IDS078 - Les bonnes pratiques de l'expert en affaires réglementaires des dispositifs médicaux
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

ELHARTI Houda 
IDIHYA Kawtar 
BOUSHABA Salma 
ESSAAID Imane
Contacts
Citation
A rappeler pour tout usage : ELHARTI Houda, IDIHYA Kawtar, BOUSHABA Salma, ESSAAID Imane « Les bonnes pratiques de l'expert en affaires règlementaires des dispositifs médicaux », Mémoire de projet, Master Ingénierie de la Santé « IDS », Parcours Dispositifs Médicaux et Affaires Réglementaires (DMAR). Université de Technologie de Compiègne (France), Décembre 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids078/
Résumé
Ce Mémoire d’Intelligence Méthodologique a été réalisé dans le cadre d’une unité d’enseignement nommée « Ingénierie de projet » encadrée par l’Université de Technologie de Compiègne pour le parcours Master dispositif médical et affaires réglementaires.
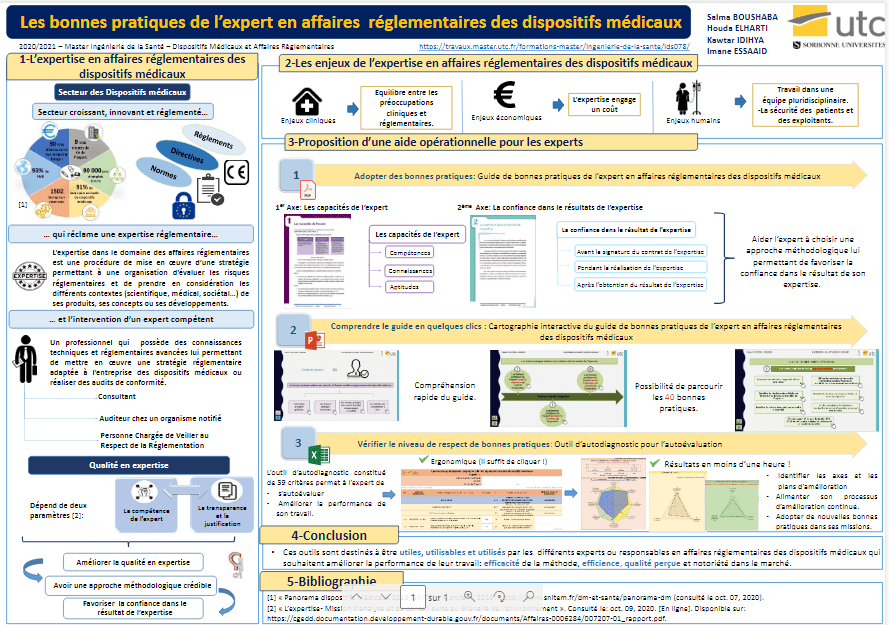
Tous les experts en affaires réglementaires des dispositifs médicaux, notamment les consultants, les personnes chargées de veiller au respect de la réglementation (PCVRR) chez le fabricant et les auditeurs chez un organisme notifié, étant confrontés à l’alimentation de leur processus d’amélioration continue et la livraison d’un rendu de qualité, se doivent de respecter les normes en relation avec la qualité en expertise. Le projet consiste à étudier ces normes afin d’aider les experts en affaires réglementaires et les prescriptions générales de compétence pour une expertise afin d’aider les experts en affaires réglementaires des dispositifs médicaux à aborder une approche méthodologique permettant de favoriser la confiance dans le résultat fourni à leurs clients. Pour cela, trois outils ont été réalisés :
- Un guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux ;
- Une cartographie interactive d’aide à l’utilisation du guide.
- Un outil d’autodiagnostic pour permettre aux experts en affaires réglementaires des dispositifs médicaux d’évaluer leur respect aux bonnes pratiques.
Les trois outils ont pour but de mettre en œuvre facilement les recommandations des normes de la qualité en expertise.
Mots clés : Dispositifs médicaux, affaires réglementaires, expert, qualité en expertise.
Abstract
This Methodological Intelligence Memory was carried out within the framework of a teaching unit called “Project Engineering” supervised by the University of Technology of Compiègne for the Master’s degree course in medical devices and regulatory affairs.
All experts in medical device regulatory affairs, including consultants, persons in charge of ensuring compliance with regulations at the manufacturer’s and auditors at a notified body, being faced with the challenge of feeding their continuous improvement process and delivering a quality rendering, must comply with standards related to quality in expertise.
The project consists in studying the standards to help regulatory affairs experts and the general requirements of competence for an expertise in order to help medical device regulatory affairs experts to have a methodological approach for foster confidence in the result provided to their clients. For this purpose, three tools have been developed :
- A Medical Device Regulatory Affairs Expert Good Practice Guide.
- An interactive map to help users use the guide.
- A self-diagnostic tool to enable medical device regulatory affairs experts to assess their compliance with good practices.
The three tools are designed to easily implement the recommendations of the quality standards in expertise.
Keywords : Medical devices, regulatory affairs, expert, quality in expertise.
Téléchargement

Les bonnes pratiques de l'expert en affaires réglementaires des dispositifs médicaux
Poster

Guide des bonnes pratique de l'expert en affaires réglementaires des dispositifs médicaux

Cartographie d'aide à la compréhension du guide des bonnes pratiques
Outil d'autodiagnostic (basé sur Excel)
Mémoire complet :
Les bonnes pratiques de l'expert en affaires réglementaires des dispositifs médicaux
Introduction
L’expertise dans le domaine des affaires réglementaires des dispositifs médicaux consiste à choisir et concevoir une méthode adaptée aux besoins du client et mettre en œuvre une procédure réglementaire permettant à une entreprise de dispositifs médicaux de se certifier par rapport à une norme, d’obtenir la conformité UE, d’appliquer sa stratégie de mise sur marché d’un dispositif médical et d’évaluer les risques réglementaires relatifs à son domaine d’activité.
Les experts en affaires réglementaires des dispositifs médicaux sont confrontés à plusieurs défis dans l’expertise qu’ils conduisent. Un de ces défis est l’amélioration de l’efficacité de leurs méthodes de travail, l’efficience de leurs processus et donc l’amélioration de la qualité de leurs services et plus précisément dans le résultat de leurs missions. Plusieurs normes et textes réglementaires ont été élaboré pour guider les experts dans leurs activités et pour définir les exigences de compétence pour une expertise.
La problématique qui se pose est alors : comment aider les experts en affaires réglementaires des dispositifs médicaux à adopter des bonnes pratiques dans leurs projets réglementaires en répondant aux exigences normatives.
Ce mémoire d’intelligence méthodologique sera en conséquence rédigé afin d’aider les experts en affaires réglementaires des dispositifs médicaux à pouvoir améliorer leur méthode de travail d’où la favorisation de la confiance dans l’expertise qu’ils fournissent en se référant à un certain nombre de critères. Une synthèse des normes et des exigences sera présentée avec :
- Des alternatives possibles pour anticiper les risques liés au métier de l’expert en affaires réglementaires des dispositifs médicaux ;
- Un guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux
- Une cartographie interactive d’aide à l’utilisation du guide ;
- Et, un outil d’autodiagnostic élaboré pour permettre aux experts en affaires réglementaires des dispositifs médicaux de se situer et d’évaluer leur conformité par rapport aux exigences des normes de la qualité en expertise et donc leur respect aux bonnes pratiques
Chapitre 1 : Contexte et enjeux de l’expertise en affaires réglementaires des dispositifs médicaux
I Contexte
1. Le secteur des dispositifs médicaux
Selon le règlement européen 2017/745, un dispositif médical est tout instrument, équipement, appareil, implant, réactif, logiciel, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- Diagnostic, contrôle, prévention, pronostic, prédiction, traitement ou atténuation d'une maladie
- Diagnostic, traitement, contrôle, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci
- Investigation, modification ou remplacement d'une structure ou fonction anatomique ou état physiologique ou d'un processus ou pathologique
- Communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons de sang, d'organes, et de tissus,
Et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens immunologiques ou pharmacologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens »[1].
En Europe, le secteur du dispositif médical (DM) se divise en différents grands secteurs, celui des DM, des DMIA (implantables actifs) ainsi que celui des DM DIV (diagnostic in vitro) et il est constitué des entreprises (PME) qui conçoivent, fabriquent et distribuent. Ces entreprises développent des technologies variées et font face à des problématiques différentes et c’est grâce à leur rôle actif que l’UE dispose d’un secteur de DM compétitif, innovant et présentant un acteur important de l’économie européenne.
En France, la filière des dispositifs médicaux est très diversifiée et demeure essentiellement composée de PME (93%), et est génératrice d’emplois : près de 90 000 emplois sur le territoire national. Pour cela le secteur du dispositif médical qui compte dans le milieu industriel français est considéré comme un des acteurs majeurs dans les industries de santé [2].
Le secteur du dispositif médical en France affiche une dynamique considérable ; en 2019, le nombre d’entreprises du DM recensées a augmenté de plus de 200 par rapport à 2017 pour atteindre le chiffre de 1502 sociétés avec un chiffre d’affaires de 30 Milliards € [2].
Ce secteur est soutenu par un cadre réglementaire qui vise à assurer le bon fonctionnement du marché intérieur, en prenant pour base un niveau élevé de protection de la santé des patients et des utilisateurs et est soumis plusieurs textes réglementaires :
Directives :
- Directive 90/385/CEE concerne le rapprochement des législations des États membres relatives aux dispositifs médicaux implantables actifs [3].
- Directive 93/42/CEE du Conseil relative aux dispositifs médicaux (DDM) : présente les rôles et les obligations de chaque acteur dans le domaine des dispositifs médicaux [4].
- Directive 98/79/CE du parlement européen et du conseil, relative aux dispositifs médicaux de diagnostic in vitro (DMDIV)[5].
Règlements :
Ils sont entrés en vigueur le 25 mai 2017 pour renforcer la sécurité sanitaire et harmoniser l’application des règles au sein de l’Union européenne. Ces règlements seront d’application obligatoire à partir de Mai 2021.
- Le règlement européen 2017/745 sur les équipements médicaux qui a mis à jour les règles relatives à la mise sur marché et à la mise en service dans l’Union européenne (UE) des dispositifs médicaux destinés à l’homme, ainsi que de leurs accessoires. Il contient aussi les règles portant sur la conduite des évaluations de ces mêmes dispositifs (ou de leurs accessoires) dans l’UE [1] .
- Le règlement européen 2017/746 qui vise la garantie du bon fonctionnement du marché intérieur des dispositifs médicaux de diagnostic in vitro, sur la base d'un niveau élevé de protection de la santé pour les patients ainsi que les utilisateurs [6] .
2. Les affaires réglementaires des dispositifs médicaux
Les affaires réglementaires sont une profession née de la volonté des gouvernements de protéger la santé publique en contrôlant la sécurité et l'efficacité des produits dans différents domaines tels que les produits pharmaceutiques, les médicaments et les dispositifs médicaux.
Du moment où les entreprises responsables de la découverte, des essais, de la fabrication et de la commercialisation des dispositifs médicaux ont voulu s'assurer qu'elles fournissent des produits sûrs et qui apportent une contribution utile à la santé et au bien-être publics ; une nouvelle catégorie de professionnels est apparue pour traiter ces questions réglementaires : Les professionnels des affaires réglementaires. Ces derniers remplissent une fonction essentielle tout au long du cycle de vie du dispositif médical, en menant une stratégie de pré commercialisation de premier plan, en rédigeant des soumissions réglementaires et en assurant la conformité après la mise sur marché[7].
Les affaires réglementaires des dispositifs médicaux sont une industrie qui supervise la façon dont les produits médicaux sont développés, testés, fabriqués, commercialisés et distribués afin de certifier qu'ils répondent aux normes réglementaires pour l'utilisation humaine.
Grâce à une stratégie réglementaire efficace du développement d’un dispositif médical, les problèmes potentiels peuvent être identifiés et corrigés, ce qui permet d'éviter des efforts de correction plus tard, de rationaliser le développement du dispositif et, dans les cas les plus extrêmes, empêcher la nécessité d'une nouvelle conception[8].
3. L’expertise
3.1 Généralités
Le domaine juridique représente le premier domaine qui a connu la naissance de l’expertise à partir de la moitié du XIXe siècle[9].
L'expertise est une méthode souvent utilisée pour solliciter des opinions, des explications et des suggestions afin d’expliquer l'origine d'un événement ou d'une catastrophe, notifier la résolution de conflits et évaluer toute forme de dommage, de biens ou de services. Elle doit être basée sur des preuves et des jugements scientifiques [10]. Elle a l’objectif de donner à tous les décideurs les paramètres dont ils auront besoin : les options avec détermination de leurs risques, leurs coûts et bénéfices, ainsi que les mesures d’accompagnement ou de restriction concevables.
L’expertise peut être conduite selon différentes modalités [10]:
-Expertise individuelle : Réalisée par un seul expert sous sa propre responsabilité.
-Expertise collégiale : Réalisée par un collège d’expertise sous leur responsabilité collective.
-Expertise institutionnelle : conduite par une institution qui fait intervenir un ou plusieurs experts, sous leur propre responsabilité.
3.2 La différence entre expertise et compétence
La compétence définit l’aptitude d’agir et de mettre en œuvre des savoir-faire. Compte tenu de ce qui précède, la différence entre la compétence et l’expertise est que la compétence définit la qualité ou l'état d'être compétent, alors que l’expertise se définit par l’association de l’ensemble de compétences d’un ou de plusieurs experts sur un domaine précis.
3.3 L’expert
C'est un professionnel qui dispose d'une expérience spécifique et qui se caractérise par sa pratique avérée. Ces deux éléments lui permettant d’avoir une large base de données, lui accordent une bonne perception et une rapidité de diagnostic et de résolution et, le rend plus apte que d'autres professionnels à donner des avis ou des conseils précieux aux personnes qui en ont besoin. L’expert doit avoir une expérience élargie et de haut niveau dans un domaine spécifique, c’est la rareté de son expérience qui en fait de lui un expert[11].
Il existe une approche qui présente la définition de l’expert en le comparant avec le non-expert. Cette approche détermine l'expertise comme le niveau de compétence que les non-experts peuvent atteindre. Donc les personnes ayant des niveaux de connaissances élevées seront considérées comme des experts, et les personnes ayant des niveaux de connaissances inférieurs seront considérées comme des non-experts.
Dans le domaine des dispositifs médicaux, chaque fabricant doit disposer d’un ou de plusieurs experts veillant au respect des règlements et des normes d’expertise dans le domaine des dispositifs médicaux[12].
3.4 La qualité de l’expertise
La qualité d’une expertise dépend de deux principaux paramètres :
Compétences de l’expert : selon la norme NF X50-110 « Qualité en expertise-prescriptions générales de compétence pour une expertise », l’expert doit être :
- Honnête, juste et respectueux des exigences de son métier.
- Ouvert d’esprit, pensif, ouvert dans la considération des avis différents mais avec esprit critique.
- Persévérant et adaptable facilement aux situations rencontrées.
- Observateur et capable d’établir un raisonnement et une analyse logique.
- Capable de simplifier des situations complexes sous forme écrite ou verbale [10].
La transparence et la justification : elles sont indispensables pour la qualité de l’expertise parce que d’une part, elles participent à la construction de la confiance et à la favorisation de la bonne compréhension des demandeurs, et d’autre part, ces deux atouts contribuent à la qualité en lisibilité et probité d’où la crédibilité de l’expertise [12].
3.5 La différence entre un expert et un auditeur
Selon la norme NF EN ISO 19011 « Lignes directrices pour l'audit des systèmes de management », l’auditeur a plusieurs connaissances et aptitudes requises pour répondre aux besoins du programme d’audit. Les comportements professionnels des auditeurs souhaités consistent à être :
- Intègres, sincères, attachés à la vérité, honnêtes et discrets.
- Ouverts d’esprit, c’est-à-dire soucieux de prendre en considération des idées ou des points de vue différents
- Observateurs, c’est-à-dire activement attentifs à l’environnement physique et aux activités associées
- Capables de tirer des conclusions fondées sur un raisonnement et analyse logique et prendre des décisions adaptées.
- Capables d’agir de manière responsable et déontologique.
Il existe un ensemble de compétences communes entre l’auditeur et l'expert. En outre, la confiance accordée au processus d'audit, d'expertise et à la capacité de réaliser leur objectif dépend de la compétence de l'auditeur et l'expert. Il convient d’évaluer régulièrement leur compétence au moyen d’un processus tenant compte des comportements personnels et de la capacité à appliquer les connaissances et aptitudes obtenues par leur formation initiale et aussi leur expérience professionnelle [13].
3.6 Les normes liées à l’expertise
Les normes relatives à la qualité en expertise sont :
La norme NF X50-110 « Qualité en expertise - Prescriptions générales de compétence pour une expertise ». Elle définit les exigences générales d'aptitude et de compétence requises pour exercer une expertise, dans le but d’améliorer la maîtrise des points clés de l’expertise et de permettre une reconnaissance de la capacité à conduire des expertises [10].
Le fascicule de documentation FD X50-046 « Recommandations pour l'application de la norme NF X 50-110 ». Il développe l’article 7 « Prescription pour une expertise » de la norme précédentes, pour préciser les modalités de traitement d'une expertise. Il a comme objectif de fournir aux organismes d’expertise des éléments essentiels pour faciliter l'exploitation opérationnelle de la norme NF X50-110 [14].
Le fascicule de documentation FD X50-045 « Utilisations possibles de la norme NF X 50-110 ». Il concerne tous les domaines d’expertise et a pour objet d’illustrer des utilisations possibles de la norme NF X 50-110, de formuler des recommandations pour le choix d’une expertise en fonction des niveaux de garantie souhaités, afin de permettre aux clients de l'expertise et aux organismes d’expertise d’avoir des bases communes pour leurs relations [15].
La norme NF EN 16775 « Services d'expertise — Exigences générales relatives aux services d'expertise ». Elle traite les exigences minimales relatives aux services d'expertise fournis par des experts individuels ou des groupes d'experts, à un client. Son objectif est de normaliser les services d'expertise afin d’amener la réponse la plus précise et la plus fiable possible à une question posée lors de l’expertise [16].
3.7 L’expertise en affaires réglementaires
L’expertise dans le domaine des affaires réglementaires est une procédure de mise en œuvre d’une stratégie permettant à une organisation d’évaluer les risques réglementaires et de prendre en considération les différents contextes (scientifique, médical, sociétal…) de ses produits, ses concepts ou ses développements.
L’expertise dans le domaine des affaires réglementaires des dispositifs médicaux consiste à choisir et concevoir une méthode adaptée aux besoins du client et mettre en œuvre une procédure réglementaire permettant à une entreprise de dispositifs médicaux de se certifier par rapport à une norme, d’obtenir la conformité UE, d’appliquer sa stratégie de mise sur marché d’un dispositif médical et d’évaluer les risques réglementaires relatifs à son domaine d’activité.
En effet, l’expertise en affaires réglementaires des dispositifs médicaux est en relations avec plusieurs parties prenantes dans le domaine de la santé comme par exemple, le patient qui est l’axe principal de cette expertise, les professionnels de la santé, les administrateurs de la santé, les fabricants des dispositifs médicaux et les autorités de la santé.
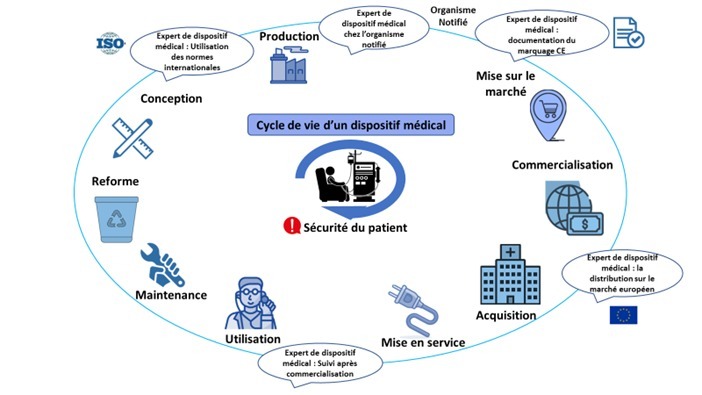
L’expert en affaires réglementaires des dispositifs médicaux intervient dans les différentes étapes du cycle de vie d'un dispositif médical (Figure 1):
Figure 1 : Schéma d'intervention de l'expert pendant le cycle de vie du DM [source auteurs]
II Les enjeux de l’expertise en affaires réglementaires des dispositifs médicaux
Les exigences réglementaires en matière de dispositifs médicaux augmentent actuellement de façon spectaculaire, leur renforcement et complexification ont augmenté les enjeux, le spectre d’intervention des experts et les activités de préparation et de suivi des dossiers réglementaires qui permettent à une entreprise d’être en phase avec les directives et les normes européennes et internationales.
1. Enjeux humains
Les experts en affaires réglementaires doivent mettre en œuvre une stratégie efficace afin de s’assurer que les DM mis sur le marché sont parfaitement conformes aux réglementations en vigueur tout en étudiant et évaluant les éventuels impacts réglementaires d’une modification sur un dispositif médical. De plus, ces experts doivent apporter leur contribution et leur expertise à la négociation des conditions de la mise sur marché des DM, afin que celle-ci se fasse dans les meilleures conditions et dans les meilleurs délais possibles.
L'expertise en affaires réglementaires exige que l’expert soit bien familiarisé avec la gestion de ses relations avec les autorités administratives compétentes et les autres services de l’entreprise, qui nécessite par conséquent des réelles qualités relationnelles et de communication. En outre, la communication professionnelle au cours de l’expertise avec les parties prenantes est essentielle pour assurer la transparence de l’expertise.
En plus, dans son domaine d’activité, l’expert en affaires réglementaires des dispositifs médicaux est à l’interface des différents services de l’entreprise (R&D, production, qualité, vente, Marketing...) ; il est donc amené à travailler dans une équipe pluridisciplinaire et leur apporter le conseil et l’assistance. Aussi, au-delà de la coordination des différents projets à différentes phases d’avancement, il doit veiller à la prise en connaissance et la compréhension des réglementations normatives en vigueur par tous les services.
Cette expertise est aussi dans l’intérêt du patient et de l’utilisateur du dispositif médical. D’une part, elle garantit la fiabilité du dispositif et la protection continue de la santé publique en respectant les exigences de la réglementation et de la performance. D’une autre part, la mission de l’expert à l’égard de la vérification et la validation des manuels d’utilisation des DM l’implique dans l’assurance d’un niveau de sécurité élevé pour l’exploitant final.
2. Enjeux économiques
L’absence de l'expertise en affaires réglementaires des DM dans une organisation a un fort impact économique. En effet, en favorisant l’intégration de cette expertise dans une entreprise des dispositifs médicaux, les pièges et les risques oubliés ou arriérés peuvent être évités ou corrigés, et ceci est équivalent à un gain financier pour l’entreprise.
Ajouter à cela, le chiffre d’affaires augmente en améliorant l’expertise dans le domaine des affaires réglementaires, dans le sens où les experts poussent cette entreprise à aborder de nouveaux marchés internationaux (chine, japon, brésil, USA…). Dans ce cas, si l’expert dispose des éléments clés pour le bon accompagnement de l’entreprise dans toutes les démarches réglementaires, il pourra avoir les autorisations de vente des DM par exemple, ces autorisations de vente présentent un gain d’argent et automatiquement une croissance de chiffre d’affaires de l’entreprise.
Aussi, lorsque les responsables des affaires réglementaires utilisent leur expertise pour obtenir un marquage CE et le maintenir à jour, ceci va permettre de faire circuler librement le DM en Europe et de le garder dans le marché, ainsi l’entreprise va constituer un gage de confiance et avoir une notoriété dans le secteur des dispositifs médicaux, ce qui génère un profit économique à l’entreprise.
3. Enjeux cliniques
L’enjeu majeur de l’expertise en affaires réglementaires des dispositifs médicaux est d’atteindre l’équilibre entre les préoccupations cliniques et réglementaires et disposer des preuves cliniques approfondies sur la sécurité, la fonctionnalité et les performances d’un dispositif médical.
En effet, pour les démarches avant et post commercialisation d’un DM, un accompagnement réglementaire est primordial. Il s’agit du contrôle et de la vérification de la conformité des études cliniques aux règlements et politiques applicables, cela se concrétise par les évaluations, les enquêtes cliniques et la préparation de l’ensemble des demandes réglementaires spécifiques : autorisations d’essais cliniques et technologiques, le rapport d’évaluation clinique, le plan de suivi clinique après commercialisation, autorisation de mise sur marché (AMM), dossier de marquage CE.
L’évaluation et la gestion des incidents de vigilance sanitaire s’inscrivent parfaitement dans les principaux enjeux de l’expertise en affaires réglementaires. En effet, pour promouvoir le bon usage du produit et garantir la sécurité de l'utilisateur, il faut diagnostiquer et anticiper les risques cliniques liés à l’exploitation d’un dispositif médical et formuler des recommandations utiles à toutes les parties prenantes.
Chapitre 2 : Problématique et risques de l’expertise en affaires réglementaires des dispositifs médicaux
I Problématique
1.1 L’expert au cœur de son métier
L’expert en affaires réglementaires des dispositifs médicaux peut être :
- Un expert dans un organisme notifié : Chargé de la réalisation des audits de conformité dans le cadre des directives européennes actuelles et les futures réglementations.
- Un expert consultant : Chargé d'accompagner le fabricant de dispositifs médicaux dans la stratégie réglementaire, le marquage CE, la construction ou l’amélioration du système qualité et même les formations aux différentes exigences actuelles du secteur de dispositif médical.
- Une personne chargée de veiller au respect de la réglementation chez un fabricant selon l'article 15 des nouveaux règlements européens 2017/745 pour les DM et 2017/746 pour les DM DIV : Chargé de veiller au respect de la réglementation des DM et des DM DIV(diagnostic in vitro) pour garantir la conformité des produits mis sur le marché en EUROPE.
Son expertise se manifeste par le choix et la conception d’une méthode adaptée aux besoins du client et la mise en œuvre d'une procédure réglementaire permettant à une entreprise de dispositifs médicaux de se certifier par rapport à une norme, d’obtenir la conformité UE, d’appliquer sa stratégie de mise sur marché d’un dispositif médical et d’évaluer les risques réglementaires relatifs à son domaine d’activité.
2. La qualité en expertise
Indépendamment de son profil, la qualité en expertise proposée par l’expert en affaires réglementaires des dispositifs médicaux dépend de plusieurs paramètres. En effet, l’expert doit identifier et analyser le besoin du client et formuler une méthode adaptée permettant de répondre au mieux à sa question et pour cela cet expert doit avoir un certain nombre de compétences et de qualifications lui permettant de conduire son travail en préservant une indépendance des jugements. Aussi, pour favoriser la confiance dans le résultat de l’expertise et livrer une meilleure qualité au client, l’expert doit avoir une manière de conduite de la problématique, basée sur un ensemble de méthodes utiles et éprouvés ou ce qu’on appelle une « approche méthodologique ».
3. La pertinence d’une approche méthodologique
L’approche méthodologique doit être robuste et crédible pour cela il faut qu’elle respecte les exigences de la transparence, la traçabilité et l’implication dans un contexte qualité.
En effet, grâce à cette approche, l’expert en affaires réglementaires des dispositifs médicaux peut contribuer à l’évolution et à l’alimentation du processus d’amélioration continue mis en place pour la conduite de sa mission d’expertise au sein d’un organisme. Cette approche donc, agit sur la performance d’où l’efficacité, l’efficience et la qualité perçue du travail de l’expert (figure 2).
Figure 2 : Pertinence de l'approche méthodologique [source auteurs]

II Les risques liés à l’expertise en affaires réglementaires des dispositifs médicaux
Comme tout autre domaine, il y’a toujours des risques liés au métier notamment concernant la responsabilité en rapport avec l’activité exercée. Pour cela, être conscient des risques de son activité et pouvoir les anticiper sont des obligations de l’expert en affaires réglementaires des dispositifs médicaux dans le but d’améliorer et maintenir la performance et garantir un niveau de sécurité élevé pour le patient, qui est au cœur des préoccupations cliniques réglementaires, tout au long de la phase de réalisation de l’expertise.
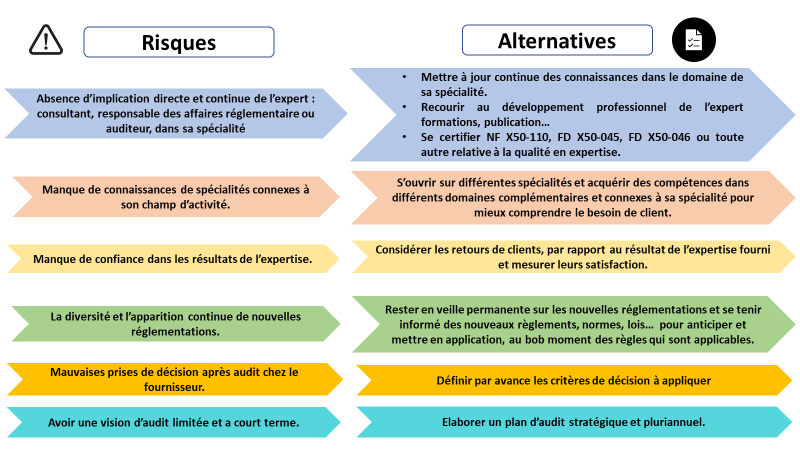
La figure ci-dessous présente les risques en rapport avec l’expertise en affaires réglementaires des dispositifs médicaux et les alternatives possibles (figure 3).
Figure 3 : Risques et alternatives du métier de l’expert en affaires réglementaires des dispositifs médicaux [source auteurs]
Chapitre 3 : Proposition d’une aide opérationnelle pour les experts
Afin d’aider chaque expert en affaires réglementaires des dispositifs médicaux de bien développer ses pratiques liées à la qualité de son expertise, une aide opérationnelle basée sur plusieurs outils praticables va être proposé à l’attention des experts.
I Une enquête à l’attention des experts
a/ Contexte
Afin d'évaluer l’existant et dans le but de proposer des solutions praticables, un questionnaire sur le site Survey Monkey a été soumis à des professionnels en affaires réglementaires des dispositifs médicaux par mail et partagé sur le réseau professionnel LinkedIn.
Ce questionnaire qui demeure l’outil essentiel pour les enquêtes, il a comme but principal de bien savoir l’état actuel du travail des experts en affaires réglementaires des dispositifs médicaux, afin de voir quels sont les avantages et les limites de chaque outil proposé vis-à-vis des réponses.
Le profil de l'expert y est renseigné en s’intéressant à son domaine : expert consultant, expert chez le fabricant ou expert chez l’organisme notifié.
En outre, ce questionnaire destiné aux professionnels de l’expertise en affaires réglementaires des dispositifs médicaux se concentre sur les six axes pertinents ci-dessous :
- Pourcentage d’experts connaissant ou appliquant les normes liées à l’expertise en affaires réglementaires des dispositifs médicaux : afin de lister tous les normes utilisées et appliquées dans ce domaine.
- Les difficultés rencontrées lors de l’application de ces normes : Pour identifier tous les défis auxquels sont confrontés les professionnels en affaires réglementaires des dispositifs médicaux en rapport avec l'application des normes qui devront être appliquées.
- Pourcentage d’experts connaissant ou appliquant les normes liées à la qualité en expertise : Dans le but d’identifier le niveau de connaissance et d’application de ces normes dans le domaine des affaires réglementaires des dispositifs médicaux.
- Méthodes utilisées pour considérer les retours des clients par rapport au travail fourni : afin de déterminer les différentes alternatives possibles pour envisager et gérer les retours défavorables et les réclamations des clients.
- L'existence et l’utilisation d’un outil pour évaluer le travail de l’expert : Pour s’informer de toutes les techniques existantes utilisées dans le processus de positionnement et d’évaluation du travail de l’expert par lui-même.
- L'utilité et les foncions attendus de l’outil d’autodiagnostic et du guide des bonnes pratiques.
b/ Résultats
Ce questionnaire a été envoyé à 115 professionnels dans le domaine des affaires réglementaires des dispositifs médicaux.
Seulement 26 réponses sur l'ensemble des professionnels contactés (Consultants affaires réglementaires, responsables affaires réglementaires, Directeurs qualité et affaires réglementaires) ont été reçues dont 13 sont complètes.
Afin de répondre aux axes cités au-dessus, les réponses des professionnels ont été présentés comme suit :
- La connaissance de différentes normes pertinentes appliquées dans le domaine des affaires réglementaires des dispositifs médicaux, tel qu’ISO 13485, ISO 14971, ISO 60601-1, ISO 16142, etc... Ainsi que l’application des directives 93/42/CEE des dispositifs médicaux et 98/79/CEE des dispositifs médicaux de diagnostic in vitro et du règlement européen 2017/745.
- Certains experts font face à de nombreuses difficultés lors de l’application et la compréhension de ces normes comme : le manque de formation et d'information par rapport à ces normes, la difficulté à définir les normes récentes appliquées (l’état de l’art), le manque de guide d’application, la difficulté d'interprétation de certaines exigences, les incohérences entre les normes et les règlements, les écarts d'interprétation entre les différents pays pour une même norme, l'existence des guides non claires pour l'harmonisation des interprétations, le manque d’expertise.
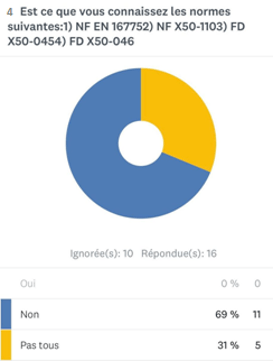
- Sur 13 réponses, 69% des experts ne connaissent pas les normes liées à la qualité en expertise (annexe 1).
- Les réclamations et les retours des clients sont pris en compte par : des systèmes de traitement des réclamations, des bilans d’année, les registres et les fiches de réclamation.
- Les experts en affaires réglementaires des dispositifs médicaux utilisent de nombreuses approches méthodologiques et des outils afin d’évaluer leur travail : les enquêtes et les formulaires de satisfaction, les indicateurs de performance et les audits externes et internes.
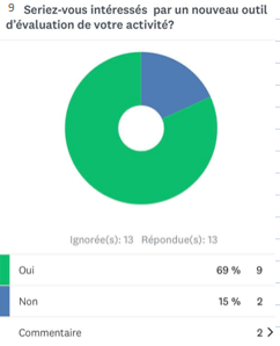
- Sur 13 réponses, 69% des experts sont intéressés par l’élaboration d’un nouvel outil d’autodiagnostic pour l’évaluation de leur travail (annexe 2).
c/ Analyse
Le manque de connaissance des normes liées à la qualité en expertise est dû au manque d’informations, à la difficulté d’identifier les normes applicables ainsi qu’à la domination des autres normes relatives au domaine des dispositifs médicaux comme l’ISO 13485 liée au management de qualité, l’ISO 14971 liée à la gestion des risques aux dispositifs médicaux et bien d’autres normes. Ce taux de connaissance peut favoriser le recours à un guide à la destination des experts qui va leur permettre de bien identifier les bonnes pratiques liées l’expertise dans leur profession. En outre, l’utilisation d’un outil d’autodiagnostic est nécessaire afin d’évaluer leur travail par rapport aux exigences réglementaires relatives à la qualité en expertise.
D’après tout ce qui précède, l’amélioration de la qualité de l’expertise en affaires réglementaires des dispositifs médicaux repose sur :
- La disposition et la mise à niveau des capacités de l’expert :
La capacité d’un expert se traduit par son aptitude et son efficience en termes de mobilisation de ses compétences, ses connaissances et son savoir être, dans la réponse à une situation professionnelle donnée.
Donc, l’expert en affaires réglementaires des dispositifs médicaux doit avoir une combinaison de connaissances et aptitudes (savoirs et savoir-faire) lui permettant de mener un projet réglementaire en respectant la législation en vigueur, superviser ou réaliser les dossiers techniques de l’entreprise et effectuer une veille réglementaire sur les dispositifs médicaux dans le pays de commercialisation. Aussi, cet expert doit certainement disposer des savoirs-être qui se traduisent dans son autonomie dans la conduite de son projet, son esprit d’analyse et de raisonnement, son sens de responsabilité et son attitude de communication en interne et en externe avec les parties intéressées.
En plus, une mise à niveau de ces capacités est essentielle pour tout expert souhaitant élargir son champ d’activité, améliorer son efficacité professionnelle et gagner de la performance dans ses missions d’audit ou technico-réglementaires.
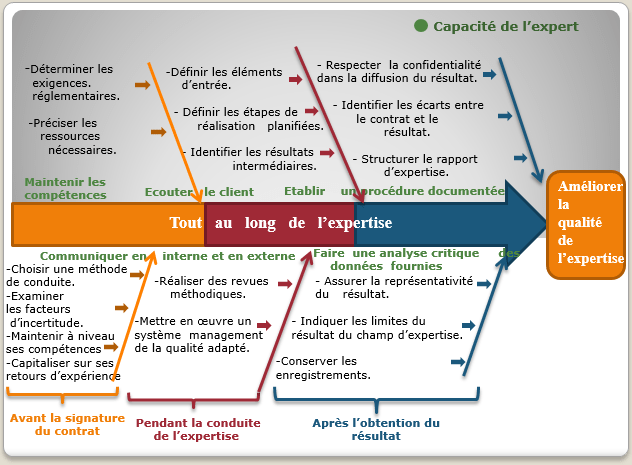
- La confiance dans le résultat de l’expertise :
La relation entre l’expert et son client relève de la confiance et plus précisément dans le résultat de l’expertise. En effet, cette confiance s’établit durant toutes les étapes de réalisation de l’activité de l’expertise ; l’expert doit analyser la question du client avec toute objectivité, en se basant sur des preuves tangibles, et en conservant les enregistrements des résultats.
Figure 4 : Visualisation des critères de l’amélioration du processus de l’expertise [source auteurs]
II Outils proposés
Les experts ont besoin de suivre des méthodes crédibles, et efficaces pour améliorer leur capacité et donner des preuves qui donnent la confiance dans les résultats qu’ils fournissent tout au long de la démarche de l’expertise. Dans ce cadre, l’élaboration d’un guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux et une cartographie pour la compréhension de ce guide ainsi que le développement d’un outil d’autodiagnostic pour l’évaluation des activités de l’expert constitueront une aide accompagnatrice aux personnes chargées du respect de la réglementation pour améliorer la reconnaissance de leurs métiers auprès de leurs clients, et pour augmenter la confiance dans leurs résultats.
1. Guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux
Le développement rapide en termes de normes et de réglementations ainsi que la complexité des sujets abordés dans le domaine des affaires réglementaires des dispositifs médicaux posent aux personnes chargées de la veille réglementaire un défi d’appropriation et d’intégration de nouvelles connaissances dans leurs pratiques. Par l’élaboration d’un guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux, des finalités particulières basées sur différentes préoccupations sont visées. Ces préoccupations comprennent :
- La réduction des variations dans les pratiques de l’expert en affaires réglementaires des dispositifs médicaux.
- Le contrôle de l’usage inapproprié ou inefficace des référentiels réglementaires
L’objectif principal de l’élaboration du guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux est d’aider cet expert à choisir une approche méthodologique lui permettant de favoriser la confiance du client dans le résultat de son expertise.
Ce guide est un travail de synthèse réglementaires d’un ensemble de normes relatives à la qualité en expertise et des articles 15 des deux règlements européens :
- La norme NF X50-110 relative aux prescriptions générales de compétence pour une expertise.
- La norme EN 16775 relative aux exigences générales du service de l’expertise.
- La norme FD X50-045 relative aux utilisations possibles de la norme NF X 50-110.
- La norme FD X50-046 relative aux recommandations pour l'application de la norme NF X50-110.
- Le règlement européen 2017/745 sur les dispositifs médicaux.
- Le règlement européen 2017/746 sur les dispositifs médicaux de diagnostic in vitro.
La synthèse de ces textes réglementaires a permis de tirer un nombre considérable de critères de bonnes pratiques dont ceux cités dans la figure 4. Ces critères ont été divisés en 2 parties : des critères relatifs aux capacités de l’expert en affaires réglementaires des dispositifs médicaux et d’autres concernant la confiance dans le résultat de l’expertise.
Sur la base de cette répartition, les axes du guide ont été définis :
- Le premier axe du guide comprend 25 bonnes pratiques relatives aux capacités de l’expert. Ces bonnes pratiques sont présentées de la manière suivante dans le guide :
- Des bonnes pratiques générales présentant les valeurs que l’expert doit démontrer sur différentes dimensions : le professionnalisme, le service du client, la transparence, la confidentialité.
- Des bonnes pratiques techniques telles que l’étude de faisabilité technique et réglementaire du projet du client et la réalisation des revues et des bilans de conduite de l’expertise.
- Les bonnes pratiques pour la construction d’un réseau pour le cas d’un consultant.
- Les bonnes pratiques pour maintenir les compétences à jour.
- Le deuxième axe traite les éléments qui permettent de donner confiance dans les résultats fournis par l’expert et il contient 15 bonnes pratiques divisées en trois sous axes :
- Avant la signature du contrat de l’expertise.
- Pendant la conduite des missions d’expertise.
- Après l’obtention du résultat comme le certificat de marquage CE, une autorisation de mise sur marché, le plan de suivi après commercialisation, etc….
2. Cartographie interactive du guide de bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux :
Afin de faciliter la compréhension du guide pour des utilisateurs novices ou pour des experts qui n’auront pas assez de temps pour lire tout le guide, une cartographie d’aide à la compréhension du guide a été réalisée. Ceci dans le but de faciliter l’identification des bonnes pratiques de l’expert en affaires réglementaires des dispositifs médicaux. En effet, il s’agit d’un outil interactif qui donne la possibilité de parcourir l’ensemble des bonnes pratiques.
La cartographie est constituée des mêmes axes du guide de bonnes pratiques et elle est utilisée de la manière suivante :
Il suffit de cliquer sur les icônes spécifiées dans le mode d'emploi, pour naviguer dans la cartographie
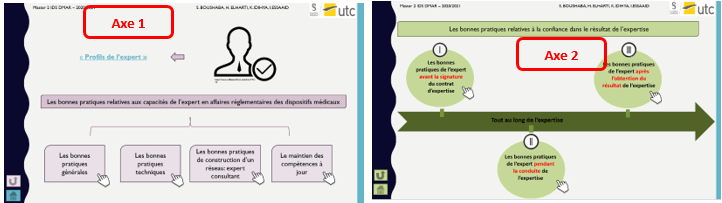
Les bonnes pratiques sont divisés en deux grands axes qui se présentent comme suit (figure 5):
Figure 5 : Les axes de la cartographie interactive [source auteurs]

Il est possible aussi d’obtenir les sous catégories de bonnes pratiques de chaque axe (figure 6):
Figure 6 : Représentation détaillée des 2 axes de la cartographie [source : auteurs]
Pour accéder aux bonnes pratiques, il suffit de cliquer sur la sous-catégorie (figure 7) :
Figure 7 : Visualisation des bonnes pratiques d’un axe [source : auteurs]

En cliquant ensuite sur la bonne pratique, il est possible d’obtenir plus d’informations qui permettent d’expliciter sa pertinence (figure 8) :
Figure 8 : Visualisation détaillée d'une bonne pratique [source : auteurs]

3. Outil d’autodiagnostic pour l’évaluation des activités de l’expert
L’outil d’autodiagnostic est un élément central qui va permettre à chaque personne qui travaille dans le domaine des affaires réglementaires des dispositifs médicaux d’évaluer son travail, de comparer sa conformité par rapport aux exigences réglementaires relatives à la qualité en expertise, d’identifier les capacités requises pour un expert en affaires réglementaires des dispositifs médicaux, et ainsi reconnaître les points critiques et les limites de ses activités.
Sous format Excel, l’outil permet de regrouper l’ensemble des exigences des normes liées à la qualité en expertise d’une manière claire sous forme de critères de bonnes pratiques, et de les décomposer par la suite en deux grands axes précis. En se basant sur le résultat de l’outil d’autodiagnostic, l’expert a la possibilité d’établir un plan d’action pour améliorer la performance de son travail.
Cet outil d’autodiagnostic est simple à utiliser, dynamique et il offre une évaluation rapide et efficace axe par axe. Il contient 5 onglets : le mode d’emploi, la grille d’évaluation, les résultats globaux, les résultats par axe et une déclaration ISO 17050 selon le guide (figure 9).
Figure 9 : Les onglets de l'outil d'autodiagnostic [source : auteurs]
Tableau 1 : Description détaillée des onglets de l'outil d’autodiagnostic [source : Auteurs]

Les différents onglets comportent un en-tête demandant des informations à remplir par l’expert (figure 10) : identité de l’expert, de l’organisme d’expertise, ainsi que leurs coordonnées.
Figure 10 : « En-tête » d’un onglet de l’outil d’autodiagnostic [source : auteurs]

Dans le mode d’emploi en figure 11, les seules cases modifiables par l’expert ont un fond blanc et une police bleu vif, standard défini dans l’objectif de capter l’attention lors de l’usage. Les objectifs du taux de respect peuvent être personnalisés dans l'onglet “Mode d’emploi” en fonction de l’objectif de conformité visé.
Figure 11 : Onglet du mode d'emploi de l'outil d'autodiagnostic [Sources auteurs]

L’évaluation se déroule autour de 59 critères de bonnes pratiques tirés des articles 15 des règlements européens (2017/745, 2017/746) et des normes liées à la qualité en expertise. Ils sont représentés sous forme de phrases affirmatives pour lesquelles l’utilisateur (expert consultant, chargé/responsable des affaires réglementaires chez le fabricant, expert auditeur) donne son évaluation. Ils sont répartis en catégories : capacités de l'expert et la confiance dans le résultat de l'expertise, et bien structurés pour faciliter leur compréhension et leur exploitation (figure 12).
L’axe de la capacité de l’expert est composé de 3 sous axes :
- Connaissances de l’expert.
- Compétences de l’expert.
- Aptitudes de l’expert.
L’axe de la confiance dans le résultat de l'expertise est composé aussi de 3 sous axes :
- Avant la signature du contrat.
- Dans la conduite de l’expertise.
- Après l’obtention de l’expertise.
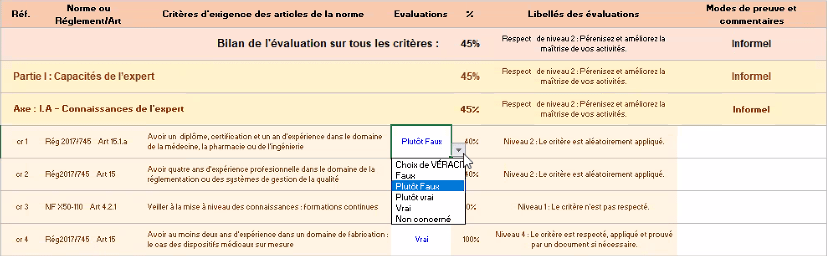
L’évaluateur doit alors choisir entre les différents niveaux de véracité :
- Vrai : le critère est respecté et appliqué
- Plutôt Vrai : le critère est appliqué, mais des améliorations sont possibles)
- Plutôt Faux : le critère n’est respecté que partiellement
- Faux : le critère n’est pas respecté du tout
- Non-concerné : l’expert n’est pas concerné par ce critère.
Figure 12 : Onglet d’évaluation de l’outil d’autodiagnostic [source : auteurs]
Pour donner suite à l’évaluation, deux onglets permettent d’obtenir une appréciation globale et détaillée des résultats : résultats globaux et résultats par axe ainsi que des graphiques permettent d'illustrer le respect des différents critères de bonnes pratiques et de rendre l’information rapidement lisible par un lecteur candide.
L’onglet « résultats globaux » permet d’obtenir un graphique global du respect des critères des 6 axes, une visualisation claire des niveaux de véracité des 59 critères (figure 13).
Figure 13 : Onglet des résultats globaux de l’outil d’autodiagnostic [source : auteurs]

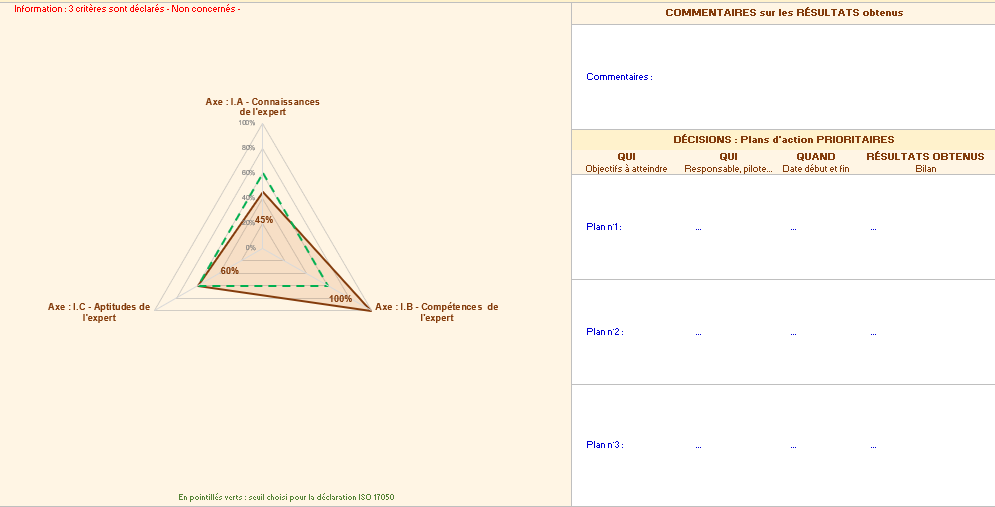
L’onglet « résultats par axe » permet d’obtenir deux graphiques, axe par axe détaillant le respect de chaque axe. Il est possible d’utiliser les feuilles pour définir des plans d’actions en identifiant des axes d’améliorations (figure 14).
Figure 14 : Onglet des Résultats par axe de l’outil d’autodiagnostic [source : auteurs]
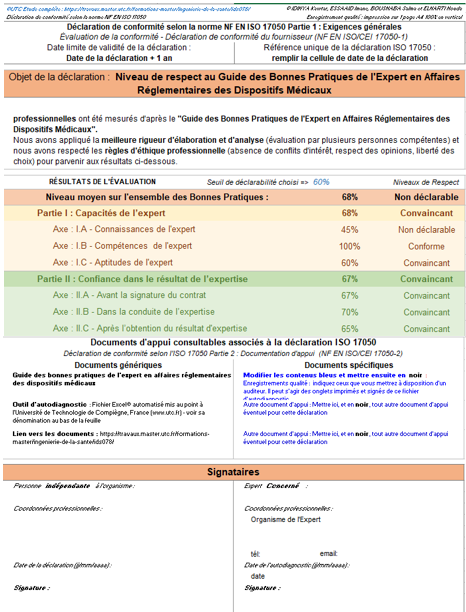
Enfin l’onglet « Déclaration ISO 17050 » permet de réaliser une auto déclaration selon le guide des bonnes pratiques qui est déjà indiqué dans la partie précédente. Cette auto déclaration est réalisée en fonction des résultats de l’évaluation, l'expert a la possibilité aussi de choisir son seuil de déclarabilité. En cas de conformité satisfaisante à tous les axes, l’auto déclaration est un moyen de preuve permettant d’identifier le niveau de respect des bonnes pratiques tirées des normes liées à la qualité en expertise et qui sont déjà traitées dans le guide des bonnes pratiques (figure 15).
Figure 15 : Onglet de la déclaration ISO 17050 selon le guide [source : auteurs]
Conclusion
L’expert en affaires réglementaires des dispositifs médicaux a de plus en plus besoin d’alimenter son processus d’amélioration continue en adoptant une méthodologie crédible lui permettant de donner confiance dans le résultat de l’expertise qu’il fournisse. Pour cela, il a besoin d’utiliser des référentiels pertinents en relation avec la qualité en expertise.
Avec ce mémoire d’intelligence méthodologique et l’ensemble des outils développés, l’expert en affaires réglementaires dans tous les profils : personne chargée de veiller au respect de la réglementation, expert consultant et expert auditeur, a la possibilité d’intégrer l’ensemble des exigences des normes relatives à la qualité en expertise, dans sa méthode de travail, sous forme de bonnes pratiques. Aussi, cet expert peut vérifier son respect à ces bonnes pratiques et mettre en œuvre un plan d’action pour améliorer ses pratiques et favoriser la confiance dans le résultat de son expertise.
Certains points ont pu être améliorés et d’autres nécessitent encore plus de travail pour être mis en place. Aussi, il est nécessaire d’avoir plus d’avis des experts sur l’utilisation des outils.
Références bibliographiques
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra.
[2] « Panorama dispositifs médicaux 2019, SNITEM », snitem, 2019. https://www.snitem.fr/dm-et-sante/panorama-dm (consulté le oct. 07, 2020).
[3] « Directive 90/385/CEE du Conseil, du 20 juin 1990, concernant le rapprochement des législations des États membres relatives aux dispositifs médicaux implantables actifs », Journal officiel de l’Union européenne, Document 31990L0385, Journal officiel n° L 189 du 20/07/1990 p. 0017-0036. [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1990/385/oj/fra.
[4] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux », Journal officiel de l’Union européenne, Document 31993L0042, Journal officiel n° L 169 du 12/07/1993 p. 0001-0043. [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1993/42/oj/fra.
[5] « Directive 98/79/CE du Parlement européen et du Conseil du 27 octobre 1998 relative aux dispositifs médicaux de diagnostic in vitro », Journal officiel de l’Union européenne, Document 31998L0079, Journal officiel n° L 331 du 07/12/1998 p. 0001-0037. [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1998/79/oj/fra.
[6] « Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/746/oj/fra.
[7] Jeff Shiffman, « Le rôle critique des affaires réglementaires dans l’industrie des dispositifs médicaux », Northeastern University Graduate Programs, mars 13, 2018. https://www.northeastern.edu/graduate/blog/regulatory-affairs-in-medical-device-industry/ (consulté le oct. 07, 2020).
[8] « La stratégie réglementaire des fabricants des dispositifs médicaux », www.tuvsud.com. https://www.tuvsud.com/en/industries/healthcare-and-medical-devices/medical-devices-and-ivd/regulatory-strategy (consulté le oct. 07, 2020).
[9] G. Calafat, « Expertise et compétences », Hypotheses, vol. 14, no 1, p. 95‑107, juin 2011.
[10] « Norme NF X50-110-Qualité en expertise - Prescriptions générales de compétence pour une expertise », Ed. Afnor, Paris, www.afnor.org, mai 01, 2003.
[11] « Qu’est ce qu’un expert ? », topformation.fr. https://www.topformation.fr/guide/articles/expert-technique-management-animation-12367 (consulté le oct. 09, 2020).
[12] « L’expertise- Mission d’analyse et de conseil suite au Grenelle de l’environnement ». Consulté le : oct. 09, 2020. [En ligne]. Disponible sur : https://cgedd.documentation.developpement-durable.gouv.fr/documents/Affaires-0006284/007207-01_rapport.pdf.
[13] « norme NF EN ISO 19011 - Lignes directrices pour l’audit des systèmes de management (Tirage 2 (2018-08-14)) », Ed. Afnor, Paris, www.afnor.org, juill. 04, 2018.
[14] « Norme FD X50-046 Qualité en expertise - Recommandations pour l’application de la norme NF X50-110:2003 (Prescriptions générales de compétences pour une expertise) », Ed. Afnor, Paris, www.afnor.org, févr. 01, 2011.
[15] « Norme FD X50-045 Qualité en expertise - Utilisations possibles de la norme NF X 50-110 (Prescriptions générales de compétence pour une expertise) », Ed. Afnor, Paris, www.afnor.org, janv. 01, 2010.
[16] « Norme NF EN 16775-Services d’expertise - Exigences générales relatives aux services d’expertise », Ed. Afnor, Paris, www.afnor.org, janv. 16, 2016.
Annexes
Annexe 1 : schéma représentant le pourcentage des experts connaissant les normes de la qualité en expertise [source auteurs]
Annexe 2 : schéma représentant le pourcentage des professionnels intéressés par l'outil d'autodiagnostic [source auteurs]