ISO 13485 [8] et de l’élaboration de la documentation technique selon le règlement IVDR [2].

Le résultat du cycle CAPD est présenté sous forme de cartographie en annexe document (voir annexe I.1). Cela a permis d’avoir une vue globale afin de planifier les tâches à réaliser pendant le stage.
II.3.2. Aspects réglementaires
II.3.2.1. Time Lines de transition
Les échéances de mise en application des règlements européens se rapprochent. Malgrés la crise sanitaire due au COVID 19, la date prévue initialement pour l’application du règlement relatif aux dispositifs médicaux de diagnostic in vitro n’a pas été impactée (voir figure 5).
Passé cette date, il ne sera plus possible aux fabricants de demander le marquage CE selon la directive, mais uniquement selon les règlements. A partir de 2024 aucun produit marqué CE selon la directive ne devra encore être présent sur le marché. Inspiré des articles 110, 112 et 113 de règlement IVDR, La figure ci-dessus donne toutes les dates importantes à retenir pour la transition de la directive IVDD au nouveau Règlement IVDR.

II.3.2.2. Stratégie réglementaire à mettre en place
Le règlement mentionne à l’article 11 que, les fabricants établis hors UE et qui souhaitent commercialiser dans l’UE, doivent désigner un mandataire unique dans l’UE pour les représenter. La suisse ne fait pas partir de l’UE mais il existe des accords de coopération bilatéraux entre l’UE et la suisse qui permettent de se passer des services d’un mandataire [9]. Ce qui fait que BAP commercialise dans l’UE sans avoir besoin d’un mandataire.
Pour commencer, il faut définir l'utilisation prévue du dispositif, car c’est en fonction de cette utilisation, que sera déterminé la classe de risque du dispositif ainsi que la stratégie à suivre pour démontrer sa conformité. Ces preuves de conformité (conception du dispositif, gestion des risques, validation des procédés spéciaux, évaluation biologique et clinique, évaluation des performances, suivi post production....) permettent d’obtenir le marquage CE (figure 6).
Pour continuer à avoir accès au marché européen et commercialiser ses produits en toute liberté même après le 26 mai 2022 (date d’application du règlement), BAP devra absolument avoir obtenu le marquage CE. BAP devra donc suivre et respecter les étapes suivantes :

a) Les produits développés sont-ils des dispositifs médicaux de diagnostic in vitro ?
Il faut s’assurer que les produits répondent à la définition de dispositif médical de diagnostic in vitro visée à l’article 2 section 2 du règlement IVDR [2]. Les produits BAP sont des dispositifs médicaux de diagnostic in vitro car ce sont des trousses utilisées seules et destinées par BAP à être utilisées in vitro dans l'examen d'échantillons provenant du corps humain, dans le but de fournir des informations concernant un état pathologique.
b) La classe de risque de ces dispositifs
Tous les dispositifs de l’entreprise sont de classe C d’après la règle 3 section C de l’annexe VIII du règlement 2017/746 [2] car les dispositifs développés par l’entreprise BAP ont pour but de détecter la présence d'agents infectieux.
c) Exigences générales applicables
Pour démontrer la conformité aux exigences générales en matière de sécurité et de performances applicables aux produits BAP, la nouvelle procédure d’analyse d’écart réglementaire développée pendant ce stage a été utilisée (voir annexe II.5). Ce qui a rendu l’analyse des différences et équivalences entre les Exigences essentielles (EE) de l’annexe I de l’IVDD [1] et les exigences générales en matière de sécurité et de performances (EGSP) de l’annexe I de l’IVDR [2] plus aisée, avec à l’appui des plans d’action pour la mise en conformité des produits BAP aux nouvelles exigences apportées par l’annexe I de l’IVDR.
d) Evaluation clinique
Les évaluations cliniques des produits BAP ont été réalisées, selon l’IVDD et la norme ISO 13612 [10] (voir Annexe II.6). Pendant la dernière veille réglementaire effectuée, une nouvelle norme sur l’étude des performances clinique a été décelée, il s’agit de la norme IS0 20916 [11]. Une amélioration continue a donc été ouverte, pour vérifier que les évaluations cliniques existantes sont conformes aux exigences du règlement IVDR.
BAP devra réaliser une évaluation des performances selon les exigences de l'article 56 et de l'annexe XIII (SPAC) dans le but de respecter les exigences de l’article 10 du règlement IVDR.
Les changements entre les exigences pour l’évaluation clinique IVDD et IVDR, implique juste de mettre à jour annuellement les rapports d’évaluation avec les données de surveillance après commercialisation (SPAC). Et ce rapport actualisé devra être mis à disposition de l’ON.
Le fabricant élabore son plan d’évaluation en impliquant des participants (investigateur, laborantins) et une fois les essais réalisés, ces derniers rédigent le rapport avec les résultats analysés. Et c’est sur cette base que sera élaborée la déclaration de performance et la déclaration de conformité.
e) Identification et enregistrements sur EUDAMED
BAP devra se conformer aux exigences concernant le système IUD (Identification Unique des Dispositifs) énoncées à l'article 24 ainsi qu’aux exigences liées à l'enregistrement énoncées aux articles 26 et 28. EUDAMED est une base de données publique où les opérateurs économiques devront s’enregistrer ainsi que leurs produits. L’IUD devra être mis en place et enregistré dans la base de données EUDAMED pour identifier chaque lot produit. Une amélioration continue a été ouverte, pour se conformer à ces exigences.
Les produits BAP ne disposent que d’un seul conditionnement externe, ce qui va simplifier la mise en place de cette IUD. BAP possède déjà des codes-barres sur ses produits mais sous un format simple (GTIN), donc il n’aura plus qu’à demander à son fournisseur de code barre, de lui générer les IUD en fonction des numéros de lot et dates (péremption et fabrication).
f) Documentation technique et Système de management de la qualité
1. Documentation technique :
L’entreprise BAP devra élaborer et tenir à jour une documentation technique relative à ses différents produits, dans le but de respecter les exigences de l’article 10 du règlement IVDR [2].
Avant d’entamer l’élaboration de la documentation technique, l’entreprise doit d’abord trouver un organisme notifié selon le nouveau règlement IVDR puis signer avec ce dernier, un devis de certification. BAP possède déjà un organisme notifié dans le cadre de l’évaluation de son système qualité (Organisme notifié français : GMED). Bien que ce dernier ne soit pas encore accrédité selon le règlement IVDR, BAP n’envisage pas de le remplacer et va patienter jusqu’à son accréditation.
Pour l’élaboration de cette documentation technique, vu qu’il s’agit d’une première soumission de dossier (demande initiale de marquage CE), BAP devra valider un devis pour une demande initiale auprès du GMED. Après cette étape, il remettra la liste des éléments à fournir pour constituer la documentation (trame des documents attendus).
Le GMED n’étant pas encore accrédité, nous avons suivi les annexes II et III du règlement IVDR et le guide de constitution de dossier technique de BSI [12] (ON qui est déjà accrédité selon le règlement IVDR [2]) pour anticiper l’élaboration de cette documentation. De plus, nous avons utilisé le guide de soumission ToC de L’IMDRF [13] pour la structure de ce dossier. Ce guide fournit un format de dossier universel valable quelle que soit la zone réglementaire où l’on souhaite commercialiser les produits, afin de faciliter l’enregistrement des produits hors UE.
Les documents d’accompagnement sont une partie importante de la documentation technique (étiquettes, notices…). BAP a déjà revu ses documents en 2018, pour se mettre en conformité à la section 20 de l'annexe I de l’IVDR
La Documentation technique n’est pas un document figé, il doit être mis à jour en fonction des résultats des activités de SAC (surveillance après commercialisation) décrit dans l’annexe III du règlement IVDR.
2. Système de Management de la Qualité
Conception et fabrication du produit
BAP devra s’assurer que les phases de conception et de fabrication des produits s’effectuent conformément aux exigences de l’article 10 du règlement IVDR, avant de libérer le produit. La conformité de ces phases est assurée par l’application d’un système de gestion de la qualité selon la norme ISO 13485 : 2016. En effet, le règlement IVDR s’appuie sur des normes harmonisées comme cette norme pour prouver la conformité. Les modifications qui peuvent influencer la conformité du produit doivent prises en compte dans l’entretien du SMQ (modifications de la conception, des normes harmonisées et des caractéristiques du produit).
BAP est certifié ISO 13485 : 2016 depuis 2018 pour une validité qui ira jusqu’en 2021.
Gestion de risque
BAP devra établir, documenter, mettre en œuvre et maintenir un système de gestion des risques conformément à l’article 10, section 3 du règlement IVDR [2]. La conformité de cette phase est assurée par l’application de la norme ISO 14971 : 2012 [14].
Suite à l’analyse d’écart normative de la nouvelle version 2019 de la norme ISO 14971 [12], la mise à jour du système de gestion de risque (analyse, plan, rapport et dossier de gestion de risque) a été planifié conformément aux nouvelles exigences (voir annexe II.7). La norme ISO/TR 24971 [16] nous servira de guide pour l’application de la norme ISO 14971 : 2019.
Une revue annuelle de l’analyse des risques a également été effectuée afin de :
- Mettre à jour les critères et seuils de la matrice d’évaluation
- Identifier de nouveaux dangers et situations dangereuses sur la base de l’analyse des activités de production et de post-production.
- Effectuer une nouvelle cotation des probabilités et des gravités des dommages
- Effectuer une nouvelle évaluation de risque (comparer les scores (probabilité * gravité) de chaque risque par rapport aux critères définis dans la matrice d’évaluation, dans le but de déterminer l’acceptabilité de chaque risque).
- Rechercher de nouvelles mesures de maîtrise de risques pour qu’elles réduisent au mieux les risques résiduels globaux. La figure suivante donne un aperçu du tableau final.
Gestion du rapport bénéfice / risque
Concernant la gestion du rapport bénéfice / risque, la norme expérimentale XP S99-223 [17] a été publié en février 2020. Une amélioration continue a été ouverte pour prendre en compte les exigences de cette norme dans la gestion du rapport B/R de l’analyse de BAP. Pour le moment, la norme ISO 14971 : 2012 [14] est celle utilisée pour la gestion B/R.
g) Evaluation de la conformité
Au vue du changement de classe des 14 produits BAP, l’entreprise devra choisir une méthode d’évaluation de la conformité parmi celles énoncées aux annexes IX, X et XI du règlement IVDR. Les produits de l’entreprise étant de classe C et sur conseil de son ON, BAP a choisi une évaluation de son SMQ par un organisme notifié selon l'annexe IX, chapitres I et III avec évaluation de la documentation technique selon la même annexe chapitre II.
h) Marquage CE et Déclaration de conformité
Une fois la démonstration de la conformité aux exigences applicables établit par BAP conformément à la procédure d'évaluation de la conformité choisie, il devra élaborer une déclaration de conformité UE selon l'article 17 et apposer le marquage de conformité CE selon l'article 18. Par cette déclaration de conformité UE, BAP s’engage à respecter les exigences du règlement IVDR et s’assure de la mise à jour cette déclaration à chaque modification significative sur les produits.
Tous les produits BAP respectant les exigences de la directive IVDD portent le marquage de conformité CE conformément à cette directive. Apres évaluation de la conformité selon le règlement IVDR par le GMED, ce dernier délivrera un certificat de conformité qui donnera le droit à BAP d’apposer le marquage de conformité CE sur les dispositifs. Ce marquage CE devra être accompagné du numéro d'identification du GMED.
i) Surveillance post-marché et réactovigilance
BAP devra appliquer et mettre à jour le système de surveillance après commercialisation selon l'article 78. Cela consiste en la collecte d’information auprès des utilisateurs et l’analyse de celles-ci afin de s’assurer que le produit est sûr et performant. BAP possède déjà une procédure selon le règlement IVDR pour cette surveillance.
En cas de non-conformité ou d’incident grave lié à un dispositif déjà mis sur le marché, BAP a mis en place des mesures correctives nécessaires pour le conformer, le retirer ou le rappeler ainsi qu’informer les autorités compétentes des états membres où est commercialisé le dispositif, l'organisme notifié ayant délivré le certificat et tous les distributeurs de ce dispositif.
II.4. Résultats escomptés et obtenus
Le résumé des taches réalisées pendant ce stage (voir annexe II.3).
II.4.1. Résultats escomptés
- Réaliser l’analyse d’écart règlementaire entre les EE (IVDD) et EGSP (IVDR) et proposer un plan de mise en conformité
- Mise à jour de la liste des normes, guides, règlements applicables aux produits BAP
- Elaborer la documentation technique selon les annexes II et III de l’IVDR
II.4.2. Résultats obtenus
a. Analyse d’écart réglementaire
La procédure d’analyse d’écart réglementaire a été conçue pour identifier de façon claire, les différences et les équivalences entre les exigences de deux versions d’une norme, d’un règlement ou d’un guide (voir annexe II.4). La procédure existante étant non adaptée à une analyse approfondie, une revue a été réalisée du durant le stage.
La procédure en question prend en compte :
- Les exigences anciennes et nouvelles avec un code couleur selon le statut (identique, différent ou nouveau)
- Leurs applicabilités aux produits BAP ou une justification en cas de non-applicabilité
- La référence aux normes ou guides pour répondre aux exigences
- La conformité actuelle des produits Bordier aux exigences
- Les références aux preuves de conformité
- En cas de non-conformité, les actions correctives ou préventives à mettre en place
Cette procédure a été utilisée plusieurs fois durant le stage :
- Analyse d’écart entre les exigences essentielles (EE) annexe I (IVDD) et les exigences générales en matière de sécurité et de performance (EGSP) annexe I (IVDR).
- Analyse d’écart entre les exigences de deux versions de la norme de gestion de risques ISO 14971 : 2012 et ISO 14971 : 2019.
- Analyse des correspondances entre les exigences de l’annexe 1 de l’IVDR et celles des guides N47 et N52 de l’IMDRF.
A la suite de chacune de ses analyses, un rapport d’amélioration continue a été ouvert et un plan d’action de mise en conformité a été élaboré.
b. Liste des normes, guides, règlements applicables
Afin de remplir les objectifs du stage, une mise à jour des normes, guides et règlements applicables aux produits BAP s’est avérée nécessaire. Une revue des normes harmonisées applicables et des guides applicables (IMDRF, MEDDEV, MDCG…) a été réalisée. Une fois la revue de la liste des normes, règlements et guides applicables réalisée, il a été observé qu’il n’existait pas de moyen pour prouver la conformité à ces référentiels.
La procédure de preuve de conformité a donc été créée pour apporter de façon claire, la preuve de conformité aux exigences contenues dans une norme, un règlement ou un guide.
La procédure en question prend en compte :
- Les exigences du référentiel utilisé
- Leurs applicabilités aux produits BAP ou une justification en cas de non-applicabilité
- La conformité actuelle des produits Bordier aux exigences
- Les références aux preuves de conformité
- En cas de non-conformité, les actions correctives ou préventives à mettre en place
Cette procédure a été utilisée et prévoit d’être utilisé :
- Pour apporter la preuve de conformité aux exigences contenues dans la norme ISO 13485 : 2016 concernant les exigences à des fins réglementaires du système de management de la qualité pour les dispositifs médicaux.
- Pour apporter la preuve de conformité aux exigences contenues dans la norme ISO 13641 : 2002 concernant l’élimination ou la réduction du risque d'infection relatif aux réactifs de diagnostic in vitro.
- Pour apporter la preuve de conformité aux exigences contenues dans la norme XP S99-223 concernant la gestion du rapport bénéfice/risque des dispositifs médicaux.
A la suite de chacune de ses analyses, un rapport d’amélioration continue a été ouvert et un plan de mise en conformité élaboré.
c. Élaboration de la documentation technique
Ce document a été conçu pendant le stage pour présenter les preuves de conformité des produits BAP auprès des organismes notifiés et autorités compétentes. C’est un formulaire qui résume toutes les exigences de l’annexe II et III de l’IVDR de façon à être adaptable à tous les produits BAP (voir annexe I.2). Il sert de table des matières pour l’organisme notifié qui réalise la revue de la documentation d’un produit. Il doit donc faire références à toutes les informations ou documents apportant les preuves de conformité d’un produit aux exigences de l’annexe II et III de l’IVDR. Ce formulaire a été réalisé à environ 80% et il devrait être appliqué à un produit d’ici la fin du stage. L’audit interne de BAP est planifié en septembre 2020 par le consultant MEDIDEE Suisse. La moitié de l’audit sera consacré à la revue de ce dossier technique. En fonction des résultats de l’audit, des améliorations pourront être apportées au formulaire et à la documentation pour se préparer à l’audit externe avec le GMED prévu pour décembre 2020.
d. Autres résultats obtenus
Gestion des risques :
La revue annuelle de l’analyse des risques a permis d’identifier de nouveaux dangers à partir des activités de production et de post-production. Lors de cette revue, toutes les situations dangereuses ont été reformulées pour être plus claires et plus compréhensibles. Une nouvelle cotation des probabilités et gravités des dommages a été effectuée, les critères de la matrice d’évaluation ont été redéfinis et une nouvelle évaluation des risques a été effectuée dans le but de déterminer l’acceptabilité de chaque risque. Les mesures de maitrise de risques ont également été revues dans le but de réduire au mieux les risques résiduels globaux et identifier de nouvelles mesures à mettre en place.
Veille réglementaire :
La veille réglementaire permet de se tenir informer des évolutions de toutes les normes, guide et règlements applicables aux produits BAP. La procédure existante étant obsolète, une revue a été réalisée du durant le stage. Deux veilles trimestrielles ont été réalisées durant le stage. Elles ont permis d’identifier des documents intéressants et de déterminer si ces documents nécessitaient un plan de mise en conformité.
Une nouvelle communication de l’UE portant sur la liste des normes harmonisées a été détectée. L’analyse de cette communication a permis de s’assurer qu’il n’y avait pas de nouvelles normes applicables aux produits BAP. La norme expérimentale de la balance bénéfice / risques (XP S99-223) a été détectée et une amélioration continue a donc été ouverte.
II.5. Difficultés rencontrées et solutions envisagées
La nécessité d’un visa pour venir en suisse a retardé la date de début du stage. Et un peu avant l’obtention du visa, la crise sanitaire du COVID 19 a forcé la Suisse à geler toutes demandes de visa. Il a été décidé de démarrer le stage à distance, avec des solutions de travail comme Teamviewer et Skype. Bien que la formation n’ait pas été idéale de cette manière, le travail a pu bien évoluer avec des suivis réguliers. Au bout d’un moment, le travail à distance était devenu trop contraignant et il était clair que la demande visa n’allait pas être accordé, le déplacement vers une ville française frontalière à la suisse (Thonon-les-Bains) a été nécessaire. Cela a permis d’atteindre les objectifs du stage.
La formation nécessaire à la réalisation du travail, impliquait une compréhension du mécanisme d’action biologique des produits. Le manque de formation de base dans ce domaine a nécessité de nombreux efforts.
En cas de difficulté à interpréter une exigence d’un règlement ou d’une norme, des actions proactives auprès de divers connaissances du domaine (réseau LinkedIn, réseau UTC, consultant externe MEDIDEE…) était menés afin d’avoir plus d’éclaircissement. Cette méthode a permis d’obtenir des informations utiles pour l’avancement du travail.
III. Apports du stage
III.1. Compétences et comportements acquises
Ce stage m’a permis de :
- Acquérir des connaissances spécifiques liées au domaine de la biologie médicale.
- Découvrir le monde professionnel d’entreprise suisse
- Développer une capacité d’adaptation aux situations difficiles
- Développer une capacité d’autonomie
- Développer une aisance rédactionnelle (rédaction de documents réglementaires de BAP)
- Maitriser le cycle de vie d’un produit depuis la conception jusqu’à la mise sur le marché
- Acquérir des connaissances dans l’élaboration des documentations techniques
- Maitriser le processus de veilles règlementaires
- Maîtriser le processus de gestion des risques
- Découvrir le système de management de la qualité
III.2. Compétences et comportements à acquérir
Ce qu’il y’a à acquérir pour être plus complet dans le domaine :
- Développer une capacité de manager ou responsable d’équipe et de service
- Développer une capacité à répondre aux audits et à mener des audits
- Gérer les communications avec les ON et AR pour la soumission de dossiers techniques ou rapport de vigilances
- Développer une capacité relationnelle pour animer les équipes et collaborer avec les services
III.3. Liens avec la formation théorique
Les enseignements théoriques reçus pendant la formation ont été utiles pendant ce stage :
- Le cours d’IDCA intitulé Management de la Qualité et des Organisations Biomédicales a apporté les connaissances concernant le système de management de la qualité, ce qui a permis d’établir des plans d’améliorations continues pour l’entreprise.
- Le cours d’IDCB intitulé Ingénierie de projet a permis d’élaborer et mettre en place des solutions pour résoudre les différents problèmes rencontrés pendant le stage.
- Le cours d’IDCC intitulé Communication professionnelle de projet a permis d’être plus efficace dans la rédaction et le rendu des documents demandés (Rapport, cartographie, procédures, processus…), ainsi que dans la rédaction de mon rapport au format WordPress.
- Les cours d’IDCD, IDCL et IDCE intitulé respectivement Projet d'intégration et Dispositifs Médicaux, Affaires Règlementaires etCycle de vie d'un DM ont permis d’arriver dans la société en connaissant la définition et cycle de vie du DM, ainsi que les règlements et normes applicables au DM. Ces connaissances ont permis une prise en main rapide des produits BAP.
- Le cours d’IDCF intitulé Organisation du système de santé, a donné une première connaissance générale sur le système de santé, ce qui a été utile pour comprendre et situer les clients BAP dans ce système. Cela a également permis de comprendre l’intérêt des produits BAP pour la santé des patients.
- Le cours d’IDCH intitulé Traitements et soins, n’a pas vraiment servi pendant ce stage caril concernait essentiellement les activités du bloc-opératoire, de l'anesthésie-réanimation et la dialyse alors que le stage concernait les activités du laboratoire d’analyse médical.
- Le cours d’IDCK intitulé Audit et évaluation des organisations : normes et processus, a permis de comprendre les notions clés et le déroulement d’un d’audit. Il a également permis de préparer et anticiper les deux audits à venir à savoir : l’audit interne qui sera effectué en début septembre 2020 par le consultant (MEDIDEE) et l’audit externe qui sera effectué par le GMED en décembre 2020.
A l’issue de ce stage, mon projet
professionnel a été revu, j’ai développé des compétences à la fois d’Affaires
Règlementaires et d’Assurance Qualité grâce à la taille de l’entreprise BAP.
Donc j’envisage exercer dans une entreprise à taille humaine comme BAP où je
pourrai occuper le poste d’Ingénieur Assurance Qualité et Affaires
Règlementaires et contribuer dans l’évolution et la croissance de l’entreprise.
Conclusion
Le monde des dispositifs médicaux évolue très rapidement ainsi que les référentiels normatifs et réglementaires qui s’y appliquent. Pour s’assurer de mettre sur le marché des dispositifs toujours sûrs et performants pour les patients et utilisateurs, BAP met en place, maintien et améliore des mesures de maîtrise des risques durant tout le cycle de vie du produit. L’arrivée du nouveau règlement entraine un besoin de revue des dossiers techniques, d’où le sujet de ce stage.
Malgré toutes difficultés rencontrées, les objectifs de stage ont été remplis. Le formulaire est rédigé à 80% et la documentation technique est en cours d’élaboration pour un produit. Ce travail va permettre de présenter des dossiers de documentations techniques de qualité aux organismes notifiés en vue d’obtenir le marquage CE selon le règlement IVDR.
Des tâches annexes ont été réalisées pendant le stage, comme l’analyse d’écart réglementaire IVDD - IVDR et le plan d’action de mise en conformité, l’élaboration d’une procédure de preuve de conformité, la revue de la procédure de veille réglementaire et de la liste des normes, guides et règlements. Toutes ces tâches ont permis l’amélioration du système de management de la qualité dont la conformité est également nécessaire pour l’obtention du marquage CE.
Ce stage a été
bénéfique à la fois pour l’entreprise BAP et pour le stagiaire car il
a permis de découvrir les métiers du laboratoire d’analyse médical, de gagner
en expérience et de développer les compétences, d’apprendre auprès de
professionnels qualifiés et de se constituer un réseau professionnel. Ce stage
m’a également permis d’apprendre à travailler en équipe et de mettre en
pratique les cours théorique reçus pendant ma formation ainsi que d’enrichir mon CV.
Bibliographie
[6] « Organisme notifiés selon le règlement MDR 2017/745 », consulté le 09.09.2020.
[7] « Organisme notifiés selon le règlement IVDR 2017/746 », consulté le 09.09.2020
[9] « Accord de collaboration bilatéraux entre la suisse et UE », consulté le 09.09.2020
[12] BSI, « Guide de bonne pratique pour la soumission de DT ». mai 2020, consulté le 09.09.2020
Annexes I
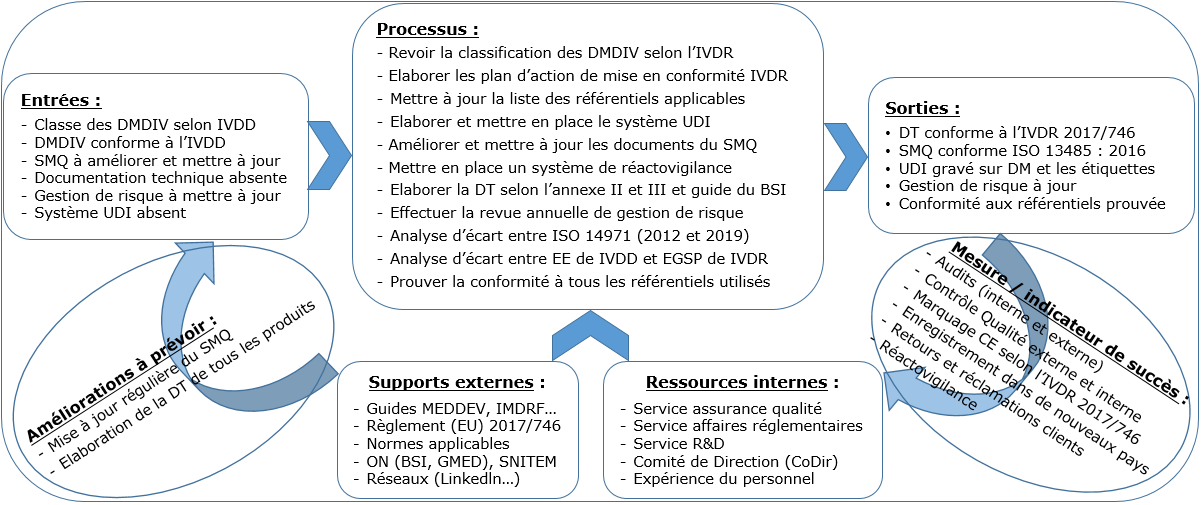
Annexe I.1 : Cartographie de conformité au règlement IVDR 2017/746 (Source : Auteur)

Annexe I.2 : Sommaire de la documentation technique élaborée (Source : Auteur)
