
IDS064 - FABRICANTS : Maîtrise réglementaire d'une innovation en échographie
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Citation
A rappeler pour tout usage : Olivier Donald NYAGAM KEMAJOU « FABRICANTS : Maîtrise réglementaire d'une innovation en échographie », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositif Médical et Affaires Réglementaires (DMAR), Mémoire de stage, Juillet 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids064/
Contact
Résumé
Assurer la sécurité du patient est une démarche qui vise à éviter à un usager toute atteinte évitable liée aux soins qui lui sont administrés. C’est dans ce contexte que s’inscrit l’innovation en Echographie qui fait l’objet de ce mémoire : les technologies « Point of Care UltraSound (POCUS) » qui sont sollicitées aux chevets des patients à des fins de dépistage ou de diagnostic. L’innovation, la recherche et le développement ne suffisent pas comme stratégie pour concevoir un produit sûr.
Pour que cette démarche soit efficace à court et à long terme, avec un rapport bénéfice/risque des soins optimal pour le patient, elle doit être encadrée par les aspects réglementaires qui viennent donner de l’éthique, de la valeur et du sens à l’innovation. Raison pour lesquelles des normes, les textes réglementaires, et des guides de bonnes pratiques existent, pour maîtriser les risques lors la conception de tout dispositif électro médical, améliorer leurs performances essentielles pendant leur durée de vie prévue, et garantir un niveau élevé de sécurité des patients.
Mots clés : Sécurité du patient, Echographie, POCUS, Risque, Dispositif électro médical, Performances essentielles, Durée de vie prévue.
Abstract
Ensuring patient safety is an approach that aims to prevent a patient from suffering any avoidable harm related to the care they receive. It is in this context that the ultrasound innovation that is the subject of this report is being developed : Point of Care UltraSound (POCUS) technologies that are used at the bedside for screening or diagnostic purposes. Innovation, research and development are not enough as a strategy to design a safe product.
For this approach to be effective in the short and long term, with an optimal risk/benefit ratio for patient care, it must be framed by regulatory aspects that give ethics, value and meaning to innovation. For this reason, standards, regulatory texts and good practice guides exist to control risks during the design of all electro-medical devices, to improve their essential performance during their expected lifetime, and to guarantee a high level of patient safety.
Keywords : Patient safety, Ultrasound, POCUS, Risk, Electro-medical device, Essential performance, Expected lifetime.
Téléchargements


Mémoire complet :
FABRICANTS : Maîtrise réglementaire d'une innovation en échographie
Remerciements
La réalisation de ce mémoire a été possible grâce au concours de plusieurs personnes à qui je témoigne toute ma reconnaissance.
Merci, à DIEU d’avoir donné la motivation, le courage, et la santé durant toutes ces années d’études, c'est grâce à lui que ce travail a pu être réalisé ;
Sincères remerciements aux responsables de master Mme. Isabelle CLAUDE, Mme. Françoise MERESSE, et M. Jean Mathieu PROT pour avoir permis d’effectuer ce stage, merci pour leur disponibilité, leurs conseils, et leur soutien lors des démarches administratives ;
Merci à M. Gilbert FARGES, pour l’encadrement durant ce stage, il a été une source d’inspiration pour moi durant toute ma formation. Ses conseils judicieux ont contribué à alimenter ma réflexion ;
Merci à M. David SAVERY, pour l’accueil chaleureux au sein de l’entreprise, ses précieux conseils, sa clairvoyance, et ses compétences multidisciplinaires m’ont été d’une aide inestimable dans toutes mes activités, et à la réalisation de ce mémoire ;
Merci à M. Antoine DURAND et toute l’équipe « Business Unit Active Probe », l’agréable interaction avec chaque personne a aidé à réaliser mes missions, leur bienveillance m’a permis de m’intégrer rapidement au sein de l’équipe et de l’entreprise ;
Merci à tous les enseignants qui ont eu l’aimable attention de partager leurs connaissances nécessaires à résoudre les problèmes dans les tâches qui m’étaient confiées ;
Merci à toute la Famille KEMAJOU, SA’A NYAGAM, NOUNGA et TCHANKOUO, pour leur soutien inconditionnel et leur amour à mon égard ;
Je remercie également tous mes ami(e)s, camarades de la promotion et aîné(e)s pour leur encouragement, leur aide, et leur dévouement tout au long de cette formation.
Introduction
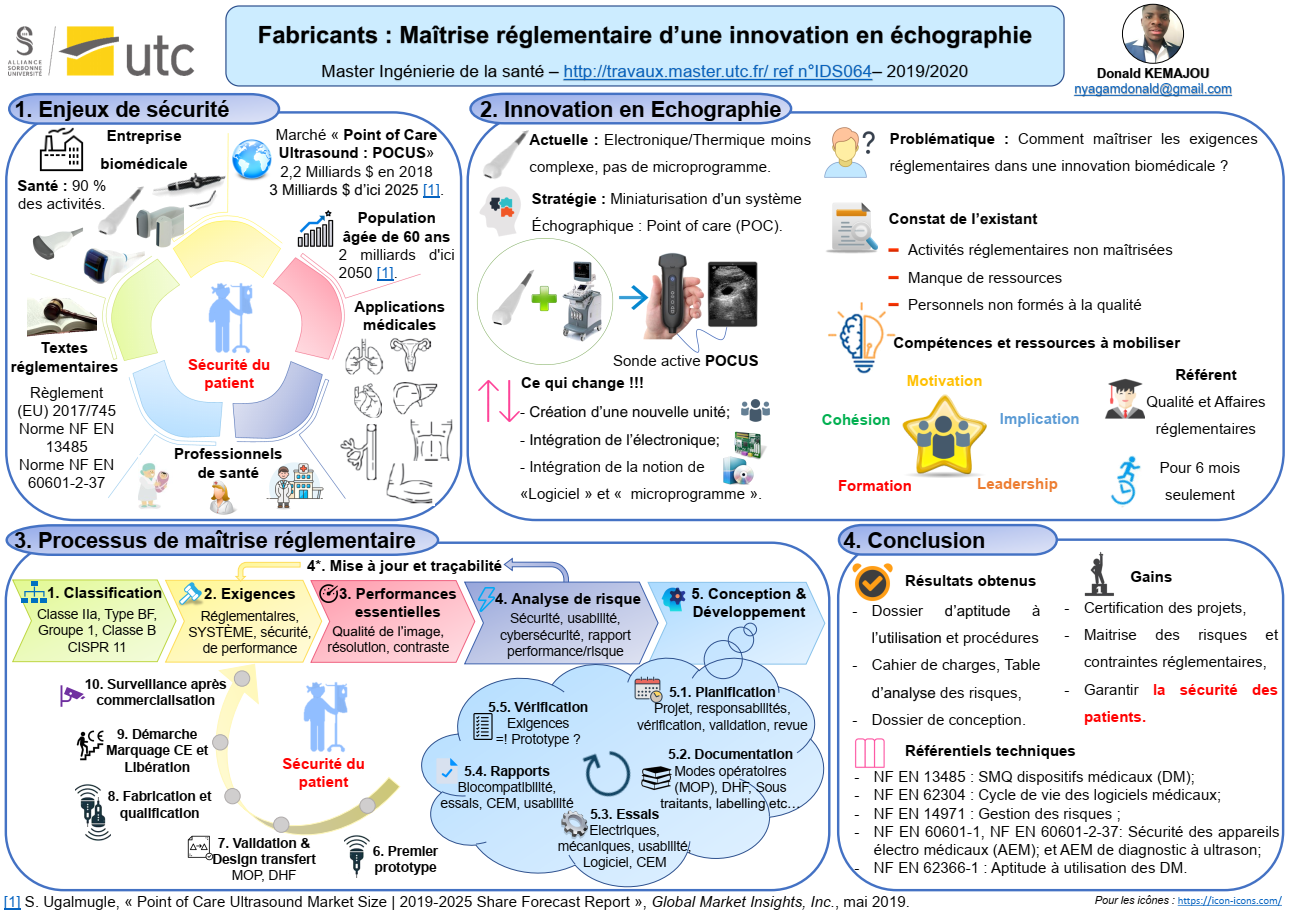
J’ai eu l’opportunité de réaliser mon stage de fin d’études d’une durée de six mois dans une entreprise biomédicale, où j’ai occupé le poste de référent « Assistant Qualité/Affaires réglementaires des dispositifs médicaux ».
Dans le monde de la santé, toujours aussi très évolutif et innovant, le marché mondial des dispositifs électro médicaux de diagnostic ultrasonore connait actuellement des avancées remarquables, à l’instar de la technologie « Point of Care Ultra Sound (POCUS) », qui se définit comme une échographie de chevet, permise par la miniaturisation des systèmes d’imagerie échographique. Intégrée dans les domaines de la pratique clinique au début des années 1990, cette technologie vient révolutionner la pratique de la médecine, en influençant la manière dont les soins sont administrés [1]. Le marché mondial de cette technologie est estimé en 2018 à 2,2 milliards de dollars US, et pourra atteindre la valeur de 3 milliards de dollars US d’ici 2025. Avec la population âgée de 60 ans, qui devrait dépasser 2 milliards d’ici 2050, ces appareils ultraportables sont de plus en plus sollicités aux chevets des patients à des fins de diagnostic, de dépistage, ou de suivi de thérapie [2].
Qui dit évolutions technologiques, dit également aspects et évolutions réglementaires à prendre en compte dans la conception et le développement des dispositifs médicaux afin de garantir la sécurité des patients. C’est ainsi que l’entreprise, dans le but d’augmenter son chiffre d’affaire, envisage d’adopter cette technologie et d’aborder ce marché. Raison pour laquelle, j’interviens dans le support à la certification de ses futurs produits. Afin de retracer les activités effectuées tout au long de ce stage, un mémoire est élaboré et porte dans un premier temps sur le projet de développement du nouveau système d’échographie, les enjeux et les objectifs réglementaires à atteindre, pour assurer la qualité des nouveaux produits et garantir un niveau élevé de sécurité du patient. Puis, sont établis la démarche opérationnelle utilisée, et les aspects réglementaires pris en compte dans le processus de développement et la conception du nouveau produit. Enfin, l’ensemble des difficultés rencontrées, les perspectives d’avenir, ainsi que les risques et opportunités liés au projet sont abordés.
I. Projet de développement du nouveau système échographique
A. Contexte et enjeux réglementaires
Les systèmes d’imagerie sont en pleine expansion et deviennent de plus en plus miniaturisés. C’est le cas des technologies « POCUS » qui arrivent sur le marché des dispositifs médicaux, avec l’idée de brancher les sondes non plus à des gros systèmes d’imagerie encombrants et lourds (~150 kg), mais plutôt à des tablettes, ordinateurs, ou appareils dédiés légers et compacts.
Ces technologies et produits se développent exponentiellement, rendant de plus en plus facile les procédures et examens thérapeutiques et de diagnostic, grâce à son ultra portabilité, son prix raisonnable, et sa qualité de l’image qui devient meilleure. Dans le but de se développer, la société décide de se lancer dans la conception et la fabrication des sondes actives dites « POCUS ».
Actuellement, les sondes dites classiques (voir Figure 1) commercialisées par la société sont destinées à être utilisées sur des systèmes d’imagerie des clients, ce qui rend leur conception plus simple et moins complexe (pas de pilotage de l’émission/réception des éléments de transducteurs, pas d’échantillonnage ni de filtrage des signaux, pas de formation de faisceaux dans la sonde, et gestion de la thermique plus légère) qu’un système POCUS.

Figure 1 : Sondes classiques de l’entreprise (Source : Auteur d’après [7])
Les nouvelles sondes « POCUS »(voir Figure 2) viennent avec des fonctionnalités supplémentaires, mentionnées dans le Tableau 1 :
Tableau 1 : Enjeux réglementaires Technologie POCUS (Source : Auteur)
| Fonctionnalités supplémentaires | Contraintes et Enjeux réglementaires |
| L’intégration des modules de contrôle des éléments du transducteurs (FPGA, AFE, TX), l’échantillonnage des signaux et la formation des faisceaux (Beamformer) dans la sonde | Management de la thermique à l’intérieur et à la surface de la sonde => Essais et tests de sécurité suivant la NF EN 60601-1 [8], la NF EN 60601-2-37 [9], la NF EN 60601-1-2 [10]. |
| Modalité d’interface sur la sonde (bouton, LED, batterie), la connectivité (USB ou Wi-Fi) | Aptitude à l’aptitude à l’utilisation => La NF EN 62366-1 [11] & guides FDA [12]. |
| Driver pour la communication avec le système hôte, microprogramme VDHL (FPGA) pour le contrôle des composants électroniques, logiciel de contrôle sonde-système hôte | Procédure de conception et développement des logiciels, Vérification et Validation des éléments et unités logiciels, Intégration des logiciels => NF EN 62304 [13], Guidance Cybersécurité FDA [14] & ANSM [15], MDCG [16]. |

Figure 2 : Sondes actives POCUS de l’entreprise (Source : Auteur d’après [7])
Qui dit nouveau produit, dit nouvelles ressources humaines à mobiliser. C’est dans cette logique qu’a été créée la nouvelle unité « BUAP » dédiée aux activités de conception et de développement des sondes actives POCUS. Elle est constituée d’une douzaine d’ingénieurs (acousticiens, électroniciens, informaticiens, mécaniciens, etc…) travaillant dans un environnement de type « Start-up ».
D’après l’étude du marché, plusieurs entreprises commercialisent déjà les sondes ultrasonores munies de cette technologies « POCUS ». Sans être exhaustive, il y a les produits représentés dans la Figure 3.

Figure 3 : Benchmarking des sondes POCUS (Source : Auteur)
Comme applications de ces technologies POCUS, il y a les spécialités du Tableau 2[17]:
Tableau 2 : Spécialités et applications médicales (Source : Auteur d’après [17])
| Spécialités | Applications médicales des ultrasons |
| Anesthésie | La surveillance peropératoire |
| Cardiologie | Echocardiographie intracardiaque |
| Soins intensifs | Echocardiographie pulmonaire ciblée |
| Dermatologie | Diagnostic des lésions et des tumeurs cutanées |
| Urgence | *FAST, diagnostic ciblé des situations d'urgence |
| Chirurgie endocrine et endocrinologie | Diagnostic de la thyroïde et de la parathyroïde |
| Chirurgie générale | Échographie du sein, diagnostic peropératoire |
| Gynécologie | Diagnostic du col de l'utérus, de l'utérus et des annexes |
| Néonatalogie | Diagnostic de la grossesse, détection des anomalies du fœtus |
| Néphrologie | Diagnostic crânien et pulmonaire |
| Neurologie | Accès vasculaire pour la dialyse Doppler transcrânien, diagnostic des nerfs périphériques |
| Ophtalmologie | Diagnostic de la cornée et de la rétine |
| Chirurgie orthopédique | Applications musculosquelettiques |
| ORL | Diagnostic des masses de la thyroïde, de la parathyroïde et du cou |
| Pédiatrie | Diagnostic de la vessie |
| Médecine pulmonaire | Diagnostic pulmonaire trans-thoracique, diagnostic endo-bronchique |
| Radiologie interventionnelle et radiologie | L’échographie est effectuée sur le patient accompagnée d’une interprétation |
| Rhumatologie | Surveillance de la synovite |
| Chirurgie traumatologique | *FAST |
| Urologie | Diagnostic des fonctions rénale, vésicale et prostatique |
| Chirurgie vasculaire | Diagnostic de la carotide, de l'artère et de la veine |
*FAST dénote un diagnostic ciblé avec l’échographie pour les traumatismes (Focused Assessment With Sonography in Trauma)
B. Problématique et objectifs
Deux projets de développement forment le quotidien de la nouvelle unité « BUAP » à savoir :
- DKPOC (Development Kit Point Of Care)
C’est le projet qui fait l’objet de ce travail, et qui se rapproche de la description des technologies POCUS ci-dessus. L’objectif de ce projet est de développer un kit de démonstration qui consistera à fournir à un client potentiel (systémier), une sonde « intelligente » , des accessoires contractualisés (boitier d’alimentation et câble USB), et un pilote pour la communication de la sonde avec le système hôte du client.
Pour garder ce statut de « Fournisseur de dispositifs médicaux », le logiciel de contrôle de la sonde, d’affichage des images échographiques et de gestion des données patient n’est pas fourni au client. C’est plutôt au client de développer ce logiciel.
Toutefois un logiciel démonstrateur est produit par l’unité BUAP pour démontrer la performance et valider la fonctionnalité de la sonde. Cette stratégie a aussi pour avantage de réduire les contraintes réglementaires relatives aux dispositifs médicaux telles que la démarche d’obtention du marquage CE, qui est plutôt sous la responsabilité du systémier (l’entreprise cliente de la société qui conçoit et commercialise le dispositif médical final).
- MDK432 (Matrix Development Kit 432 channels)
C’est une sonde constituée des matrices bidimensionnelles de transduction, qui concerne l’imagerie 3D ou 4D temps réel. Elle se différencie des sondes classiques à 128 éléments par sa matrice, qui offre la possibilité d’avoir 3000 à 60 000 éléments. Actuellement ce projet est encore en maturation technologique.
Avec un système de management de la qualité (SMQ) existant certifié ISO 13485 [6], la nouvelle unité se doit de développer ses futurs produits suivant les activités mises en place dans la norme NF EN ISO 13485 [6] et le Règlement (EU) 2017/745 relatif aux dispositifs médicaux et dispositifs médicaux implantables [18]. C’est sur cette base référentielle que les missions suivantes m’ont été confiées :
- Vérifier la documentation de conception et produit ;
- Assurer le support pour la certification des projets ;
- Rédiger les plans qualité ;
- Sensibiliser de l’équipe actuelle aux exigences qualité.
II. Processus de maîtrise réglementaire
A. Démarche opérationnelle
Afin de bien agir dans l’exécution et dans la hiérarchisation des tâches, une démarche opérationnelle est élaborée sur le principe de l’amélioration continue. C’est la démarche CAPD qui consiste dans un premier temps à :
- Check : Vérifier
Cette étape permet d’analyser le système de management de la qualité (SMQ) existant de la société, afin de savoir comment sont rédigés et documentés les différents documents qualité de l’entreprise par rapport à la norme NF EN ISO 13485 [6], mais aussi de comprendre les différents processus organisationnels de l’entreprise. Une attention particulière est portée à la procédure de conception et de développement des dispositifs de la société, et aux différentes procédures annexes (achats, sélection des fournisseurs, gestion des risques, etc…).
- Act : Agir
A l’issue de cette analyse du SMQ, les procédures qui sont impactées par la conception du nouveau produit sont identifiées. Il s’agit notamment de la procédure de conception, de la gestion des risques, et la procédure de développement des logiciels, qui sont déjà existantes et qui sont cependant mises à jour. En raison des aspects réglementaires cités ci-dessus à prendre en compte, plusieurs autres documents/procédures sont créés. Il s’agit notamment de la procédure de cybersécurité, et la procédure d’aptitude à utilisation.
Une interaction avec les ingénieurs logiciels, électroniciens, mécaniciens et acousticiens de l’unité est indispensable pendant cette phase, afin de comprendre l’activité de chaque membre de l’équipe, et d’évaluer l’impact de son activité sur le prototype ou sur le produit final.
Une sélection des sociétés de conseil en affaires réglementaires est également effectuée pendant cette phase, suivant les critères bien définis à savoir :
- Compétences dans le domaine, qualifications, expériences, crédibilité ;
- Contacts références , portfolio ;
- La compréhension des besoins et objectifs ;
- Le nombre de jours d’accompagnement et le coût de l’offre ;
- La capacité à communiquer, à s’adapter aux ressources internes de la société.
- Plan : Planifier
Après identification des activités à réaliser, le but de cette phase est de planifier chaque activité dans le temps, sur toute la durée prévue du stage. Une sensibilisation de la nouvelle équipe à la qualité est planifiée, ainsi qu’une formation sur aux référentiels IEC 62304 [13] sur le cycle de vie des logiciel, la NF EN 60601-1 [8] et la NF EN 60601-2-37 [9] sur la sécurité et les performances essentielles des appareils électro médicaux (AEM), et de AEM de diagnostic à ultrason.
- Do : Faire
Cette étape recense toutes les activités qui sont réalisées pour le développement du produit. Il s’agit des activités de la Figure 4.

Figure 4 : Activités réalisées (Source : Auteur)
B. Aspects réglementaires pris en compte
i. Classification du dispositif
Un appareil électro médical est un « appareil électrique qui possède une partie appliquée ou qui transfère de l'énergie vers le patient ou à partir de celui-ci ou qui détecte un tel transfert d'énergie vers le patient ou à partir de celui-ci et qui est :
- Équipé au plus d'un moyen de raccordement à un réseau d’alimentation donné ; et
- Destiné par son fabricant à être utilisé :
- Pour le diagnostic, le traitement ou la surveillance d'un patient ou
- Pour la compensation ou l'atténuation d'une maladie, d'une blessure ou d'une incapacité » [8].
Le produit faisant l’objet de ce travail répond à cette définition car il transfère de l’énergie mécanique (ultrason) au patient via la tête de sonde (partie appliquée), il est équipé d’un moyen de raccordement à un réseau d’alimentation (de manière indirecte et temporaire) et est utilisé à des fins de diagnostic d’un patient.
La classification du dispositif est de la responsabilité du fabricant. Cette classification se fait en fonction du niveau de risque lié à son utilisation. D’après le Règlement (EU) 2017/745 et selon la règle n°10, le dispositif « sonde ultrasonore active » faisant l’objet de ce travail est de classe IIa avec un niveau de risque potentiel modéré car « il est destiné à fournir de l’énergie qui est absorbée par le corps humain à des fins de diagnostic » [18].
Dans son ensemble, le dispositif entre en contact non invasif avec le patient, et ses parties appliquées (tête de sonde par exemple) doivent également être classées afin de garantir une utilisation sûre du dispositif. Cependant, selon la norme particulière NF EN 60601-2-37, les parties appliquées du dispositif sont classées de type BF (Body Flottant) pour assurer un niveau moyen de protection électrique, et protéger le patient dans le cas où plusieurs équipements non invasifs sont connectés.
Des phénomènes qui représentent généralement dans tout système échographique un risque inacceptable, sont les artéfacts ou bruits issus des perturbations électromagnétiques. Ils entraînent une mauvaise qualité de l’image en échographie et sont directement liés à l’environnement du dispositif et à la conception interne du dispositif en lui-même.
En fonction de cet environnement, les exigences réglementaires en comptabilité électromagnétique sont plus ou moins durcies. Pour ce dispositif, selon la norme NF EN 60601-2 [10], il est du Groupe 1 car il utilise de l’énergie à fréquence radioélectrique pour son fonctionnement interne et de la classe B du CISPR 11 [19]car il est utilisé dans des établissements de santé, les cabinets médicaux y compris ceux situés en zones résidentielles.
ii. Exigences générales en matière de sécurité et de performances du Règlement Européen 2017/745 et de la NF EN 60601-2-37
Pour tout fabricant ou fournisseur de dispositif médical qui souhaite commercialiser son dispositif dans l’Union Européenne, et quelle que soit la classe de risque du dispositif, a pour obligation de concevoir et de fabriquer son dispositif conformément aux exigences générales en matière de sécurité de performance de l’annexe I du Règlement Européen 2017/745 [18].
Extrait de mon travail précédent sur la maîtrise des évolutions réglementaires entre les directives 90/385 [20], 93/42 [21] et du Règlement Européen 2017/745 [18] relatif aux dispositifs médicaux (DM) et aux dispositifs médicaux implantables actifs (DMIA) [22], un outil de diagnostic personnalisé est élaboré et mis à disposition dans l’entreprise, pour suivre la conformité aux exigences générales en matière de sécurité et de performances de l’annexe I du Règlement Européen 2017/745 [18].
Dans cet outil, une correspondance entre les exigences du Règlement Européen 2017/745 et les essais recommandés de la norme générale NF EN 60601-1 [8], de ces normes collatérales, et particulières est établie. Sous la responsabilité de la Personne Chargée de Veiller au Respect de la Réglementation (PCVRR), après avoir remplir cet outil, un graphe est généré, lui permettant de visualiser les résultats de l’évaluation sur l’ensemble des exigences générales de l’annexe I. Dans ce même outil, elle peut rajouter toutes les références de preuves documentaires pour rendre la conformité à l’exigence encore plus factuelle. Cependant, il existe une feuille de résultats dédiée aux preuves documentaires, qui a pour but de comptabiliser toutes les preuves documentaires fournies et de les matérialiser par un graphe radar.
Pour une stratégie marketing, les résultats obtenus à partir de cet outil sous forme de graphe radar peuvent être une preuve visuelle, objective et factuelle de la conformité aux exigences générales en matière de sécurité et de performances de l’annexe I du Règlement (EU) 2017/745 [18], et permettre à l’entreprise d’être plus compétitive sur le marché des sondes « POCUS ».
En ce qui concerne les exigences « SYSTEME » elles constituent toutes les spécifications relatives à la sonde en elle-même, au microprogramme (firmware), au driver de communication, au logiciel de démonstration et aux accessoires tels que boitier d’alimentation.
Ces exigences « SYSTEME » sont groupées en deux catégories :
- Les spécifications internes, de haut niveau, plus proche du langage client et de l’usage prévu : ces exigences servent de base pour la phase de validation du produit. Il y a les exigences sur la connectivité, la compatibilité avec système hôte, la performance, l’alimentation, la transmission et la réception du signal, l’interface utilisateur, et les éléments du transducteur ultrasonore.
- Les spécifications externes, de bas niveau, plus proche du langage technique des ingénieurs : ces exigences servent de base pour la phase de vérification des activités de la conception.
Chaque exigence « SYSTEME » est identifiée par un numéro d’identification unique nécessaire pour suivre l’exigence de sa définition jusqu’à son adéquation. C’est ainsi qu’une matrice de traçabilité est mise en œuvre, et suit le schéma de la Figure 5.

Figure 5 : Traçabilité (Source : Auteur)
iii. Identification des performances essentielles du dispositif
Une performance essentielle est une performance nécessaire pour assurer l'absence d'un risque inacceptable [8]. Le fabricant a l’obligation d’identifier les performances essentielles de son dispositif, et ces performances doivent être maintenues à la suite d’un essai technique. Grâce au Tableau 201.102 la norme NF EN 60601-2-37 [9], les performances essentielles du dispositif sont identifiées de manière à éviter qu’un risque soit inacceptable.
Tableau 3 : Performances essentielles identifiées (Source : Auteur d’après [9])
| Risques inacceptable | Performances essentielles |
| Absence d’affichage d’indications incorrectes liée à la sécurité | Alarme en cas de batterie à plat |
| Absence de production d’une sortie non intentionnelle ou excessive d'ultrasons. | Puissance acoustique |
| Absence de bruit sur une forme d'onde ou artefacts ou déformation dans une image ou erreur d'une valeur numérique affichée ne pouvant pas être attribuée à un effet physiologique et qui peut altérer le diagnostic. | Qualité de l’image Résolution axiale et latérale Contraste etc.… |
| Absence de production d'une température de surface de la sonde non intentionnelle ou excessive. | Température à la surface de la sonde Message d’alerte |
| Absence d’affichage de valeurs numériques incorrectes associées au diagnostic à effectuer | Indice mécanique et thermique Interface Homme – Machine (Ecran) |
Cette étape est réalisée en parallèle avec l’analyse des risques car
c’est sur la base de ces performances essentielles que sont définis les
critères d’acceptabilité du risque.
iv. Analyse des risques et rapport performance / risque
Un risque est un danger éventuel plus ou moins prévisible pouvant causer un dommage. Généralement, il est défini comme la combinaison de la probabilité de la survenue d’un dommage et de sa gravité [23]. L’analyse des risques est une activité du processus de gestion de risques, qui consiste alors à identifier et à évaluer tous les dangers potentiels liés à l’utilisation d’un produit durant tout son cycle de vie : c’est à dire de la conception jusqu’à sa mise au rebut.
Dans le cadre de ce projet, les risques électriques, mécaniques, chimiques, de cybersécurité, d’usabilité, électromagnétiques etc… sont identifiés et évalués dans un rapport d’analyse de risques. Cette analyse des risques est initiée (analyse des risques préliminaire) en répondant au questionnaire de l’annexe C de la norme de gestion des risques NF EN ISO 14971 v2013 [23] pour identifier au maximum les risques relatifs au produit.
Ensuite, ces risques sont analysés dans une table où sont estimés de manière quantitative la probabilité d’occurrence, la gravité, la criticité, le facteur de réduction de risque, et où sont déduits tous les moyens de maîtrise des risques, qui deviennent les éléments d’entrée (une partie des exigences systèmes) du processus de conception et de développement [6]. Après cette analyse, les exigences « SYSTEME » sont mises à jour.
En ce qui concerne le calcul du rapport performance / risque, la norme expérimentale XP S99 223 [24] est utile pour évaluer ce rapport. Grâce à l’annexe E de ladite norme, la criticité, l’importance de la performance, la performance et les différents niveaux d’acceptabilité du rapport performance / risque sont évalués.
iv. Procédure de conception et dossier de conception du dispositif médical
La procédure de conception et de développement est un document qui décrit comment réaliser les activités liées (planification, vérification, validation, enregistrements etc…) à la conception et au développement du produit. Avec l’intégration de composantes « SYSTEME » relativement nouvelles pour les produits de l’entreprise, comme l’électronique, la thermique, ou la notion de logiciel de dispositif médical, il faut mettre à jour la procédure déjà existante pour mentionner comment sont effectuées les tâches relatives à ces nouveaux domaines.
De manière générale, la procédure de conception décrit les grandes étapes à effectuer, de l’analyse du besoin jusqu’à la fabrication en série du produit. Le schéma de la Figure 6 illustre ces grandes étapes. Une revue est nécessaire à la fin de chaque phase.

Figure 6 : Les étapes grandes de la procédure de conception (Source : Auteur)
*Généralement réclamés par les auditeurs, les DHF, DHR, et DMR sont les dossiers exigés par la réglementation sur le système de management de la qualité aux Etats Unis [25]. C’est pour cette raison qu’il est fortement recommandé de commencer à initier ces documents depuis le début de la conception et du développement d’un produit. De manière brève :
- Le DHF (Design History File) est une compilation d'enregistrements (documents d’entrée et de sortie de la conception, revue de la conception, validation et vérification de la conception, enregistrement du change control etc. …) qui décrit l'historique de conception d'un appareil fini ;
- Le DHR (Device History Record) est une compilation d'enregistrements (Date de fabrication, quantité fabriquée, quantité libérée pour la distribution, étiquetage, de chaque unité, numéro de contrôle des dispositifs, les non conformités, les opérations de sous-traitances, traçabilité etc…) contenant l'historique de production d'un dispositif fini ;
- Le DMR (Device Master Record) est une compilation d'enregistrements (Liste des matériaux achetés, les factures et instructions, les schémas, les diagrammes, identification de produits, familles, numéro de modèles, description du produit, procédure de packaging, procédure de fabrication, procédure de stérilisation, procédure d’étiquetage, procédure d’expédition, etc…) contenant les procédures et les spécifications d'un appareil fini.
Pendant la phase 0, le chef de projet documente la charte du projet qui identifie le périmètre du projet, ces objectifs, les normes applicables au projet, les besoins des clients, le planning global du projet dans temps, ainsi que tous les rôles et responsabilités (le responsable qualité, responsable de la conception, responsable de la validation, la vérification, de l’électronique, l’acoustique, logiciel, du packaging, etc…) des personnes intervenant dans le projet.
Durant la phase 1, le DHF est initialisé et l’analyse des risques détaillée du produit est réalisée. Par la suite les exigences « SYSTEME » sont mises à jour, en prenant en compte les exigences du logiciel « Démonstrateur », du « Microprogramme » développé sur FPGA, et du logiciel « Driver ». Dès lors, le cycle de vie du logiciel suivant la norme NF EN 62304 [13] s’impose du moment où un logiciel est développé et destiné à fonctionner sur un futur dispositif médical.
Pour ce produit, la NF EN 62304 [13] est applicable au logiciel « Démonstrateur » car il est nécessaire à la validation des performances du produit final même s’il n’est pas fourni aux clients. Le logiciel « Driver » utilisé à cet effet est considéré comme SOUP (source d’origine inconnue) au sens de la NF EN 62304 [13] car il n’est pas développé par l’équipe et encore moins suivant ladite norme.
Ce qu’il faut faire pour ce logiciel SOUP est de rechercher sur le site du constructeur les résultats du bug tracker du logiciel SOUP, afin d’évaluer l’impact de bugs résiduels sur l’utilisation sûre du produit final, et de justifier l’acceptabilité ou non de certains bugs. Quant au « Microprogramme » développé sur FPGA avec les outils VHDL, la norme NF EN 62304 [13] n’est pas appliquée [26] toutefois, ce « Microprogramme » est développé, vérifié et validé en respectant les règles de de bonnes pratiques.
Pendant cette phase, tous les exigences bas niveau (proche du langage technique) « Hardware » sont identifiées (PCB, Flex, les cartes et composants électroniques, les isolations etc… ) de manière à répondre aux exigences de haut niveau (proche du langage client).
Au cours de la phase 2 et 3, la planification des tests, essais « Software » et « Hardware » et « Microprogramme » est effectuée. En ce qui concerne les essais à effectuer sur le produit conformément à la norme générale NF EN 60601-1 [8] et la norme particulière NF EN 60601-2-37 [9] , le plan de vérification mis en place concerne les essais de la Figure 7.

Figure 7 : Essais NF EN 60601-1 (Source : Auteur)
Ces essais sont mentionnés dans un tableau, accompagné des critères d’acceptation de chaque essai, ainsi que la référence du rapport d’essai. L’initialisation du processus d’ingénierie à l’aptitude à l’utilisation fait également partir de cette phase.
Quant à la sélection des sous-traitants, étant certifiée ISO 14001, l’entreprise a pour obligation de respecter sa politique environnementale. Ceci étant, toutes les matières premières utilisées dans les produits de la société ne doivent pas figurer dans la liste des matériaux/substances interdites de l’Annexe II et III de la Directive RoHS 2011/65/UE [27]mais des exemptions peuvent être prises en compte.
La sélection des sous-traitants, requière de connaître leur statut réglementaire en ce qui concerne ces restrictions. A long terme, pendant et après la fabrication en masse du produit, il est fondamental pour l’entreprise de prendre également en compte les exigences du Règlement REACH [28], et de la Directive D3E 2012/19/UE [29].
v. Processus d’ingénierie à l’aptitude à l’utilisation et dossier d’accompagnement du dispositif
C’est un processus qui traite de l’ensembles des facteurs humains avec pour but d’optimiser l’interface utilisateur, de maximiser la facilité d’utilisation d’un produit donné, d’être efficace et d’améliorer la satisfaction des utilisateurs finaux.
Les éléments d’entrée de ce processus sont :
- Des spécifications d’interface,
- Les informations générales du dispositif (indication médicale, population visée, partie en interaction avec le corps humain, environnement d’utilisation, etc…),
- Le principe de fonctionnement du produit, et
- Le profil utilisateur.
Les éléments de sortie sont :
- Le rendement, l’efficacité, et la satisfaction de l’utilisateur final dans l’environnement d’utilisation prévue,
- Le dossier d’ingénierie à l’aptitude à l’utilisation, démontrant que les erreurs liées à l’utilisation sont maitrisées.
Ce processus vient alors transformer les éléments d’entrée, en éléments de sortie, en identifiant tous les phénomènes, les erreurs d’utilisation, et les situations dangereuses liées à l’utilisation du dispositif médical.
Comme cité plus haut, ces risques sont traités dans le processus de gestion des risques du dispositif, et les moyens de maîtrise des risques sont évalués par une évaluation formative (pendant la conception), et une évaluation sommative (après la conception / validation du processus d’usabilité), planifiées pour évaluer leur efficacité, le rendement, et la satisfaction de l’utilisateur dans l’environnement d’utilisation prévue.
A cette issue il est nécessaire de mettre à jour les spécifications d’interface du dispositif.
Ce processus est piloté par l’équipe qualité, et les ressources humaines nécessaires sont : les ingénieurs de la R&D, l’équipe responsable de l’étiquetage et de l’emballage du produit, et un ou plusieurs utilisateurs finaux.



Figure 8 : Ingénierie à l’aptitude à l’utilisation (Source : Auteur d’après [11])
Dans le processus d’ingénierie à l’aptitude à l’utilisation, les notions d’étiquetage et de d’emballage sont prises en compte car même avant l’utilisation physique du dispositif, il est important que toutes les informations nécessaires telles que les marquages, symboles de sécurité de la NF EN 15223-1[30], les alarmes de la NF EN 60601-1-8 [31], soient visibles par l’utilisateur, et durables tout au long de la durée prévue du dispositif. Cela fait donc partir des informations à fournir par le fabricant mentionnées dans la norme NF EN 1041+A1 [32], et son projet de révision PR NF EN 20417 [33]. Pour tout fabricant qui souhaite ou qui évolue dans le domaine de l’échographie avec la technologie « POCUS », les informations suivantes peuvent au minimum se retrouver sur son étiquette :
Tableau 4 : Listes des symboles utilisés (Source : Auteur d'après [8], [30], [33] )

Une fois les symboles marqués sur l’étiquette / emballage ( pour certains) du dispositif, ou sur le dispositif en lui-même, ces marquages doivent réussir le test de lisibilité, et durabilité comme mentionné dans la norme NF EN 60601-1 [8].
Au niveau des alarmes visuelles, les couleurs jaune, orange, et verte sont utilisées pour matérialiser les différents statuts de la batterie (par exemple), et le statut de fonctionnement de l’appareil. A ces alarmes visuelles, sont attribuées une fréquence de clignotement, et un rapport cyclique comme mentionnés dans la norme NF EN 60601-1-8 [31].
La signification des symboles , des marquages, les instructions d’utilisation, les avertissements, les signes de sécurité, et la description technique du dispositif [8] figurent dans la documentation d’accompagnement du dispositif.
vi. Cybersécurité du dispositif médical
Tout dispositif destiné à être connecté (par câble ou par Wi-Fi) à d’autres dispositifs médicaux ou non médicaux, ou même à un réseau internet doit être conçu et fabriqué de manière à limiter tout incident de cybersécurité qui pourrait compromettre son utilisation en toute sécurité. Dans le cas du produit faisant l’objet de ce travail, c’est un dispositif qui est connecté à un appareil ( de type ordinateur, ou smartphone).
Ce dispositif, dans les spécifications fonctionnelles n’est pas normalement conçu pour fonctionner ou pour partager des données sur un cloud ou sur un site Internet. Mais étant donné que l’ordinateur, ou le smartphone sur lequel il est connecté pourrait être connecté à Internet, alors il subsiste un risque que le dispositif soit potentiellement exposé à des attaques.
C’est la raison pour laquelle, une analyse de risque est effectuée, pour identifier tous les risques (accès non autorisé, modification non autorisée, etc. ) auxquelles est exposé le dispositif, et de prévoir des mesures de maîtrise de ces risques.
Selon les recommandations du NIST (National Institute of Standards and Technology), pendant la conception et le développement des dispositifs médicaux, le fabricant doit identifier les menaces les évaluer, protéger le système contre les attaques, détecter les utilisations non autorisées et les intrusions, répondre aux attaques et récupérer les capacités ou les services qui sont altérés en raison d'un incident de cybersécurité.

Figure 9 : Roue de la cybersécurité (Source : Auteur)
Les référentiels ou guides utilisés pour cette activité de cybersécurité sont : le guide de FDA [14], [34], le guide de l’ANSM [15], et le guide du MDCG sur la cybersécurité [16].
Il est recommandé d’avoir au début du processus de cybersécurité, une architecture comportant [14]:
- Les interfaces, les composants actifs, les voies de communication, les protocoles de réseaux utilisés par le dispositif ;
- Le mécanisme d’authentification et les contrôle pour chaque communication ou composant du système (y compris les sites web, cloud etc…) ;
- Le rôle et responsabilité des utilisateurs s’ils interagissent avec les voies de communication ;
- La méthode de cryptographie utilisée, le type et niveau d’utilisation des clés cryptographiques (longueur de la clé, norme utilisée, etc…) ;
- Les détails sur la protection cryptographique des mises à jour de microprogrammes et de logiciels.
Une grande partie des moyens de maîtrise issus de la gestion des risques est prise en compte dans le logiciel de contrôle du dispositif qui est développé par le systémier.
Pour suivre la roue mentionnée ci-dessus il y a comme solutions dans le Tableau 5 :
Tableau 5 : Solutions proposées en cybersécurité (Source : Auteur d’après [14])
| Étapes | Solutions proposées |
| Identifier | Accès non autorisé Modification non autorisée du microprogramme |
| Protéger | Authentification : pour empêcher l’accès non autorisé aux fonctions du dispositif médical, et à empêcher également l’exécution non autorisée du logiciel de contrôle Autorisation : pour donner des privilèges pour l’accès aux données sensibles Confidentialité : Renforcement de la protection par mot de passe Cryptage : pour protéger les informations sensibles de la divulgation non autorisée Les appareils sont identifiés électroniquement par les utilisateurs autorisés Numéro de modèleNuméro de série Les dispositifs sont être conçus pour "refuser par défaut", c'est-à-dire que ce qui n'est pas expressément autorisé par un dispositif est refusé par défaut. L’appareil rejette toutes les connexions non autorisées Les connexions TCP, USB, Bluetooth Intégrité : Vérifier que l'intégrité des logiciels soit validée avant leur exécution, Vérifier l'intégrité de toutes les données entrantes. |
| Détecter | Prévoir des balayages de routine de la sécurité et d'antivirus de sorte que la sécurité et les performances essentielles du dispositif ne soient pas affectées Créer et stocker des fichiers journaux pour les événements de sécurité. |
| Répondre | Avertir les utilisateurs en cas de détection d'une éventuelle faille de cybersécurité. Anticiper le besoin de correctifs et de mises à jour des logiciels pour faire face aux futures vulnérabilités de la cybersécurité. Faciliter la vérification, la validation et l'essai rapide des correctifs et des mises à jour |
| Récupérer | Mise en place des dispositifs qui protègent les fonctionnalités essentielles et données, même lorsque la cybersécurité de l'appareil est compromise. Prévoir des méthodes de conservation et de récupération de la configuration du dispositif par un utilisateur privilégié authentifié. |

Figure 10 : Processus de maîtrise réglementaire ( Source : Auteur )
II. Difficultés rencontrées, solutions préconisées et risques dans projet
Difficultés rencontrées et solutions envisagées
L’entreprise étant « Fournisseur de Dispositifs médicaux », il est parfois difficile de définir le périmètre en ce qui concerne le respect de certaines exigences réglementaires, également dans l’identification des risques (usabilité, cybersécurité etc. ...), puisque les responsabilités se partagent entre le fournisseur et le systémier.
La question de savoir si la norme NF EN 62304 [13] est applicable dans le développement du microprogramme développé en VHDL sur le FPGA est un vrai débat. Mais avec l’aide de la société de conseil en affaires règlementaires, le problème est résolu grâce aux réponses de l’association Team-NB à la foire aux questions, où figure une question dans le même sens qui est résolue [26].
Parfois les exigences du produit étaient incomplètes, ou même les membres de l’équipe de conception avaient du mal à se décider sur certains de choix de la conception. Par exemple définir l’environnement d’utilisation du dispositif, n’est pas un choix simple car en fonction de cet environnement, les exigences de performance et de sécurité (compatibilité électromagnétique) sont plus ou moins durcies, d’autres normes pouvant entrer en jeu [35], [36], et d’autres ressources sont à mobiliser en conséquence. Le marché visé étant très compétitif, le bon compromis a été choisi.
Risques et opportunités du projet
Durant la
période de stage, la crise sanitaire
liée au Covid-19 a eu un impact sur les activités liées au produit, dans le
sens où certaines manipulations et
essais relatifs aux normes NF EN 60601-1 [8] et NF EN 60601-2-37 [9] n’ont pas eu lieu à des moments prévus avant la crise ou bien parce
que l’organisation du travail ou de la communication entre employés ont pu être
affectées. Mais grâce au télétravail, et aux outils de
communication mis en place par l’entreprise, et par la capacité d’adaptation
de l’équipe, les activités et, les
réunions périodiques ont été maintenues et facilitées. Cette expérience a
ainsi permis de découvrir les vertus du télétravail, et de comprendre qu’en
présentiel ou à distance, l’efficacité
dans le travail peut être maintenue, voire peut s’améliorer.
IV. Perspectives d’avenir
Avec le statut de « Fournisseur de dispositif médicaux », et selon la stratégie de l’entreprise qui est de commercialiser ses produits à des systémiers (client assembleur) et non à des utilisateurs définitifs, le démarche d’obtention du marquage CE ne s’applique pas. Mais toutefois, dans les perspectives futures, le schéma qu’il faut emprunter pour l’obtention du marquage CE est illustré sur la Figure 11.

Figure 11 : Démarche d'obtention du marquage CE (Source : Auteur)
Dans le cas où l’entreprise veut commercialiser son produit au Etats Unis, elle soumet un dossier 510 K (car le dispositif a déjà été classé aux Etats-Unis) et prend en compte l’autorisation de mise sur le marché des appareils ultrasonores et transducteurs du CDRH [37]. Il est également pertinent de prendre en compte les exigences du programme d’audit MDSAP [38] pour envisager commercialiser son dispositif dans les pays adhérents que sont : les Etats-Unis, le Canada, le Japon, la Chine, le Brésil, l’Argentine, la Corée du sud, et l’Australie [39].
Tableau 6 : Démarche d'obtention du marquage CE (Source : Auteur d’après [18])
| Étapes | Activités conformément au Règlement (EU) 2017/745 |
| Mon produit est-il un dispositif médical ? | Pour que le dispositif actuel soit entièrement un dispositif médical, la société doit concevoir elle-même le logiciel de contrôle de la sonde car sans ce logiciel, la sonde ne peut pas atteindre la finalité médicale revendiquée (diagnostic des organes). |
| Classe du dispositif médical | Une fois le logiciel de contrôle conçu, la société attribue une classe de risque au logiciel (Annexe VIII règle 11), et au dispositif (sonde intelligente) lui-même. D’après le point 3.3, Chapitre II Annexe VIII du Règlement (EU) 2017/745, « Le logiciel commandant un dispositif ou agissant sur son utilisation relève de la même classe que le dispositif. » [18]. |
| Exigences générales applicables | Une très grande partie des exigences générales est déjà respectée à ce stade. Mais une attention particulière aux informations à fournies sur la notice d’utilisation et l’étiquetage (UDI) du dispositif + le logiciel (Annexe I, Annexe VI). |
| Référentiels techniques | Les référentiels/normes techniques ne changent pas, mais une attention particulière à la protection des données personnelles [40], au dossier technique (Annexe II) du dispositif qui doit être bien formalisé, et à la documentation technique de surveillance après commercialisation (Annexe III) qui doit être initiée. |
| Enregistrement EUDAMED | Surveiller les modules d’EUDAMED et s’enregistrer (le fabricant, ses dispositifs, distributeurs, et mandataire etc.) dans la base de données EUDAMED au moment où elle est déployée [41]. |
| Evaluation clinique | Approfondir l’évaluation clinique qui est quand même une très grande responsabilité du systémier. Pas d’investigations cliniques car le dispositif existe déjà, par conséquence une attention particulière à la notion d’équivalence sur le plan biologique, technique, et clinique de l’annexe XIV Partie A point 3 du Règlement (EU) 2017/745 [18]. |
| Evaluation de la conformité par un Organisme Notifié | Une fois que la société est prête, alors elle choisit son Organisme Notifié sous le règlement [42], qui l’évalue suivant les annexes IX, X, ou XI du Règlement (EU) 2017/745 [18] en fonction de que la société est capable de prouver. |
| Marquage CE | A la suite de cette évaluation, si elle est positive alors la société obtient son Marquage de conformité CE et fait une déclaration de conformité CE suivant l’article 19 contenant les informations de l’annexe IV du Règlement (EU) 2017/745 [18], et pose le marquage de conformité CE conformément à l’annexe V de ladite réglementation. |
| Mise sur le marché et distribution | Le marquage de conformité CE autorise la libre circulation des dispositifs médicaux au sein de l’Union Européenne. Désigner les distributeurs, mandataires et importateurs si nécessaire pour les dispositifs, et les soumettre à leur obligation conformément aux articles 10, 14 et 15 du Règlement (EU) 2017/745 [18]. |
| Matériovigilance & Surveillance post-marché | Vendre un dispositif médical c’est bien mais le suivre durant tout son cycle de vie c’est mieux. La société doit mettre en place un plan de surveillance après commercialisation (Annexe II), pour collecter les informations post commercialisation, les analyser et les traiter dans un rapport périodique actualisé de sécurité (PSUR avec une mise à jour au minimum tous les deux ans dans ce cas), communiquer avec les autorités compétentes, et préparer les rapports de tendances (Article 85 et 88). |
NB : Récemment le Règlement (EU) 2017 / 745 a eu
des amendements. Il est fondamental de prendre en compte ses corrigendums I
[43] , II [44] et le Règlement (EU)
2020/561 [45]. Pour tout fabricant déjà
conforme à la directive 93/42, les travaux sur la maîtrise des évolutions
réglementaires entre les directives 90/385 [20], 93/42 [21] et du Règlement Européen
2017/745 [18] relatif aux dispositifs
médicaux (DM) et aux dispositifs médicaux implantables actifs (DMIA) [22] peuvent être utiles.
Conclusion
En somme, le marché de la technologie « POCUS » est très évolutif et très concurrentiel ; cependant la réglementation évolue aussi. Il faut donc bien mobiliser des ressources non seulement sur le plan technique avec de la Recherche et du développement, mais aussi sur le plan qualité et réglementaire. Dans le cadre de ce travail, au regard du processus de maîtrise réglementaire mis en place, les ressources techniques et réglementaires se sont mobilisées.
Malgré les points critiques et difficultés rencontrées, la motivation, l’implication et l’adhésion du personnel sont des compétences nécessaires à exploiter pour le bon déroulement des activités de ce processus. La finalité est d’avoir un dispositif sûr et performant, qui respecte les normes qui lui sont applicables, tout ceci afin de garantir un niveau élevé de la sécurité du patient.
Pour toute Personne Chargée de Veiller au Respect de la Réglementation (PCVRR) traitant un appareil électro médical, les thématiques abordées dans mémoire sont nécessaires pour porter les projets vers la certification, maîtriser les risques et les contraintes réglementaires applicables, et assurer la sécurité des patients.
Références bibliographiques
[1] N. J. Soni, R. Arntfield, et P. Kory, Point of Care Ultrasound E-book. Elsevier Health Sciences, 2014.
[2] S. Ugalmugle, « Point of Care Ultrasound Market Size | 2019-2025 Share Forecast Report », Global Market Insights, Inc., mai 2019. https://www.gminsights.com/industry-analysis/point-of-care-ultrasound-market (consulté le juill. 07, 2020).
[3] « Norme NF EN ISO 9000 - Systèmes de management de la qualité - Principes essentiels et vocabulaire ». Editions Afnor, Paris, www.afnor.org, oct. 15, 2015.
[4] « Norme NF EN ISO 9001 - Systèmes de management de la qualité - Exigences ». Edition Afnor, www.afnor.org, oct. 15, 2015.
[5] « Norme NF EN ISO 14001 - Systèmes de management environnemental - Exigences et lignes directrices pour son utilisation ». Editions Afnor, Paris, www.afnor.org, oct. 15, 2015.
[6] « Norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires ». Editions Afnor, Paris, www.afnor.org, juill. 2012.
[7] T. L. SZABO, Diagnostic Ultrasound Imaging : Inside Out. Elsevier, 2014.
[8] « Norme NF EN 60601-1 : Exigences générales pour la sécurité de base et les performances essentielles ». Editions Afnor, Paris, www.afnor.org, janv. 2007.
[9] « Norme NF EN 60601-2-37 : Exigences particulières pour la sécurité de base et les performances essentielles des appareils de diagnostic et de surveillance médicaux à ultrasons ». Editions Afnor, Paris, www.afnor.org, avr. 2008.
[10] « Norme NF EN 60601-1-2 : Exigences générales pour la sécurité de base et les performances essentielles – Norme collatérale : Perturbations électromagnétiques – Exigences et essais ». Editions Afnor, Paris, www.afnor.org, déc. 09, 2016.
[11] « Norme NF EN 62366-1 : Application de l’ingénierie de l’aptitude à l’utilisation aux dispositifs médicaux ». Editions Afnor, Paris, www.afnor.org, déc. 18, 2015.
[12] C. for D. and R. Health, « Applying Human Factors and Usability Engineering to Medical Devices », U.S. Food and Drug Administration, sept. 02, 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/applying-human-factors-and-usability-engineering-medical-devices (consulté le juill. 07, 2020).
[13] « Norme NF EN 62304 : Logiciels de dispositifs médicaux Processus du cycle de vie du logiciel ». Editions Afnor, Paris, www.afnor.org, oct. 2006.
[14] C. for D. and R. Health, « Content of Premarket Submissions for Management of Cybersecurity in Medical Devices », U.S. Food and Drug Administration, mai 14, 2019. http://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-premarket-submissions-management-cybersecurity-medical-devices (consulté le juill. 07, 2020).
[15] « Recommandations pour la cybersécurité des dispositifs médicaux - ANSM : Agence nationale de sécurité du médicament et des produits de santé ». https://www.ansm.sante.fr/ (consulté le juill. 07, 2020).
[16] Medical Device Coordination Group, « Guidance on Cybersecurity for medical devices », déc. 16, 2019. https://ec.europa.eu/docsroom/documents/38941 (consulté le juill. 07, 2020).
[17] C. L. Moore et J. A. Copel, « Point-of-Care Ultrasonography », New England Journal of Medicine, vol. 364, no 8, p. 749‑757, févr. 2011, doi : 10.1056/NEJMra0909487.
[18] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) ». Ed. JO L 117, mai 05, 2017, [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra.
[19] « Norme CISPR 11 : Appareils industriels, scientifiques et médicaux - Caractéristiques de perturbations radioélectriques ». Editions Afnor, Paris, www.afnor.org, janv. 2019.
[20] « Directive 90/385/CEE du Conseil, du 20 juin 1990, concernant le rapprochement des législations des États membres relatives aux dispositifs médicaux implantables actifs ». Ed. JO L 189, juill. 20, 1990, [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1990/385/oj/fra.
[21] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux ». Ed JO L 169, juill. 12, 1993, [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1993/42/oj/fra.
[22] O. D. NYAGAM KEMAJOU, S. MHAMDI, et K. KABBABI, « IDS041 - Fabricants : Passage de la Directive 93/42 au Règlement Européen 2017/745 relatif aux dispositifs médicaux », Bibliothèque des travaux Master. https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids041-fabricants-de-la-directive-93-42-au-reglement-2017-745/ (consulté le juill. 07, 2020).
[23] « Norme NF EN ISO 14971 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux ». Editions Afnor, Paris, www.afnor.org, déc. 2019.
[24] « Norme XP S99-223 : Dispositifs médicaux — Gestion du rapport bénéfice/risque ». Editions Afnor, Paris, www.afnor.org, févr. 2020.
[25] « CFR - Code of Federal Regulations Title 21 ». https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=820&showFR=1&subpartNode=21:8.0.1.1.12.3 (consulté le juill. 07, 2020).
[26] Team-NB, « Frequently Asked Questions related to the Implementation of EN 62304:2006 with respect to MDD 93/42/EEC ». avr. 05, 2013, Consulté le : juill. 07, 2020. [En ligne]. Disponible sur : https://www.team-nb.org/wp-content/uploads/2015/05/documents2013/FAQ_62304_Final_130804.pdf.
[27] « Directive 2011/65/UE du Parlement européen et du Conseil du 8 juin 2011 relative à la limitation de l’utilisation de certaines substances dangereuses dans les équipements électriques et électroniques Texte présentant de l’intérêt pour l’EEE », vol. 174. Ed OJ L 174, juill. 01, 2011, [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/2011/65/oj/fra.
[28] « REACH - Produits chimiques - Environnement - Commission européenne ». https://ec.europa.eu/environment/chemicals/reach/reach_en.htm (consulté le juill. 07, 2020).
[29] « DIRECTIVE 2012/19 / UE DU PARLEMENT EUROPÉEN ET DU CONSEIL du 4 juillet 2012 sur les déchets d’équipements électriques et électroniques (DEEE) », vol. 197. Ed JO L 197, juill. 24, 2012, [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/2012/19/oj.
[30] « Norme NF EN ISO 15223-1 : Symboles à utiliser avec les étiquettes, l’étiquetage et les informations à fournir relatifs aux dispositifs médicaux ». Editions Afnor, Paris, www.afnor.org, janv. 2017.
[31] « Norme NF EN 60601-1-8 : Exigences générales pour la sécurité de base et les performances essentielles - Norme collatérale:Exigences générales, essais et guide pour les systèmes d’alarme des appareils et des systèmes électromédicaux ». Editions Afnor, Paris, www.afnor.org, oct. 2007.
[32] « Norme NF EN 1041+A1 : Informations à fournir par le fabricant ». Editions Afnor, Paris, www.afnor.org, nov. 02, 2013.
[33] « Norme PR NF EN ISO 20417 : Informations à fournir par le fabricant ». Editions Afnor, Paris, www.afnor.org, mai 2019.
[34] C. for D. and R. Health, « Postmarket Management of Cybersecurity in Medical Devices », U.S. Food and Drug Administration, juill. 03, 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/postmarket-management-cybersecurity-medical-devices (consulté le juill. 07, 2020).
[35] « Norme NF EN 60601-1-11 : Exigences générales pour la sécurité de base et les performances essentielles -Norme Collatérale : Exigences pour les appareils électromédicaux et les systèmes électromédicaux utilisés dans l’environnement des soins à domicile ». Editions Afnor, Paris, www.afnor.org, avr. 2015.
[36] « Norme NF EN 60601-1-12 : Exigences générales pour la sécurité de base et les performances essentielles -Norme collatérale : Exigences pour les appareils électromédicaux et les systèmes électromédicaux destinés à être utilisés dans l’environnement des services médicaux d’urgence ». Editions Afnor, Paris, www.afnor.org, avr. 2015.
[37] C. for D. and R. Health, « Marketing Clearance of Diagnostic Ultrasound Systems and Transducers », U.S. Food and Drug Administration, juin 27, 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/marketing-clearance-diagnostic-ultrasound-systems-and-transducers (consulté le juill. 07, 2020).
[38] C. for D. and R. Health, « Companion Document - Medical Device Single Audit Programm », U.S. Food and Drug Administration, juin 27, 2019. https://www.fda.gov/media/123467/download (consulté le juill. 07, 2020).
[39] Rachida, « MDSAP Program Is Now Utilized by 2 Other Countries : South Korea and Argentina », avr. 02, 2020. https://lne-america.com/library/news/mdsap-program-is-now-utilized-by-2-other-countries-south-korea-and-argentina (consulté le juill. 07, 2020).
[40] « Règlement (UE) 2016/679 du Parlement européen et du Conseil du 27 avril 2016 relatif à la protection des personnes physiques à l’égard du traitement des données à caractère personnel et à la libre circulation de ces données, et abrogeant la directive 95/46/CE (règlement général sur la protection des données) (Texte présentant de l’intérêt pour l’EEE) ». Ed OJ L 119, mai 04, 2016, [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2016/679/oj/fra.
[41] P. INGLING, « European database on medical devices (EUDAMED) », Internal Market, Industry, Entrepreneurship and SMEs - European Commission, mai 14, 2019. https://ec.europa.eu/growth/sectors/medical-devices/new-regulations/eudamed_en (consulté le juill. 07, 2020).
[42] « EUROPA - Commission européenne - Croissance - Politique réglementaire - NANDO ». https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34 (consulté le juill. 07, 2020).
[43] « Corrigendum to Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (OJ L 117, 5.5.2017) ». Ed OJ L 117, mai 03, 2019, Consulté le : juill. 07, 2020. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/corrigendum/2019-05-03/oj/eng.
[44] « Corrigendum to Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (OJ L 117, 5.5.2017) ». Ed OJ L 334, déc. 27, 2019, Consulté le : juill. 07, 2020. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/corrigendum/2019-12-27/oj/eng.
[45] « Règlement (UE) 2020/561 du Parlement européen et du Conseil du 23 avril 2020 modifiant le règlement (UE) 2017/745 relatif aux dispositifs médicaux en ce qui concerne les dates d’application de certaines de ses dispositions (Texte présentant de l’intérêt pour l’EEE) ». Ed OJ L 130, avr. 24, 2020, [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2020/561/oj/fra.