
IDS076 - Optimisation du processus de gestion du rapport bénéfice/risque des dispositifs médicaux selon la norme XP S99-223
DOI mémoire
https://doi.org/10.34746/n21z-0x80Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

Ayadi Mohamed Aziz 
Lemoine Alexandre 
Pierre-Louis Woodeline 
Rossin Valériane
Contacts
- AYADI Mohamed Aziz : ayadimedaziz@gmail.com
- LEMOINE Alexandre : alexlemoine56860@gmail.com
- PIERRE-LOUIS Woodeline : woodeline@hotmail.fr
- ROSSIN Valériane : valeriane.rossin@outlook.fr
Citation
A rappeler pour tout usage : Valériane ROSSIN, Woodeline PIERRE-LOUIS, Alexandre LEMOINE et Mohamed Aziz AYADI," Optimisation du processus de gestion du rapport bénéfice/risque des dispositifs médicaux selon la norme XP-S99 223", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de projet, https://travaux.master.utc.fr/, réf n° IDS076, https://doi.org/10.34746/n21z-0x80, janvier 2021, url directe : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids076/
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_10_idsap
Résumé
La nouvelle règlementation européenne 2017/745 marque un tournant dans l’industrie des dispositifs médicaux en Europe. Son arrivée introduit de nouveaux concepts qui n’étaient pas présents dans les directives jusque-là. Parmi eux, la notion de la gestion du rapport bénéfice/risque, qui vise à assurer la sécurité des utilisateurs lors de l’utilisation d’un dispositif, est présente. Ce concept, souvent difficile à saisir, a fait l’objet d’une norme expérimentale XP S99-223 publiée 2020 dont l’objectif est de fournir un cadre technique et méthodologique pour la gestion du rapport bénéfice/risque. Ainsi, des outils, comprenant une cartographie interactive et un outil de positionnement permettant d’aider les fabricants à comprendre et utiliser cette norme, ont été développés.
Abstract
The new European Regulation 2017/745 marks a turning point in medical devices industries. Its arrival introduces new concepts that were not present in Directives until then. Among them is the notion of benefit/risk management, which aims to ensure the safety of users with respect to the devices they use. Often hard to grasp, this concept was the object of a new experimental standard XP S99-223 published in 2020 which gives a technical and methodological framework for benefit/risk management. Then, tools to help users understand this new standard have been developed, including interactive mapping and a tool to help users position themselves in relation to the standard.
Remerciements
Nous souhaiterions tout d’abord adresser toute notre gratitude à M. Gilbert Farges, enseignant-chercheur à l’Université de Technologie de Compiègne (UTC), pour la qualité de ses conseils et sa disponibilité qui nous ont permis de mener à bien ce projet.
Nous tenons à remercier Mme Isabelle Claude, enseignant-chercheur et responsable de formation à l’UTC et M. Jean-Matthieu Prot, ingénieur de recherche et responsable de formation à l’UTC pour leurs remarques lors des soutenances orales qui nous ont aidées pour l’amélioration de nos livrables.
Enfin, nous remercions Mme Beatrice Konig, responsable veille et recherche documentaire à l’UTC pour ses remarques pertinentes concernant notre bibliographie.
Téléchargements

Outil d'appropriation de la norme XP S99-223

Evaluer le rapport Bénéfice/Risque de votre dispositif médical selon la loi XP-S99 223
Introduction
Dans toute décision relative à la santé du patient, il est important de s’assurer que le bénéfice de l’action effectuée l’emporte sur le risque éventuel auquel il est exposé.
Dans le cadre du règlement européen 2017/745 relatif aux dispositifs médicaux, il est impératif de fournir un rapport bénéfice/risque jugé acceptable.
La gestion, l’évaluation et le suivi de ce rapport permettent de prouver que le dispositif répond bien aux exigences réglementaires applicables et prouvent sa conformité dans le cadre du marquage CE. Pour ce faire, il est important de prendre en considération toutes les données réunies lors des évaluations cliniques, mais également les facteurs humains, sociaux et économiques qui y sont associés. Cela permet d'assurer la continuité de la gestion du rapport bénéfice/risque tout au long du cycle de vie du dispositif médical et par conséquent, une meilleure prise en charge du patient [1].
La norme XP S99-223 est une norme expérimentale publiée en février 2020. Elle permet aux organismes, pour chaque application définie au préalable, d’établir des méthodes d’évaluation et d’acceptabilité du rapport bénéfice/risque. Destinée aux fabricants de dispositifs médicaux et aux instances réglementaires, elle permet d’établir un cadre et de rassembler les données essentielles pour établir et maintenir la conformité réglementaire d’un dispositif [2].
Face aux enjeux importants que représentent la gestion du bénéfice/risque, bien appréhender la rme est essentiel pour les fabricants de dispositifs médicaux.
Ceci amène donc la question suivante : Comment optimiser la gestion du rapport du bénéfice/risque selon la norme XP S99-223 au sein d’une entreprise ?
L’objectif de ce mémoire d’intelligence méthodologique est de définir et de situer la gestion du rapport bénéfice/risque dans le cadre règlementaire actuel.
Il présente également la méthode utilisée pour créer et proposer un outil d’autodiagnostic permettant aux fabricants de dispositifs médicaux, de pouvoir évaluer l’acceptabilité du rapport bénéfice risque de leurs dispositifs médicaux selon la norme XP S99-223. Il détaille également les différentes propositions et la solution la plus pertinente relevée.
Les fabricants vont pouvoir comprendre aisément le contenu de la norme, se situer rapidement par rapport à leurs niveaux de respect de la norme pour visualiser les axes étant à développer ainsi que les différents plans d’action à entreprendre pour y arriver.
1. La gestion du rapport bénéfice risque des dispositifs médicaux
1.1 Le secteur des dispositifs médicaux
1.1.1 Définitions
Le règlement (UE) 2017/745 définit les dispositifs médicaux comme « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par l’organisme à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- Diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie ;
- Diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci ;
- Investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique ;
- Communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus ;
- Et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Les produits ci-après sont également réputés être des dispositifs médicaux :
- Les dispositifs destinés à la maîtrise de la conception ou à l'assistance à celle-ci ;
- Les produits spécifiquement destinés au nettoyage, à la désinfection ou à la stérilisation des dispositifs médicaux ; » [3].
Il existe une grande variété de dispositifs médicaux, allant des consommables aux équipements d’imagerie, plusieurs centaines de milliers d’équipements différents. Le marquage CE permet la commercialisation de ces dispositifs médicaux dans toute l’Union Européenne. Elle assure leurs conformités vis-à-vis des exigences règlementaires essentielles [4].
Le bénéfice clinique d’après le règlement 2017/745 (UE) est « l'incidence positive d'un dispositif sur la santé d'une personne physique, se traduisant par un (des) résultat(s) clinique(s) significatif(s), mesurable(s) et pertinent(s) pour le patient, y compris le(s) résultat(s) en matière de diagnostic, ou une incidence positive sur la prise en charge de la santé du patient ou sur la santé publique ».
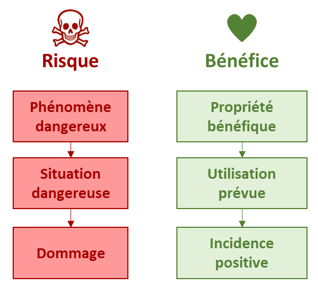
Le risque est défini d’après le règlement 2017/745 (UE) comme « la combinaison de la probabilité de survenance d'un dommage et de la sévérité de celui-ci » [3] (Figure 1).
Figure 1 : Approche Bénéfice/Risque, Source : d'après [5]
1.1.2 Un marché croissant
Figure 2 : Caractéristiques du secteur des DM, source : Snitem, « Panorama 2019 », Source d’après : [6]

D’après une étude du Syndicat National de l’Industrie des Technologies Médicales (SNITEM) réalisée sur un échantillon de 237 entreprises, le secteur des dispositifs médicaux représente en France un chiffre d’affaires de 30 milliards d’euros en 2019 contre 28 milliards d’euros en 2016 et un nombre d’entreprises recensées en France en constante augmentation qui reflète une image dynamique du secteur, pour atteindre le chiffre de 1502 sociétés en 2019 (Figure 2).
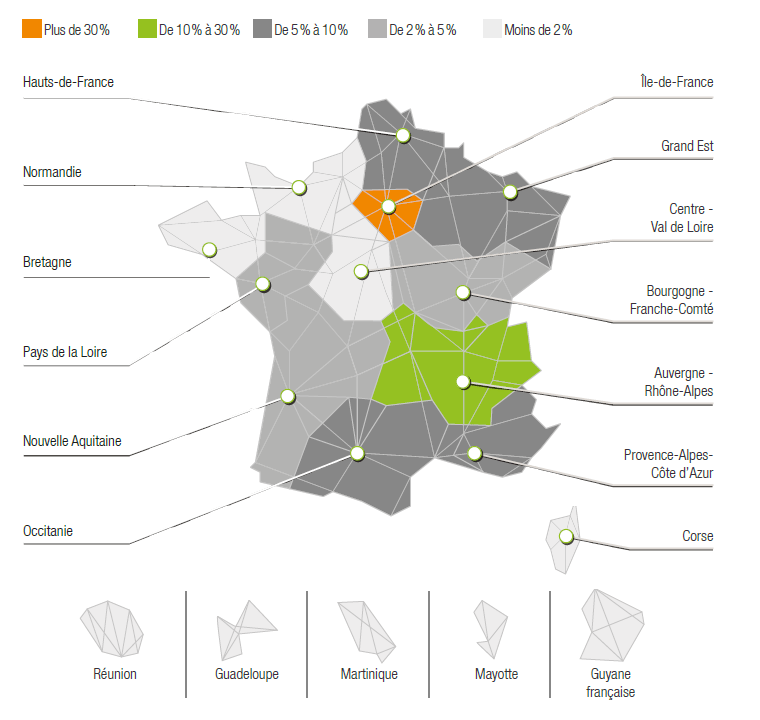
La filière se caractérise par une très forte dominante de PME (93 %) et est génératrice d’emplois (plus de 90 000 emplois) sur le territoire français. Néanmoins, la répartition géographique des entreprises de dispositifs médicaux est assez inégale avec un établissement plus important dans les régions Ile-de-France et Auvergne/Rhône-Alpes (Figure 3).
Figure 3 : Répartition géographique des PME en France, Source : Snitem, « Panorama 2019 », Source : d’après [6]
Ces entreprises ne limitent pas leur activité à la frontière Française, elles se caractérisent par une importante internationalisation (9 milliards de chiffre d’affaires). Plus d’un tiers des recettes du secteur sont destinées à l’export, ainsi selon les structures interrogées « 70% de la croissance anticipée serait générée par l’export ». Les trois marchés privilégiés sont l'Europe, l'Asie et l'Amérique du Nord.
Les entreprises actives en recherche et développement (R&D) sont de l’ordre de 13%. Le nombre de brevets par an dépasse les 3750 soit plus de 10 brevets par jours. Alors que le secteur déploie des capitaux pour ses activités de R&D, il doit également surveiller les produits et les risques inhérents à ceux-ci au vu des enjeux patients, soignants et utilisateurs : ainsi la matériovigilance a pour objectif d’éviter que ne se reproduisent des incidents et risques graves [6].
1.1.3 La sécurité du patient, au cœur des préoccupations
Plus de 18000 incidents liés aux dispositifs médicaux ont été déclarés en matériovigilance au cours de l’année 2017 auprès de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) et environ 158 000 incidents en dix ans, selon « le Monde » [7].
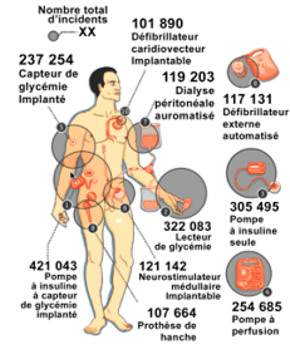
Des journalistes ont établi à partir de la base de données de l’Agence de santé américaine (FDA) un classement des dix dispositifs les plus souvent mis en cause dans la survenue d’incidents entre 2008 et 2017. En tête de listes des incidents, les pompes à insuline à capteur de glycémie implantées (421.000), suivie des lecteurs de glycémie (322.000) et pompes à insuline seule (305.000). Viennent ensuite les pompes à perfusion (254.000), les capteurs de glycémie implantée (237.000) et les neurostimulateurs médullaires implantables (121.000) [8], [9].
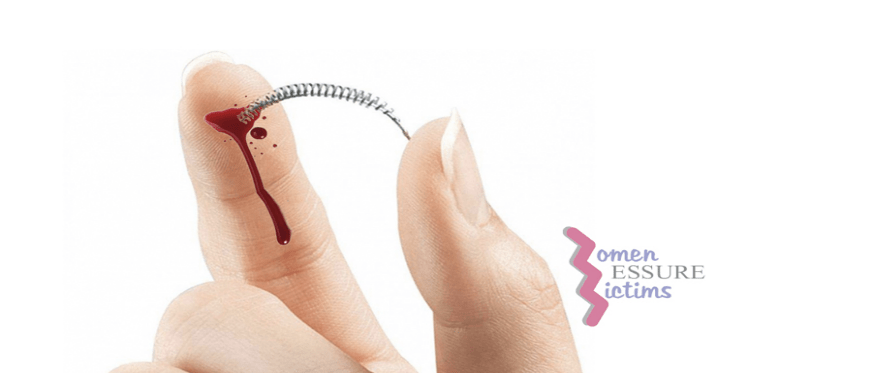
Par ailleurs, de nombreux scandales comme celui du dispositif ESSURE commercialisé par la société Bayer en Europe de 2002 à 2017 ont émergé. Présenté comme la solution sûre, sans effet secondaire et à la pointe de la technologie, cet implant de contraception définitive a attiré l’attention notamment en 2018 avec l’enquête des Implant files. Plus de 175 000 femmes en France ont été implantées. ESSURE ou « la bombe qui nous détruit chaque jour un peu plus » a été retiré du marché européen pour des effets indésirables graves (Figure 4).
Figure 4 : A) Nombre total d'incidents au niveau du corps humain, Source : d'après [7] B) Implants ESSURE®: Dispositif médical de contraception définitive , Source : d'après [10]
Ce n’est pas le seul scandale que « Implant files » relève. En effet en 2019, l'ANSM a annoncé, l'interdiction des implants mammaires macrotexturés en silicone et des implants en polyuréthane. Il a été établi que le port de ces implants dits « macrotexturés » pouvaient créer une inflammation chronique qui déclencherait des Lymphomes Anaplasique à Grandes Cellules (LAGC) [11].
Une autre étude publiée en 2015 montrait que les cathéters étaient la quatrième cause d’infections liées aux soins à l’hôpital en France. Aussi, plusieurs travaux ont été publiés dans la Revue Francophone des Laboratoires, ces derniers dénonçaient les risques infectieux associés aux dispositifs médicaux invasifs. Ces travaux estiment que plus de 60% des infections associées au soin auraient pour origine un dispositif invasif [12].
Ces scandales et bien d’autres ont pu mettre en exergue le lobbying des industriels du secteur des DM [13], [14].
Pour finir, depuis une dizaine d’années le monde scientifique s’est beaucoup intéressé à la perception et l’étude du rapport bénéfice/risque suite à ces incidents. Le changement de la règlementation européenne est notamment né de ces derniers.
1.1.4 Le cycle de vie du dispositif médical
Figure 5 : Cycle de vie du dispositif médical, Source : auteurs
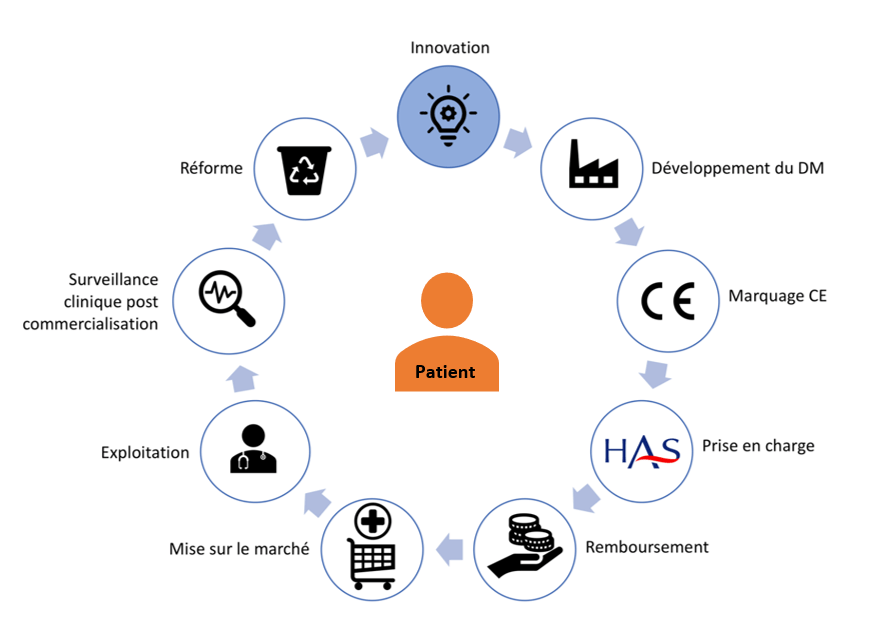
La première étape du cycle de vie du dispositif médical [15] est l’innovation. Il s’agit de faire naître une idée dans un laboratoire de recherche publique, dans le département Recherche & Développement d’entreprises privées grâce aux retours d’utilisateurs, aux rapprochements de deux concepts ou encore aux investigations cliniques.
Ensuite s’en suit une phase de développement de l’idée [15] :
- Définition des exigences de conception selon la règlementation, les normes techniques, les risques identifiés, les exigences médicales et d’ergonomie ;
- Prototypage ;
- Etudes pré-cliniques : expérimentation animale, test in vitro ;
- Etudes cliniques : Etude de faisabilité et Etude du bénéfice clinique ;
- Démonstration de la fiabilité des procédés de fabrication ;
- Réalisation de la documentation technique.
La phase suivante est le marquage CE. L’étape de prise en charge (et tarification en France) par la solidarité nationale du dispositif médical selon l’usage du DM (en ville ou en établissement de santé) est réalisée par la Haute Autorité de Santé. Le remboursement du dispositif médical est, par la suite, évalué par le ministère de la santé et la Commission Nationale d’Evaluation Des Dispositifs Médicaux et Des Technologies De Santé (CNEDiMTS) [16]. Les phases de mise sur la marché et d’exploitation du dispositif médical par les établissements de Santé et centrales d’achat sont les suivantes dans le cycle. Enfin, la surveillance clinique post-commercialisation termine pratiquement ce cycle pour garantir toujours une sécurité des patients et une traçabilité des effets secondaires apparaissant. La dernière phase, la réforme peut engendrer une élimination du dispositif médical du marché si les nouvelles normes et règlementations ne sont pas respectées.
1.2 La norme Bénéfice/Risque
1.2.1 Enjeux règlementaires
L’article 2 du règlement 1025/2012 définit la norme comme « une spécification technique, approuvée par un organisme reconnu de normalisation pour application répétée et continue, dont le respect n’est pas obligatoire […]. » [17]. Ces règles ont évolué de manières considérables au cours des dernières années en suivant les innovations technologiques.
D’après le site internet français vie-publique.fr monitoré par la Direction de l’information légale et administrative [18], la règlementation est « un acte juridique européen, de portée générales, obligatoire dans toutes ses dispositions […]. Le règlement est donc directement applicable dans l’ordre juridique des Etats membres. ». Ladirective quant à elle, est « un acte juridique européen pris par le Conseil de l’Union européenne avec le Parlement ou seul dans certains cas. […] Les Etats membres doivent donc transposer la directive dans leur droit national. » [19].
Il s’agit maintenant d’aborder les « normes harmonisées ». Elles sont catégorisées dans les normes européennes particulières et élaborées par un Organisme Européen de Normalisation (OEN) qui peut être le Comité Européen de normalisation (CEN), le Comité Européen de Normalisation Electrotechnique (CENELEC) ou l’Institut Européen des normes de Télécommunication (ETSI). Dans leur cas, la Commission Européenne mandate un OEN pour réaliser la norme harmonisée. Des spécifications techniques permettent le respect des exigences techniques de la législation européenne [20]. Comme pour les autres normes, leur application est facultative, cependant, elles offrent une preuve de qualité et fiabilité du produit. Ainsi, le respect de ces normes harmonisées permet de répondre de manière efficace aux exigences d’une directive ou règlementation européenne. Généralement, il est présent dans leur annexes Z des liens permettant de relier les exigences règlementaires liées à la législation européenne et les réponses liées à la mise en œuvre de la norme harmonisée.
Suite au développement de nouvelles technologies dans les années 80-90, la directive 93/42/CEE [21] fait son entrée en Europe afin de donner un cadre à la mise sur le marché des nouveaux dispositifs médicaux en France.
Au niveau normatif, en 1996, la norme ISO 13485 apparaît à destination notamment des industriels. Elle fixe toutes les bases d’un système management de la qualité dans les organisations biomédicales. La conformité du fabricant est donc démontrée à ce moment par la preuve d’un SMQ robuste en s’appuyant sur la norme ISO 9001 : 2000. En 1998, c’est la gestion des risques qui est à son tour cadrée par la norme ISO 14971-1. Le texte MEDDEV 2.7/1 définit lui en 2003 comment mener à bien les évaluations cliniques.
Enfin, le règlement 2017/745 rentrant en application en mai 2021 reprend majoritairement toutes les exigences de la directive 93/42/CEE en insistant désormais sur la surveillance clinique après commercialisation (SCAC), l’investigation clinique, l’identification unique du dispositif et la gestion du rapport Bénéfice/Risque (B/R) [1], [3]. Il abroge par ailleurs, la directive 93/42/CEE.
Au cœur du règlement 2017/745, se trouve la gestion du rapport bénéfice/risque, dans toutes les étapes du cycle de vie du dispositif médical. En effet, les données cliniques obtenues doivent pouvoir prouver un rapport acceptable. Par ailleurs, l’investigation clinique ne peut être effectuée si et seulement si le rapport bénéfice risque est favorable. Enfin, la SCAC est indispensable pour démontrer la continuité d’un rapport favorable. Toute modification du dispositif médical doit se faire en accord avec une gestion positive du bénéfice/risque. Ce rapport est ainsi très surveillé dans le nouveau règlement.
Le texte du MEDDEV et l’ISO 14971 définissaient déjà l’importance de l’analyse de ce rapport mais n’expliquaient pas concrètement comment mettre œuvre les exigences à ce niveau. De plus, l’ISO 14971 présentait quelques incohérences avec le nouveau règlement notamment au niveau d’une approche de l’évaluation du rapport bénéfice risque qui n’est pas systématique mais post-maîtrise des risques [1].
Le 10 décembre 2019, la nouvelle version de l’ISO 14971 est publiée : l’EN ISO 14971 harmonisée selon les exigences des règlements (UE) 2017/745 et (UE) 2017/746. Les changements effectués s’appuient notamment sur les nouveaux règlements 2017/745 et 2017/746, sur l’EN ISO 13485 : 2016 (harmonisée par rapport aux directives 93/42/CEE, 98/79/CE et 90/385/CEE) et permet d’avoir un outil à jour de transition pour les fabricants [22]. L’obtention du marquage CE est conditionnée par l’audit de cette norme. C’est pourquoi, de nombreux fabricants se font certifier ISO 13485 : 2016, ce qui leur facilite l’entrée sur le marché européen [23].
C’est dans cette période de transition entre la directive et la règlementation que la course au marquage CE est lancée depuis peu. Le marché européen est en effervescence avec de nombreuses entreprises qui sont dans l’espoir d’obtenir le nouveau marquage CE.
1.2.2 Norme XP S99-223
C’est en 2020 que la norme expérimentale XP S99-223 fait son apparition et propose un cadre technique de la gestion du rapport bénéfice risque [2] (Figure 6). Cette norme française n’est en aucun cas imposable par les auditeurs. Elle est donc uniquement un guide de bonnes pratiques pour les fabricants du secteur des dispositifs médicaux.
Figure 6 : Balance Bénéfice/Risque, Source : d’après Auteurs
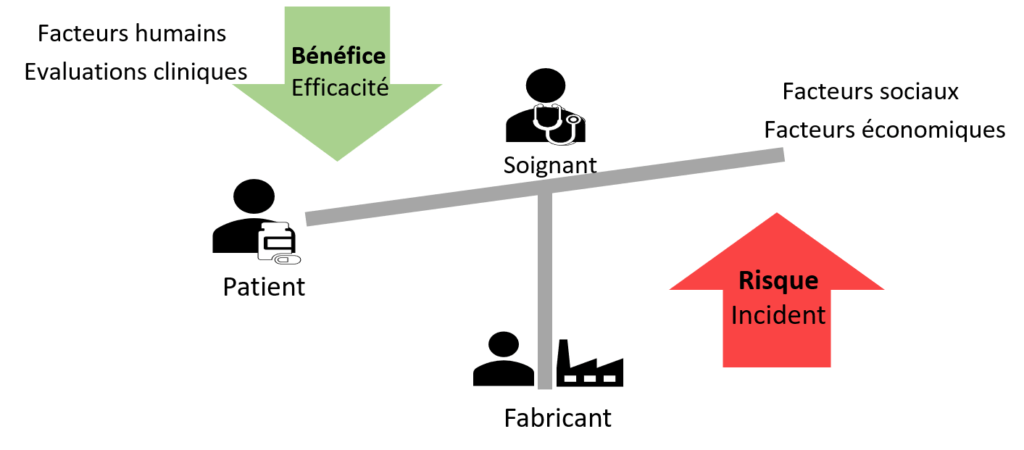
Pourquoi cette norme a-t' elle été mise en place [24] ? La gestion du rapport bénéfice risque est au cœur et à l’interaction de toutes les activités de cycle de vie du DM (cf partie 1.1.4 et Figure 7).
Figure 7 : Gestion du rapport bénéfice risque au cœur du cycle des dispositifs médicaux, Source [2]
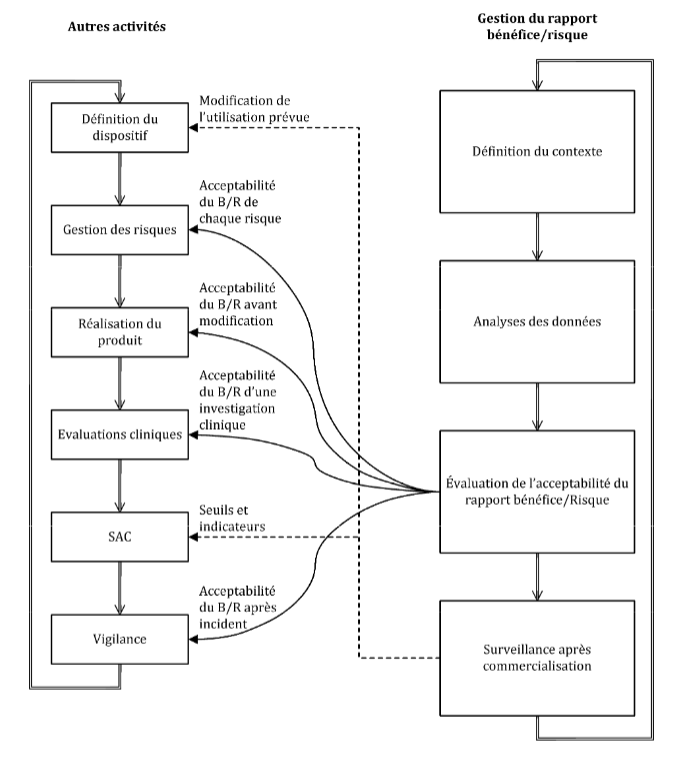
C’est pourquoi, il est difficile d’en déterminer un réel processus. La démonstration de conformité liée au règlement 2017/745 se fait par la maîtrise du rapport bénéfice risque. La nécessité d’un cadre, d’objectifs, de moyens d’évaluation de ce rapport est devenue primordiale. La maîtrise du bénéfice/risque a longtemps été mise de côté par le monde du dispositif médical. Ces exigences règlementaires, apparues clairement dans le règlement 2017/745, sont très jeunes. Cette norme a permis de cadrer techniquement et méthodologiquement une gestion du rapport bénéfice/risque qui paraissait encore floue à la sortie du nouveau règlement. La visualisation, pondération, comparaison, estimation ou identification de données sont définies dans cette norme expérimentale. La méthode de traitement de l’opinion des patients et de ces incertitudes est aussi explicitée. Le patient est mis au cœur de l’évaluation du rapport bénéfice risque afin d’obtenir un rapport représentatif de la réalité. Pour terminer, cette norme se repose sur le système de management de la qualité (ISO 13485), la norme ISO 14971, la surveillance (pré)clinique et toutes les étapes clefs du cycle du dispositif médical.
Ainsi, l’importance de cette nouvelle norme a pu être démontrée. Les fabricants et constructeurs de dispositifs médicaux ont alors l’obligation d’évaluer le rapport bénéfice risque de leurs dispositifs médicaux s’ils souhaitent obtenir le marquage CE nécessaire pour une mise sur le marché de leurs produits.
Cependant, ils rencontrent souvent des difficultés à évaluer leur rapport bénéfice/risque du fait des nombreux éléments à prendre en compte.
1.3 Maîtriser la norme Bénéficie/Risque
Démontrer que les bénéfices sont supérieurs aux risques nécessite des compétences clés que les fabricants n’ont pas toujours. Par ailleurs, cette approche doit se faire centrée sur les futurs utilisateurs/patients du dispositif médical. Une méthodologie claire est explicitée dans la norme XP S99-223 en annexe afin d’éclairer les fabricants sur la démarche pas toujours très simple permettant l’évaluation du rapport du Bénéfice/Risque. Les compétences techniques indispensables relèvent des domaines suivants :
- Identification des risques et bénéfices ;
- Estimation de la probabilité, gravité pour en calculer les risques et bénéfices ;
- Pondération des données ;
- Comparaison des données ;
- Visualisation des données ;
- Communication des données ;
- Suivi des données.
Grâce à ses huit annexes, la norme bénéfice/risque guide les fabricants dans le calcul du rapport bénéfice/risque :
- Annexe A : Approche par les risques ;
- Annexe B : Principes de gestion du rapport bénéfice/risque ;
- Annexe C : Dossier de gestion du rapport bénéfice/risque ;
- Annexe D : Identification des opinions ;
- Annexe E : Estimation des risques, des bénéfices et du rapport bénéfice/risque et mesure après commercialisation ;
- Annexe F : Visualisation des données ;
- Annexe G : Évaluation de l’acceptabilité du rapport bénéfice/risque.
L’annexe ZA permet d’identifier comment la norme permet de répondre aux exigences sur le rapport bénéfice/risque du règlement 2017/745. Ainsi, si elle voit le jour, elle pourrait devenir une norme harmonisée par la suite puisqu’elle contient une annexe Z.
L’acceptabilité du rapport bénéfice/risque est détaillée par un cheminement clair dans la norme. Tout d’abord, l’identification du contexte autour du dispositif étudié est importante. Il permet de rassembler tout le cadre règlementaire et normatif applicable, autorités de santé, professionnels de santé, cible dans la population etc. Ensuite, l’étape sur l’identification de l’opinion du patient (Annexe D) est fondamentale. Ceci permet de posséder tous les éléments clés pour une évaluation du rapport bénéfice/risque correcte. Il s’agit donc de se baser sur des groupes représentatifs hétérogènes concernés par l’utilisation prévue du dispositif. C’est à ce stade que par des moyens d’entretien, de questionnaires, d’internet qu’on recueille des informations sur la perception des risques, des bénéfices, l’acceptabilité de la balance bénéfice/risque et sur le contenu et la forme des informations à fournir (Cf norme IEC 62366 : Aptitude à l’utilisation). Les méthodes d’identification de l’opinion sont décrites dans l’Annexe D et permettent d’obtenir une opinion non biaisée en laissant toujours le patient exprimer ses avis et ses craintes.
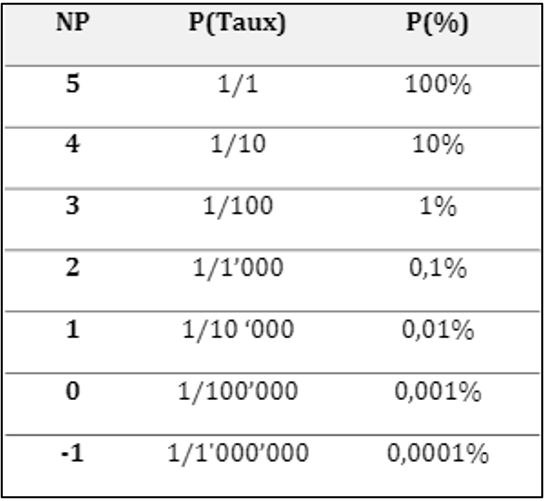
L’étape de l’analyse des risques est la suivante. Le risque est caractérisé par une probabilité d’occurrence et une gravité. Dans le règlement 2017/745, il est mentionné que les risques doivent être autant que possible limités et sans altérer le rapport bénéfice/risque. Cette étape demande donc une preuve que toutes les exigences applicables aux produits, que tous les dispositifs équivalent techniquement et/ou cliniquement, que toutes les alternatives technologiques et leurs limites ont été prises en compte dans la maîtrise du risque. L’altération du rapport bénéfice/risque peut se faire soit en augmentant les risques soit en diminuant les bénéfices. Ainsi, prendre en compte les bénéfices et risques clés qui vont jouer sur la balance bénéfice/risque est primordial. L’estimation du risque se fait donc de la manière suivante : Probabilité (P) x Gravité (G). Les estimations quantitatives de la probabilité et de gravité sont basées sur l’estimation qualitative et sont détaillées dans l’annexe E. Pour les niveaux de probabilité on peut utiliser le tableau suivant (Figure 8) :
Figure 8 : Niveau de probabilité, Extrait de la norme XP S99-223, Source : d’après [2]
Concernant la gravité, de nombreuses échelles existent pour estimer la souffrance, l’atteinte sur la santé du patient, la norme propose cette méthodologie (Figure 9) :
Figure 9 : Niveau de gravité, extrait de la norme XP S99-223, Source : d’après [2]
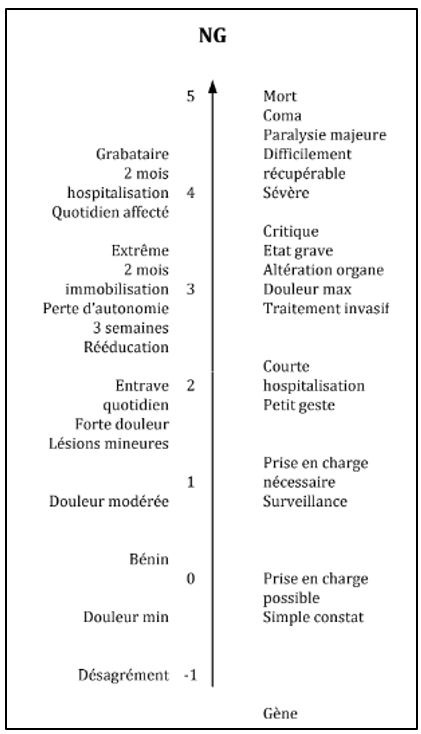
Les niveaux négatifs sont des dommages que l’on peut à peine ressentir. Une fois l’estimation du couple P et G faite, on peut alors estimer le niveau de risque. L’acceptabilité du risque ne pourra se faire qu’uniquement avec la matrice de risque mais avec la remise en contexte aussi (Figure 10).
Figure 10 : Matrice de risque, Extrait de norme XP S99-223, Source : d’après [2]
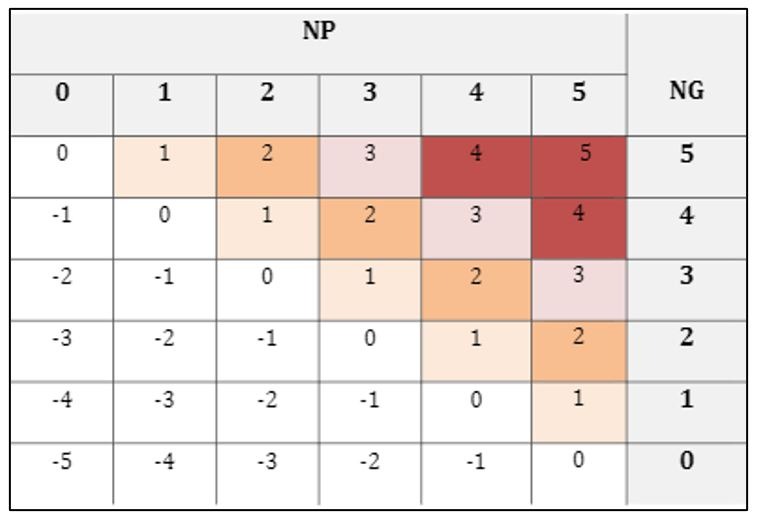
Par exemple, ici le niveau 5 correspond au libellé suivant : A chaque fois qu’on allume le dispositif on tue le patient. La réalisation de l’estimation quantitative du risque permet grâce à l’utilisation du logarithme de retomber sur cette matrice avec des valeurs de niveau de risque se trouvant dans l’intervalle [-5 ;5]. Cette matrice permet le suivi dans le temps, une estimation quantitative issu de l’évaluation clinique et de retour des patients ainsi que la comparaison de plusieurs dispositifs. Les risques combinés peuvent aussi être calculés.
Par la suite, l’analyse des bénéfices est à réaliser. Les bénéfices se calculent en mesurant l’importance de l’incidence positive sur le patient et la probabilité d’occurrence. L’échelle pour la probabilité d’occurrence des bénéfices est la même que celle utilisée pour les risques. L’importance, quant elle, peut se mesurer aussi en utilisant l’échelle de la gravité des risques en comprenant que l’importance de l’incidence positive sur le patient est équivalente à la gravité de la pathologie traité, diagnostiqué ou prise en charge ou encore le niveau de gravité atteint en l’absence du dispositif. La même matrice de niveau de bénéfice peut être obtenue.
Une fois l’estimation du niveau de bénéfices et de risques réalisée, l’estimation du niveau du rapport bénéfice/risque (NBR) peut se faire.
NBR = log(B/R) = Niveau de bénéfice combiné – Niveau de risque combiné
Figure 11 : Matrice de niveau de bénéfice/risque, Extrait de la norme XP S99-223, Source : d’après [2]
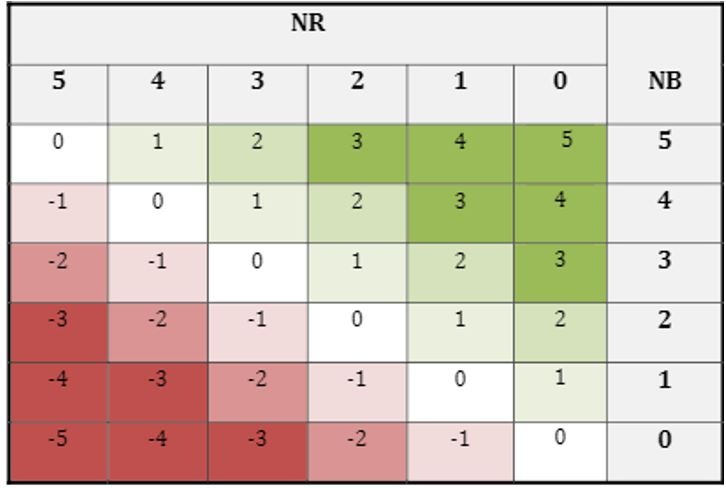
Cette matrice (Figure 11) permet d’identifier qualitativement si le niveau du rapport bénéfice/risque est défavorable, mauvais, faible, moyen, élevé ou très élevé. Ceci est un indicateur et nécessite d’être remis dans un contexte et comparé aux alternatives médicales du dispositif actuellement sur le marché.
Différentes situations possibles, tel que lorsque tous les risques sont nuls, sont décrites dans l’annexe E, les consulter est indispensable pour ne pas estimer un rapport bénéfice/risque biaisé.
Par ailleurs, l’annexe G décrit l’évaluation de l’acceptabilité du rapport bénéfice/risque du produit. Cette évaluation se réalise selon un contexte technique, règlementation et normatif déjà identifié. Elle doit se réaliser en prenant compte à la fois de l’acceptabilité individuelle et de l’acceptabilité globale. Ceci est à évaluer pour chaque utilisation prévue du dispositif. L’acceptabilité de chaque risque doit se faire de la manière suivante : chaque niveau de risque est composé au niveau de risque des dispositifs comparables et la mesure du rapport bénéfice/risque est systématiquement faite et se doit d’être favorable. L’acceptabilité globale s’assure que le niveau du rapport bénéfice/risque est favorable et au moins aussi bon que les alternatives présentes sur le marché. Le maintien de l’acceptabilité globale de ce rapport après la commercialisation est détaillé dans l’annexe E partie Mesure après commercialisation. Une conclusion sur l’acceptabilité de ce rapport est nécessaire. Elle doit contenir, les éléments favorables, défavorables, limitations des groupes représentatifs étudiés, incertitudes, surveillance après commercialisation en introduisant les notions d’aptitude à l’utilisation avec les informations à fournir, les communications externes (autorités, patient, etc.) et l’acceptabilité du rapport du bénéfice/risque. Les cas difficiles comme les femmes enceintes ou personnes incapables doivent avoir été pris en compte.
Enfin, les différentes techniques de visualisation des données sont précisées en annexe F. Les échelles log/log, tableaux, arbre de probabilité, histogramme, infographies et d’autres y sont décrites.
La finalité est la surveillance après commercialisation qui va nécessiter la surveillance constante des données clés, risques clés, bénéfices clés et incertitudes notables. Suivre et estimer le nombre final prévu du dispositif la durée de vie de vie et la fréquence de recours au dispositif par leur utilisateur est nécessaire.
En conclusion, la maîtrise du rapport du bénéfice/risque demande une intégration dans les processus de l’entreprise (SAC, gestion des risques etc.) afin de réaliser une analyse globale et temporelle pertinente.
1.4 Etat de l’art des solutions techniques existantes
La gestion du rapport bénéfice risque est trop récente pour que des outils aient pu être développés dans cet intervalle de temps. Cependant, certains outils publiés permettent la gestion du risque dans les dispositifs médicaux.
1.4.1 Le guide pratique : Gestion des risques des dispositifs médicaux (version 2018) du Snitem
Un guide pratique : « Gestion des risques des dispositifs médicaux » [25] a été créé en 2014 (revue en 2016 puis 2018) par le Centre technique des industries mécaniques (Cetim) en collaboration avec le Syndicat national de l’industrie des technologies médicales (Snitem). Il permet aux fabricants de pouvoir mettre en application au mieux la norme NF EN ISO 14971 et la norme NF EN ISO 62304. Il est disponible en deux langues français et anglais. Cette aide opérationnelle propose un outil prenant en compte les risques des dispositifs médicaux tout au long de son cycle de vie. Il établit des critères d’acceptabilité reconnus par les professionnels et utilisables pour des analyses liées à la matériovigilance. De nombreux exemples concrets permettent d’accompagner le lecteur.
1.4.2 Le guide pratique de la Food and Drug Administration (FDA)
Le guide pratique : « Prise en considération de l’incertitude lors de la détermination du rapport bénéfice/risque dans les approbations avant mise sur le marché, classifications de novo et exemptions pour usage humanitaire » [26] a été créé en 2019 par la FDA. Il décrit l’approche de bénéfice/risque pour les dispositifs médicaux très innovants ou concernant des populations très réduites.
1.4.3 Logiciel de gestion du risque : ERM by Knowllence (Enterprise Risk management)
Disponible sur abonnement le logiciel « Medical Device Suite » est dédié à l’analyse de la norme ISO 14971 : 2019. Le module RM 14971 permettant la gestion du risque élabore un dossier complet de validation à l’issu de l’utilisation du logiciel [27]. Les avantages principaux sont la structuration des démarches, la sécurisation des audits, le gain de temps, réalisation d’analyse de gestion des risques selon la norme ISO 14971 et ainsi une cohérence globale dans les données de l’entreprise.
1.4.4 Outil d’autodiagnostic ISO 14971 : 2019
Cet outil d’autodiagnostic de la norme NF EN ISO 14971 : 2019 a été réalisé en 2019 par R. Cheng, F.Gandar et L. Zaghdoudi [28]. Il est à destination des entreprises de dispositifs médicaux souhaitant évaluer leur conformité à cette norme. Cet élément central est une aide précieuse qui contribue à garantir l’obtention du marquage CE par la suite.
1.4.5 Autres outils
D’autres outils professionnels peuvent être utilisés comme :
- L’Analyse des Modes de Défaillances, de leurs Effets et de leur Criticité (AMDEC) est un outil utilisé dans une démarche de qualité dans une organisation ou bien un produit. Il permet de limiter les risques de défaillances qui pourraient impacter le bon fonctionnement de notre produit, les processus de conception et de production. Il vise à l’amélioration continue de la satisfaction du client en anticipant les possibles dysfonctionnements du produit. Il se base sur les techniques d’analyses de la fiabilité du système comme décrites dans l’ISO 60812. Cette norme détaille les causes et effets des modes de défaillance et en fait l’analyse [29] ;
- L’IEC 31010 permet une approche inductive des risques : Analyse Préliminaire des Risques (APR), arbres des conséquences, HAZard et OPerability (HAZOP) etc [30]. Elle établit des techniques d’appréciation du risque pour différentes situations afin d’aider à prendre les décisions qui conviennent ;
- Approche déductive : Arbre des causes.
1.4.6 Méthode de développement de l’outil
Comme expliqué précédemment (partie 1.3), un outil de positionnement par rapport à la norme expérimentale XP S99-223 n’existe pas mais d’autres outils abordant notamment la gestion des risques sont déjà en accès libre actuellement.
Le tableau suivant permet de détailler les avantages et inconvénients des différents outils (Tableau 1):
Tableau 1 : Comparaison des outils existants
| Ergonomie | Outil Compatible | Temps passé | Visualisation de résultats obtenus | Evaluation de la maîtrise documentaire | Solution traitant le bénéfice/risque | |
| Guide pratique du SNITEM | + | ++ | -- | - | - | -- |
| Guide pratique de la FDA | + | ++ | -- | - | - | -- |
| Logiciel de gestion de risque | - | + | - | + | - | -- |
| Outil d’autodiagnos-tic de l’ISO 14971 | ++ | + | + | + | + | -- |
| Autres outils | -- | + | ++ | - | -- | -- |
Légende :
++ : Répond parfaitement au critère ;
+ : Répond de manière convaincante au critère ;
- : Répond moyennement au critère ;
-- : Ne répond pas au critère.
Cette étude critique de ces articles a permis d’amener les résultats suivants :
- L’ergonomie du mode d’emploi est primordiale pour la création d’un guide performant ;
- L’utilisation d’un outil compatible pour tous utilisateurs est essentielle ;
- Le temps passé sur cet outil doit être le plus court possible ;
- La visualisation claire et intuitive des résultats est primordiale pour une meilleure compréhension et appréhension des axes d’amélioration ;
- Une évaluation de la maîtrise documentaire doit être présente car elle permet d’apporter des détails pertinents quant à la conformité de la gestion documentaire à la norme ;
- Ergonomie globale intuitive de l’outil afin de pouvoir naviguer entre les onglets d’une manière fluide.
Ces différents points constituent les spécifications du cahier des charges permettant la réalisation d’un nouvel outil d’autoévaluation répondant aux besoins actuels.
2. Solution d’aide opérationnelle sur la gestion du bénéfice/risque
2.1 Cartographie intuitive de la norme XP S99-223
Afin de faciliter la compréhension de la norme, une cartographie détaillée a été mise au point. Cette dernière, accessible sous format PDF, est un outil interactif permettant d’appréhender au mieux chaque axe de la norme de manière rapide et simple. Elle permettra à tous les fabricants souhaitant respecter les exigences règlementaires relatives au rapport bénéfice/risque de se repérer dans cette norme. Elle donne une vulgarisation des éléments essentiels à prendre en compte ainsi que tous les documents à rédiger pour une conformité à 100%.
Pour une évaluation complète et compréhensible de tous, une analyse opérationnelle de la norme a été effectuée. Celle-ci permet de vulgariser et de synthétiser les aspects associés au rapport bénéfice/risque. Chaque élément essentiel de la norme a été ensuite retranscrit dans ce guide.
Le guide s’articule ensuite selon trois niveaux de profondeur :
- Vue générale
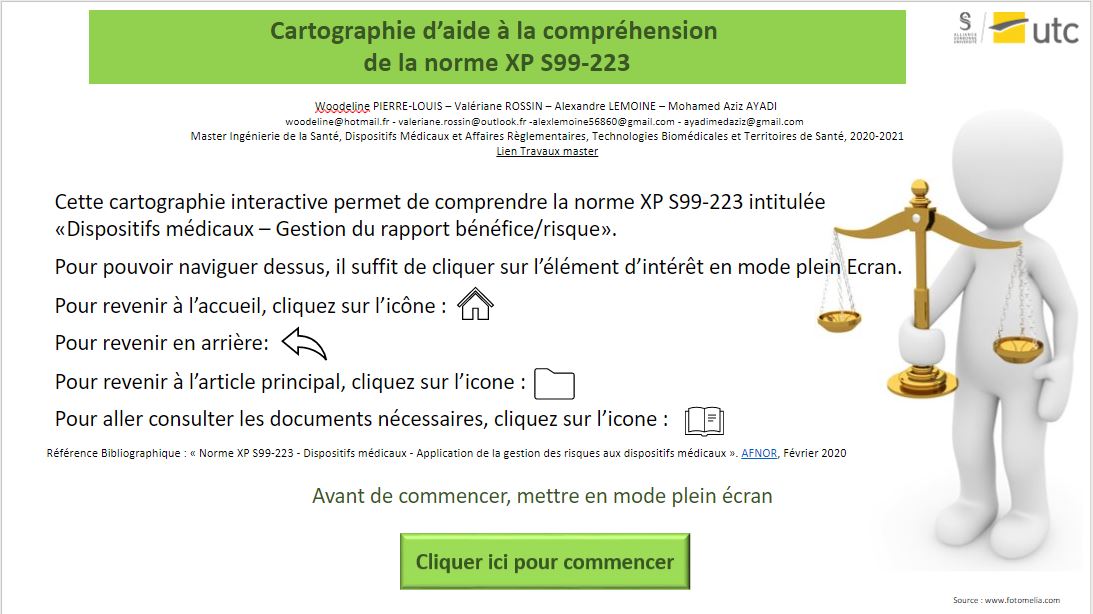
L’interface d’accueil présente la norme ainsi qu’un mode d’emploi (Figure 12).
Figure 12 : Mode d’emploi de la cartographie interactive, Source : auteurs
En appuyant sur le bouton « Cliquer ici pour commencer », l'utilisateur accède aux différents articles de la normes (Figure 13). Pour accéder au détail, il suffit de cliquer sur la zone correspondante et l’utilisateur est automatiquement redirigé vers la page de son choix. Le bouton sous format de balance permet d’accéder à une explication détaillée permettant de comprendre les acteurs et la finalité de cette norme.
Figure 13 : Vue globale de la norme XP S99-223, Source : auteurs
- Vue par article
Il est alors possible, de manière intuitive, d’accéder aux sous-articles (Figure 14) et à une visualisation de leurs sous-parties si existantes.
Figure 14 : Second niveau de profondeur – Article 9.6, Source : auteurs

- Vue par sous-articles
Les critères y sont explicités de manière compréhensible, avec, si besoin, des ressources supplémentaires afin de mieux les assimiler (Figure 15). Cette interface permet également de connaître les documents à fournir si exigée par la norme.
Figure 15 : Troisième niveau de profondeur – Article 9.3, Source : auteurs
Une page récapitulative des documents à fournir est également disponible (Figure 16) :
Figure 16 : Maîtrise documentaire, Source : auteurs
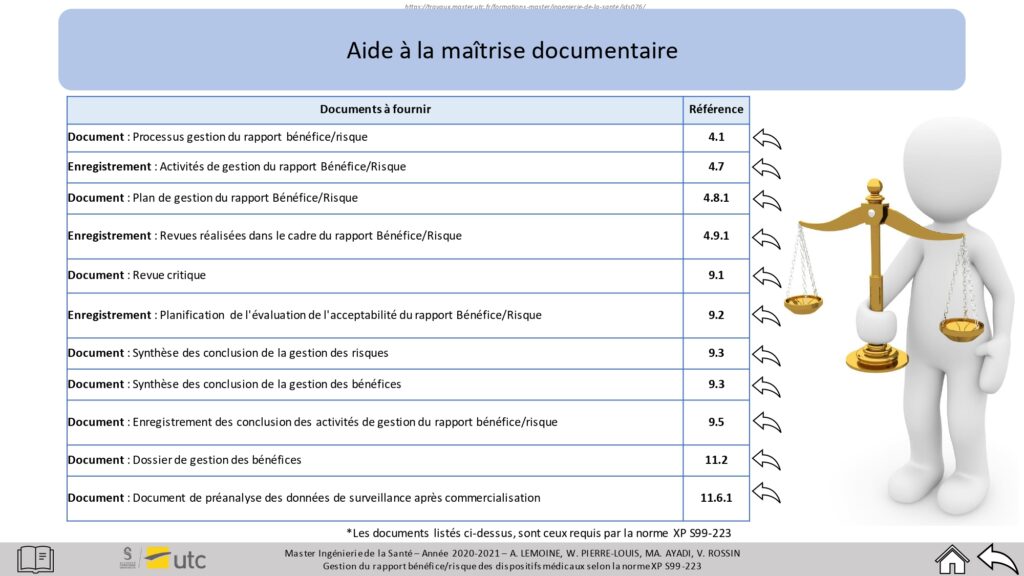
2.2 Outil d’autodiagnostic de la norme XP S99-223
2.2.1 L’outil d’autodiagnostic
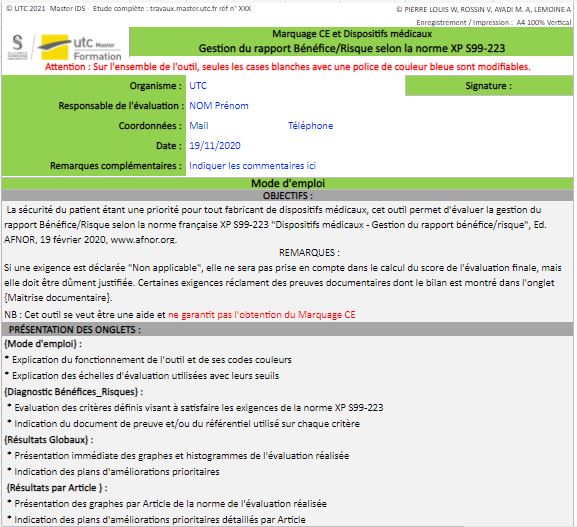
L’outil d’autodiagnostic proposé permettra aux différents fabricants de dispositif médicaux de pouvoir évaluer l’acceptabilité du rapport bénéfice risque de leurs dispositifs médicaux selon la norme XP S99-223. Cela permettra également à celui-ci d’identifier les axes d’améliorations à prioriser. Cet outil se présente sous format Excel et est composé des onglets suivants visibles sur la Figure 17 :
- Mode d’emploi, expliquant le fonctionnement de l’outil ainsi que les échelles d’évaluation utilisées avec leurs seuils ;
- Diagnostic, où sont présenté les exigences essentielles des articles de la norme XP-S99-223 que le fabricant évalue sur 66 critères ;
- Résultats globaux, affichant sous forme graphique les résultats des articles, pour avoir une vision globale de la conformité du fabricant et de viser les articles à améliorer en priorités ;
- Résultats par article, donnant le résultat sous forme graphique de chaque critère de l’article et ainsi pouvoir établir un plan des d’actions prioritaires ;
- Maîtrise documentaire, il fait un bilan et une synthèse des différents preuves documentaires et montre la maîtrise des évolutions ;
- Déclaration 17050, permettant d’éditer une déclaration de conformité selon la norme ISO 17050 et de communiquer son niveau de conformité à la norme XP-S99-223, spécifique à la gestion du rapport Bénéfice/Risque suite à son évaluation.
Figure 17 : Onglets de l’outil d’autodiagnostic, Source : Auteurs

2.2.2 Onglet « Mode d’emploi »
Figure 18 : Métadonnées, Onglet Mode d’emploi, Source : auteurs

Cet onglet de l’outil est clef pour une utilisation correcte. Ainsi, un mode d’emploi clair et précis permet à l’utilisateur de comprendre comment fonctionne l’outil. L’objectif, la présentation des onglets et des remarques pertinentes sur l’outil y sont détaillés (Figure 19).
Dans un souci d’ergonomie, les cases modifiables par l’utilisateurs sont de couleurs blanches avec une police bleue. L’objectif principal est d’attirer l’œil de l’utilisateur. Une partie en haut permet d’annoter les métadonnées relatives à l’évaluation (Figure 18). Ainsi, les données suivantes sont mentionnées : la date d’évaluation, les coordonnées de l’évaluateur et le nom de la société à laquelle il appartient.
Les niveaux de véracité sont indiqués ainsi que les taux de véracité correspondant. Les objectifs de conformité sont modifiables par l’utilisateur afin de l’adapter à ses besoins.
L'utilisateur a la possibilité de modifier les seuils de conformité (Taux moyens minimaux) ce qui permet d'être dans un processus qualité par une amélioration continue des performances et des objectifs de l'organisme utilisant l’outil. Les taux moyens maximaux se calculent automatiquement pour être pertinent dans le diagnostic.
Figure 19 : Extrait de l’onglet « Mode d'emploi », Source : auteurs
2.2.3 Onglet « Diagnostic XP-S99-223 »
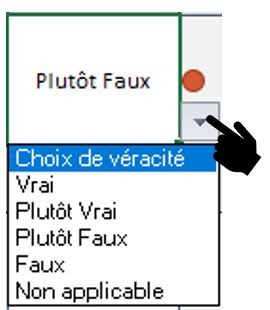
Pour évaluer le rapport Bénéfice/Risque de son dispositif médical le fabricant va devoir répondre à 64 critères. Ces critères regroupent les exigences essentielles présentes dans la norme XP-S99-223. La personne en charge de l’évaluation aura la possibilité de choisir entre les niveaux de véracité suivant (Figure 20) :
- Vrai à L’exigence est respectée ;
- Plutôt Vrai à La conformité à l'exigence est en cours ;
- Plutôt Faux à L'exigence n'est pas respectée ou alors très aléatoirement ;
- Faux à L'exigence n'est pas respectée ;
- Non Applicable à L'exigence est non applicable.
Figure 20 : Choix de véracité, Sources : auteurs
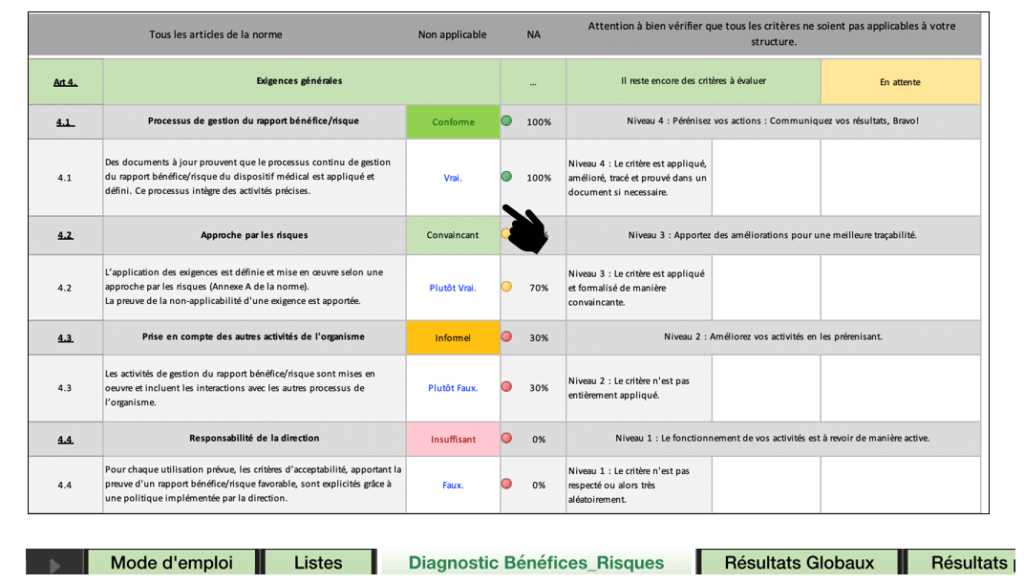
De plus, un code couleur est identifiable en fonction de la véracité d’évaluations des articles, le vert pour ''Vrai'', le vert clair pour "Plutôt vrai", exprimant une non-conformité partielle, l'orange pour "Plutôt faux" exprimant une non-conformité majeure et enfin la couleur rouge pour ''Faux". Une fois l’évaluation de l’article terminée, un pourcentage représentant le taux de véracité est affiché ainsi qu’un niveau de conformité qui lui est associé. Plus l'utilisateur se rapproche du respect de l'exigence plus le taux de véracité sera haut, jusqu'à atteindre 100% quand l'exigence est respectée (Figure 21).
Figure 21 : Extrait de l’onglet « Diagnostic Bénéfices_Risques », Source : auteurs
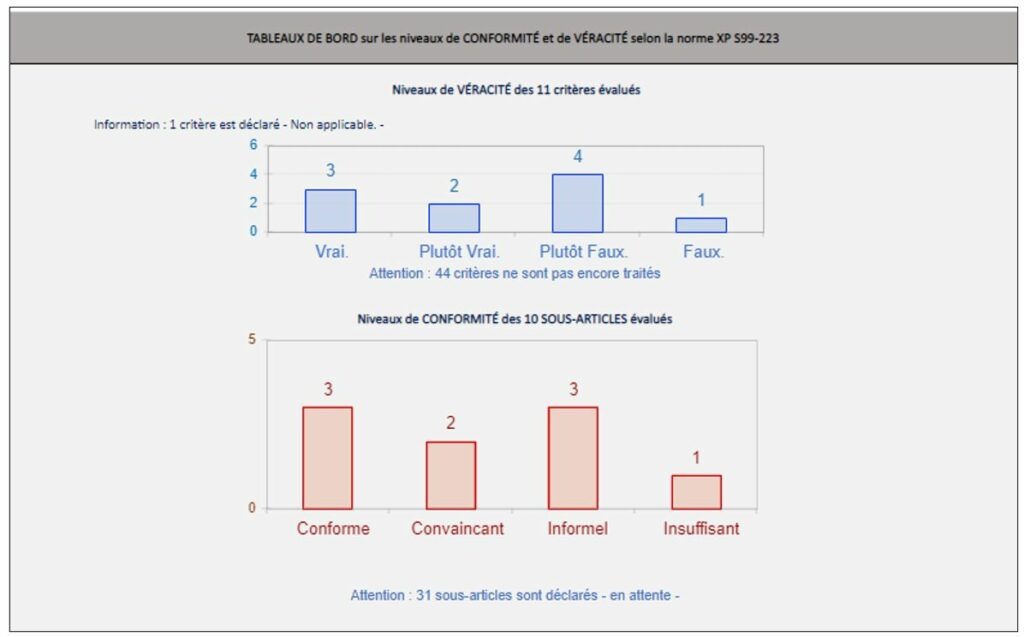
2.2.4 Onglet « Résultats Globaux »
L’onglet des résultats globaux (Figure 22) permet à l’évaluateur une fois son diagnostic terminé, de pouvoir visualiser d’une manière globale et détaillé le niveau de conformité par article. Différents graphiques informent le lecteur de la conformité des critères. Celui-ci peut également apporter des commentaires sur les résultats obtenus en établissant un plan d’actions prioritaires, en indiquant :
- Les objectifs à atteindre ;
- La personne responsable de l’objectif ;
- Le deadline.
Enfin, un tableau récapitulatif expose les sous articles ainsi que leurs niveaux conformités pour que l'utilisateur puisse avoir une vision globale.
Figure 22 : Extrait de l’onglet « Résultats globaux », Sources : auteurs
2.2.5 Onglet « Résultats par articles »
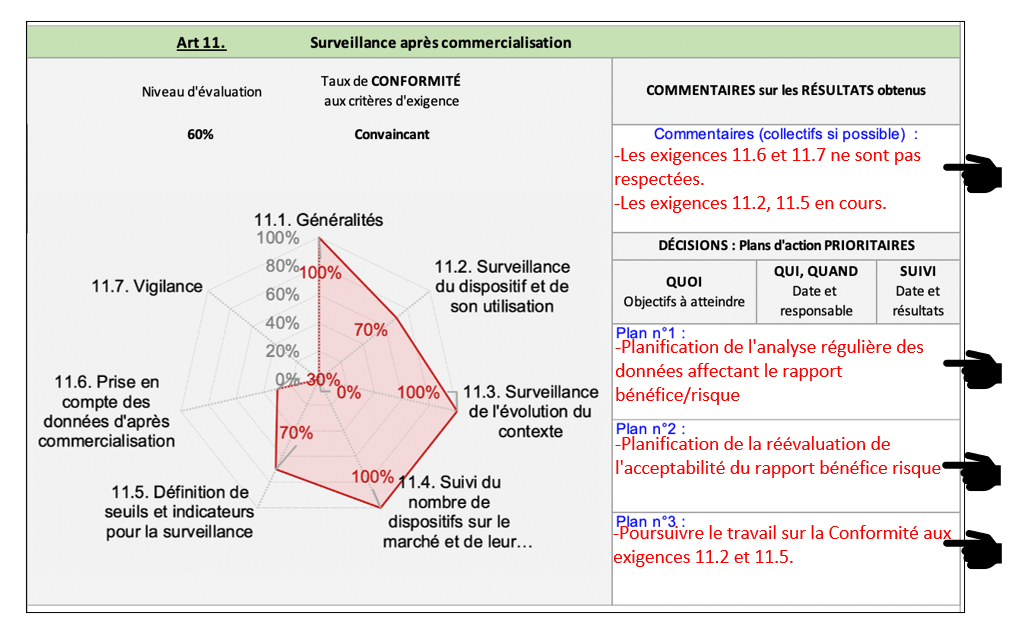
Dans cet onglet les résultats (Figure 23) par articles sont présentés sous forme de graphique radar. Ces graphiques permettent une visualisation directe des points à améliorer, des points où il faut maintenir les efforts et des points à urgence critique. La compréhension du lecteur néophyte est facilitée par un design mettant en exergue les éléments clefs comme le niveau de conformité de l’article ainsi que son intitulé. L’utilisation de cette partie permet également de définir un plan d’action associé à chaque article en déterminant les principaux axes d’amélioration notable.
Figure 23 : Extrait de l’onglet « Résultats par Article », Source : auteurs
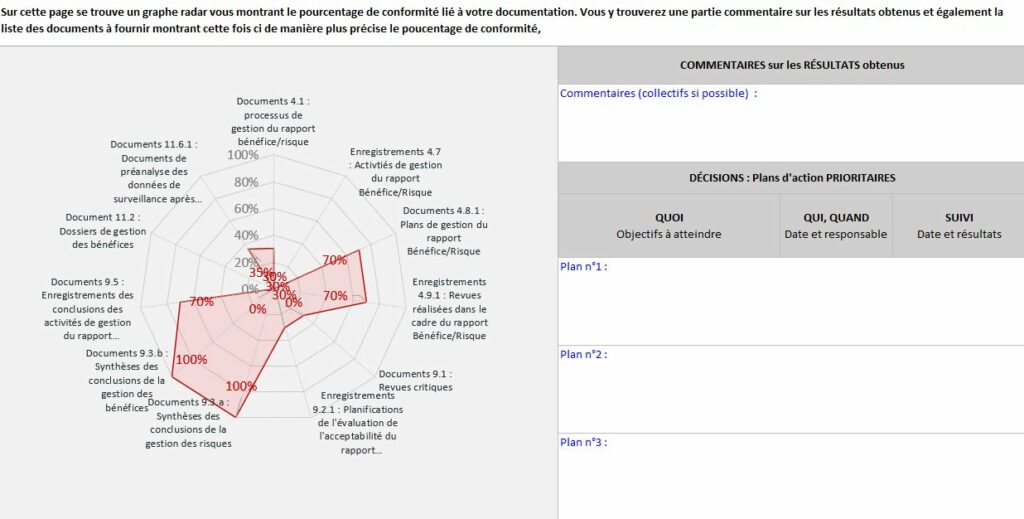
2.2.6 Onglet « Maitrise documentaire »
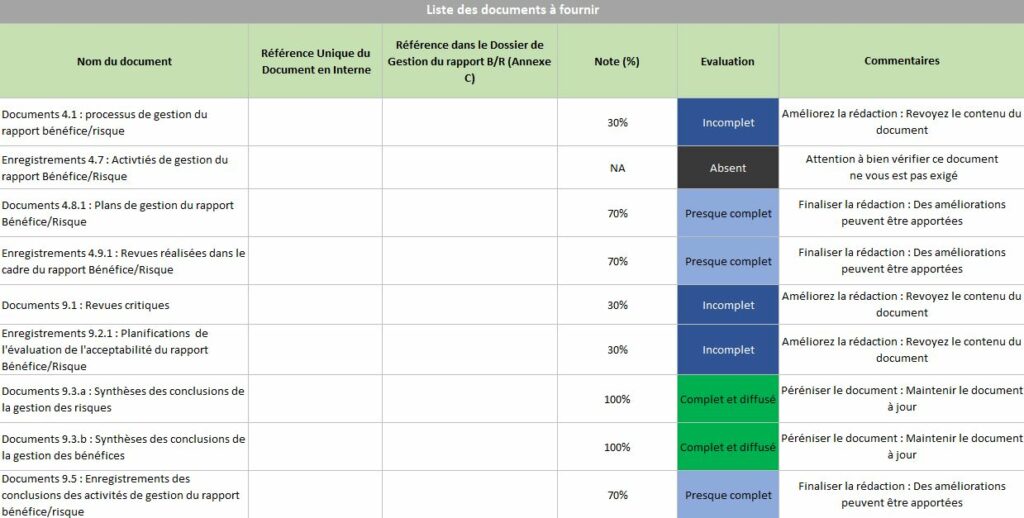
La Maitrise documentaire de l’outil donne une aide à l’organisme (Figure 24). Elle montre les justificatifs documentaires manquants pour être conforme aux exigences des critères liés à la documentation. Grace à un tableau et un graphe radar, l’organisme identifie les problèmes documentaires présents liés à leurs niveaux de véracité et d’acceptabilité. Dans cet onglet, une partie est également réservée pour commenter les résultats obtenus et établir un plan des actions à prioriser. Il est possible d’indiquer les objectifs à atteindre, le responsable des objectifs ainsi qu’une deadline.
Figure 24 : Extrait onglet « Maîtrise documentaire », Source : auteurs
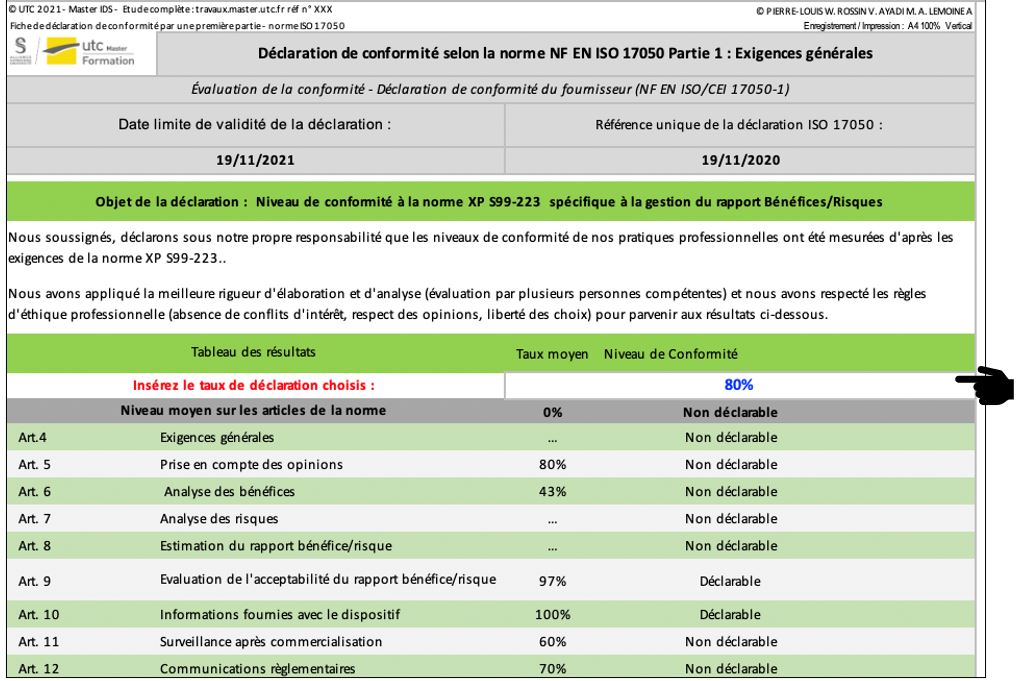
2.2.7 Onglet « Déclaration 17050 »
Cette partie contribue à la réalisation d’une pré-édition de l’auto-déclaration selon la norme ISO 17050 (Figure 25). Cette auto-déclaration est remplie par l’évaluateur dans le but de communiquer ses résultats sur la conformité aux exigences règlementaires de la norme XP S99-223. Si les résultats sont satisfaisants, alors, elle permet uniquement de faire présomption de conformité aux exigences règlementaires applicables. La preuve de conformité est, quant à elle, apportée par une certification. L’utilisateur pourra au préalable choisir le taux de conformité minimum lui permettant de communiquer ses résultats en réalisant cette auto-déclaration.
Figure 25 : Extrait de l’onglet « Déclaration 17050 », Source : auteurs
Conclusion
L’analyse bénéfice/risque, comme décrite dans la norme XP S99-223 intitulée « Dispositifs médicaux – Gestion du rapport bénéfice/risque », est désormais une exigence clé de la règlementation 2017/745. L'évaluation de ce rapport pour un fabricant est donc primordiale s’il souhaite obtenir le marquage CE nécessaire à la mise sur le marché de son dispositif médical.
L’objectif de ce projet est de guider les fabricants dans la compréhension de la gestion du rapport bénéfice/risque.
En effet, les apports essentiels de ce dernier, sont les deux outils présentés dans ce mémoire, accessibles gratuitement sur le web :
- Un outil Excel permettant de s’autoévaluer conformément à la norme bénéfice/risque ;
- Une cartographie interactive guidant les fabricants dans la navigation des différents articles de normes.
Ils apportent des solutions de familiarisation destinées aux fabricants afin de faciliter le respect aux exigences de la norme.
Ces solutions se veulent être ergonomiques, en conformité avec la norme et également rapides à prendre en main par les futurs utilisateurs. In fine, les fabricants sont guidés dans leur processus de gestion du rapport bénéfice/risque. Les notions relatives aux bénéfices et aux risques sont alors mieux maitrisées.
L’incidence positive des dispositifs médicaux permet de réduire les incidents de matériovigilance. A l’aide des solutions développées dans ce projet, les fabricants sont en mesure d’assurer la sécurité de leurs dispositifs médicaux, ce qui a pour conséquence d’améliorer la sécurité du patient.