
IDS092 - Mise en place d'un système de management de la qualité conforme à la norme ISO 13485 : 2016 et son amendement A1 pour une innovation technologique en néphrologie.
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Contact
Paul Brochet : paul.brochet@icoud.com
Citation
À rappeler pour tout usage : Paul BROCHET, « Mise en place d’un système de management de la qualité conforme à la norme ISO 13485 : 2016 et son amendement A1 pour une innovation technologique en néphrologie. », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositif Médical et Affaires Règlementaires (DMAR), Mémoire de stage réf°IDS092, juill. 2021. [En ligne]. Disponible sur : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids092/
Résumé
Si la commercialisation du premier dispositif médical est l’un des objectifs majeurs de toutes les jeunes startups de l’écosystème medtech, l’atteinte de cet objectif nécessite le passage obligé de nombreux jalons. En effet, les nouveaux règlements européen 2017/745 [1] et 2017/746 [2] demandent la mise en place d’un système de management de la qualité et l’élaboration d’une documentation technique complète et robuste. Les startups, en général fragile économiquement, peu familier au parcours règlementaires et au management de la qualité, ont donc besoin de mettre en place un système de management de la qualité conforme à la norme ISO 13485 :2016 [3] ainsi qu’à son amendement A1 et ce le plus tôt possible afin d’éviter une perte de ressources matérielles et immatérielles difficilement récupérables après plusieurs mois de développement.
Ce mémoire de stage a pour objectif de présenter une stratégie agile de mise en place d’un système de management de la qualité durable et performant conforme à la norme ISO 13485 :2016 ainsi que plusieurs livrables afférents qui ont été générés : une feuille de route technico-règlementaire, un manuel qualité, des procédures documentées, les premiers éléments de la documentation technique et des outils opérationnels.
Mots-clés : Système de Management de la Qualité - Approche processus – Management des risques - Start-up - Réglementation - Documentation technique – Innovation – Dispositif médical
Abstract
While the commercialization of the first medical device is one of the major goals of all the young startups in the medtech ecosystem, achieving this goal requires the passage of many milestones. Indeed, the new European regulations 2017/745 [1] and 2017/746 [2] require the establishment of a quality management system and the elaboration of a complete and robust technical documentation. Startups, which are generally economically fragile and unfamiliar with the regulatory process and quality management, therefore need to set up a quality management system that complies with ISO 13485:2016 [3] and its amendment A1 as soon as possible in order to avoid the loss of material and intangible resources that are difficult to recover after several months of development.
The aim of this internship is to present an agile strategy for implementing a sustainable and efficient quality management system in compliance with ISO 13485:2016 and several related deliverables that have been generated : a technical roadmap regulatory, quality manual, documented procedures, initial technical documentation and operational tools.
Keywords : Quality management system, Process approach, Risk management, Start-Up, Regulation, Technical Documentation – Innovation – Medical Devices
Téléchargement

Mise en place d’un système de management de la qualité conforme aux références NF EN ISO 13485 : 2016 et son amendement A1 pour une innovation technologique en néphrologie.
Mémoire complet :
Mise en place d'un système de management de la qualité conforme à la norme ISO 13485 : 2016 et son amendement A1 pour une innovation technologique en néphrologie
Glossaire
| Accélérateur : | « Un accélérateur est un dispositif qui aide les entrepreneurs à faire prospérer leur entreprise plus vite. Il les accompagne dans les étapes clés du développement de leur activité comme l'internationalisation ou la transformation numérique. » |
| Bioimpédance | La bioimpédance traite de certaines propriétés électriques passives des tissus : la capacité à s'opposer (entraver) au flux de courant électrique [29] |
| Deep tech : | Cette expression désigne les startups de la « Deep Tech » qui proposent des produits ou des services sur la base d’innovations de rupture. Leur ambition ? S'attaquer à la résolution des grands défis du XXIe siècle : une nouvelle technique pour lutter contre le cancer ou le changement climatique, par exemple. Et tous les domaines sont concernés [30]. |
| Incubateur : | « Acteurs de l'innovation, les incubateurs jouent un rôle essentiel dans la maturation d'un projet innovant. Présents à la fois en amont de la création et au cours de la vie de l'entreprise, ils mettent à disposition des porteurs de projet une multitude de services leur permettant de se lancer dans les meilleures conditions. Ils concourent ainsi à la formation d'un écosystème propice à l'émergence et au développement de startups. » [31] |
| La French Tech : | « Communauté de ceux qui travaillent dans ou pour les startups françaises, en France ou à l’étranger. Les entrepreneurs en premier lieu, mais aussi les investisseurs, ingénieurs, designers, développeurs, accélérateurs, incubateur, …qui s’engagent pour la croissance des startups et leur rayonnement international. » [13] |
| Medtech : | « La Medtech regroupe donc toutes les technologies destinées à l’environnement de soin et désigne aussi bien un site de prise de rendez-vous en ligne qu’un organe artificiel ou un robot chirurgical. L’écosystème de startups de la Medtech a donc pour ambition d’inventer la médecine de demain. » [32] |
| POC : | Proof of concept en anglais. « Une preuve de concept (que nous appellerons POC pour Proof of Concept) est une démonstration de faisabilité, c’est à dire une réalisation expérimentale concrète et préliminaire, courte ou incomplète, illustrant une certaine méthode ou idée afin d’en démontrer ou pas la faisabilité, avec un budget accessible à un chef de projet. Situé très en amont dans le processus de développement d’un produit ou d’un process nouveau, le POC est habituellement considéré comme une étape importante sur la voie d’un prototype pleinement fonctionnel. »[33] |
| Startup : | Jeune entreprise innovante, à la recherche d’un modèle économique, qui lui assurerai une croissance très forte et très rapide, avec un développement international [13] |
| MDN/MDA : | MDA / MDN-codes reflect the design and intended purpose of the device and hence are mostly relevant for the allocation of personnel involved in the review of technical documentation [25]. |
| MDS : | MDS codes are horizontal codes that are applicable to devices with specific characteristics [25]. |
| MDT : | MDT codes relate to the technologies and processes that are used in the manufacturing and making. available of the devices [25]. |
Introduction
Les reins jouent un rôle essentiel dans l’homéostasie général de l’être humain. Richement vascularisé, les reins ont pour fonction principale la purification et l’équilibration des constituants du sang. Le néphron est l’élément structural et fonctionnel des reins, il est à l’origine de la fabrication de l’urine [4]. L’excrétion des déchets métaboliques dans les urines se réalise selon trois étapes bien définies. La filtration glomérulaire, la réabsorption et la sécrétion [4]. Lors de la première étape, il y a formation de l’urine dite primitive, puis lors de la seconde étape, l’urine primitive devient l’urine définitive. La dernière étape est le stockage de l’urine définitive dans la vessie avant d’être excrétée par miction [4].
Le débit de filtration glomérulaire (DFG) permet d’apprécier de façon mesurable la capacité du rein à réaliser sa fonction [5]. Le DFG se calcule grâce à la clairance d’une substance, généralement par l’insuline ou la créatinine [4]. Selon la Société Francophone de Néphrologie, Dialyse et Transplantation (SFNDT) les reins filtrent environ un litre de sang par minutes [6].
Selon la Haute Autorité de Santé (HAS), « la maladie rénale chronique (MRC) est une diminution progressive des fonctions rénales déterminée par une diminution permanente du débit de filtration glomérulaire. Elle est définie par la présence, pendant plus de 3 mois, de marqueurs d’atteinte rénale ou d’une baisse du DFG au-dessous de 60 mL/min/1,73m². » [7], elle est également définie par « la présence, pendant plus de 3 mois, de marqueurs d’atteinte rénale qui peuvent être des anomalies morphologiques, histologiques, urinaires » [8].
Il existe 5 stades de sévérité définis selon la valeur du DFG. Les premiers stades intègrent un DFG supérieur à 60 mL/min/1,73m² avec présence de marqueur d’atteinte rénale. L’insuffisance rénale chronique (IRC) est définie à partir des stades 3 et 4 lorsque la valeur de DFG est comprise entre 15 et 59 mL/min/1,73m². Au stade 5, il s’agit d’insuffisance rénale chronique terminale (IRCT) déclaré à un DFG inférieur à 15 mL/min/1,73m². A ce niveaux de sévérité, un traitement par suppléance, greffe rénale ou dialyse est inévitable pour la survie du patient [7].
Le tableau ci-dessous résume les 5 stades de la maladie rénal chronique selon, leurs valeurs de DFG, le pourcentage de fonction rénale prévalent au stade ainsi que leur traitement associé.
Tableau 1 : Les différents stades d'évolutions de la maladie rénale chronique (source : auteur inspiré de [6], [7],[9])
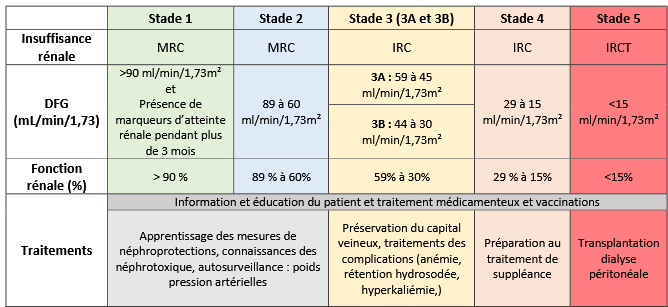
La maladie rénale chronique touche toutes les tranches d’âges [10]. D’après le rapport SFNDT, la maladie rénale chronique touche environ 850 millions de personnes dans le monde. En France, la MRC concerne 10% des adultes soit environ 5,7 millions de personnes [6]. Selon ce même rapport, la Maladie Rénale Chronique (MRC) est passé de la 27e à la 11e cause de mortalité en moins de 30 ans [6].
L'insuffisance rénale chronique terminale est une priorité de santé publique mondiale [6]. Le parcours de soin du patient dialysé, qui se caractérise par des allers-retours hebdomadaires au centre de dialyse, à l’hôpital et du suivi par son néphrologue peut être amélioré.
C’est en partant de témoignages de patients dyalisé, de ce constat et de plus d’une vingtaine d’années d’expérience dans le secteur de la dialyse que Julien Gautier a décidé de développer une solution innovante technologique qui a pour ambition d’améliorer la qualité de vie des patients atteints d’insuffisance rénale chronique.
Comme pour toutes les startups de dispositif médical, Home Habilis doit obtenir le certificat marquage CE pour pouvoir commercialiser son dispositif médical. Une préparation opérationnelle pour l’obtention de ce marquage CE est essentielle. En effet, la mise en place d’un Système de Management de la Qualité (SMQ) permet, d’éviter la perte de ressource parfois irrécupérable, une anticipation d’éléments réglementaires requis par le marquage CE et l’amélioration continue des activités de son entreprise fondée sur un management de processus efficient. La norme ISO 13485 :2016 et son amendement A1 sont les standards de système de management de la qualité pour les dispositifs médicaux.
Comment implémenter le management de la qualité selon les référentiels ISO 13485 :2016 et son amendement A1 pour une innovation technologique en néphrologie ?
Dans un premier lieu, une présentation de l’entreprise et de ses partenaires sera donnée, dans la partie suivante, le management de l’inconnu dans l’univers de la Medtech à travers une nouvelle méthodologie sera développé. Dans une troisième partie la conformité à la réglementation sera abordée et finalement les compétences développées, à acquérir et un bilan sur ce stage.
1. La société Home Habilis.
La société Home Habilis est une jeune startup de la Medtech, elle a été créée le 8 avril 2021 à Beauvais. Ses bureaux sont situés dans la pépinière d’entreprise StartLab de Beauvais. Elle a été fondée par Julien Gautier, son président avec l’appui de ses deux associés Marlène Grégoire et Guillaume Coffe. Home Habilis développe un dispositif médical et une application destinés aux patients atteints d’insuffisance rénale ainsi qu’à leur néphrologue.
Ses fondateurs soucieux, de promouvoir des valeurs tournées vers les patients, se sont donné comme objectifs, l’amélioration de la qualité de vie, un gain d’autonomie et une optimisation de la prise en charge des patients atteints d’insuffisance rénale. Home Habilis cherche à offrir aux patients dyalisés et au système de santé, une nouvelle définition du parcours patient.
Nonobstant cet engagement, Home Habilis choisit la qualité de vie au travail par l’écoute constructive, le développement continu, la création collaborative et la responsabilisation de ses collaborateurs dans les défis technologique d’aujourd’hui et de demain.
Pour atteindre ses objectifs, Home Habilis développe un Dispositif Médical (DM) et une application destinée aux patients atteints d’insuffisance rénale ainsi qu’à leur néphrologue. Ce dispositif se veut comme une amélioration du parcours patient dialysé. En effet, ses nouveaux capteurs de bioimpédence vont permettre un meilleur suivi du patient dialysé à domicile.
Pour atteindre ses objectifs, Home Habilis développe un Dispositif Médical (DM) et une application destinée aux patients atteints d’insuffisance rénale ainsi qu’à leur néphrologue. Ce dispositif innovant, de par ses nouveaux capteurs de bioimpédence, apport une réelle amélioration du parcours patient dialysé en permettant un suivi précis au domicile et au quotidien.
Ce projet se développe au sein de l’incubateur ITerra© et une entrée en accélération avec Eurasanté© est envisagé pour la suite. Avec sa récente labélisation DeepTech ainsi que le soutien de BpiFrance par la French Tech Emergence, Home Habilis rentre dans l’écosystème French Tech.
Selon le communiqué de presse du Ministère de l’économie des finances et de la relance, « la France passe devant l’Allemagne et devient, en raison du Brexit, le 1er écosystème tech de l’Union Européenne » [11]. Cette première subvention permettra à Home Habilis l’élaboration de sa preuve de concept (POC) et de capitaliser d’éventuelles autres financements que Home Habilis recherche activement
1.1 L'esprit entrepreneurial et l'esprit startup
Les activités des entrepreneurs sont partagées entre la recherche de fonds, le développement et la protection de leur innovation technologique. Ces acteurs de l’innovation, fragile économiquement, interagissent dans un environnement incertain ou la concurrence est élevée, ils doivent trouver les ressources nécessaires pour survivre. Qu'elles soient matérielles, immatérielles ou humaines toutes les ressources disponibles comptent.
Le choix des partenaires est une étape importante dans la réussite du projet. En combinant les savoirs d’acteurs d’horizons différents, les savoir-faire et les expériences spécifiques de chaque collaborateur, les jeunes entreprises peuvent espérer une croissance positive. L’entreprenariat ou le management de l’innovation est donc un exercice complexe qui demande des compétences en management de projet, financière et humaine ainsi que de respecter des principes fondamentaux de création de valeur, de ciblage vers l’avenir, d’orientation stratégique, de culture, d’idées exploitables, de gestion d’incertitude, d’adaptabilité et d’approche systématique [12]
C’est du partage de cet esprit entrepreneurial aux autres collaborateurs ou associés que naît l’esprit start-up.
Selon le rapport d’activité de L’Agence du Numérique, une startup se définie comme une « Jeune entreprise innovante, à la recherche d’un modèle économique, qui lui assurerait une croissance très forte et très rapide, avec un développement international » [13]. Selon ce même rapport, 9 400 start-up ont été recensées [13] en France entre 2015 et 2017.
1.2 Les partenaires
Conscient de la nécessité d’avoir des partenaires de qualité pour produire un dispositif médical qui respect les exigences clients ainsi que les exigences réglementaires et normatives, Home Habilis a décidé de s’entourer de partenaires d’innovation, scientifiques, techniques et financiers. La Figure 1 présente les différents partenaires de Home Habilis.
Figure 1 : Les différents partenaires de Home Habilis (source : CEO de Home Habilis)
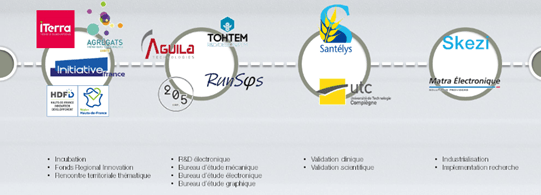
1.2.1 Partenaires d'innovations.
Depuis le 11 mai 2020, Home Habilis a intégré l’incubateur iTerra. Ce partenariat a permis d’obtenir des infrastructures, des formations régulières pour développer les compétences individuel et collectives au sein de Home Habilis, participer à des événements et ainsi faire rayonner le projet. Des co-accélérations avec un incubateur spécialisé en santé sont envisagées cluster Santé à Amiens ou Eurasanté à Lille.
1.2.2 Partenaires techniques.
Home Habilis travail aujourd’hui avec trois bureaux d’études :
- Aguila Technologie : Apportant une expertise DM, d’ingénierie des dispositifs médicaux notamment sur les logiciels ainsi qu’un support qualité et affaires réglementaires.
- Runsys : Apportant une expertise et des solutions électroniques et logiciels.
- Tohtem Makerz : Apportant une expertise et des solutions de conception mécanique.
- Bureau 205 : Apportant des solutions de design.
Le travail en commun avec ses différents bureaux d’études permettra la réalisation et la validation d’un Proof of Concept (POC) du dispositif médical de Home Habilis.
D’autres partenariats techniques sont en discussion :
- Matra Electronique : Pour la fabrication du prototype et des premières séries.
- Skezi : Pour l’hébergement des données de santé et l’exploitation en recherche clinique.
1.2.3 Partenaires scientifiques.
Pour assurer la validité et l’orientation médicale de son dispositif médical, Home Habilis a consolidé un bord médical composé de plusieurs néphrologues, ce bord médical intervient également lors de comité où des patients atteints d’insuffisance rénale chronique. Ces comités permettent d’avoir des échanges sur les besoins des patients.
Home Habilis entretient également une relation privilégiée avec l’Université de Technologie de Compiègne (UTC), notamment par le suivie des travaux sur l’impédance appliquée à l’insuffisance rénale de l’unité de recherche BMBI, mais également pour le recrutement de thésards en traitement du signal et en ingénierie des données de santé.
1.3 Les missions du stage
Pour atteindre les objectifs de la société Home Habilis, plusieurs missions en liens avec la qualité et les affaires réglementaires ont été définis :
- Mettre en place le système de management de la qualité conforme aux références ISO 13485 :2016 et PR NF EN ISO 13485 :2020.
- Déterminer des processus d’analyse et de gestion du risque.
- Documenter un manuel qualité et rédiger des procédures.
- Mettre en place des mesures et enregistrements nécessaires en phase de développement de produit.
- Mettre en place un système de contrôle des documents et des enregistrements.
- Mettre en place un système d’amélioration continue.
- Identifier d’autres normes applicables ou recommandées.
- Rédiger la documentation technique
L’atteinte de ces objectifs a été réalisé en respectant une stratégie, méthodologie basée sur de l’amélioration continue, du management de processus, la gestion de projet. Cette méthodologie est explicitée dans la partie suivante.
2. Manager l’inconnu dans l’univers de la Medtech.
2.1 Les exigences normatives et réglementaires
Comme énoncé dans l’introduction, les fabricants de dispositifs médicaux ont l’obligation de se conformer aux règlements européens 2017/745 ou 2017/746 pour mettre sur le marché leur dispositifs médicaux. Dans ce mémoire seules les exigences du règlements 2017/745 sont abordées. En effet, l’article 10 du règlement 2017/745 relatif aux obligations du fabricants, définit 16 alinéas que les fabricants doivent satisfaire pour se conformer afin obtenir le marquage CE qui leur permettra de mettre sur le marché leur dispositif médical.
C’est dans l’alinéa 9 que se définissent les exigences en terme de système de gestion de la qualité,
« Les fabricants de dispositifs, autres que des dispositifs faisant l’objet d’une investigation, établissent, documentent, appliquent, maintiennent, mettent à jour et améliorent en permanence un système de gestion de la qualité qui garantit la conformité avec les dispositions du présent règlement et ce, de la façon la plus efficace possible et d’une manière qui soit proportionnée à la classe de risque et au type de dispositif. » [1]
Cet alinéa 9 définit également 13 aspects que doit aborder le système de gestion de la qualité au minimum. Ces 13 aspects sont partiellement couvert par la norme NF EN ISO 13485 :2016 comme le met en lumière le projet de révisions PR NF EN ISO 13485 :2020 dans son annexe ZB [14].
La qualité et son management jouent donc un rôle central dans la mise sur le marché des dispositifs médicaux.
Les standards de la qualité comme les normes ISO 9001 [15] et ISO 9004 [16] démontrent que la qualité peut se définir par l’évolution de l’efficacité, l’efficience et la qualité perçu, en un mot la performance. Celle-ci est obtenue dans les entreprises par l’application de l’amélioration continue qui se traduit par, la planification, la réalisation, le contrôle et l’amélioration des activités plus communément appelé PDCA.
La matérialisation de la qualité s’observe de manière plus globale dans le mouvement de la valeur ajoutée obtenu grâce l’application de l’approche processus à tous les niveaux de l’entreprise. Pour rappel un processus est la transformation d’un élément d’entrée en un élément de sortie avec de la valeur ajoutée par le biais d’activités corrélés entre elles [17].
L’approche processus se traduit par une vision de l’entreprise comme une suite de processus bien définis. Son application permet d’acquérir « la compréhension et la satisfaction en permanence des exigences ; la prise en compte des processus en termes de valeur ajoutée ; l’obtention d’une performance effective des processus ; l’amélioration des processus sur la base d’une évaluation de données et d’informations » [15].
La gestion des risques, exigence faisant partie des aspects de l’alinéa 9 que doit couvrir le système de management de la qualité, se concrétise par une méthodologie appelée approche par les risques. Elle se caractérise par un processus de management du risque qui se compose généralement de 3 activités majeures, l’analyse du contexte et la définition des critères, l’appréciation du risque, le traitement du risque [18]. Le management du risque vu comme un processus récurrent permet la création et la préservation de valeurs [18], et de répondre de façon qualitative aux exigences de sécurité et de performance du règlement 2017/745.
La mise en place du système de management de la qualité, étape obligatoire chez les fabricants de dispositifs médicaux, cette étape peut être un réel parcours du combattant pour les startups de la Medtech.
Basé sur un PDCA (Plan, Do, Check, Act), d’une analyse de travaux similaires chez des fabricants de dispositifs médicaux différents ainsi que sur le fascicule de documentation FD X50-176 qui est un guide de mise en œuvre du management des processus [19], une nouvelle méthodologie a été développée lors de ce stage. Cette méthodologie est développée dans la partie suivante.
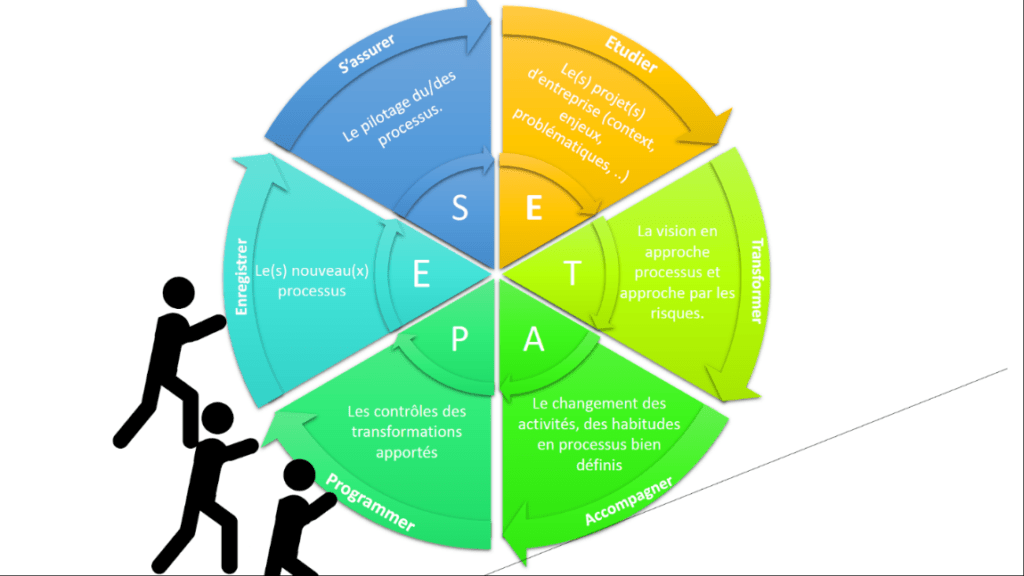
2.2 La méthodologie
La méthodologie nommée ETAPES, acronyme de Etudier, Transmettre, Accompagner, Programmer, Enregistrer, S’assurer. Elle permet par des cycles itératifs d’implémenter dans les startups medtech un système de management de la qualité performant et durable. Les différentes étapes de cette méthodologie sont détaillées dans les parties ci-dessous :
2.2.1 Etudier : le projet d'entreprise
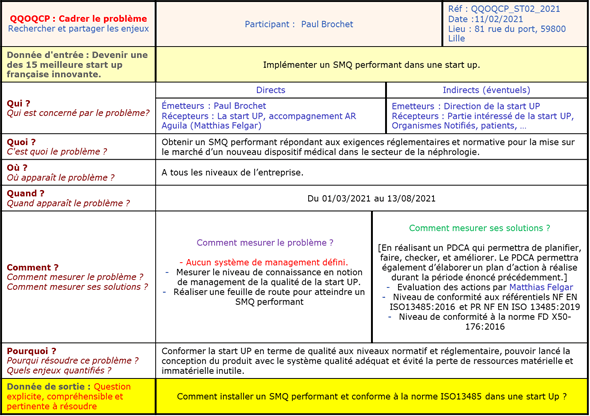
La première étape de cette méthodologie a pour objectif de définir une stratégie d’action pour implémenter le système de management de la qualité. Plusieurs outils sont nécessaires pour atteindre cet objectif. Dans un premier temps, l’entreprise doit cadrer le problème en utilisant par exemple un QQOQCP tel qu’illustré en Figure 2.
Figure 2 : QQOQCP Réalisé avant le stage et vérifié lors de la première semaine de stage (source : auteur)
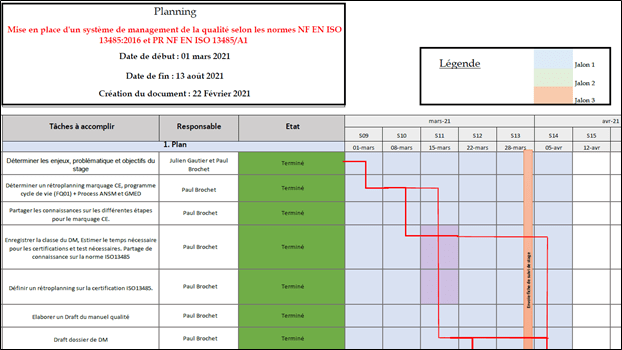
Il faut par la suite élaborer un planning d’actions en plusieurs jalons respectant les différentes phases du PDCA. Pour ce stage la Figure 3 et la Figure 4 illustrent le plan d’action pour la mise en place du système de management de la qualité.
Figure 3 : Répartitions des phases du PDCA pour les 6 mois de stage (source : auteur).
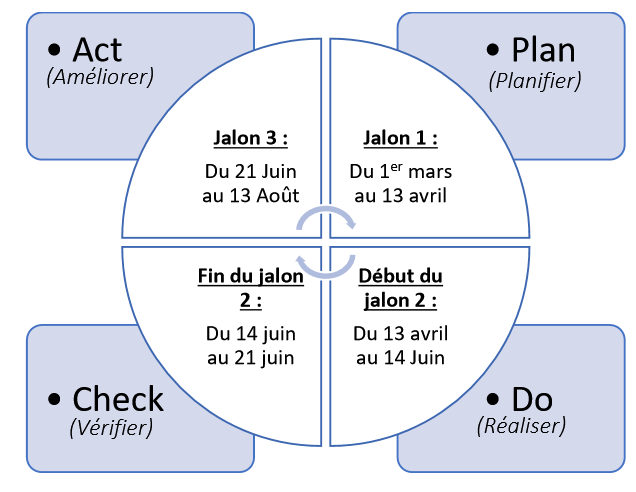
Figure 4 : Extrait du planning d’actions de mise en place du système de management de la qualité chez Home Habilis (source : auteur).
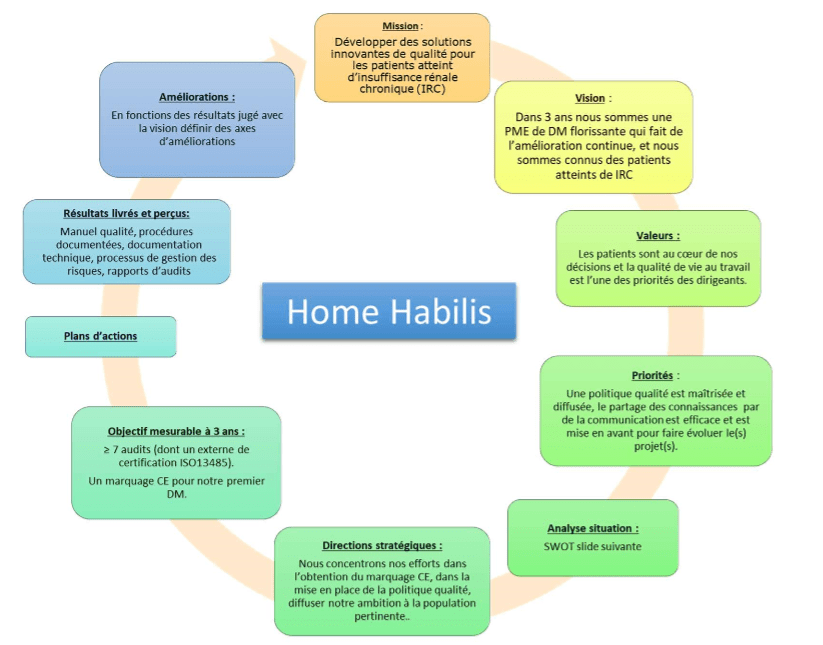
Pour terminer cette phase d’étude, l’équipe de projet peut réaliser un Plan Dynamique Stratégique (PDS). Cet outil permet de déterminer une stratégie de développement à court et long terme en déterminant, une mission, une vision, des valeurs, un bilan des forces et faiblesses de l’entreprise sous la forme d’un SWOT (Strenghts, Weaknesses, Opportunities, Threats), ainsi que des priorités dans les actions à réaliser. La Figure 5 illustre un PDS élaborer selon le projet de Home Habilis.
Avec ce dernier outil, l’entreprise définit les premiers éléments de réponse quant à l’engagement de la direction exigé dans l’article 5 de la norme ISO 13485 :2016 ainsi que les premiers éléments de réponse aux exigences d’amélioration continue de l’entreprise, notamment en termes d’audit interne défini dans son article 8.
Figure 5 : PDS de Home Habilis (source : auteur)
Cette première étape permet de cadrer le projet de l’entreprise et de répartir dans un temps limite, les tâches à réaliser ainsi que les responsabilités. Il est essentiel dans la mise en place d’un système de management de la qualité que tous les acteurs de l’entreprise comprennent et assimilent les notions de qualité, management de la qualité, de processus, d’approche processus et d’approche par les risques introduit dans la partie 2.1. Le partage des connaissances sur ces notions essentielles est développé dans l’étape suivante.
2.2.2 Transmettre : L'approche processus et l'approche par les risques
Comme défini précédemment, l’objectif de cette étape et de sensibiliser les acteurs à la qualité et à la réglementation. Transmettre les notions clés d’approche processus, d’approche par les risques, contrôle qualité, de management de la qualité mais également aux exigences réglementaires, notamment, en termes de sécurité de performance des dispositifs médicaux, d’étude pré-clinique, clinique et de Suivie Après Commercialisation (SAC).
Ces partages de connaissances sont nécessaires à une mise en place de la qualité et à la conformité aux réglementations applicables. Une compréhension complète par les acteurs dirigeants de l’entreprise de ces termes feront l’objet d’une vérification et validation dans une étape prochainement décrite.
La réalisation d’ateliers participatifs permet de partager les notions clés citées précédemment de manière performante et de prendre des décisions. Les Figures 6 et Figure 7 illustrent un exemple d’atelier réalisé pour la transmission et l’appropriation de l’approche par le risque.
Figure 6 : Extrait de l’atelier réalisé sur l'approche par les risques (1) (source : auteur)
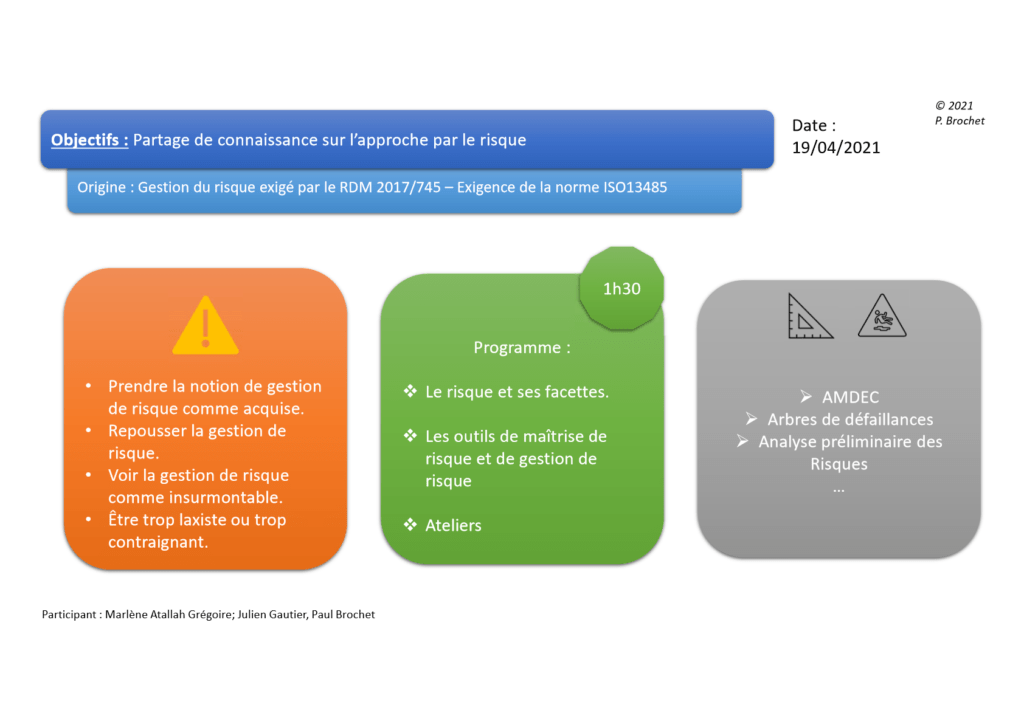
Figure 7 : Extrait de l’atelier réalisé sur l'approche par les risques (2) (source : auteur)

Le partage des connaissances sur ces notions essentielles de management de la qualité permet une implication des acteurs interne de l’entreprise et facilite le passage à la phase suivante qui est l’accompagnement vers le changement.
Cette étape de transmission de notions permet de faciliter la réorganisation de l’entreprise en adoptant la vision processus de ses activités actuelles et futures. Des outils opérationnels d’aide à la compréhension de ces différentes notions ainsi que des normes phares du domaine de la qualité et du dispositif médical sont disponibles dans la bibliothèque travaux master.
2.2.3 Accompagner : Le changement des activités au sein de l'entreprise
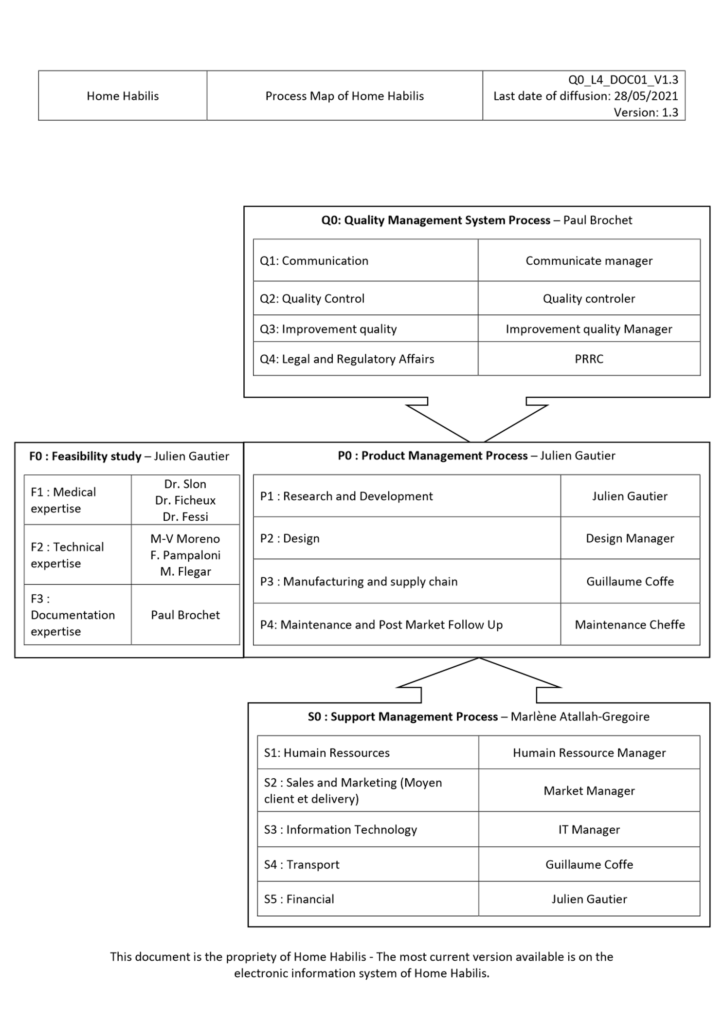
Une fois que les notions essentielles ont bien été transmises et assimilées et que l’ensemble des activités actuelles et futures de l’entreprise ont été revues, l’entreprise peut réaliser les premiers drafts que sa cartographie de processus. L’ANNEXE I présente une première version de la cartographie de processus de Home Habilis. Cette cartographie se compose de quatre processus généraux, un processus de management de la qualité nommé Process Q, un processus de faisabilité nommé process F, un processus de réalisation nommé process P et un processus support nommé process S.
Pour rappel, la mise en place d’un système de management de la qualité est un processus qui nécessite l’implication de tous les acteurs, mais également celle de la direction de l’entreprise
celle-ci se matérialise généralement par une lettre d’engagement de la direction. L’ANNEXE V présente une première version de l’engagement de la direction de Home Habilis.
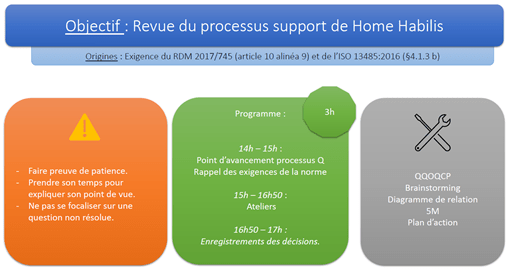
Il est essentiel de réaliser des revus de processus avant de les enregistrer, pour vérifier et valider les éléments les composant. La Figure 8 illustre la revue de processus support de Home Habilis.
Figure 8 : Extrait de l’atelier de revue du processus support de Home Habilis (source : auteur).
Une fois ce processus revu, un enregistrement de ce processus peut être réalisé avec le responsable qui a été défini en amont. En utilisant l’annexe B du fascicule de documentation FD X50-176 [19] L’entreprise documente une fiche descriptive de processus comprenant, les objectifs du processus, la personne en charge, les clients du processus, les acteurs internes du processus, son mode de pilotage, ces données d’entrées de sorties, les exigences auxquelles il a vocation de satisfaire également, les objectifs en lien avec la politique qualité ces méthodes de mesures et de surveillances, les ressources nécessaires ainsi que les risques et opportunités identifiés. L’ANNEXE II présente la fiche descriptive du processus S de Home Habilis.
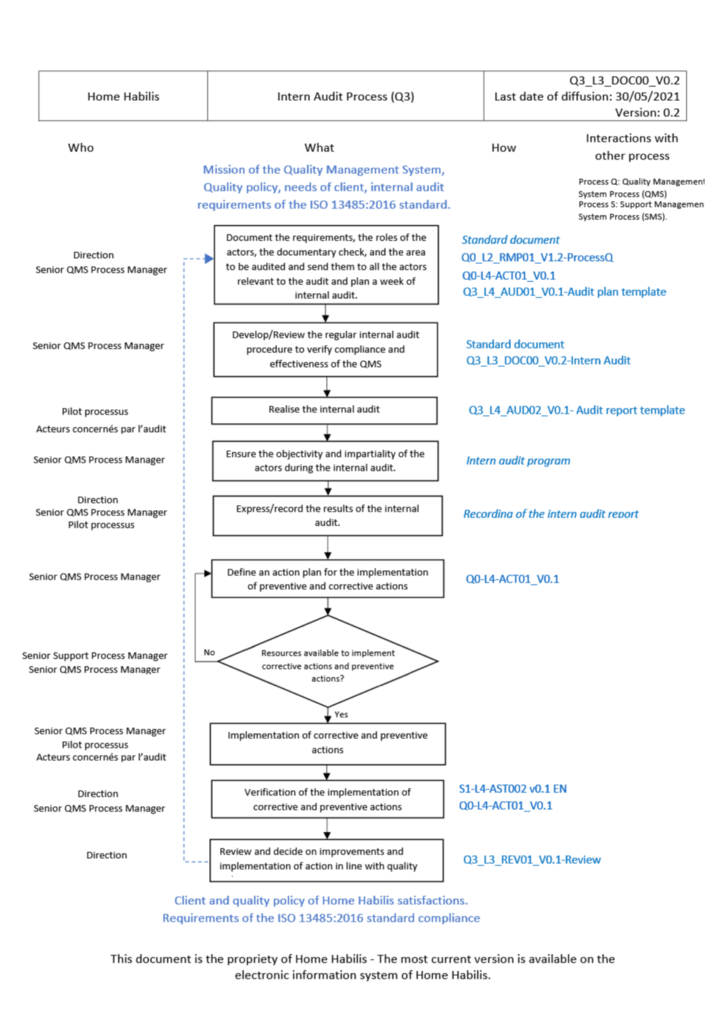
Dans cette étape les premières procédures ou « manière spécifiée de réaliser une activité ou un processus » [17] peuvent être documentées. Tout comme les processus, ces procédures nécessitent une vérification et une validation lors de revu. Ces revues doivent être réalisées à un intervalle défini. Une procédure documentée doit contenir des activités, des personnes responsables ainsi que des documents supports. L’ANNEXE III présente la procédure d’audit interne de Home Habilis.
Cette étape permet de définir la structure du système de management de la qualité de manière documentée, elle doit s’arrêter au minimum 3 semaines avant la prochaine étape qui permettra d’évaluer les changements, transformations apportées.
2.2.4 Programmer : le contrôle des transformations apportées.
Cette étape a pour objectif de préparer, programmer et réaliser un contrôle des changements apportés. Comme défini dans le dernier paragraphe de la première étape, une période de vérification (Check dans le PDCA) doit être effectuée pour vérifier et valider les objectifs définis dans cette même partie.
La réalisation d’un audit interne du système de management de la qualité sur la norme ISO 13485 :2016 ainsi que sur le règlement européen applicable au dispositif médical développé, selon la norme NF EN ISO 19011 [20] qui définit les lignes directrices pour l’audit des systèmes de management, est un moyen pertinent de vérifier la conformité et la correcte mise en place du système de management au sein de l’entreprise. La figure ci-dessous présente un processus d’audit selon la norme NF EN ISO 19011 :2018.
Figure 9 : Extrait de l’atelier sur l’audit qualité et la norme NF EN ISO 19011 :2018
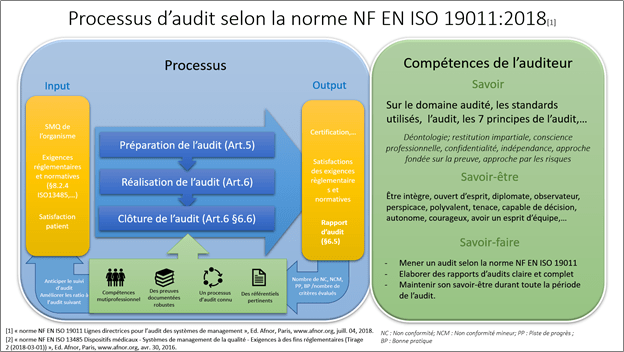
Comme l’illustre la Figure 9 et tel qu'introduit à la fin de l’étape précédente, un audit nécessite un temps de préparation d’environ 3 à 4 semaines. Ce temps nécessaire a pour but, de planifier une semaine de réalisation d’audit avec toutes les parties prenantes à l’audit selon un programme bien définit, élaborer les modèles de plan d’audit et de rapport d’audit, réaliser une revue des documents qualité et les placer dans un référentiel pour faciliter le travail de l’auditeur.
Cet audit interne ne peut pas se faire par un acteur interne de l’entreprise. En effet, la réalisation de cet audit doit être menée par un tiers familier à la norme ISO13485 :2016 et possédant des compétences confirmées à la réalisation d’audit, de cette manière l’objectivité qu’exige les normes NF EN ISO19011 et ISO 13485 :2016 seront respectées.
« Le choix des auditeurs et la réalisation des audits doivent assurer l’objectivité et l’impartialité du processus d’audit. Les auditeurs ne doivent pas auditer leur propre travail. » [3]
Une fois l’audit réalisé, une analyse du rapport d’audit doit être réalisé pour déterminer les actions correctives et préventives (CAPA) prioritaires à réaliser.
Cette étape permet de mesurer de façon objective l’avancement dans la mise en place du système de management de la qualité, ainsi que de définir des CAPA pour poursuivre le développement du système de management de la qualité. L’ANNEXE VI présente la conclusion de l’audit interne réalisée lors de ce stage.
2.2.5 Enregistrer : Les nouveaux processus, procédures et enregistrements.
Cette étape a pour objectif d’enregistrer les changements validés par l’audit réalisé dans l’étape précédente ainsi qu’enregistrer les actions correctives et préventives prioritaires à réaliser dans des enregistrements (preuve documentaire).
Enregistrer ces actions correctives et préventives permet de démontrer l’amélioration continue du système de management de la qualité par la prise en compte du rapport d’audit. Le rapport d’audit doit lui également être enregistré dans la documentation qualité car il sera demandé ainsi que les enregistrements des actions correctives et préventives lors de l’audit suivant. La Figure 10 illustre un enregistrement d’actions correctives et préventives après l’audit interne de Home Habilis.
Figure 10 : Fiche d'enregistrement d'actions corrective et d'action préventives. (source : Auteur inspiré de [21])
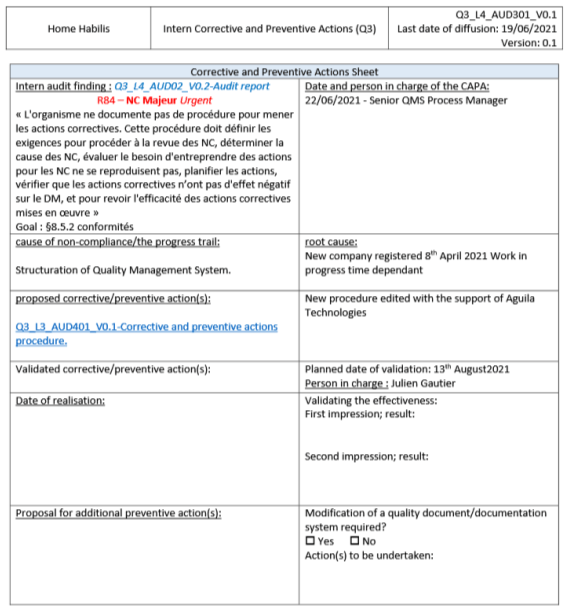
Le suivie de la réalisation des actions préventives et des actions correctives, fait partie des exigences de la norme ISO 13485 :2016 dans son paragraphe §8.4.2 :
« La direction responsable du domaine audité doit assurer que toutes les corrections et actions correctives nécessaires sont entreprises sans délai pour éliminer les non-conformités détectées et leurs causes. Les activités de suivi doivent inclure la vérification des actions entreprises et le compte rendu des résultats de cette vérification. » [3]
Pour s’assurer l’adéquation de ces actions correctives et préventives, l’entreprise peut réaliser lors de l’étape suivant un suivi de l’audit. Elle peut également s’autoévaluer à l’aide d’outils d’autodiagnostique présentés dans la dernière étape et la partie 3.1.2
2.2.6 S'assurer : Le pilotage du/des processus
Cette dernière étape a pour objectif l’autopilotage du système de management de la qualité. Une fois le suivi d’audit réalisé, les périodes de revue de direction définies.
Les responsables de processus ainsi que ces pilotes processus peuvent grâce à un outil Excel d’autoévaluation, évaluer la maturité de leur processus selon 73 recommandations du fascicule de documentation FD X50-173. Cet outil est disponible, gratuitement, sur le site de travaux master UTC avec la référence IDS074. La figure ci-dessous illustre l’autodiagnostic au début du stage du processus de management de la qualité Q de Home Habilis. La Figure 11 et Figure 12 illustrent l’autodiagnostique du processus de management de la qualité avant et après 6 mois en suivant la méthodologie ETAPES.
Figure 11 : Résultat d'autodiagnostique avant le stage (source : Auteur)
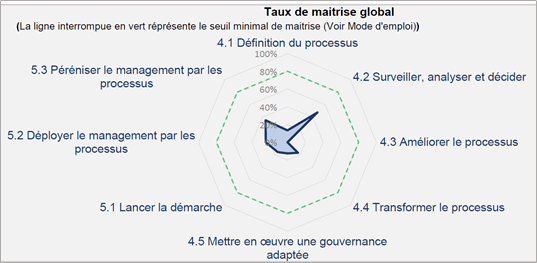
Figure 12 : Autodiagnostic après 6 mois de stage (source : auteur).
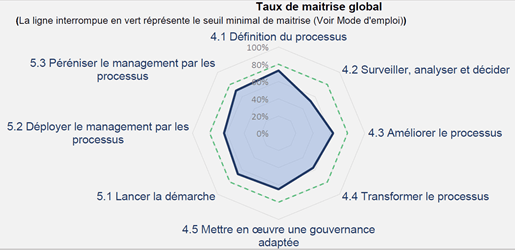
La réalisation d’autodiagnostics par chaque responsable de processus permet une amélioration du système de management de la qualité de façon autonome. En effet, chaque responsable identifie les éléments les procédures, enregistrements manquant pour la satisfaction aux exigences de la norme ISO13485 :2016 et détermine les méthodes pour y parvenir avec le soutien la direction.
2.3 Les résultats
Grace à cette méthodologie ETAPES illustré dans la figure ci-dessous et l’implication de tous les acteurs participant à la mise en place du système de management de la qualité, les entreprises de dispositifs médicaux peuvent de manière agile se conformer au référentiel ISO 13485 :2016.
Figure 13 : La roue ETAPES (source : auteur)
Au commencement de ce stage la documentation qualité n’était pas encore réalisée, à l’issue de ce stage et en suivant cette méthode, plusieurs documents qualité ont été livrés :
- Un manuel qualité (CONFIDENTIEL)
- Une cartographie des processus (ANNEXE I)
- 4 fiches processus détaillés selon la structure de l’annexe B du FD X50-176 (voir ANNEXE II)
- Plus de 5 procédures documentées (voir ANNEXE III).
- Processus de management des risques basée sur la norme ISO 31000 (voir ANNEXE IV)
- Outils de management des risques basés sur la norme et ISO 14971.
- L’engagement de la direction (ANNEXE V)
- Un modèle de plan et un modèle de rapport d'audit interne.
- Un rapport d’audit comportant 99 constats (voir ANNEXE VI)
- De nombreux enregistrements CAPA (ANNEXE VII)
La mise en place du système de management de la qualité n’est pas la seule obligation du fabricant de dispositif médical. En effet comme introduit dans la partie 2.1, l’article 10 du règlement 2017/745 exiges des fabricants une documentation technique. La conformité à cette exigence est abordée dans la partie suivante.
3. Conformité réglementaire
Comme introduit précédemment, les fabricants de dispositifs médicaux doivent également répondre à des obligations en termes de documentation technique, d’évaluation clinique, de Suivi Clinique Après Commercialisation (SCAC), de façon plus générale à des exigences en terme sécurités et performances du dispositif médical, de gestion des risques et de traçabilité, afin d’assurer la sécurité du patient et toute personne en interaction direct ou indirect avec le dispositif médical. Pour répondre à ces obligations le fabricant de dispositif médicaux doit faire appel à un nouvel acteur, la Personne Chargée de Veiller au Respect à la Réglementation (PVCRR).
Selon l’alinéa 1 de l’article 15 du règlement 2017/745 « Les fabricants disposent au sein de leur organisme d’au moins une personne chargée de veiller au respect de la réglementation possédant l’expertise requise dans le domaine des dispositifs médicaux. » [1]
Les points a) et b) de cet alinéa, définissent les domaines de formations et d’expérience requis afin de désigner une Personne Chargée de Veiller au Respect de la Réglementation (PCVRR). Les missions de cet acteur sont d’assurer que « la conformité des dispositifs soit correctement vérifiée, conformément au système de gestion de la qualité dans le cadre duquel les dispositifs concernés sont fabriqués, avant la libération d’un dispositif, la documentation technique et la déclaration de conformité UE soient établies et tenue à jour, les obligation en matière de surveillance après commercialisation soient remplies, conformément à l’article 10 paragraphe 10, … » [1].
Des dispositions particulières peuvent être prises pour les jeunes startups de la medtech, en effet, selon l’alinéa 2 de l’article 15, « Les micro et petites entreprises au sens de la recommandation 2003/36/CE de la Commission ne sont pas tenues de disposer, a sein de leur organisation, une personne chargée de veiller au respect de la réglementation mais une telle personne est en permanence et sans interruption à leur disposition. » [1]. Les recommandations de la commission indiquent que les entreprises comportant « moins de 250 personnes et dont le chiffre d’affaire n’excède pas 50 millions d’euros ou dont le total du bilan annuel n’excède pas 43 millions d’euros » [22] constituent ce que l’on peut définir comme micro, petites et moyenne entreprises.
La PCVRR introduit par la réglementation 2017/745, prend également le rôle de point de contact entre l’entreprise et les autorités de santé comme l’Agence National de Santé du Médicament et des produits de santé (ANSM) et la Haute Autorité de Santé (HAS). La PCVRR est également le point de contact entre le fabricant et l’Organisme Notifiés (ON), organisme qui évalue un fabricant de dispositif médical et détermine in fine si le fabricant peut déclarer sa conformité au règlement qui lui a été attribué sous la forme d’un certificat de marquage CE.
La conformité réglementaire est un processus complexe qui requiert des compétences et des ressources. Les fabricants de dispositifs médicaux doivent, dans un premier temps s’assurer un positionnement réglementaire en définissant une feuille de route technico-réglementaire et dans un second temps, débuter la rédaction de la documentation technique. La partie suivante présente les éléments qui constituent la feuille de route technico-réglementaire.
3.1 Positionnement réglementaire
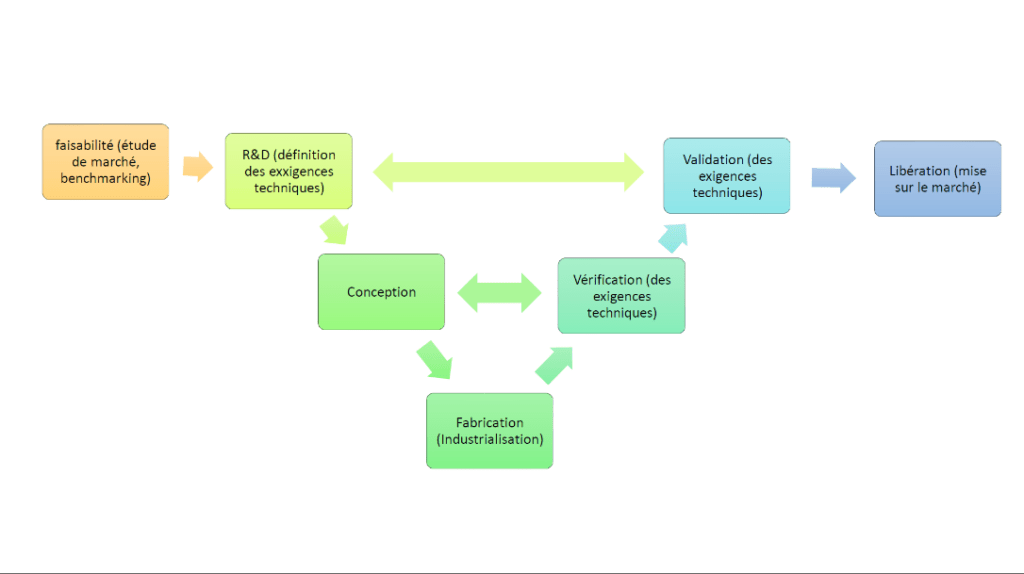
La mise sur le marché des dispositifs médicaux doit suivre des étapes bien définie. Ces étapes doivent à la fois suivre les étapes classiques de fabrication industrielle tel qu’illustré dans la figure ci-dessous, mais également les différentes étapes réglementaires, tel que l’identification des exigences normatives et réglementaires, la réalisation de test pré-clinique, clinique, la préparation au suivie après commercialisation, l’évaluation de la conformité à la réglementation par un Organisme Notifié (ON) et finalement, la réalisation du suivie après commercialisation.
3.1.1 La feuille de route technico-réglementaire.
La mise sur le marché des dispositifs médicaux doit suivre des étapes bien définies. Ces étapes doivent à la fois suivre les étapes classiques de fabrication industrielle tel qu’illustré dans la figure ci-dessous, mais également les différentes étapes réglementaires, tel que l’identification des exigences normatives et réglementaires, la réalisation de test pré-cliniques, cliniques, la préparation au suivi après commercialisation, l’évaluation de la conformité à la réglementation par un Organisme Notifié (ON) et finalement, la réalisation du suivi après commercialisation.
Figure 14 : les différentes phases de fabrications industrielle (source : auteur).
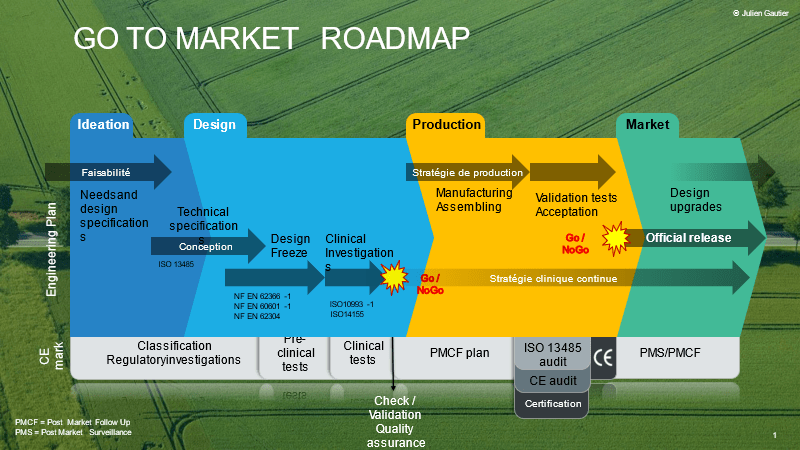
La figure ci-dessous illustre la feuille de route technico-réglementaire de Home Habilis, cette carte indique en un regarde la localisation du projet de Home Habilis dans les phases de fabrications industrielle et les différentes étapes réglementaires.
Figure 15 : Feuille de route technico-réglementaire de Home Habilis (source : Home Habilis)
Pour accompagner les fabricants de dispositifs médicaux dans leur chemin vers la mise sur le marché de leur innovation, plusieurs outils opérationnels peuvent être utilisés. Ces outils sont présentés dans la partie suivante.
3.1.2 Des guides réglementaires
Il existe plusieurs outils opérationnels pour accompagner les fabricants ainsi que leur PCVRR vers la conformité au règlement. Un groupe, le Medical Device Coordination Group (MDCG) publie régulièrement des guides pour satisfaire les exigences des règlements européens [23].
Lors de ce stage plusieurs de ces guides ont été utilisés :
- Le MDCG 2019-11 relatif à la qualification et à la classification des logiciels de dispositifs médicaux [24] en classe I, IIa, IIb ou III. ce guide aide à définir quelle est la réglementation applicable au dispositif médical développé ainsi que de définir la classe du dispositif médical.
Avec l’aide de ce guide, et l’expertise de Aguila technologie, l’innovation de Home Habilis a été définie comme dispositif médical de classe IIa sous le règlement 2017/745.
- Le MDCG 2019-14 qui est une note explicative sur les codes MDR [25] a également été utilisée afin de définir les codes MDA, MDS et MDT applicable au dispositif médical de Home Habilis, ces différents codes permettent la sélection de l’Organisme Notifié (ON) qui évaluera la conformité au règlement.
Avec l’aide de ce guide, les codes MDA, MDN et MDS applicables et potentiellement applicables à l’innovation de Home Habilis sont :
- MDA0204 : Other active non-implantable devices for monitoring and / or diagnosis [25]
- MDS1010 : Devices with a measuring function [25]
- MDT2010 : Devices manufactured using electronic components including communication devices [25]
Pour rappel la définition de ce code permet de choisir l’organisme notifié certificateur pour le marquage CE.
D’autres MDCG, comme le MDCG 2020-10/1 sur la réalisation des rapports de sécurité dans les enquêtes cliniques [26], ou encore le MDCG 2019-16 relative à la cybersécurité pour les dispositifs médicaux [27], sont disponibles sur le site de la Commission européen.
Il existe aussi plusieurs outils opérationnels pour accompagner les fabricants ainsi que leur PCVRR vers la conformité au règlement. Comme définit en partie 2.2.2, il existe des outils d’aide à la compréhension des normes incontournables dans le domaine des dispositifs médicaux, ces outils prennent la forme de cartographies interactives et sont disponibles gratuitement sur le site de bibliothèque travaux master formation Ingénierie de la santé. Une cartographie interactive offrant une représentation claire et précise de la démarche marquage CE pour des dispositifs médicaux de type IIa est disponible par exemple, un outil Excel d’aide au positionnement sur la démarche du marquage CE spécialisé pour cette même classe de dispositifs médicaux est également disponible sur cette plateforme [28].
Ces différents outils aident les fabricants de dispositifs médicaux à se positionner, s’évaluer ainsi qu’enregistrer leur progression au niveau de la conformité au règlement 2017/745. Les Figures 16 et Figure 17 illustrent l’avancement réglementaire de Home Habilis pour son dispositif médical de classe IIa.
Figure 16 : Autodiagnostic d'avancement du marquage CE d'un dispositif de classe IIa avant le stage (source : auteur).
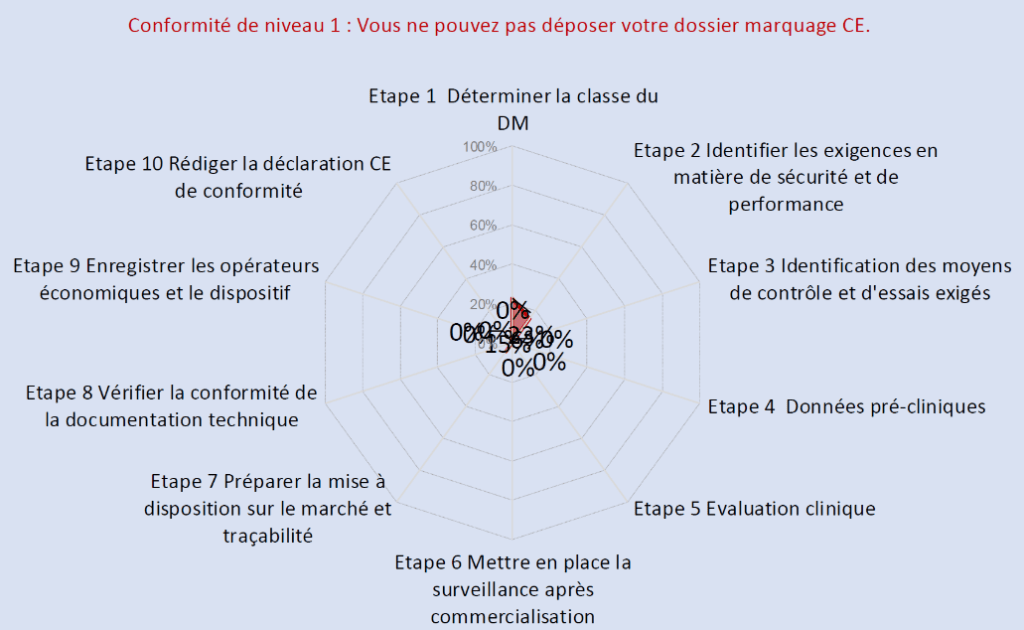
Figure 17 : Autodiagnostic d'avancement du marquage CE d'un dispositif médical de classe IIa après 6 mois de stage (source : auteur).
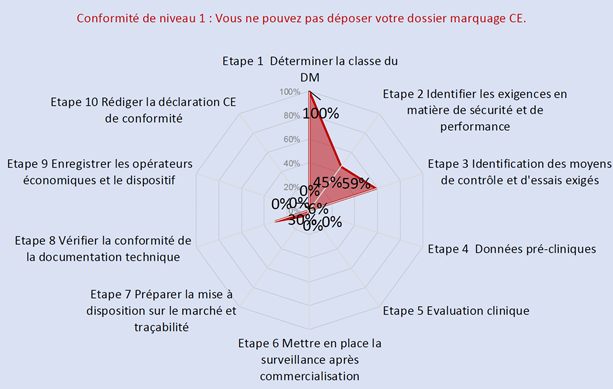
Grace à ces outils réglementaires, les entreprises de dispositifs médicaux comme Home Habilis possèdent des ressources opérationnelles à la conformité à la réglementation européenne, et pourront présenter à l’O.N. une documentation technique robuste et ainsi par la suite obtenir le certificat de marquage CE.
3.2 La rédaction de la documentation technique
L’élaboration de la documentation technique doit suivre un processus d’ajout d’information. Tel qu’énoncé dans la partie précédent l’accès à la certification marquage CE suis une route bien précise, il est important que lors de chaque étape de cette route les informations récoltées soient enregistrées dans la documentation technique.
Selon l’alinéa 4 de l’article 10 du règlement 2017/745 « Les fabricants de dispositifs autres que des dispositifs sur mesure établissent et tiennent à jour la documentation technique relative auxdits dispositifs. La documentation technique est de nature à permettre l'évaluation de la conformité du dispositif avec les exigences du présent règlement. Cette documentation technique contient les éléments prévus aux annexes II et III. La Commission est habilitée à adopter des actes délégués conformément à l'article 115 pour modifier, eu égard aux progrès techniques, les annexes II et III. » [1]
L’outil Excel d’avancement du marquage CE pour des dispositifs médicaux de classe IIa présenté dans la partie précédente, propose également une maîtrise documentaire tirée de l’analyse des annexes II, III, IX, X, XI du règlement 2017/745. La figure ci-dessous illustre cette méthode de maîtrise documentaire.
Figure 18 : Maitrise documentaire étape par étape pour un dispositif médical de type IIa (source : [28])
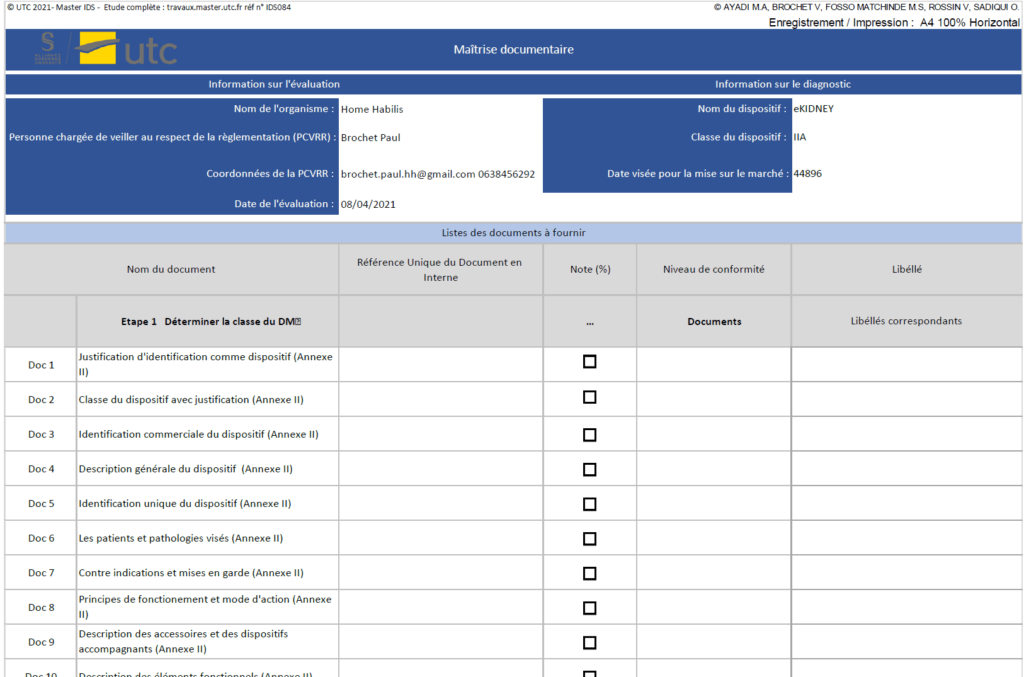
De cette manière les fabricants de dispositifs médicaux s’assurent la présentation des informations essentielles dans la documentation technique requise pour l’obtention du marquage CE.
3.3 Les résultats
Au commencement de ce stage la rédaction de la documentation technique n’était pas encore réalisée, à l’issue de ce stage les premiers éléments qui la composent ont été implémentés, ces éléments sont :
- L’identification du rôle réglementaire de Home Habilis (fabricant de dispositif médical)
- La qualification en dispositif médical de l’innovation de Home Habilis grâce à l’article 2 du règlement 2017/745 et du guide MDCG 2019-11.
- La classification du dispositif médical en classe IIa en suivant les règles de l’annexe VIII du règlement 2017/745.
- L’identification de normes et réglementation applicable à Home Habilis (liées à la sécurité et la performance du dispositif médical)
4. Bilan personnel
4.1 Compétences acquises
A la fin de ce stage en qualité de responsable qualité et affaires réglementaire dans la startup Home Habilis, j’ai eu l’opportunité confirmer mes compétences professionnelles qui sont :
- Savoir : Connaissances théoriques en gestion de projet, ingénierie de projet, en leadership, planification et anticipation.
- Savoir-faire : Elaboration du cadre projet (identifier les enjeux, les parties prenantes, les problématiques, etc.), de rétroplanning jalonné avec des tâches bien définies et y ait associé les responsabilités. Contrôler l’état d’avancement des projets, livrer des résultats mesurables et opérationnels dans les temps impartis, etc.
- Savoir-être : Capacité d’adaptation, a géré la pression et l’incertitude. Respecter les délais et communication les résultats.
- Savoir : Connaissances théoriques en contrôle, assurance et management de la qualité, en analyse, gestion et management du risque. Connaissances normatives (ISO 13485, ISO 9001, ISO19011, FD X50-176, etc.)
- Savoir-faire : Elaboration de documentation qualité (manuel qualité, processus, procédures et enregistrements). Développer une méthodologie innovante de mise en place de système de management de la qualité (méthode ETAPES). Définition d’indicateurs de performance de processus (KPI), d’actions correctives et préventives (CAPA). Réalisation d’audit interne et transfert de compétences (ateliers qualité)
- Savoir-être : Aisance rédactionnelle en qualité (preuves documentaires), expertise qualité, capacité pédagogique, capacité à communiquer les enjeux, la motivation, etc.
- Savoir : Connaissances théoriques en analyse, gestion et management du risque. Connaissances normatives (ISO 31000, ISO 14971, ISO 31010)
- Savoir-faire : Réalisation d’Analyses des Modes de Défaillances, de leurs Effets et leur Criticité (AMDEC), Apporter un processus de management des risques, développer des outils Excel de management des risques
- Savoir-être : Expertise en management des risques, approche méthodologique de la gestion des risques.
- Savoir : Connaissances théoriques en affaires réglementaires dans les secteurs des dispositifs médicaux, sur la réglementation 2017/745 et 2017/746, exigences réglementaires en termes de sécurité et performance, test pré-clinique, clinique et en suivi clinique après commercialisation, veille normative et réglementaires, connaissances normatives (ISO62304, ISO 62366, ISO 60601-1, etc.).
- Savoir-faire : Qualification et classification selon les guides européen (MDCG) du dispositif médical, positionnement réglementaire (feuille de route technico-réglementaire), recherche de normes, règlements, directives, ordonnances, décret et arrêtés applicables, partage de connaissance sur des normes du secteur des dispositifs médicaux (ISO 62304, ISO62366-1, ISO 60601-1, etc.).
- Savoir-être : Expertise en affaires réglementaires, point de contact avec les organismes notifiés, aisance rédactionnelle
Lors de ce stage j’ai également développé de nouvelles compétences professionnelles qui sont :
- Savoir : Découverte de l’environnement startup, de l’entreprenariat, de la dialyse connaissance normative (ISO 56000).
- Savoir-faire : Application de méthodologie théorique (ETAPES) en pratique, participation dans des rendez-vous innovation (Snitem : journée startup innovantes du dispositif médical), à la présentation d’ouverture de nouveau site de production (usine 2.0 de Matra électronique).
- Savoir-être : Capacité à innover, à trouver des alternatives, à anticiper.
- Savoir : Connaissances théoriques dans le domaine de l’insuffisance rénale chronique et de la Bio-impédance. Découverte du parcours patients atteint d’insuffisance rénale chronique, des associations de patients dyalisé et des prescripteurs.
- Savoir-faire : Synthèse de veille scientifique (synthèse de lecture de rapports de sociétés savantes en néphrologie, rapport de la Haute Autorité de Santé (HAS) et d’articles scientifiques sur la néphrologie et la Bio-impédance).
- Savoir-être : Capacité de synthèse de connaissances, de restitution de connaissance et d’esprit critique.
4.1.1 Difficultés rencontrées.
Travailler dans des petites structures comme les startups est une véritable opportunité, il faut une organisation minutieuse, rigoureuse et une communication bienveillante, cependant
Comme développé dans la partie 1.1, le manque de ressources est une réalité dans le milieu des startups avant les premières levées de fonds. Evoluer dans le milieu de la startup, c’est évoluer dans un environnement d’incertitude où la peur, le doute, la frustration, la réalité du quotidien sont présent à chaque instant.
La quantité d’éléments à aborder, pour la mise en place du système de management de la qualité, la conformité à la réglementation, sont conséquentes et le manque de temps créer un sentiment de peur, celle-ci a été maîtrisé, grâce à des échanges constructifs et en priorisant les notions essentielles à transmettre.
Il est important de garder un contact avec ses professeurs, ancien intervenant de formation, des experts du domaine pour pouvoir poser des questions. Le partage de leurs expériences ainsi que leurs retours ont été de réelles ressources et ont participé de manière significative à dissiper les questionnements d’ordre stratégique, opérationnel ou encore de communication interprofessionnelle.
Enfin les frustrations qui se développent dans les désaccords, le manque de temps ou l’indisponibilité ont été maîtrise par l’application de savoir-être simple qui sont :
- La prise de recul : Ce sont les situations qui sont complexes, il ne faut pas en faire une affaire personnelle.
- Privilégier la communication : L’intériorisation ne permet pas de répondre à la frustration, il faut privilégier une communication constructive.
- Proposer des compromis : La situation ne pouvant pas être résolue à l’immédiat, des alternatives sont toujours possibles.
le manque d’accompagnement à temps plein, due aux couts élevés de cette prestation, peut être également une véritable source de stress dans l’atteinte de ces objectifs complexes.
4.2 Compétences à acquérir
Pour atteindre mon objectif de devenir responsable qualité et affaires réglementaires, des compétences doivent être approfondies, notamment en management d’investigations cliniques, d’évaluation clinique, de surveillance après commercialisation.
Mes compétences linguistiques doivent continuer à être développée pour, parler de manière courante (fluente en anglais) et pour utiliser l’anglais de manière courante comme langue de travail. Cela m’aidera à ouvrir des opportunités de carrière à l’international.
Développer une culture dans les domaines ingénieries informatiques, électronique et mécanique et financière afin d’obtenir, une aisance inter-fonctionnelle dans mes futures responsabilités, communication avec des experts des domaines cité précédemment facilité et fluide et ainsi faire évoluer mon travail.
Ces compétences se développeront en acceptant dans un premier temps, un poste de consultant en qualité et affaires réglementaires afin, d’étudier directement des situations complexes sur site et de démontrer une expérience minimum pour devenir PCVRR selon l’article 25 du règlement 2017/745. Dans un second temps devenir PCVRR pour acquérir l’expérience nécessaire pour faire face à toutes les situations qui se présenteront en qualité de responsable qualité et affaires réglementaires.
4.3 Liens avec le Master
Le Master Ingénierie de la Santé, m’a amené à me questionner sur plusieurs thématiques différentes, la santé, l’innovation, l’économie, le social et l’environnement. En effet, les enseignements théoriques ainsi que les multiples projets que j’ai menés au sein de ma formation à l’UTC mon dirigé vers trois questions principales qui sont :
Comment faire pour que notre génération, corrige les travers de la génération précédent et prépare l’avenir tout en grandissant ?
Comment allier écologie, économie et social dans un monde en perpétuelle mouvement ?
Comment puis-je contribuer à changer le monde ?
La crise sanitaire qui a débuté au commencement de ma formation et semble s’atténuer à la fin de mon stage à mis en lumière l’importance de la santé publique, de l’innovation et de l’économie. Depuis près de 50 ans l’Université de Technologie de Compiègne (UTC) place une attention particulière à placer l’évolution de ces thèmes dans ses formations et ainsi donné l’envie à ses étudiants d’y participer.
En effet, en passant par des enseignements théoriques sur l’économie globale et maîtrise de la qualité (FQ01), de management des organisations biomédicale (IDCA), maîtrise des risques (TS01), d’ouverture à la recherche et innovation en santé (IDC6), l’audit et évaluation des organisations : normes et processus (IDCK), à l’ingénierie de projet (IDCB) m’ont aidé à développer une casquette de qualiticien, en délivrant une expertise sur la qualité et son management, des documents qualité et d’une nouvelle méthodologie d’implémentation de management de la qualité. Cette casquette que j’ai utilisé lors de ce stage, me permet de comprendre les situations complexe présentes et me permet d’anticiper les solutions futures et de répondre à ma première question.
En continuant sur des enseignements théoriques sur l’introduction à l’instrumentation biomédicale santé (IDC8), l’Initiation au droit (SOO6), le cycle de vie d’un dispositif médical (IDCE), les affaires réglementaires (IDCL), l’organisation du système de santé (IDCF), la communication professionnelle de projet (IDCC)pour terminer avec projet d’intégration (IDCD). Ces enseignements m’ont aidé à développer ma seconde casquette, celle de l’expert affaires réglementaires. Cette seconde casquette que j’ai également utilisé lors de mon stage en apportant une expertise réglementaire, en livrant les premiers éléments de la documentation technique et en ayant été le point de contact avec des Organismes Notifiés. Cette casquette va me permettre d’aider à faire évoluer la santé en accompagnant les acteurs de la santé à atteindre leurs objectifs. Cette seconde casquette me permet de répondre à ma seconde question.
L’utilisation quotidienne de mes deux casquettes me permettent de répondre à l’ensemble de mes questions.
La réponse commune a ses trois questions est donc :
Mon travail ainsi que ses évolutions en qualité de responsable qualité et affaires réglementaires dans le secteur de la santé et plus précisément dans le domaine des dispositifs médicaux pour le moment.
Pour conclure, mon stage au sein de Home Habilis m’a aidé à confirmer cette réponse et de continuer dans cette voie. Ce stage fut pour moi une opportunité incroyable pour moi et une véritable expérience professionnalisante.
Conclusion
La Maladie Rénale Chronique (MRC) est passé de la 27e à la 11e cause de mortalité en moins de 30 ans [6]. Cette pathologie impactant de manière conséquente la qualité de vie des patients qui en sont atteint, progresse de manière silencieuse [6]
C’est grâce au développement de la médecine, de la télésurveillance et de l’intelligence artificielle, que des innovations technologiques en santé voit le jour. Ces nouveaux projets se développent souvent dans des petites structures comme les startups. C’est ainsi que Home Habilis développe une innovation médicale destinée aux patients atteints d'insuffisance rénale chronique. C’est en proposant ce nouveau dispositif médical que Home Habilis souhaite améliorer la qualité de vie ces patients.
Pour mener à bien son projet Home Habilis doit se conformer, comme toutes les entreprises fabricante de dispositifs médicaux à la réglementation européenne relative à la mise sur le marché de dispositifs médicaux. Que ce soit le règlement 2017/745 relatif aux dispositifs médicaux [1] ou le règlement 2017/746 relatif aux dispositifs médicaux de diagnostic in vitro [2], les fabricants de dispositifs médicaux doivent centrer leurs activités autour d’un système de management de la qualité afin de toujours proposer aux patients des dispositifs médicaux dont la sécurité et la performance ont été démontrées.
La mise en place de ce système de management de la qualité selon les référentiels ISO 13485 :2016 et son amendement A1 dans les jeunes startups comme Home Habilis, nécessite une méthodologie rigoureuse, basé sur de l’amélioration continue ainsi que l’implication de ses acteurs. La méthodologie ETAPES, Evaluer, Transmettre, Accompagner, Programmer, Enregistrer et S’assurer permet cette mise en place de façon performante et durable.
L’anticipation du déploiement du système de management de la qualité permet aux jeunes startups, d’éviter une perte de ressource souvent irrécupérable, une anticipation de conformité aux exigences réglementaires applicables aux fabricants de dispositifs médicaux, est un argument supplémentaire de taille en faveur des startups lors de la recherche de levée de fonds. Ce qui est une réelle valeur ajoutée dans cet environnement incertain et concurrentiel.
Bibliographie
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, Règlement OJ L 117, 5.5.2017, avr. 2017. Consulté le : sept. 25, 2020. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/?uri=CELEX%3A32017R0745
[2] « Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission (Texte présentant de l’intérêt pour l’EEE. ) ». JOUE, https://eur-lex.europa.eu, mai 05, 2017. Consulté le : nov. 09, 2018. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/746/oj/fra
[3] « norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », Ed. Afnor, Paris, www.afnor.org, avr. 30, 2016. [En ligne]. Disponible sur : www.afnor.org
[4] M.-H. Canu et V. Bérézowski, Mémo visuel de physiologie humaine, Dunod. Dunod, 2018. Consulté le : mars 28, 2021. [En ligne]. Disponible sur : http://univ.scholarvox.com/catalog/book/docid/88863175?searchterm=physiologie%20humaine
[5] E. Vidal-Petiot et M. Flamant, « Mesure et estimation du débit de filtration glomérulaire », Néphrologie Thérapeutique, vol. 13, no 7, p. 560‑568, déc. 2017, doi : 10.1016/j.nephro.2017.10.001.
[6] SFNDT ( Société Francophone de Néphrologie, Dialyse et Transplantation), « Livre Blanc de la Dialyse à domicile », Ed. Société Francophone de Néphrologie, Dialyse et Transplantation, juin 2019. Consulté le : mars 19, 2021. [En ligne]. Disponible sur : https://www.sfndt.org/actualites/livre-blanc-de-la-dialyse-domicile
[7] « Évaluation du rapport albuminurie/créatininurie dans le diagnostic de la maladie rénale chronique chez l’adulte - Rapport d’évaluation », Ed.Haute Autorité de Santé, EVALUATION DES TECHNOLOGIES DE SANTÉ, avr. 2012. Consulté le : mars 28, 2021. [En ligne]. Disponible sur : https://www.has-sante.fr/jcms/c_1169049/fr/evaluation-du-rapport-albuminurie/creatininurie-dans-le-diagnostic-de-la-maladie-renale-chronique-chez-l-adulte-rapport-d-evaluation
[8] Haute Autorité de Santé, « Haute Autorité de Santé - Maladie Rénale Chronique (MRC) de l’enfant », Ed.Haute Autorité de Santé, GUIDE MALADIE CHRONIQUE, nov. 2018. Consulté le : mars 29, 2021. [En ligne]. Disponible sur : https://www.has-sante.fr/jcms/c_2889689/fr/maladie-renale-chronique-mrc-de-l-enfant
[9] « Guide parcours de soins maladie rénale chronique de l’adulte », Ed. Haute Autorité de Santé, Guide Points critiques du parcours de soin, févr. 2012. Consulté le : juin 06, 2021. [En ligne]. Disponible sur : https://www.has-sante.fr/jcms/c_1241102/fr/guide-parcours-de-soins-maladie-renale-chronique-de-l-adulte
[10] Cécile Couchoud, Emmanuel Villar, Luc Frimat, Anne Fagot-Campagna, et Bénédicte Stengel, « L’insuffisance rénale chronique terminale associée à un diabète : fréquence et conditions d’initiation du traitement de suppléance, France, 2006 », Inst. Veille Sanit., no 43, p. 5, nov. 2008.
[11] « Baromètre EY 2020 : la French Tech résiste et renforce sa compétitivité », Ministère de l’Économie, des Finances et de la Relance, Ed. Secrétariat général - Service de la Communication, Communiqué de presse 553, janv. 2021. Consulté le : mai 16, 2021. [En ligne]. Disponible sur : https://www.economie.gouv.fr/recherche-resultat?search_api_views_fulltext=553&sort_by=search_api_relevance&afficher_extraits=oui&gid=
[12] « norme NF EN ISO 56000 Management de l’innovation - Principes essentiels et vocabulaire », Ed. Afnor, Paris, www.afnor.org, janv. 13, 2021. Consulté le : juin 12, 2021. [En ligne]. Disponible sur : www.afnor.org
[13] « Rapport d’activité 2015-2016 de l’Agence du Numérique », MINISTÈRE DE L’ÉCONOMIE ET DES FINANCES, Ed. Agence Nationale de la Cohésion des Territoires, Rapport d’activité, mai 2017. Consulté le : juin 12, 2021. [En ligne]. Disponible sur : https://www.amenagement-numerique.gouv.fr/fr/qui-sommes-nous/nous-connaitre
[14] « norme PR NF EN ISO 13485/A1 - Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires (ISO 13485:2016) », Ed. Afnor, Paris, www.afnor.org, janv. 06, 2020. Consulté le : déc. 08, 2020. [En ligne]. Disponible sur : www.afnor.org
[15] « norme NF EN ISO 9001 - Systèmes de management de la qualité - Exigences (Tirage 2 (2015-10-01)) », Ed. Afnor, Paris, www.afnor.org, oct. 15, 2015. Consulté le : sept. 19, 2020. [En ligne]. Disponible sur : www.afnor.org
[16] « norme NF EN ISO 9004 - Gestion des performances durables d’un organisme - Approche de management par la qualité (annulée en mai 2018) », Ed. Afnor, Paris, www.afnor.org, déc. 01, 2009. [En ligne]. Disponible sur : www.afnor.org
[17] « norme NF EN ISO 9000 Systèmes de management de la qualité - Principes essentiels et vocabulaire (Tirage 2 (2015-10-01)) », Ed. Afnor, Paris, www.afnor.org, oct. 15, 2015. Consulté le : oct. 18, 2020. [En ligne]. Disponible sur : www.afnor.org
[18] « norme NF ISO 31000 Management du risque - Lignes directrices », Ed. Afnor, Paris, www.afnor.org, juin 09, 2018. Consulté le : avr. 28, 2021. [En ligne]. Disponible sur : www.afnor.org
[19] « norme FD X50-176 - Outils de management - Management des processus - Guide de mise en oeuvre », Ed. Afnor, Paris, www.afnor.org, août 23, 2017. [En ligne]. Disponible sur : www.afnor.org
[20] « norme NF EN ISO 19011 Lignes directrices pour l’audit des systèmes de management », Ed. Afnor, Paris, www.afnor.org, juill. 04, 2018. Consulté le : mai 03, 2021. [En ligne]. Disponible sur : www.afnor.org
[21] F. Gillet-Goinard et B. Seno, La boîte à outils de la qualité Ed. 4. Dunod, 2020. Consulté le : juin 19, 2021. [En ligne]. Disponible sur : http://univ.scholarvox.com/catalog/book/docid/88882009?searchterm=qualit%C3%A9
[22] « Recommandation de la Commission du 6 mai 2003 concernant la définition des micro, petites et moyennes entreprises », Journal officiel de l’Union européenne, OJ L 124, mai 2003. Consulté le : juin 20, 2021. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/ALL/?uri=celex%3A32003H0361
[23] EUROPEAN COMMISSION, « Guidance - MDCG endorsed documents | Public Health ». https://ec.europa.eu/health/md_sector/new_regulations/guidance_en (consulté le janv. 07, 2021).
[24] « Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR », Ed. European Commission, Guidance, oct. 2019. Consulté le : mars 17, 2021. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/37581
[25] « MDCG 2019-14 Explanatory note on MDR codes », Ed. Medical Device Coordination Group, Guidance MDCG 2019-14, déc. 2019. Consulté le : mai 14, 2021. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/38670?locale=fr
[26] « MDCG 2020-10/1 Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745 », Guidance, mai 2020. Consulté le : juin 20, 2021. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/41183?locale=fr
[27] « MDCG 2019-16 - Guidance on Cybersecurity for medical devices », Ed. European Commission, Guide, janv. 2020. Consulté le : févr. 08, 2021. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/41863
[28] Ayadi Mohamed Aziz, Brochet Paul, Fosso Matchinde Mégane Shandy, Sadiqui Oumaima, et Rossin Valériane, « Roadmap réglementaire pour une innovation technologique d’imagerie de haute résolution - Bibliothèque des travaux Master », Bibliothèque des travaux Master, janv. 2020. https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids084/ (consulté le janv. 08, 2021).
[29] S. Grimnes et Ø. G. Martinsen, « Chapter 1 - INTRODUCTION1 », in Bioimpedance and Bioelectricity Basics (Second Edition), Second Edition., S. Grimnes et Ø. G. Martinsen, Éd. New York : Academic Press, 2008, p. 1‑6. doi : 10.1016/B978-0-12-374004-5.00001-5.
[30] « Définition de Deep Tech », mai 03, 2018. https://www.bpifrance.fr/A-la-une/Actualites/DansNotreJargon-la-Deep-Tech-40252 (consulté le juin 23, 2021).
[31] Bpifrance Financement, « Les incubateurs », janv. 2020. https://bpifrance-creation.fr/encyclopedie/se-faire-accompagner/lieux-dhebergement-accompagnement/incubateurs (consulté le mai 18, 2021).
[32] « Qu’est-ce que la Medtech ? », oct. 16, 2020. https://www.bpifrance.fr/A-la-une/Actualites/Qu-est-ce-que-la-Medtech-50741 (consulté le juin 23, 2021).
[33] Le médiateur des entreprises, « De l’idée à l’industrialisation : Réussissez votre preuve de concept. », République Française, Ed. Ministère de l’Économie, des Finances et de la Relance, juin 2019. Consulté le : juin 22, 2021. [En ligne]. Disponible sur : https://www.economie.gouv.fr/recherche-resultat?search_api_views_fulltext=POC&sort_by=search_api_relevance&afficher_extraits=oui&gid=
ANNEXE I : Cartographie de processus de Home Habilis
Figure 19 : Draft de la cartographie des processus de Home Habilis (source : auteur).
ANNEXE II : Fiche Descriptive du processus S de Home habilis.
Figure 20 : Draft de processus documenté de Home Habilis (1) (source : auteur).
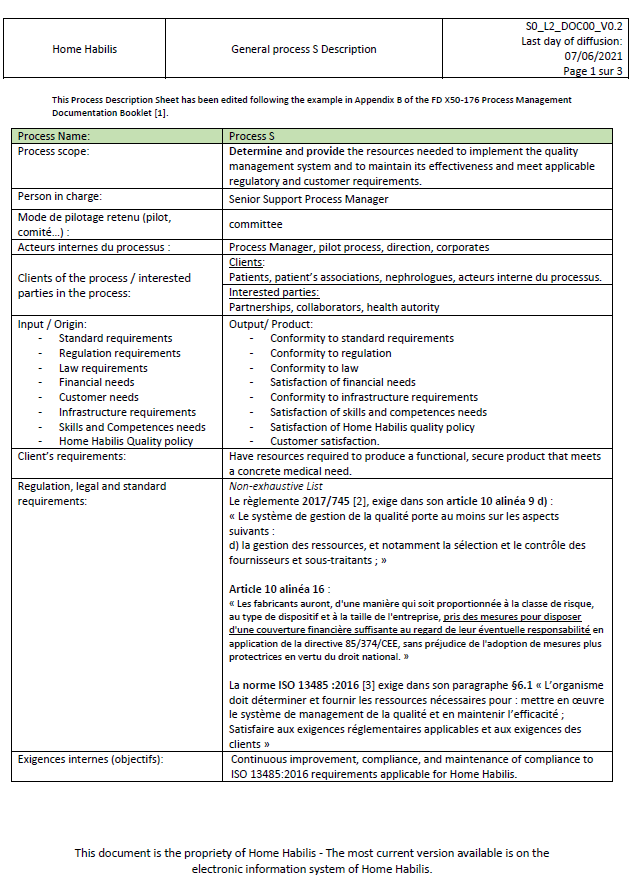
Figure 21 : Draft de processus documenté de Home Habilis (2) (source : auteur)
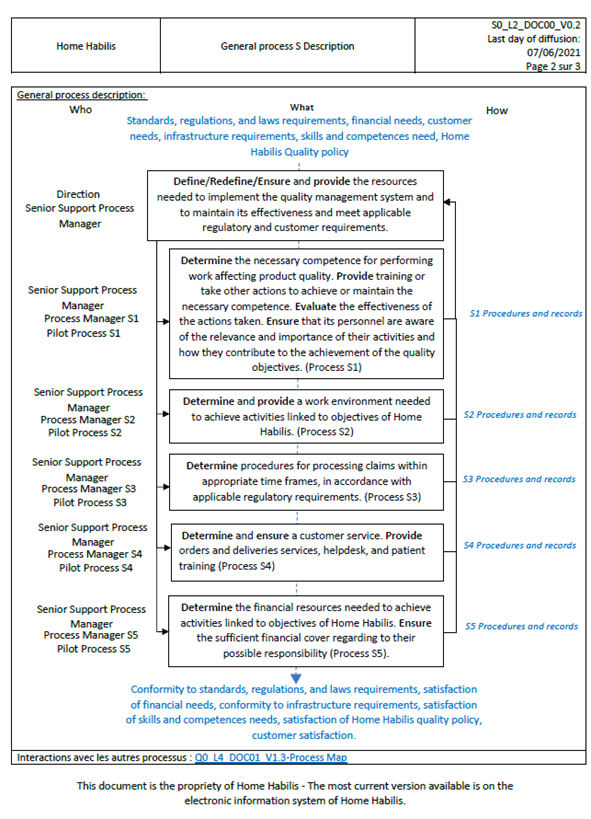
Figure 22 : Draft processus documenté de Home Habilis (3) (source : auteur).

ANNEXE III : Procédure documentée de Home Habilis.
Figure 23 : Draft de procédure documentée d'audit interne de Home Habilis (source : Home Habilis)
ANNEXE IV : Processus de management des risques basée sur la norme ISO 31000
Figure 24 : Extrait du processus de management des risques selon la norme ISO 31000:2018 (source : Home Habilis)
ANNEXE V : Engagement de la direction.
Figure 25 : Draft de Lettre d’engagement de la direction de Home Habilis (source : Home Habilis).

ANNEXE VI : Conclusion de l'audit.
Figure 26 : Extrait du rapport d'audit interne de Home Habilis (source : Home Habilis)
