
IDS084 - Roadmap réglementaire pour une innovation technologique d'imagerie de haute résolution
DOI mémoire
https://doi.org/10.34746/7csg-s841Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

Ayadi Mohamed Aziz 
Paul Brochet 
Fosso Matchinde Megane Shandy 
Rossin Valériane 
Oumaima SADIQUI
Contact
- Ayadi Mohamed Aziz : ayadimedaziz@gmail.com
- Brochet Paul : paul.brochet@icloud.com
- Fosso Matchinde Megane Shandy : shandiemegane2000@gmail.com
- Rossin Valeriane : valeriane.rossin@outlook.fr
- Sadiqui Oumaima : sadi.oumaima@gmail.com
Citation
A rappeler pour tout usage :
Valériane ROSSIN, Mohamed Aziz AYADI, Paul BROCHET, Oumaïma SADIKI et Megane Shandy FOSSO MATCHINDE, "Roadmap réglementaire pour une innovation technologique en imagerie haute résolution", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2021, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids084/ ; https://doi.org/10.34746/7csg-s841
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_09_idsap
Résumé
Start-up, Petites et Moyennes Entreprises et d’autres sociétés de dispositifs médicaux, font face à des problématiques sérieuses dans le cheminement règlementaire amenant au marquage CE comme la prise en main du règlement 2017/745, l’identification des documents nécessaires pour répondre aux exigences ou encore l’accès au marché. DeepColor Imaging, est une société parmi tant d’autres, dont le besoin principal est un accompagnement dans la démarche règlementaire pour l’obtention de la certification lui permettant la mise à disposition sur le marché Européen de son dispositif médical.
Ce mémoire a pour objectif d’éclaircir les fabricants en leur apportant des solutions pour leur roadmap règlementaire : une cartographie interactive présentant les étapes du marquage CE telles que l’identification des exigences générales, le choix de la procédure de vérification de la conformité ou encore l’évaluation clinique et un outil de positionnement permettant à l’utilisateur de suivre étape à par étape son avancée sur ce chemin règlementaire.
Ces outils sont accessibles gratuitement sur le web travaux.master.utc.fr au service des fabricants et nécessitent seulement les logiciels issus du pack Microsoft Office®.
Abstract
Start-ups, Small and Medium Enterprises and other medical device companies are facing serious issues in the regulatory process leading to CE marking, such as the handling of Regulation 2017/745, the identification of the documents needed to meet the requirements and market access. DeepColor Imaging is one of many companies whose main need is to assist them in the regulatory process to obtain the certification that will allow them to make their medical devices available on the European market.
The aim of this project is to provide manufacturers with solutions for their regulatory roadmap : an interactive map presenting the steps of the CE marking such as the identification of general requirements, the choice of conformity assessment procedure or the clinical evaluation and a positioning tool allowing the user to follow step by step his progress on this regulatory path.
These tools are available free of charge on the web for manufacturers and only require software from the Microsoft Office® package.
Téléchargements
Poster du mémoire
Cartographie interactive donnant une vision claire et précise de la démarche du marquage CE
Outil d'aide au positionnement sur la démarche du marquage CE

Mémoire d'Intelligence Méthodologique : Roadmap réglementaire pour une innovation technologique d'imagerie de haute résolution
Remerciements
L’ensemble du groupe tient à remercier en premier lieu Monsieur Fabrice RICHARD, directeur de DeepColor Imaging, porteur du projet et notre suiveur. Nos rencontres quotidiennes tout au long du projet nous ont permis d’avancer efficacement grâce à ses précieux conseils et nous as permis de nous recentrer sur le principal et d’améliorer la qualité finale de nos livrables.
Nous tenons également à remercier Monsieur Jean-Matthieu PROT qui a supervisé ce travail, et nous a accompagné dans ce projet avec des suggestions et conseils pertinents.
Nous remercions Mme Isabelle CLAUDE, responsable avec Mr PROT de l’unité d’enseignement « Projet d’intégration » et Mr Gilbert FARGES pour leurs conseils et avis qui nous ont permis d’améliorer et de rendre plus cohérent notre travail.
Introduction
DeepColor Imaging est une startup qui développe un dispositif d’imagerie médicale innovant. Ce dispositif, du nom de LightEcho Laparoscopic, vient enrichir la modalité de l’imagerie photoacoustique en lui apportant une technologie de capteur et de mesure d’ultrasons permettant d’atteindre des performances inégalées à ce jour par les techniques utilisées.
La règlementation est un frein pour de nombreuses entreprises du secteur du dispositif médical. Les startups, acteurs novices dans le domaine, en souffrent particulièrement ; notamment par la complexité des exigences réglementaires, l’accès au marché rendu difficile en conséquence et le coût élevé engendré.
La raison d’être de ce travail est donc d’aider DeepColor Imaging, et les entreprises qui se retrouvent dans la même situation, à avoir une idée plus concrète du processus réglementaire auquel sera assujetti leur produit pour arriver à l’obtention du marquage CE. Une roadmap détaillée et des outils informatiques de simplification et d'autodiagnostic ont donc été réalisés à cette fin.
1. L’innovation et la nouvelle réglementation
1.1 Définition et classification
D’après le nouveau règlement 2017/745 (Article 2), un dispositif médical est défini comme « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises […] et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. » [1].
Les dispositifs médicaux sont classés selon quatre classes principales de risque : Classe I, Classe IIa, Classe IIb et Classe III. Il existe des sous-classes notamment les classes I spéciales (Is stérile, Im mesurable et Icr chirurgicale réutilisable) et la classe IIb implantable.
La classification des dispositifs médicaux s’effectue selon l’annexe VIII du nouveau règlement (UE) 2017/745 [1]. Vingt-deux règles permettent de classer un dispositif médical. Les règles 1 à 4 sont à destination des dispositifs non invasifs, les règles 5 à 8 aux dispositifs invasifs, les règles 9 à 13 aux dispositifs actifs et le reste sont des règles particulières. Les critères permettant de classer un DM vont s’articuler autour du caractère invasif ou non, actif ou non, temps d’utilisation, dangerosité, contact avec le Système Nerveux Central ou non, à des fins chirurgicales ou non.
1.2 Le secteur des dispositifs médicaux
Le marché mondial des dispositifs médicaux est estimé à 425 milliards d’euros à l’horizon 2020. Le marché européen quant à lui est estimé à plus de 115 milliards € avec environ 675 000 employés et 27 000 fabricants dont 95 % de PME. Ce dernier a explosé en 2018 (120 milliards d’euros) en étant le deuxième marché leader dans le secteur du dispositif médical [2].
Selon le SNITEM, 1 502 entreprises ont été recensées dans le secteur, dont 93% de PME, engendrant 90 000 emplois directs. Le chiffre d’affaire généré sur le marché français atteint les 30 milliards d’euros avec 9 milliards d’euros de CA à l’export [3].
2. La société DeepColor
DeepColor Imaging est une startup nantaise, travaillant en collaboration avec un laboratoire de University College of London dans le cadre du développement d’un dispositif d’imagerie médicale innovant. DeepColor conçoit des systèmes d'imagerie médicale utilisant une technologie inédite à la frontière de l'optique et de l'échographie, la tomographie photoacoustique.
2.1 L’imagerie photoacoustique : modalité méconnue
2.1.1 Etat de l’art
Le marché de la chirurgie guidée en temps réel appliquée à l’exploration interne des muqueuses digestives comprend plusieurs dispositifs. Les équipements phares sont : l’endomicroscopie confocale à laser (ECL) qui utilise la technique de l’endoscopie pour la coupler avec un microscope miniaturisé [4]; la laparoscopie standard formé d’un tube métallique et d’un oculaire (avec caméra fixée) qui utilise de la lumière froide [5]; l’imagerie de fluorescence qui permet la détection de lésions tumorales non détectables en lumière blanche [6] et enfin la tomodensitométrie également appelée CT-Scan qui permet de réaliser des images en trois dimensions des différents organes du corps grâce aux rayons X [7].
2.1.2 La modalité d’imagerie
L’imagerie photoacoustique, inventée par Alexander Graham Bell, est une nouvelle modalité d'imagerie à la frontière de l'échographie et de l’optique. Elle délivre une image en temps-réel des tissus biologiques sans utiliser des agents de contraste [8], [9]. Il est maintenant possible grâce à cette modalité de suivre un paramètre essentiel pour anticiper les complications cliniques rencontrées dans les chirurgies complexes : la fonction vasculaire d'une zone opérée ou traitée. En effet, la technique utilisée par la société DeepColor Imaging s’appuie sur la visualisation de molécule endogène : l’hémoglobine. L’hémoglobine est un chromophore visible qui permet in fine de reconstruire les réseaux vasculaires et microvasculaires en 3D et en profondeur [9].
Figure 1 : Phénomène photoacoustique, Source : [8]
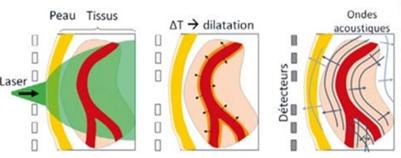
Le principe général (Figure 1) se base principalement sur une émission de lumière et réception d'un ultrason. L’intérêt principal est le fait que la lumière rentre plus profondément dans les tissus (1 à 2 cm), et que les ultrasons ne subissent pas le phénomène de diffusion biologique. Ainsi, la perte d’information notée lors de l’utilisation de lumière seule, comme il en est le cas pour les microscopes optiques, est contournée [8].
La différenciation entre les fabricants de technologies d’imagerie photoacoustique se fait sur deux éléments principaux : la manière de mesurer les ultrasons en photoacoustique et le capteur. Ainsi, C’est grâce à la mesure optique des ultrasons ainsi qu’au capteur innovant développé que DeepColor se distingue de l’offre présente actuellement sur le marché.
2.2 Contexte du produit DeepColor
2.2.1 Le cancer du colon
En France, le deuxième et le troisième cancer le plus fréquent respectivement chez la femme et l’homme est le cancer du côlon. En 2018, on relève 1.8 millions de nouveaux cas [10] dans le monde. Au niveau national, 44 872 nouveaux cas sont relevés avec 17 684 décès en 2017 [11]. La laparoscopie étant la méthode privilégiée pour le traitement chirurgical de cette condition, son importance est vite justifiée.
2.2.2 La chirurgie laparoscopique
La coelioscopie également appelée laparoscopie permet de « réaliser une opération à l’intérieur du ventre en visualisant l’extérieur des conduits biologiques sans que ce soit nécessaire de pratiquer de larges incisions »[12],[13]. Contrairement à la laparotomie [14], la laparoscopie est la technique la plus pertinente à utiliser pour le diagnostic ou le traitement de nombreuses pathologies gynécologiques, digestives, urologiques ou cancéreuses [15]. Cependant, cette technique chirurgicale, comprenant une anastomose (communication établie entre deux conduits organiques [16]), ne permet pas encore d’éviter les complications post-opératoires.
La fistule anastomotique, désignée comme une mauvaise cicatrisation de l’anastomose, est une des complications post-opératoires majeures lors des chirurgies gastro-intestinales [17]. Elle intervient entre 5 et 10% des cas de complications post-opératoires de colectomie ; et peut engendrer des conséquences sévères à long terme, dont un risque de sténose anastomotique. Une diminution de la qualité de vie et de la survie est aussi notée.
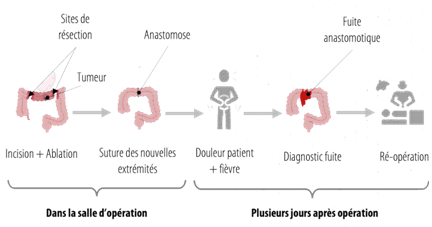
Le diagnostic précoce de la fistule anastomotique (à l’origine de la fuite anastomotique) est donc déterminant pour éviter la mise en jeu du pronostic vital du patient [17] (Figure 2) . La ré-opération, qui survient dans les cas les plus graves (Grade C), engendre des coûts supplémentaires liés à la nouvelle prestation chirurgicale, des durées d’hospitalisation allongées, des taux d’échec de suture plus importants, ainsi que des taux de mortalité croissants [18].
Figure 2 : Schéma récapitulatif d’apparition d’une fuite anastomotique dans le cadre d’un cancer du côlon, Source : d’après [19]
2.2.3 Air leak test et fluorescence
Peu de techniques en rapport avec l’évaluation de l’anastomose en peropératoire , et donc d’éventuelles fuites anastomotiques, existent à ce jour ; les deux techniques phares sont l’Air-Leak test et la fluorescence.
La méthode de vérification actuelle pour l’examen de l’intégrité anastomotique peropératoire intègre l’Air-Leak test. Facile et peu couteux, il peut être associé à de l’imagerie endoscopique, mais il est subjectif et ne permet pas la bonne évaluation de la perfusion de l’intestin [20].
Une autre technique a été récemment introduite ; il s’agit de l'angiographie peropératoire par fluorescence (API) afin d’optimiser l’évaluation de la réserve sanguine anastomotique. L’API évalue la perfusion intestinale, en administrant du vert d’Indocyanine (agent de contraste), à deux étapes critiques de l’intervention chirurgicale : juste avant la transsection colique proximale, et juste après l’anastomose. En chirurgie colorectale, cette technique peropératoire peut être utilisée pour s’assurer de la perfusion adéquate durant la formation de l’anastomose [21].
2.3 Le LightEcho Laparoscopic
La spécificité de DeepColor se traduit par la détection et/ou mesure optique des ultrasons, ce qui lui confère une meilleure résolution et sensibilité.
Cette technique de mesure optique, couplée à un capteur forment le cœur du premier produit en développement de la startup : un laparoscope photoacoustique du nom de LightEcho Laparoscopic.
Il est composé de différentes parties [19] :
- Un laser d’excitation qui envoie la lumière sur l’organe/tissu biologique. Ce laser est pulsé et accordable avec différentes longueurs d’onde possible ;
- Un capteur Fabry Perot qui réalise une mesure optique d’ondes ultrasonores ;
- Un laparoscope avec le capteur au bout d’une longueur de 265 mm ;
- Un laser à longueur d’onde fixe qui mesure l’impact de l’onde ultrason générée. Il n’a pas d’interaction avec le patient ;
- Logiciel pour reconstruire l’image et pour contrôler les différents composants cités.
Ce laparoscope est destiné à la chirurgie minimalement invasive en laparoscopie en chirurgie gastro-intestinale, plus particulièrement pour la chirurgie des cancers du côlon.
Plusieurs problématiques médico-économiques sont relevées pour cette première application clinique. La problématique majeure demeure celle des complications médicales fréquentes occasionnées par la chirurgie des cancers du côlon, notamment les fuites anastomotiques qui induisent un taux de mortalité plus élevé et une durée d’hospitalisation, et donc des coûts, plus conséquents.
C’est dans la résolution de cette problématique qu’apparaît tout le bénéfice de l'outil Deepcolor. En effet, le LightEcho Laparoscopic donne au chirurgien une vision résolue et en temps-réel de la fonction vasculaire durant l'acte chirurgical. Il permettra d'optimiser et sécuriser l'anastomose pour anticiper les potentielles fuites anastomotiques en chirurgie gastro-intestinale.
2.4 Le produit DeepColor et la règlementation
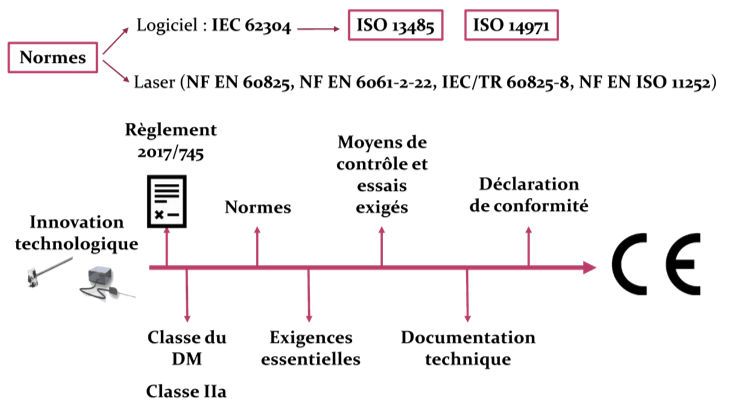
Dans le but de mettre sur le marché leur nouveau dispositif, DeepColor Imaging a besoin d’obtenir le marquage CE pour son produit. Les exigences réglementaires pour l’obtention du marquage CE diffèrent d’un dispositif à un autre, et ce selon sa classe, sa nature et son application.
Avant d’entamer donc le détail du processus réglementaire à suivre, le dispositif doit être bien défini et son titre de dispositif médical justifié, sa classe doit être déterminée et justifiée.
2.5 Contextes règlementaires
2.5.1 Définitions
« Le règlement est un acte juridique européen, de portée générale, obligatoire dans toutes ses dispositions : les États membres sont tenus de les appliquer telles qu’elles sont définies par le règlement. Le règlement est donc directement applicable dans l’ordre juridique des États membres. Il s’impose à tous les sujets de droit : particuliers, personnes morales, États, institutions. Ceci le différencie de la décision, autre acte européen obligatoire dans toutes ses dispositions, mais seulement pour les destinataires qu’il désigne. » [22].
Différents points sont à prendre en compte pour les entreprises en vue de se conformer au règlement 2017/745 [3], [23] :
- Le respect des
deadlines en tenant compte de :
- L’augmentation des dossiers complets à réaliser ;
- Les essais cliniques et suivi post-commercialisation beaucoup plus important ;
- La diminution du nombre d’organismes notifiés.
- Un besoin accru en
ressources humaines :
- Former et recruter un responsable règlementaire ;
- Former du personnel pour interagir avec les organismes notifiés.
- Un coût financier :
- Augmentation des coûts de prestation des ON ;
- Délais des ON dans la prise en considération, l’étude des dossiers et la certification du marquage CE ;
- Salaire des ressources humaines nécessaires : Il est devenu impossible d’obtenir le marquage CE sans la présence d’au moins une personne qualifiée et dédiée à la qualité et les affaires règlementaires. Utiliser un prestataire externe permettant de réaliser le marquage CE d’une entreprise est possible, mais couteux ;
- Mise à jour des dossiers techniques pour l’application de nouvelle règlementation en 2022 ;
- Mise en place de l’évaluation clinique pouvant varier de 50 000 à 100 000€ ;
- Evaluation biologique pour garantir une biocompatibilité des matériaux utilisés entraîne des coûts supplémentaires.
2.5.2 Changement règlementaire et le nouveau règlement
La directive Européenne 93/ 42/CEE apparue en 1993 donnait un cadre règlementaire à la mise sur le marché des dispositifs médicaux. Cependant, de nombreuses autres directives sont apparues pour étoffer cette dernière. Elles sont toutes applicables à l’ensemble des états membres de l’Union Européenne mais l’application dans chaque état étant laissée libre, l’interprétation des exigences concernant la mise sur le marché d’un DM diffère d’un pays à l’autre. L’ajout à ceci des nombreux scandales évoqués comme Poly Implant Prothèse (PIP) ou encore celui du dispositif Essure ont provoqué des tensions au cœur du secteur des DMs [23].
Pour pallier les dysfonctionnements des applications des directives et aux scandales naissants, une nouvelle règlementation européenne a fait son apparition. Cette dernière, comme étant « directement applicable dans l’ordre juridiques des états membres », permet de rassembler tous les acteurs du dispositif médical sous un seul et même règlement. Le règlement 2017/745 a été initié en automne 2012 par la commission Européenne.
Ce nouveau règlement garanti la transparence des informations grâce à l'identifiant unique qui est obligatoire, il permet de retrouver le DM dans la base de données (EUDAMED) nouvellement développée. Son plus grand intérêt est de surveiller le marché et de renforcer la vigilance en collectant des donnés sur le fonctionnement des DMs [1].
Ce règlement répond aussi à un réel besoin qui est d’assurer la sécurité du patient et des utilisateurs en imposant les contrôles nécessaires au fabricant. Ceci permet d’anticiper les risques et d’éviter les incidents.
Pour tout fabricant, qui aimerait mettre sur le marché son dispositif médical, il est obligatoire de respecter les exigences générales de la réglementation appliquée aux dispositifs médicaux. Avec la sortie de la nouvelle réglementation européenne 2017/745, il est important pour un nouveau fabricant d’appliquer directement cette règlementation au lieu de la directive 93/42/CEE qui sera d’application obligatoire à partir du 26 mai 2021.
En effet, à compter de cette date, tous les fabricants de dispositifs médicaux seront dans l’obligation de montrer que leurs dispositifs médicaux respectent les exigences dudit règlement s’ils souhaitent conserver leurs produits sur le marché. Ainsi, s’ils appliquent la directive au lieu du règlement, cela sera à leur désavantage car il y aura une perte de ressources humaines et ils ralentiront le temps de la mise sur le marché de leurs produits.
Le 27 mai 2024, tous les certificats délivrés par les organismes notifiés sur la base des directives prendront fin et devront avoir été obligatoirement renouvelés pour conserver la commercialisation [1].
2.5.3 Le marquage CE
Le règlement 2017/745 définit quelles sont les procédures d’évaluation de la conformité pour mettre en œuvre une procédure de marquage CE. Ce marquage est défini selon ce règlement comme : « un marquage par lequel un fabricant indique qu’un dispositif est conforme aux dispositions applicables du présent règlement et des autres actes législatifs d’harmonisation de l’Union qui en prévoient l’apposition » [1]. Les six étapes à respecter pour l’obtention du marquage CE sont les suivantes (Figure 3) :
- Identification de la règlementation applicable au produit ;
- Identification des exigences générales des règlements ;
- Identification des moyens de contrôle et d’essais exigés ;
- Rédaction de la documentation technique ;
- Rédaction de la déclaration de conformité ;
- Apposition du marquage CE.
Figure 3 : Démarche de marquage CE, Source : auteurs
Néanmoins, le marquage CE d’un produit ne garantit pas sa qualité. Il signifie tout simplement que le produit respecte les exigences en termes de performance et sécurité mais cela ne signifie pas que c’est un produit de qualité. Il n’est pas également une homologation qui est une autorisation de mise sur le marché de certains dispositifs médicaux qui a disparu depuis 1998, date à laquelle le marquage CE est devenu obligatoire pour tous les dispositifs médicaux. Pour finir, ce n’est pas non plus, un critère de remboursement [24].
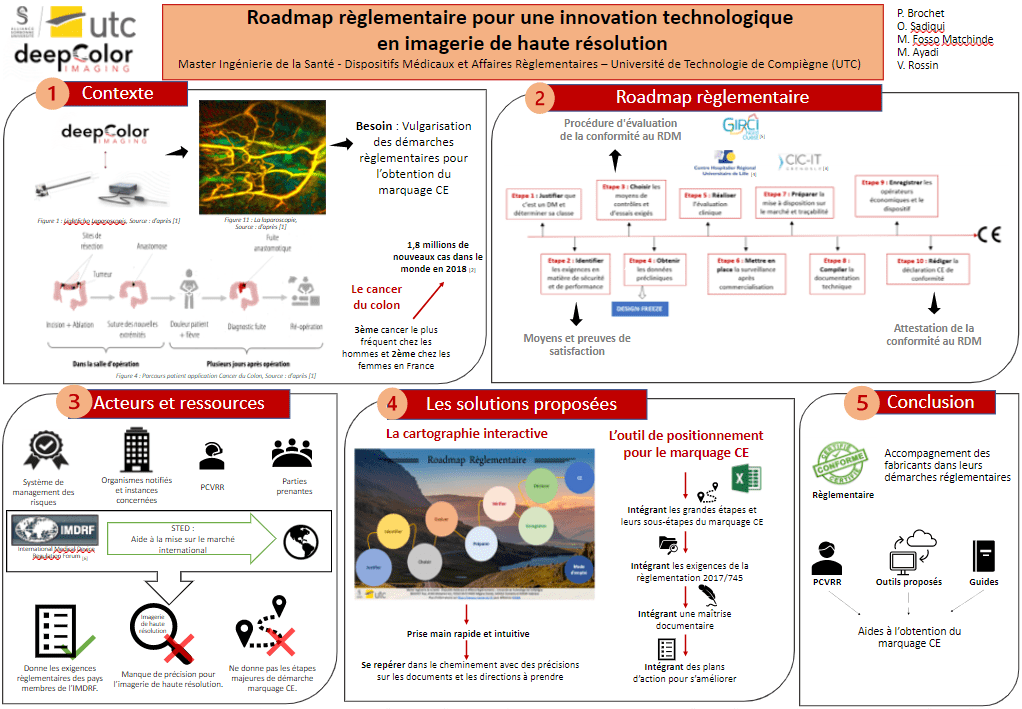
3. Un parcours règlementaire clair et intuitif
Afin d’obtenir le marquage CE, la conformité au règlement 2017/745 est une priorité. Un plan stratégique clair est donc nécessaire [25]. La Figure 4 permet de visualiser les principales étapes.
Figure 4 : Plan d’actions prioritaires, Source : auteurs
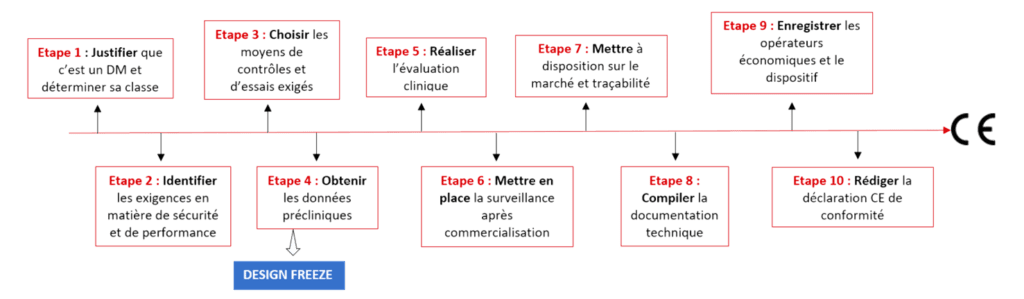
Design freeze : Conception final du prototype si est sujet à l’assurance qualité conception.
Suivant ce plan stratégique, il est important de déterminer la stratégie de mise sur le marché (internationale, nationale, etc..) afin de déterminer les textes règlementaires applicables dans le but de gagner en ressources. Il faudra également constituer une équipe accompagnante la personne chargée de veiller au respect de la règlementation.
Vis-à-vis du dispositif, il faut prouver que le dispositif est bien un dispositif médical et justifier sa classe selon les règles du règlement 2017/745. Avant la conception du dispositif, il est nécessaire de mettre en œuvre une analyse des risques, identifier les exigences en matière de sécurité et de performance applicables et choisir les moyens de contrôle et d’essais exigés. La conception doit s'arrêter (design freeze) pour permettre l’obtention des données précliniques et effectuer une évaluation clinique.
Ensuite, il est important de mettre en place d’une surveillance après commercialisation, le respect des exigences de la mise à disposition sur le marché et la traçabilité du dispositif.
C’est après toutes ces étapes que la documentation technique pourra être compilée. Et faire l’enregistrement du dispositif en lui affectant un identifiant Unique (IUD) et des opérateurs économiques.
Pour terminer, il faut réaliser une première version de la déclaration de conformité juste après l’audit de certification et l’apposition du marquage CE.
L’amélioration continue sera au cœur de la démarche règlementaire post-marquage car il faudra se préparer aux éventuelles visites des Organismes Notifiés (ON) et procéder à une surveillance après commercialisation traçable et tenue à jour.
3.1 Préparer
3.1.1 Etape 1 : Définition de la classe du dispositif médical
Il s’agit, dans un premier temps, d’appréhender la définition de dispositifs invasifs et actifs. Selon le règlement, un dispositif invasif est défini comme un dispositif qui « pénètre à l’intérieur du corps à travers la surface du corps, y compris à travers les muqueuses d’orifices du corps, à l’aide ou dans le cadre d’un acte chirurgical, et un dispositif opérant une pénétration par une voie autre qu’un orifice du corps. ». Un dispositif actif est considéré comme « tout dispositif dont le fonctionnement dépend d’une source d’énergie autre que celle générée par le corps humain à cette fin ou par la pesanteur et agissant par modification de la densité de cette énergie ou par conversion de celle-ci. Les dispositifs destinés à la transmission d’énergie, de substances ou d’autres éléments, sans modification significative, entre un dispositif actif et le patient ne sont pas réputés être des dispositifs actifs. Les logiciels sont aussi réputés être des dispositifs actifs. ».
Dans un second temps, il s’agit de clarifier les différents temps d’utilisation selon le règlement :
- Utilisation temporaire : « normalement destiné à une utilisation en continu pendant moins de soixante minutes » ;
- Utilisation à court terme : « normalement destiné à une utilisation en continu entre soixante minutes et trente jours » ;
- Utilisation à long terme : « normalement destiné à une utilisation en continu pendant plus de 30 jours »
Dans le cas de DeepColor, l’innovation LightEcho Laparoscopic est un dispositif qui sera inséré à l’intérieur du corps dans le cadre d’un acte chirurgical. Il s’agit donc d’un dispositif invasif. Le temps d’usage de ce dispositif correspond au temps de l’acte chirurgical. C’est pourquoi, LightEcho Laparoscopic est dans la catégorie « utilisation à court terme ». LightEcho Laparoscopic, fonctionne par transmission de pulse lumineux à l’intérieur du corps humain. Ainsi, il fonctionne par le biais d’une autre source d’énergie que celle du corps humain et est donc catégorisé dans les dispositifs actifs.
Tableau 1 : Justification de la classe du dispositif DeepColor
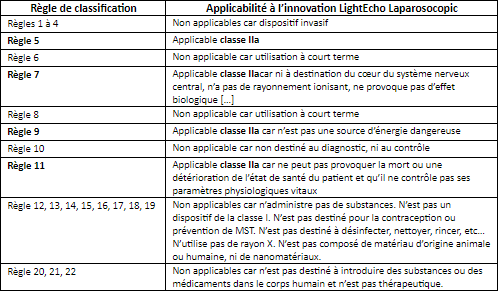
Selon à l’annexe VIII du règlement (UE) 2017/745, LightEcho Laparoscopic est classée IIa (Tableau 1). Le comparer avec un dispositif laparoscopique actuellement sur le marché permettrait de confirmer l’affirmation précédente : Le dispositif Trocart Laparoscopique est un dispositif de classe IIa [26] ce qui conforte le raisonnement précédent.
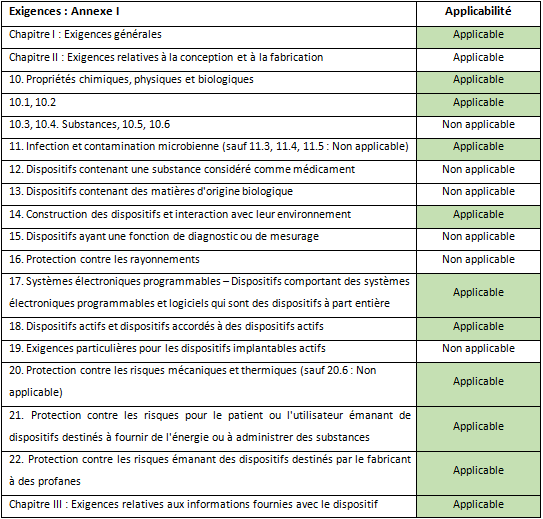
L’annexe 1 permet d’identifier les chapitres et annexes du règlement concernant les classes IIa.
3.1.2 Etape 2 : Identification les exigences en matière de sécurité et de performances
Généralités
La mise sur le marché ou la mise en service d’un dispositif médical ne peut se faire que lorsqu’il est conforme au règlement 2017/745. Lorsque celui-ci correspond à un champ d’application du règlement, le fabricant a pour obligation de s’assurer de l’existence de procédures de production conformes aux exigences en matière de sécurité et de performances.
D’après le règlement, il existe trois types d’exigences [1] :
- Les exigences générales applicables à l’ensemble des dispositifs (Annexe I, Chapitre I)
Ce sont des exigences énumérant globalement les exigences applicables à tous les dispositifs médicaux. Elles sont au total 9 exigences liées aux risques, aux erreurs d’utilisation, à la sécurité et à la performance garantie par le fabricant durant tout le cycle de vie du dispositif médical.
- Les exigences relatives à la conception et à la fabrication (Annexe I, Chapitre II)
Ce sont 13 exigences concernant les matériaux à utiliser et leur interférence avec l’environnement.
Le choix des exigences à appliquer dépendra du fabricant du dispositif. Ces choix se feront en fonction des caractéristiques techniques, de l’utilisation du dispositif et également des procédures à mettre en place pour répondre à ces exigences.
- Les exigences relatives aux informations fournies avec le dispositif (Annexe I, Chapitre III)
Ces exigences indiquent les informations qui doivent être incluses dans les instructions du produit.
Pour l’innovation LightEcho Laparoscopic de DeepColor, les exigences de l’Annexe I du règlement 2017/745 ont été répertoriée en fonction des caractéristiques d’un dispositif de classe IIa d’imagerie (Tableau 1).
Exigences de l’innovation DeepColor
Pour l’innovation LightEcho Laparoscopic de DeepColor, les exigences de l’Annexe I du règlement 2017/745 ont été répertoriées en fonction des caractéristiques d’un dispositif de classe IIa d’imagerie (Tableau 2).
Tableau 2 : Tableau de correspondance entre les exigences et l’innovation de DeepColor
Documentation à fournir pour la satisfaction aux exigences
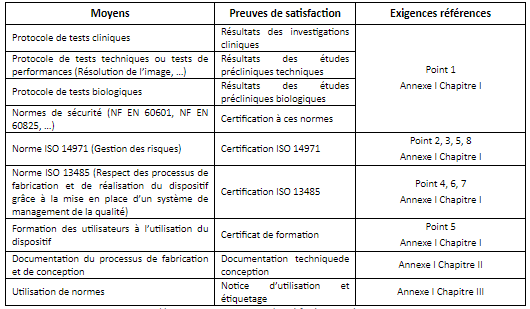
Pour satisfaire aux exigences énumérées précédemment, il est important de choisir les moyens de satisfaction qui vont aboutir à des preuves de satisfaction répondant à ces exigences. Toutes ces preuves de satisfaction seront des éléments à intégrer dans la documentation technique. Les moyens les plus importants sont cités dans le tableau suivant (Tableau 3) :
Tableau 3 : Moyens et preuves de satisfaction aux exigences
3.1.3 Etape 3 : Choix des moyens de contrôle et d’essais exigés
Pour obtenir l’apposition du marquage CE, le DM doit répondre à deux conditions : le DM doit répondre à l’ensemble des exigences qui lui sont applicables et doit, avoir fait l’objet d’une procédure d’évaluation de la conformité, prévue par le nouveau règlement. En effet, le règlement 2017/745 impose certaines procédures d’évaluation de la conformité des produits aux exigences.
Pour les dispositifs médicaux de classe I, le fabricant peut procéder à une auto-certification et ne passe pas par un ON.
Pour toutes les autres classes (classes I spéciales incluses), il doit soumettre une demande à l’organisme notifié avec tous les documents nécessaires pour l’évaluation de la conformité selon des procédures définies au niveau des annexes du règlement 2017/745.
a) Annexes définissant les procédures de vérification de la conformité des DM
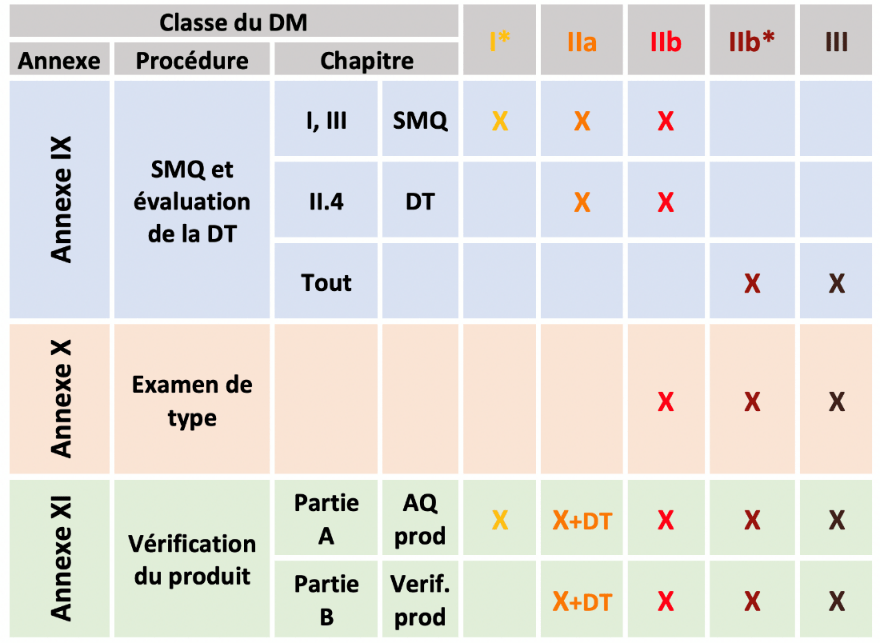
Les procédures de vérification de la conformité des DMs sont définies par les annexes IX, X, XI décrivant les différentes procédures d’essais et contrôles de conformité nécessaire à l’obtention du marquage CE, il faudra donc en choisir une parmi celles associées à une classe de dispositif (Figure 5).
Figure 5 : Procédure d'évaluation de la conformité en fonction de la classe du dispositif médical, Sources : auteurs inspiré de [27]
SMQ : Système de management de la qualité ; DT : Documentation technique ; AQ : Assurance qualité ; I*: DM stériles, intégrant une fonction de mesurage ou les instruments chirurgicaux réutilisables ; Ib*: Classe IIb visé à l'annexe VIII, section 6.4 (règle 12) du règlement.
b) Cas du LIGHTECO LAPAROSCOPIC (DM de classe IIa) :
Selon le règlement 2017/745, et suite à l’élaboration de la documentation technique selon les annexes II et III le fabricant par la déclaration de conformité́ déclare que le DM de classe IIa est fabriqué conformément à cette DT et satisfait aux exigences du règlement.
Les fabricants de dispositifs de classe IIa, autre que les DM faisant partie de cas particuliers comme les DM sur mesure, peuvent choisir entre trois procédures pour l’évaluation de la conformité (Figure 6).
Figure 6 : Procédure d'évaluation de la conformité des DMs de classe IIa, Source : auteurs
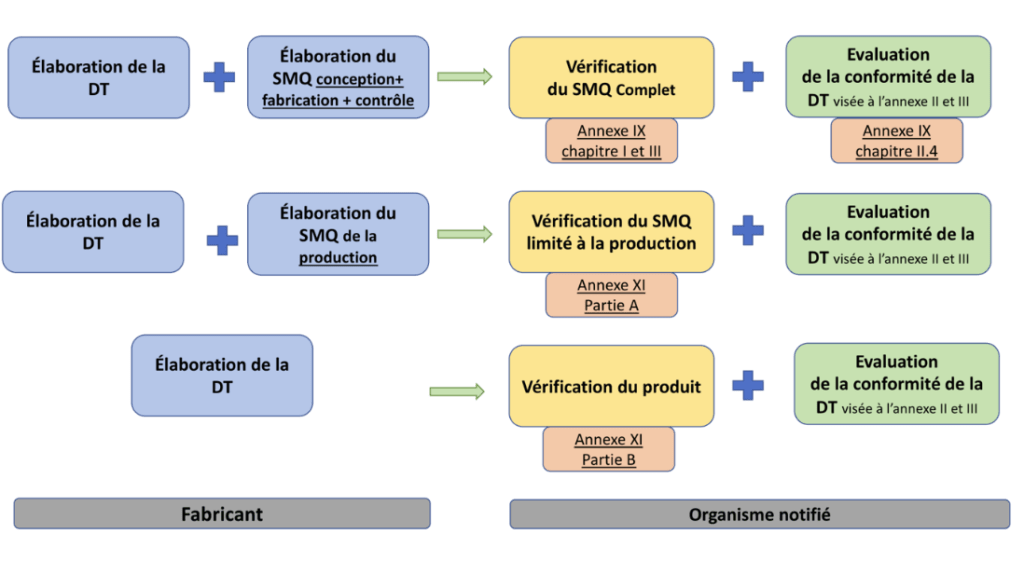
- Évaluation de la conformité sur la base d’un Système de Management de la Qualité (SMQ) et de l’évaluation de la Documentation Technique (DT) d’après l’annexe IX.
Le fabricant élabore un système complet d'assurance qualité de la conception, la fabrication et du contrôle qu'il va fournir à l’ON accompagné de la documentation technique, du dossier de conception et toutes les informations appropriées.
L'organisme notifié en procédant à des audits va vérifier la SMQ, étudier la documentation et inspecter les locaux.
- Vérification de la conformité́ de la production selon l’annexe XI partie A.
Le fabricant va élaborer un système d'assurance de qualité de la production selon la partie A de l'annexe XI qu'il va fournir à l’ON accompagné de la documentation technique. L'organisme notifié va vérifier le SMQ et étudier la documentation.
L’ON va sélectionner un ou des échantillons représentatifs pour cela il doit tenir compte selon le règlement 2017/745 : « de la nouveauté de la technologie, des similitudes dans la conception, la technologie, la fabrication et les méthodes de stérilisation, de l'utilisation prévue et des résultats de toute évaluation antérieure pertinente (concernant, par exemple, les propriétés physiques, chimiques, biologiques ou cliniques) réalisée conformément au présent règlement ».
Suite à ça l'ON pour justifier le choix des échantillons va établir un dossier justificatif. Le certificat au titre de l'annexe XI sera délivré par l'ON si l'évaluation effectuée confirme que le DM est conforme à la DT et satisfait les exigences du règlement 2017/745.
- La vérification de chaque DM fabriqué selon l’annexe XI partie B.
Après avoir contrôlé chaque dispositif fabriqué, Le fabricant déclare par la déclaration de conformité que ses dispositifs médicaux sont produits en conformité avec la documentation technique et qu'ils satisfont aux exigences du règlement.
L’ON va faire les examens et essais nécessaires sur chaque DM individuellement conformément à̀ la section 14 et 15 de l'annexe XI afin de vérifier la conformité́ des dispositifs avec la documentation technique et les exigences du règlement. Si la vérification confirme que les dispositifs sont conformes, l'organisme notifié délivre le certificat [1].
3.2 Réaliser
3.2.1 Etape 4 : Les données précliniques
Avant toute évaluation de la performance et de la sécurité de leur dispositif médical, et une fois que les études de faisabilité, l’installation du système de management de la qualité ont été finalisées et validées, les fabricants doivent réaliser des études dites précliniques.
Pour rappel, les données obtenues des études précliniques renseignent les organismes notifiés sur « des essais expérimentaux pertinents »[1].
a) Les données précliniques techniques.
Ces données sont relatives à des essais mécaniques, électriques, thermiques, chimiques et élastiques du dispositif médical. Ces études permettent de démontrer que toutes les caractéristiques précédemment énoncées se maintiennent de manière performante et en toute sécurité dans le temps. Dans cette étape le fabricant démontre qu’il maitrise les caractéristiques internes de son dispositif en appliquant des standards par exemple de sécurité électrique comme la norme NF EN 60601-1 relative aux exigences générales pour la sécurité de base et les performances essentielles pour appareils électro médicaux [28], ou encore le standard d’application de l’ingénierie de l’aptitude à l’utilisation aux dispositifs médicaux défini par la norme NF EN 62366-1 [29].
Pour le LightEco, les normes énoncées précédemment sont à considérer ainsi que des normes comme par exemple la norme NF EN 60601-2-22 [30], ainsi que la norme vue dans la partie 2.4.1 : la norme électrique IEC/TR 60825-8 [31].
b) Les données précliniques biologiques
Après s’être assuré du maintien des caractéristiques techniques, le fabricant doit faire de même pour les caractéristiques biologiques de son dispositif médical, il réalise des essais biologiques pour évaluer les risques biologiques (prolifération bactérienne, biocompatibilité, …) qui ne doivent pas être confondus avec les risques cliniques qui sont étudiés lors de l’évaluation clinique.
Comme pour les évaluations précliniques techniques, il existe des standards de réalisation de tests précliniques biologiques par exemple selon l’annexe A de la norme NF EN ISO 10993-1 : 2010 [32]. Cette norme fait l’objet d’un projet de révision [33]. Les tests à effectuer pour le LightEcho Laparoscopic sont les suivants : Test de cytotoxicité, Test de sensibilisation ainsi que le Test d’irritation et de réactivité intercutanée, Test de pyrogénicité à médiation matérielle et Test de toxicité systémique aigue. Les prérequis pour réaliser cette évaluation du risque biologique sont les informations physiques et/ou chimiques.
Après avoir récolté suffisamment de données précliniques, le fabricant peut entamer la conception de son prototype, il devra étudier attentivement quand il devra lancer la conception de prototype final et lancer l’enregistrement de son assurance conception qu’il placera dans sa documentation technique et ainsi pourra démarrer son évaluation clinique.
3.2.2 Etape 5 : L’évaluation clinique avant la mise sur le marché
a) Généralités
Le règlement 2017/745 définit l’évaluation clinique comme « un processus systématique et planifié visant à produire, collecter, analyser et évaluer en continu les données cliniques relatives à un dispositif afin de vérifier la sécurité et les performances, y compris les bénéfices cliniques, de celui-ci lorsqu’il est utilisé conformément à la destination prévue par le fabricant. » [1]. C’est une étape incontournable de la démarche marquage CE.
Le niveau de preuve clinique nécessaire est donné par le fabricant et permet de démontrer la conformité aux exigences générales pertinentes en matière de sécurité et de performances énoncées dans l’annexe I du règlement.
Pour démontrer leur satisfaction à ces exigences, les fabricants doivent démontrer la sécurité et les performances de leurs dispositifs médicaux et pour cela, ils doivent réaliser une évaluation clinique conforme aux exigences de l’article 61 et de l’annexe XIV et notamment un Suivi Clinique Après commercialisation (SCAC). Ils peuvent décider de passer par un organisme expert pour réaliser cette étape, dans ce cas-là, selon l’article 61 du règlement 2017/745, ils n’ont aucune autorité sur le groupe d’experts qui doit réaliser l’évaluation clinique [1].
Enfin, l’évaluation clinique est à réaliser tout au long du cycle de vie du DM. Trois points sont à identifier selon le MEDDEV 2/7-1 Rev.4 (faisant office d’état de l’art en attendant sa révision pour une conformité au nouveau règlement) [1] :
- Elle est effectuée pendant la phase de conception du DM. Les données identifiées permettent de justifier la mise sur le marché. Les données nécessaires à obtenir sont celles sur les performances et la sécurité clinique, l’équivalence avec d’autres DM, les lacunes identifiées sur les données de la littérature scientifique ;
- L’identification des données liées nécessaires pour le suivi post-marketing est à effectuer. Ceci permettra de déterminer les risques résiduels provenant de complications rares, des incertitudes des performances à long terme, l’utilisation à grande échelle etc. ;
- La mise à jour des documents liés à l’évaluation clinique est obligatoire. Il s’agit donc à cette étape de définir de manière justifiée la fréquence de mise à jour qui doit être pour les classes III au maximum annuelle et pour les autres de 2 à 5 ans ;
Une méthodologie précise est à suivre pour l’évaluation clinique, elle est décrite dans le MEDDEV 2/7-1 en étapes distinctes décrites dans l’Annexe 2.
Les éléments constituant cette évaluation doivent se retrouver dans un plan d’évaluation clinique, un plan de développement clinique, des documents d’analyse de données cliniques, un plan de suivi après commercialisation, et pour terminer un rapport d’évaluation clinique.
L’ensemble de ces documents se rassemblent dans un dossier d’évaluation. L’Annexe 3 de ce mémoire présente les documents que doit fournir le fabricant avec les éléments qui les composent et selon la disponibilité un guide pour réaliser cet élément.
b) Le plan d’évaluation clinique
Les fabricants doivent planifier, manager et enregistrer leur évaluation clinique. De ce fait ils doivent, en premier lieu établir un plan d’évaluation clinique. Ce plan d’évaluation représente l’ambition et la rigueur clinique, biologique, et technique des fabricants. Il est composé de : une description du DM, sa destination clinique, des informations d’équivalent avec un autre DM si c’est possible, une analyse bénéfices/risques clinique documentée, un état de l’art documenté sur le contexte scientifique où s’intègre son DM prouvé par de la littérature scientifique, les types de données cliniques ou non cliniques, ainsi que les données cliniques, non cliniques attendues.
Les fabricants doivent vérifier et confirmer ce plan d’évaluation clinique en faisant attention à tirer régulièrement de la littérature scientifique les données cliniques, biologiques et techniques utiles pour leur dispositif et dans son utilisation spécifique.
c) Evaluation clinique pour DM équivalent
Dans le cadre où une équivalence avec un autre DM sur le plan technique, biologique ou clinique, l’évaluation clinique du DM du fabricant peut s’établir sur l’évaluation clinique du DM équivalent selon les critères du règlement à l’Annexe XIV Partie A alinéa 3. Si les caractéristiques similaires sont justifiées dans la littérature scientifique alors les deux DMs pourront être définis comme ne présentant « pas de différence cliniquement significative en ce qui concerne la sécurité et les performances cliniques » [1]. Le règlement 2017/745 insiste sur l’obligation d’une preuve d’accès aux données du dispositif équivalent (documentation technique). Le fabricant sera forcé d’établir un contrat avec des concurrents pour obtenir ces données. Il faut ainsi noter que les concurrents ont tendance à garder ces informations confidentielles ce qui rend l’obtention de la documentation, compliquée.
d) Plan de développement clinique
Ce plan est défini à l’annexe XIV partie A alinéa 1. Il définit l’ensemble des investigations cliniques et non cliniques nécessaires à la confirmation aux exigences de sécurités et de performances applicables au DM du fabricant.
Selon le guide MEDDEV 2.7/1 rev4, il existe de nombreuses façons de récupérer les données cliniques et non cliniques pour prouver la conformité de sécurité et de performance du DM cependant, aucun document ne donne le plan de développement clinique pour un DM donné, ce plan de développement est à l’initiative du fabricant pour son DM. Le MEDDEV donne des recommandations sur la qualité méthodologique de recueil de données et d’analyse de ces données [34].
Grâce à la littérature scientifique et aux normes harmonisées, le fabricant pourra établir un plan de développement clinique assuré par la littérature scientifique, la Figure 7 schématise l’élaboration du plan de développement clinique.
Figure 7 : Elaboration d'un plan de développement clinique. NB : Cette méthodologie se veut explicative, elle ne donne pas la méthode qui garantit l’élaboration du plan de développement clinique. (Source : auteurs inspirés de [1],[34],[35]).
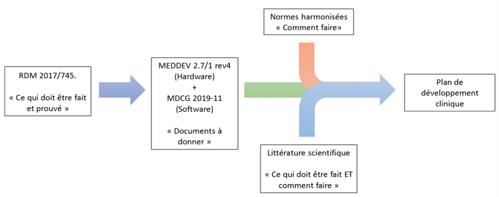
Une fois que le fabricant a déterminé son plan de développement clinique, il peut se lancer dans les investigations cliniques après avoir déterminé le ou les promoteurs des investigations cliniques.
e) Les investigations cliniques
Le règlement 2017/745 définit l’investigation clinique comme « toute investigation systématique impliquant un ou plusieurs participants humains destinée à évaluer la sécurité ou les performances d’un dispositif » [1]. Du début à la fin du processus d’investigations cliniques, le fabricant doit respecter des principes éthiques reconnus.
Les investigations cliniques doivent démontrer, dans leur méthode et en considérant la littérature scientifique en termes d’état de l’art de la technique, les avantages et inconvénients de leur DM en qualité de sécurité et performance. Ces méthodes doivent faire preuve de rigueur scientifique pour valider les résultats d’investigations cliniques, les méthodes de rigueur scientifique sont documentées et respectées pour chaque investigation clinique.
Si le fabricant ne peut pas réaliser les investigations cliniques, il doit nommer un promoteur. Il aura la charge le lancement, le management et la gestion du financement de l’investigation clinique.
Pour chaque investigation clinique, le promoteur doit auparavant faire une demande d’investigation clinique selon les exigences de l’article 70 et formulée selon l’annexe XV chapitre II du règlement via le système EUDAMED [1]. Cette investigation reçoit un numéro d’identification unique après 10 jours de sa mise en ligne et sera utilisée comme référence dans tout le territoire de l’Union.
Après avoir réalisé ses investigations cliniques prévues dans son plan de développement clinique et réalisé une analyse rigoureuse et impartiale de ses résultats cliniques et non cliniques, le fabricant conclue sur la sécurité et les performances cliniques de son DM, ainsi que sur ses bénéfices cliniques que démontre ses études. Ces informations se retrouvent dans le rapport d’évaluation clinique qui peut être élaborer avec l’aide du nouveau guide européen MDCG 2020-13 [36].
3.2.3 Etape 6 : Mettre en place la Surveillance Après Commercialisation (SAC)
Avant toute mise sur le marché, le fabricant doit fournir un système de surveillance après commercialisation planifié, maintenu, mis à jour, organisé dans un plan de Surveillance Après Commercialisation. Un tableau rassemblant des guides pratiques sur ce système est disponible en Annexe 4. Ce système fera partie intégrante du SMQ. Ces données de surveillance permettront de se conformer à l’article 83 alinéa 3 et 4 du règlement 2017/745 [1]. Ce système permet aux fabricants de mener des actions préventives ou correctives (pour améliorer la santé et la sécurité des patients, des utilisateurs et autres) à la suite de la réception d’incident ou d’accident sur leur DM sur la plateforme EUDAMED, après commercialisation de leur DM.
Après avoir récolté l’ensemble des nouvelles données de surveillance clinique d’un DM ou si le cas se présente par regroupement de DM, le fabricant de classe IIa, IIb et III éditent un rapport périodique actualisé de Sécurité (PSUR) comportant les éléments décrits dans l’article 86 alinéa 1 du règlement 2017/745 [1]. Pour les dispositifs de classe IIa, le PSUR est mis à disposition directement pour les organismes notifiés mais uniquement sur demande pour les autorités compétentes.
La documentation après commercialisation comprend donc un plan de surveillance après commercialisation et un PSUR.
a) Le plan de Surveillance Après Commercialisation
Tout comme le plan de développement clinique, il doit être élaboré par le fabricant et inclus dans la documentation technique.
Ce plan donne la méthodologie de surveillance après commercialisation ainsi qu’un processus faisant partie intégrante de ce plan (selon l’article 83 du règlement [1]) dit de Suivi Clinique Après Commercialisation (SCAC) présenté dans la partie B de l’annexe XIV du règlement 2017/745.
Les données cliniques récoltées tout au long du cycle de vie du DM marqué CE sur les humains ont trois buts distincts :
- Attester de la conformité aux exigences en matière de sécurité et de performance pendant toute la durée de vie du DM ;
- Prouver l’acceptabilité des risques identifiés et de maîtriser de nouveaux effets secondaires éventuels ;
- Identifier les possibles risques sur la base de preuves tangibles.
Les méthodes et les procédures de récolte et d’évaluation des données sont clairement définies dans un plan de SCAC. Ce dernier comprend aussi tous les éléments explicités dans le point 6.2 de l’annexe XIV partie B du règlement. Un rapport d’évaluation du SCAC est partie intégrante du rapport d’évaluation clinique. La mise en place de mesures préventives et/ou correctives découle des données générées par le SCAC. La Figure 8 ci-dessous schématise la structure de la SAC.
Figure 8 : Schéma présentant la structure de conformation à l'exigence de Surveillance Après Commercialisation (SAC), (Source : auteurs). NB : En vert le fabricant se trouve avant la certification marquage CE.
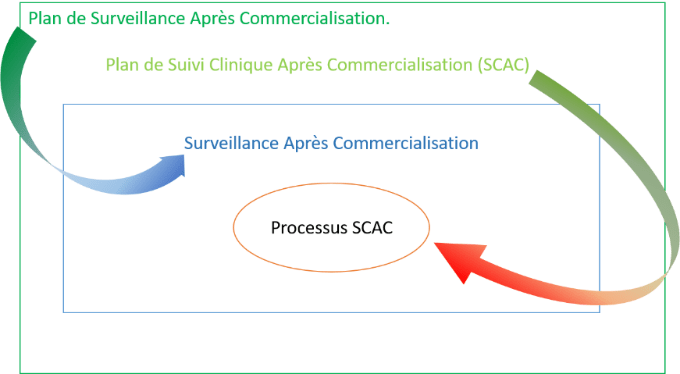
b) Vigilance
Les fabricants sont ainsi obligés de notifier des autorités compétentes des Etats membres dans lesquels leur dispositif est mis à disposition. Des délais stricts sont à respecter :
- Un incident grave doit être notifié sous 15 jours ;
- Un incident de détérioration grave de l’état de santé du patient ou un décès doit être notifié sous 10 jours ;
- Un incident menaçant la santé publique doit être notifié sous 2 jours.
Des rapports de synthèse peuvent remplacer des rapports d’incidents dans le cas où les incidents se sont déjà produits ou s’ils sont déjà documentés. Dans tous les autres cas, un rapport d’incident est à réaliser.
c) Système électronique et analyse des incidents graves
A la suite de chaque incident grave, des investigations obligatoires sont à mener afin de déterminer les risques et mesures correctives qui en découlent. L’ensemble des paramètres affectant la possibilité d’un nouvel incident et permettant la définition de mesures correctives de sécurité performantes, doivent être pris en considération. L’annexe I du règlement 2017/745 abordant le principe de sureté inhérente, peut influencer positivement la mesure corrective.
Grâce à un système électronique respectant la collecte et le traitement d’informations comme décrit dans le règlement (article 92), le fabricant communique les conclusions et les mesures correctives applicables tirées de l’investigation clinique.
Un avis de sécurité doit être diffusé et traduit dans la langue de chaque pays où une mesure corrective est appliquée. Cet avis doit faire l’objet d’une validation par l’autorité compétente coordinatrice et doit être, sauf exception, le même dans tous les pays concernés.
Cet avis indique les IUD de tous les dispositifs concernés et expose clairement les risques auxquels s’exposent les utilisateurs et les dispositions qu’ils doivent prendre.
3.2.4 Etape 7 : Préparer à la mise à disposition sur le marché et traçabilité
Dans cette étape, le fabricant devra respecter les exigences de mise en service et mise à disposition sur le marché, respecter les conditions de ventes à distance s’il y a lieu, nommer une personne chargée de veiller au respect de la règlementation, enregistrer les fabricants, mandataires et importateurs ou encore vérifier l’identification des chaînes d’approvisionnement.
3.3 Finaliser
3.3.1 Etape 8 : Compiler la documentation technique
Tous les dispositifs médicaux doivent être accompagnés d’une documentation technique claire, organisée et non-ambigüe. Pour un dispositif médical de classe IIa, la documentation technique exigée par le règlement 2017/745 telle qu’explicitée dans l’annexe II, se résume tel que suivra dans ce chapitre.
Il s’agit tout d’abord de fournir, comme il en est le cas pour tout dispositif médical, les documents permettant d’identifier ce dernier. Pour identifier convenablement un dispositif médical de classe IIa du type innovation médicale avec hardware et software comme LightEcho Laparoscopic, le fabricant doit fournir les éléments suivants :
- Le nom commercial du dispositif ;
- La description générale du dispositif en précisant le but de son utilisation et les utilisateurs auxquels il est destiné ;
- L’IUD ID qui est identifiant unique propre au dispositif que chaque fabricant est tenu de donner. A défaut de l’identification IUD, un code, une référence, un numéro de catalogue ou tout autre système permettant d’identifier le dispositif est accepté ;
- Les patients concernés par le dispositif ainsi que les pathologies sur lesquelles ce dernier agira ;
- Les contre-indications et mises en garde propres au dispositif médical ;
- Les principes de fonctionnement et le mode d’action du dispositif médical ;
- Les raisons pour lesquelles le produit est classifié comme dispositif médical ;
- La classe de risque du dispositif médical avec justification (selon l’annexe VIII) ;
- Description des accessoires et des dispositifs accompagnants le dispositif médical et/ou destinés à être utilisés en combinaison avec lui ;
- Description générale des éléments fonctionnels clés tels que les pièces ou composants ; y compris le logiciel. La formulation, la composition, la fonctionnalité et, le cas échéant, la composition qualitative et quantitative avec schémas descriptifs des éléments et composants du module ;
- Une description des matières premières intégrées dans les éléments fonctionnels clés et les éléments en contact direct ou indirect avec le corps humain ;
- Les caractéristiques, dimensions et caractéristiques de performance, du dispositif et de toute variante/configuration ou de tout accessoire qui figurent habituellement dans les spécifications du produit mises à disposition de l’utilisateur, par exemple dans des brochures, des catalogues et des publications similaires.
Le fabricant se doit également de fournir les informations suivantes :
- L’intégralité de l’étiquetage présent sur le dispositif ainsi que son conditionnement durant toutes les étapes depuis sa fabrication jusqu’à sa livraison (unité de production, vente, transport) dans les langues acceptées des états où le dispositif sera commercialisé.
- La notice d’utilisation dans les langues acceptées des états où le dispositif sera commercialisé.
Il s’agit ensuite de satisfaire les exigences de la rubrique informations sur la conception et la fabrication. Cette rubrique stipule que doivent faire partie du dossier technique des documents comprenant les données suivantes :
- Données expliquant le procédé de conception du dispositif médical et toutes ses étapes ;
- Données intégrales sur la fabrication du dispositif, ses procédés et leurs validations, les adjuvants, le contrôle continu mis en place ainsi que les tests et essais effectués sur le produit final ;
- Informations permettant l’identification de l’intégralité des sites ayant hébergé des activités de conception et de fabrication dont les sites de tout sous-traitant sollicité.
Ensuite, la documentation technique doit comprendre des éléments permettant de prouver la conformité aux exigences générales en matière de sécurité et de performance stipulées dans l’annexe I du règlement 2017/745. Ces éléments sont réunis dans ce qui est appelé la démonstration de conformité. La démonstration de conformité doit contenir :
- Les exigences générales en matière de sécurité et de performance applicables aux dispositifs médicaux de classe IIa avec justification de la non-application du reste des exigences ;
- Les méthodes utilisées pour être conformes à chacune des dites exigences ;
- Les solutions adoptées qu’ils s’agissent de normes harmonisées, de spécifications communes ou autre ;
- La référence précise des documents contrôlés servant de preuve de conformité à toute méthode appliquée ou solution retenue (norme harmonisée, spécification commune ou autre). Ces informations doivent permettre de retrouver ces preuves dans la documentation technique complète et dans son résumé le cas échéant.
Le volet suivant porte sur l’analyse bénéfice risque et gestion des risques. La documentation requise à ce niveau doit fournir les informations suivantes :
- L’analyse bénéfice-risque explicitée dans les sections 1 et 8 de l’annexe I du règlement 2017/745.
- Informations sur les solutions retenues et les résultats de la gestion des risques mentionnée au niveau de la section 3 de l'annexe I
Le dernier volet de la documentation technique pré-commercialisation est celui portant sur la vérification et validation du produit subdivisée en deux sous-groupes : les données précliniques et cliniques et les informations additionnelles dans des situations spécifiques. Dans le cas de LightEcho Laparascopic, les données à fournir sont les suivantes :
- Dans la catégorie : données précliniques et cliniques
- Les résultats d'essais (ingénierie, en laboratoire, des simulations, et des essais sur des animaux, etc.) et d'évaluations contenus dans la littérature publiée qui sont applicables au dispositif ou à des dispositifs similaires, concernant la sécurité préclinique du dispositif et le respect des spécifications ;
- Des informations détaillées relatives à la conception des essais, aux protocoles d'essai ou d'étude complets, aux méthodes d'analyse des données, accompagnées de synthèses de données et de conclusions des essais sur les points suivants :
- La sécurité électrique et la compatibilité électromagnétique ;
- La liste des matériaux en contact direct ou indirect avec le patient ou l'utilisateur et la biocompatibilité du dispositif médical ;
- La vérification et la validation du logiciel. Il s’agit de fournir une description intégrale de la conception et du processus de développement du logiciel et preuve de la validation de celui-ci avec un résumé des résultats de l'ensemble de la vérification, de la validation et des essais réalisés en interne et dans un environnement d'utilisation simulé ou réel avant la libération finale. Les données doivent également prendre en compte toutes les différentes configurations du matériel informatique et des différents systèmes d'exploitation figurant dans les informations fournies par le fabricant ;
- La stabilité y compris la durée de conservation en stock ;
- Les performances et la sécurité.
- Le rapport sur l'évaluation clinique avec ses mises à jour ainsi que le plan d'évaluation clinique visé à l'article 61, paragraphe 12, et à l'annexe XIV, partie A ;
- Le plan de suivi clinique après commercialisation (SCAC) ;
- Le rapport d'évaluation du SCAC, visés à l'annexe XIV, partie B ou une justification expliquant pourquoi un SCAC n'est pas applicable.
- Dans la catégorie : informations additionnelles dans des situations spécifiques
La seule situation spécifique en rapport avec LightEcho Laparoscopic qui est concernée par cette catégorie est celle portant sur son utilisation raccordée à un ou plusieurs autres dispositifs. La réglementation exige dans ce cas une description du raccordement et/ou de la configuration incluant la preuve qu'il/elle est conforme aux exigences générales en matière de sécurité et de performance pour tous les dispositifs concernés une fois connectés, au regard des caractéristiques indiquées par le fabricant.
- Documentation technique relative à la surveillance post-commercialisation
Le fabricant est tenu d'établir une documentation technique relative à la surveillance après commercialisation claire, organisée et non ambigüe.
Le plan de surveillance après commercialisation comprend au moins :
- Un processus proactif et
systématique qui permet de définir correctement les caractéristiques de performance
des dispositifs et d'effectuer une comparaison entre les dispositifs et des
produits similaires disponibles sur le marché ;
- Des méthodes et des processus appropriés et efficaces pour l'évaluation des données collectées ;
- Des indicateurs et des seuils adaptés à utiliser pour procéder à la réévaluation continue de l'analyse bénéfice/risque et de la gestion des risques ;
- Des méthodes et des outils appropriés et efficaces pour donner suite aux réclamations et analyser les données d'expérience en matière de commercialisation collectées sur le terrain ;
- Des méthodes et des protocoles servant notamment à établir une éventuelle progression statistiquement significative de la fréquence ou de la sévérité des incidents ainsi que la période d'observation ;
- Des méthodes et des protocoles permettant une communication efficace avec les autorités compétentes, les organismes notifiés, les opérateurs économiques et les utilisateurs ;
- Une référence aux procédures permettant aux fabricants de satisfaire aux obligations visées aux articles 83, 84 et 86 ;
- Des procédures systématiques pour définir et engager les mesures appropriées, y compris des mesures correctives ;
- Des outils efficaces permettant d'identifier et de retrouver les dispositifs susceptibles de nécessiter des mesures correctives ;
- Un plan de SCAC ou tout élément justifiant qu'un SCAC n'est pas applicable.
Le fabricant fourni également un PSUR (Periodic Updated safety report) et rapport sur la surveillance après commercialisation.
3.3.2 Etape 9 : Enregistrer les opérateurs économiques et le dispositif
Cette étape nécessite de fournir toutes les informations requises, d’enregistrer le dispositif sur EUDAMED et de vérifier que le système IUD est réalisé conformément à la règlementation, un guide est disponible pour faciliter cette étape, il s’agit du Guide de l’utilisateur d’EUDAMED [37]. Les utilisateurs d’EUDAMED ont des droits et des obligations [38], qu’ils obtiennent après avoir signée une déclaration d’information de sécurité et de responsabilité [39].
3.3.3 Etape 10 : La déclaration de la conformité UE
La rédaction de la déclaration de conformité UE est l’acte final par lequel le fabricant atteste que son dispositif est conforme aux exigences du règlement 2017/745.
La déclaration de conformité doit répondre à certains points, comme l'identification du fabricant, du produit et des références réglementaires, de même que l'indications des moyens de mise en conformité. Elle permet aussi de préciser les conditions de validité.
La déclaration de conformité́ UE doit contenir plusieurs informations énumérées au niveau de l'annexe IV du règlement :
- Nom du fabricant ou de la marque déposée ;
- Nom et code du produit ;
- L'IUD-ID du dispositif médical ;
- Classe du dispositif médical ;
- Un document attestant que seule la responsabilité du fabricant est engagée dans la déclaration de conformité ;
- Nom du règlement et autre(s) législation(s) visée(s) ;
- Procédure pour le marquage CE employée avec les noms des annexes ;
- Une attestation que le dispositif respecte règlement ;
- Le lieu et la date de la déclaration de conformité ;
- Indentification de la personne signant la déclaration ;
- Identifiant de l’ON ;
- Date de validité de la déclaration de conformité.
Voire l’Annexe 5 pour un exemple de déclaration de conformité.
3.4 Normes aidant à la conformité au règlement
Une liste de normes aidant à la conformité aux exigences du règlement 2017/745 a été établi. Il est important de savoir que les normes étant exhaustives, il a été répertorié celles qui ont été trouvé les plus importantes :
- NF EN ISO 13485 : Dispositifs médicaux —Systèmes de management de la qualité — Exigences à des fins réglementaires [40]
Cette norme traite l’application du système de management de risque dans le domaine spécifique des dispositifs médicaux. Elle spécifie les exigences d'un système de management de la qualité à une ou plusieurs étapes du cycle de vie du dispositif et ce au profit des organismes impliqués. Cela peut concerner également les activités telle la distribution, les prestations associées dont le support technique.
- NF EN ISO 14971 : Dispositifs médicaux — Application de la gestion des risques aux dispositifs médicaux [41]
Il s’agit de la norme portant sur la gestion des risques appliquée aux dispositifs médicaux. Elle permet de spécifier le processus de gestion des risques relatif à tous les types de dispositifs médicaux y compris les logiciels. Elle vise à aider les fabricants dans l’identification des risques, leurs estimations, leurs évaluations et enfin leur maîtrise et leur surveillance.
- Les logiciels de santé embarqués sont règlementés par la norme NF EN 62304 : 2006.
Ils sont considérés comme « faisant partie d’une dispositif médical » [42]. Cette norme implique que le fabricant doit répondre à des exigences générales en matière de management de la qualité, de gestion des risques et d’ingénierie de l’aptitude à l’utilisation. Le système qualité certifié ISO 13485 permet de prouver la première exigence. Enfin, la norme IEC 62366-1 de justifier l’exigence sur l’ingénierie de l’aptitude à l’utilisation [29].
Concernant les normes sur les lasers, voici les normes principales qui sont à prendre en compte :
- La norme NF EN 60825 « Sécurité des appareils à laser - Partie 1 : classification des matériels et exigences » [43] est applicable uniquement pour les lasers émettant des rayonnements compris entre les longueurs d’onde 180 nm et 1 mm. Les effets du rayonnement laser sur les tissus biologiques (annexe D.2), descriptions des classes de dangerosité des laser (annexe C) ainsi que la valeur d’exposition maximale permise (annexe A) y sont explicités.
- La norme IEC/TR 60825-8 : « Safety of laser products -part 8 : Guidelines for the safe use of laser beam on humans » [31], uniquement disponible en anglais.
- La norme NF EN 60601-2-22 : « Appareil électromédicaux – Partie 2-22 : exigences particulières pour la sécurité de base et les performances essentielles des appareils chirurgicaux, esthétiques, thérapeutiques et de diagnostic à laser » [30], applicable pour les laser médical de classe 3B ou de classe 4 selon la norme NF EN 60825.
- La norme NF EN ISO 11252 « Lasers et équipements associés aux lasers - Source laser - Exigences minimales pour la documentation » définit, quant à elle, les exigences minimales de documentations, marquage et étiquetage [44].
4. Les solutions pour aider les fabricants
4.1 Solutions existantes pour les méthodes de roadmap règlementaire
L’International Medical Device Regulation Forum (IMDRF), organisation internationale qui rassemble 11 membres comprenant les Etats-Unis, l’Union Européenne, le Japon, le Brésil, la Chine, la Russie, le Canada, l’Australie, la Corée du Sud, ainsi que Singapour [45], crée des outils qui permettent aux fabricant d’accéder plus rapidement aux marchés internationaux pour les dispositifs médicaux.
Le Summary Technical Documentation (STED) est un de leurs outils, créé en 2008 ce document standardise la mise sur le marché d’un DM pour les états membres [46].
Il existe un outil d’autodiagnostic permettant aux fabricants d’évaluer l’état d’avancement d’un STED et assure un coût réduit de mise en conformité aux pays membre de l’IMDRF [47].
Un nouveau document remplaçant le STED est arrivé en mars 2019, il est également à l’initiative de l’IMDRF, il se présente sous la forme d’une « Table de matière », il complète le STED en donnant un comparatif des exigences spécifiques des membres du IMDRF [48].
Ces deux outils peuvent être une approche envisageable pour les entreprises possédant des fonds suffisants pour assumer le respect des différentes exigences des membres de l’IMDRF, cependant pour les PME fabricant des DM d’imagerie haute résolution, ces outils ne sont pas efficients.
En effet, pour les fabricants d’imagerie haute résolution, la documentation à fournir ainsi que la démarche à suivre pour une mise sur le marché reste complexe et d’autant plus pendant la période de transition.
4.2 Les solutions proposées
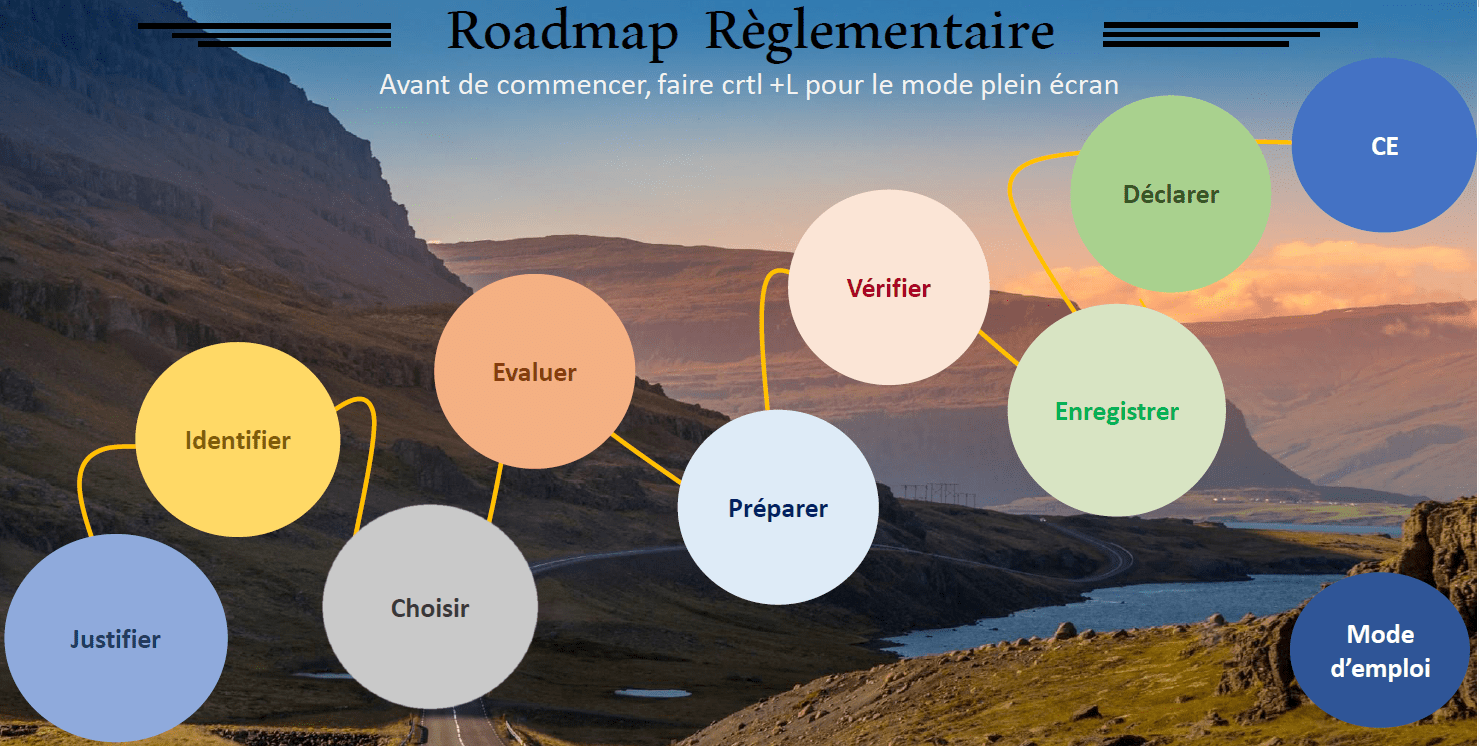
Afin de fournir une aide pertinente aux start-up et PME, une cartographie interactive en ligne gratuite offrant une compréhension du nouveau règlement et mettant en lumière les étapes majeures de la démarche de marquage CE, ainsi qu’un outil de positionnement sur cette démarche marquage CE ont été développés dans le cadre de ce travail.
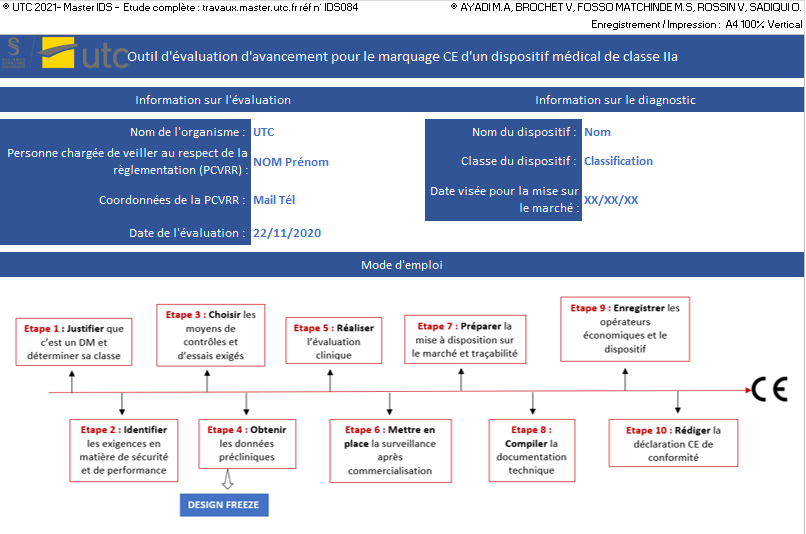
4.2.1 L’outil de positionnement
Cet outil, conçu sur Excel, permet au fabricant de dispositif de classe IIa de se positionner au niveau de la démarche marquage CE en se conformant aux exigences du règlement 2017/745. Composé de six onglets, une présentation détaillée est donnée si dessous :
- [Mode d’emploi]
Cet onglet présente le contenu de chaque feuille ainsi que les deux modes d’évaluation qui ont été choisis. Il explique comment naviguer entre les différentes feuilles de l’Excel.
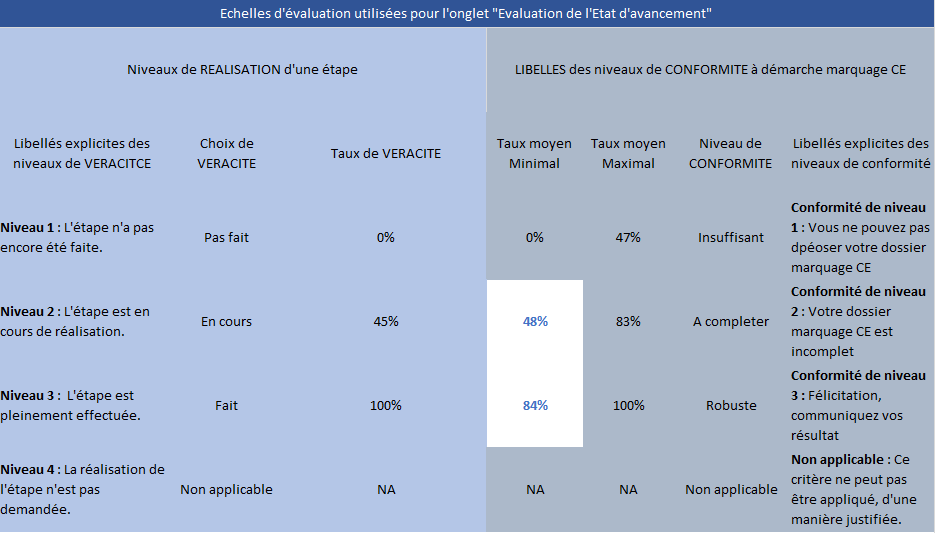
Chacune des étapes est évaluée suivant un taux de véracité de « 0% », « 45% », « 100% », « NA » correspondant respectivement à « Pas fait », « En cours », « Fait » et « Non applicable » (Figure 23).
Chacune des exigences des sous-étapes (actions) sont évaluées suivant un taux de véracité de « 0% », « 30% », « 70% », « 100% » et « NA » correspondant respectivement à « Faux », « Plutôt faux », « Plutôt vrai », « Vrai » et « Non applicable » (Figure 9).
Figure 9 : Onglet Mode d'emploi, Echelle d’évaluation pour la démarche marquage CE, Source : auteurs
Cet onglet rassemble aussi les métadonnées pour le fabricant. L’identité et coordonnées de la Personne Chargée de Veiller au Respect de la Règlementation (PCVRR), le nom et type du dispositif, les coordonnées le nom de l’entreprise y sont demandés (Figure 10).
Figure 10 : Ongle Mode d’emploi, métadonnées, Sources : auteurs

- [Evaluation_Etat_Avancement]
Cet onglet est une roadmap sous forme d’étapes clé. Une fois que toutes les actions d’une étape sont réalisées, l’étape est complète. Pour passer à l’étape suivante, il faut un avancement d’au moins 85% de chaque étape précédente. Le niveau de conformité s’affiche automatiquement une fois que l’évaluation de tous les points est faite. Il faut aussi noter que des couleurs pour attirer l’œil de l’utilisateur sont conditionnés aux cases choisies dans la colonne état. Lorsque ces cases sont blanches avec une écriture bleue selon signifie que l’utilisateur n’a pas encore évalué cette action (Figure 11).
Figure 11 : Onglet Evaluation de l'état d'avancement de la démarche marquage CE, Sources : auteurs
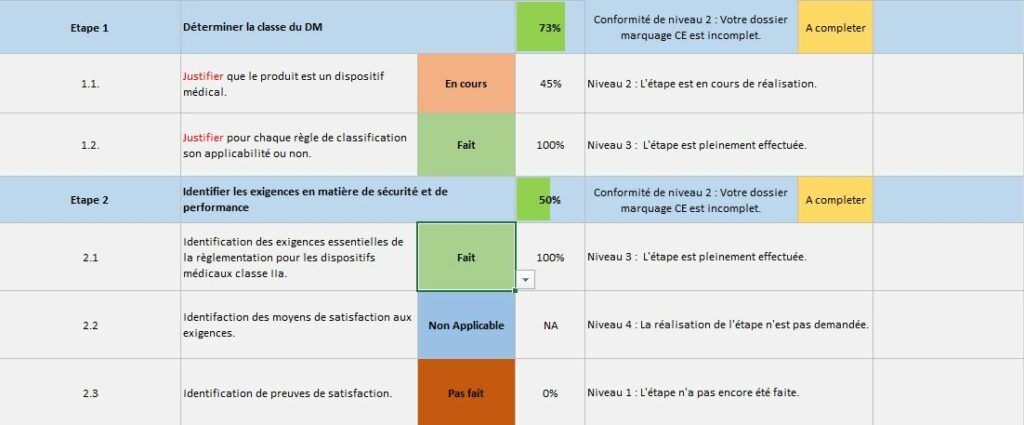
La Figure 12 introduit un point clé de cet onglet. En effet, certaines sous-étapes sont parfois compliquées à évaluer ainsi, des exigences de la règlementation 2017/745 correspondant à cette sous-étape ont été ajoutées afin de pouvoir aider le fabricant dans son évaluation et son respect aux exigences règlementaires. En appuyant sur le bouton « plus » situé sur le côté gauche une liste de critère apparaît (Figure 13).
Figure 12 : Zoom sur les critères des sous-étapes, onglet [Evaluation _Etat_Avancement], Source : auteurs
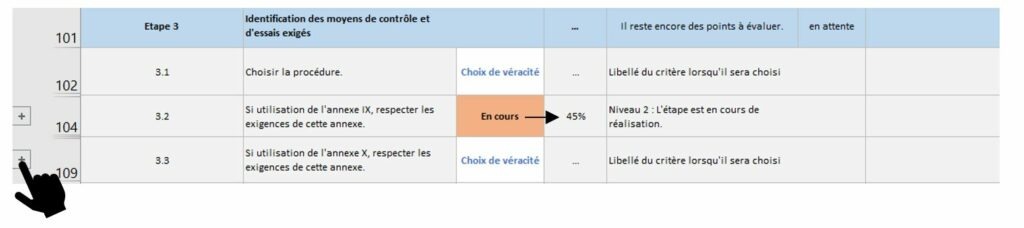
Figure 13 : Liste de critères, onglet [Evaluation_Etat_Avancement], Source : auteurs
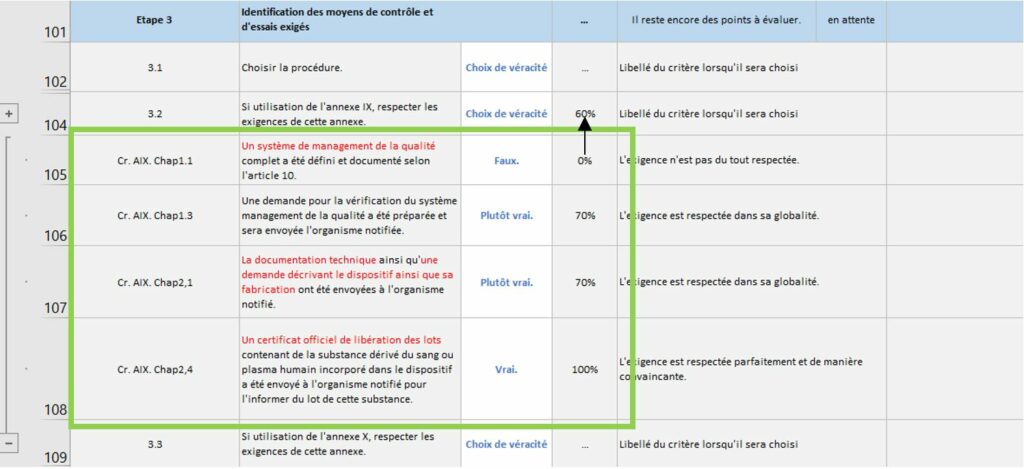
L’évaluation peut donc se faire soit par les critères de l’action soit par la sous étape directement.
- [Résultat_Marquage_CE]
Cet onglet rassemble les résultats sur l’avancement de la démarche de marquage CE pour un fabricant. Il est essentiel pour donner une vue globale et pointer les étapes critiques. Un plan d’actions prioritaires avec des commentaires peuvent être ajoutés afin de pouvoir améliorer ses résultats par la suite. Le suivi de ce tableau de bord est indispensable pour une compréhension globale et synthétique du fabricant (Figure 14).
Figure 14 : Onglet [Résultat_Marquage_CE], Source : auteurs
- [Résultat_Détaillé_Par Etape]
Cet onglet permet de détailler par étape les résultats. En effet, il rassemble toutes les sous-étapes de chaque étape afin de comparer leur avancement. Il donne une vue détaillée au fabricant afin de cibler directement les points critiques et points d’amélioration (Figure 15).
Figure 15 : Résultat détaillée étape 3, onglet [Résultat_Détaillé_Par Etape], Source : auteurs
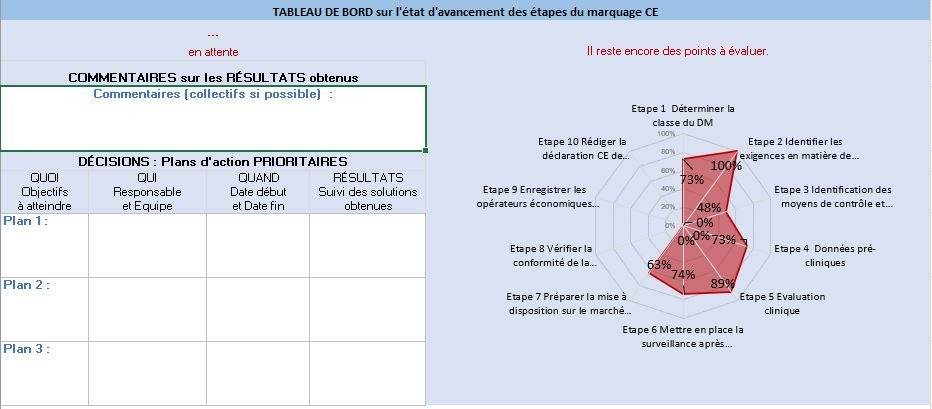
- [Maîtrise documentaire]
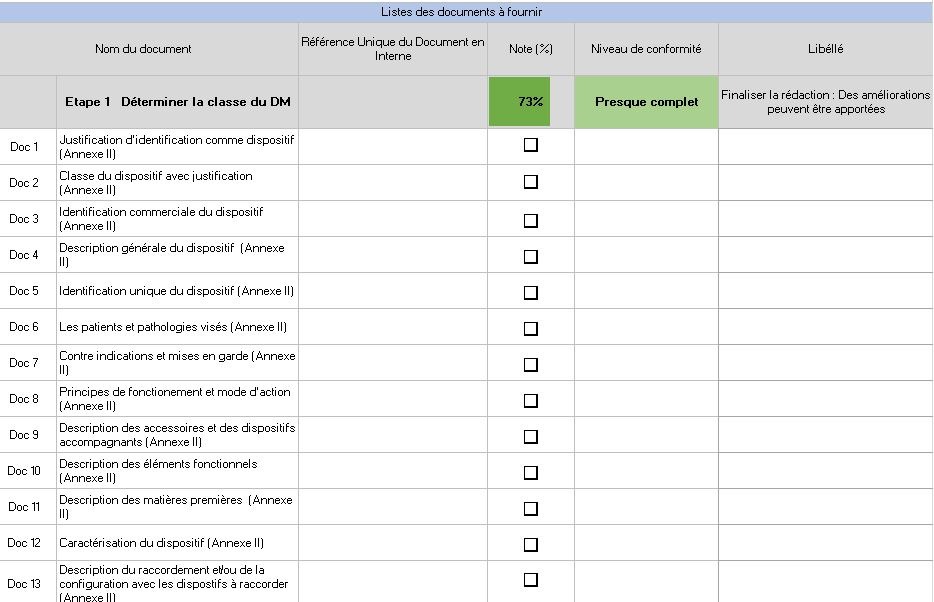
Cet onglet permet d’accentuer sur la documentation indispensable pour obtenir le marquage CE. Une évaluation globale permet de visualiser si la documentation de chaque étape est « Absente », « Incomplète » ou « Complète » en fonction de taux préétabli. Les documents nécessaires à chaque étape sont mentionnés et une colonne permet à l’utilisateur de cocher lorsque le document a été fait. Une documentation robuste est un premier pas pour l’obtention de la certification (Figure 16).
Figure 16 : Onglet [Maîtrise documentaire], Source : auteurs
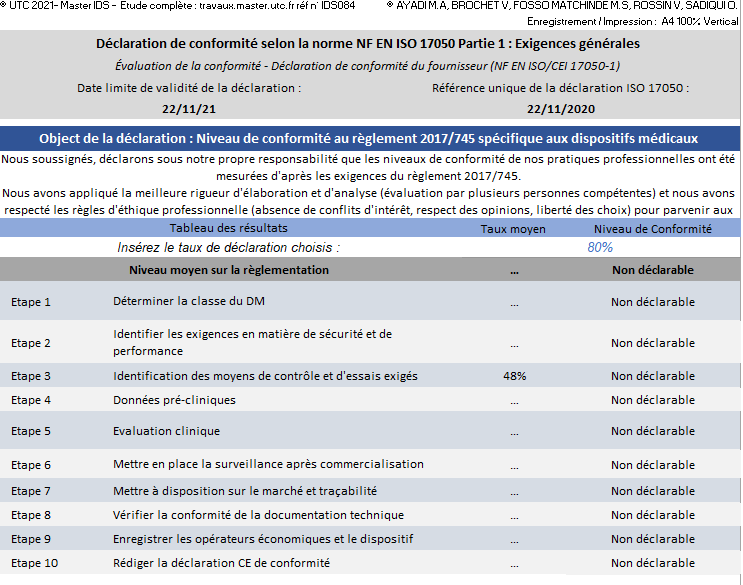
- [Déclaration_Conformité_17050]
L’autodéclaration de conformité établie selon la norme NF EN ISO 17050 est sous la responsabilité de l’entreprise. Selon un taux de conformité choisi par l’utilisateur, une étape est déclarable ou non. La communication sur la réalisation d’une étape se fait selon son taux d’avancement et donc sa conformité au règlement 2017/745 (Figure 17).
Figure 17 : Onglet Déclaration de conformité selon la norme NF EN ISO 17050, Source : auteurs
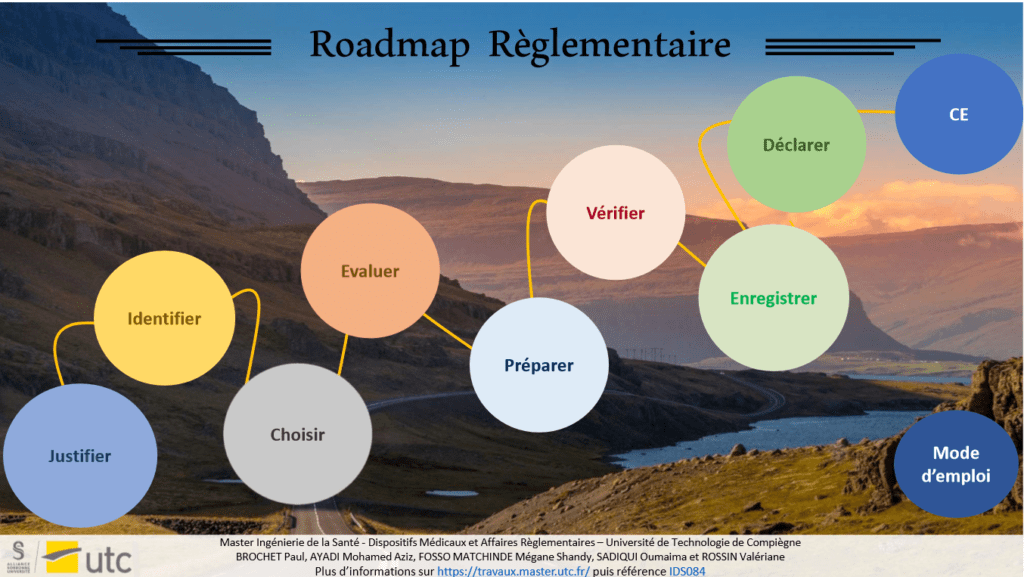
4.2.2 La cartographie interactive
Cet outil est à destination des fabricants, cette cartographie interactive (Figure 18) se veut donner une vision claire et précise de la démarche marquage CE en vue d’une mise sur le marché. Elle rassemble des documents ainsi que des éléments qui les composent afin de se conformer à la règlementation pour les dispositifs médicaux de classe IIa. L’outil aiguille le fabricant sur l’élaboration de ses documents grâce à la sélection de guides Européen.
Figure 18 : Roadmap règlementaire, Source : auteurs.
Un mode d’emploi est présent ci-dessus, il permet une compréhension de l’outil et une prise en main rapide et complète de celui-ci (Figure 19).
Figure 19 : Mode d'emploi de la cartographie interactive (Source : auteur)

4.3 Les bénéfices des solutions proposées
L’outil de positionnement marquage CE ainsi que la cartographie permettent au fabricant de s’y retrouver dans un chemin règlementaire complexe. La maîtrise documentaire de l’outil Excel permet au fabricant de fournir une documentation technique complète et conforme au règlement 2017/745. Par ailleurs, les doutes concernant la compréhension des actions de chaque étape du marquage C ne sont plus possibles grâce à la fonctionnalité de l’outil Excel utilisée permettant de déplier les critères du règlement 2017/745 sous certaines actions le nécessitant.
Enfin, la cartographie est un moyen performant offrant une vue globale de la roadmap. Très intuitive, elle guidera le lecteur dans ses démarches règlementaires.
Conclusion
L’arrivée du nouveau règlement oblige les fabricants en imagerie de haute résolution à se préparer davantage avant d’obtenir le marquage CE, en effet les obligations qui leurs incombent par exemple en termes d’évaluation clinique, doivent être étudiées et appliquées dans les plus brefs délais pour éviter une perte de ressource irrécupérable. Ils peuvent avec l’aide d’une PCVRR, des guides disponibles et des outils présentés dans ce mémoire se préparer à lancer leur démarche marquage CE pour leur dispositif médical et espérer obtenir le marquage CE de la manière la plus efficiente possible.
Bibliographie
[1] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, Règlement OJ L 117, 5.5.2017, avr. 2017. Consulté le : sept. 25, 2020. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/?uri=CELEX%3A32017R0745.
[2] MedTech Europe, « MedTech Europe’s Facts and Figures 2020 », Bruxelle, Belgique, mai 2020. Consulté le : oct. 06, 2020. [En ligne]. Disponible sur : https://www.medtecheurope.org/resource-library/medtech-europes-facts-and-figures-2020/.
[3] SNITEM, « Panorama 2019 et analyse qualitative de la filière industrielle des dispositifs médicaux en France », Ed. Syndicat National de l’Industrie des Technologies Médicales (SNITEM), déc. 2019. Consulté le : oct. 06, 2020. [En ligne]. Disponible sur : https://www.snitem.fr/dm-et-sante/panorama-dm.
[4] « Cellvizio Biopsie Ciblée - Mauna Kea Technologies », Mauna Kea Technologies, juill. 20, 2020. https://www.maunakeatech.com/fr/cellvizio (consulté le oct. 16, 2020).
[5] SNITEM, « Dispositifs Médicaux & Progrès en appareil digestif ». déc. 2016, [En ligne]. Disponible sur : https://www.snitem.fr/le-snitem-en-action/les-publications/le-livret-appareil-digestif.
[6] S. Mordon et G. Bourg-Heckly, « Photodiagnostic et chirurgie guidés par la fluorescence », L’Actualité Chimique, p. 1‑15, juin 2015.
[7] Rachel Ross, « What Are CT Scans and How Do They Work ? », Live Science, nov. 14, 2018. https://www.livescience.com/64093-ct-scan.html (consulté le oct. 16, 2020).
[8] S. Vilov, T. Chaigne, B. Arnal, et E. Bossy, « Imagerie photoacoustique biomédicale », Photoniques, no 94, p. 24‑29, nov. 2018, doi : 0.1051/photon/20189424.
[9] C. Elwell, « Photoacoustic imaging enables scientists to step up war on cancer », Ed Financial Times, Londres, www.ft.com, janv. 18, 2019.
[10] « Colorectal cancer statistics », World Cancer Research Fund, août 22, 2018. https://www.wcrf.org/dietandcancer/cancer-trends/colorectal-cancer-statistics (consulté le oct. 18, 2020).
[11] « Quelques chiffres sur le cancer colorectal », Fondation pour la Recherche Médicale, sept. 2018. https://www.frm.org/recherches-cancers/cancer-du-colon/quelques-chiffres-sur-le-cancer-colorectal (consulté le oct. 18, 2020).
[12] La Société Française de Chirurgie Endoscopique, « Fiche information Chirurgie colique FCVD », lasfce.com, juill. 10, 2013. https://www.lasfce.com/fr/fiches-informations-patients-fcvd (consulté le oct. 16, 2020).
[13] La Société Française de Chirurgie Endoscopique, « Fiche information Chirurgie rectale FCVD », lasfce.com, juill. 10, 2013. https://www.lasfce.com/fr/fiches-informations-patients-fcvd (consulté le oct. 16, 2020).
[14] Institut National du Cancer, « Laparotomie et Cœlioscopie », Accélérons les progrès face aux cancers, oct. 16, 2020. https://www.e-cancer.fr/Patients-et-proches/Les-cancers/Cancer-du-colon/Chirurgie/Laparotomie-et-Coelioscopie (consulté le oct. 16, 2020).
[15] Caisse National d’Assurance Maladie, « Comment se déroule une cœlioscopie (ou laparoscopie) ? », ameli.fr, nov. 14, 2019. https://www.ameli.fr/oise/assure/sante/examen/exploration/deroulement-coelioscopie (consulté le oct. 16, 2020).
[16] Centre National de la Recherche Scientifique, « Définition de ANASTOMOSE », Centre National de Ressources Textuelles et Lexicales. https://www.cnrtl.fr/definition/anastomose (consulté le oct. 13, 2020).
[17] E. Girard et al., « Diagnostic et prise en charge d’une fistule anastomotique en chirurgie digestive », Journal de Chirurgie Viscérale, vol. 151, no 6, p. 455‑465, déc. 2014, doi : 10.1016/j.jchirv.2014.08.006.
[18] American Medical Association, « In this Issue of JAMA Surgery », JAMA Surg, vol. 151, no 9, p. 789‑884, sept. 2016, doi:10.1001/jamasurg.2015.2940.
[19] Richard Fabrice, « Document entreprise DeepColor ». 2020.
[20] J. Martin Rutegård et R. Rutegård, « Anastomotic leakage in rectal cancer surgery : The role of blood perfusion », World Journal of Gastrointestinal Surgery, vol. 7, no 11, p. 289‑292, nov. 2015, doi : 10.4240/wjgs.v7.i11.289.
[21] F. Ris, T. Yeung, R. Hompes, et als, « Enhanced Reality and Intraoperative Imaging in Colorectal Surgery », Clin Colon Rectal Surg, vol. 28, no 03, p. 158‑164, sept. 2015, doi : 10.1055/s-0035-1555007.
[22] « Actes juridiques de l’UE : le règlement », Vie Publique, nov. 11, 2018. https://www.vie-publique.fr/fiches/20370-actes-juridiques-de-lue-le-reglement (consulté le oct. 05, 2020).
[23] Mangeol Cyril, « Enjeux et exigences de la nouvelle règlementation européenne des dispositifs médicaux », Pharmacie, Université d’Aix-Marseille - Faculté de Pharmacie, Marseille, 2019.
[24] Association Française de Normalisation (AFNOR), « La nouvelle approche », France Normalisation. https://www.francenormalisation.fr/les-acteurs-de-la-normalisation/la-nouvelle-approche/ (consulté le oct. 03, 2020).
[25] « The New EU Medical Device Regulation », Maetrics, déc. 19, 2020. https://maetrics.com/whitepaper/new-medical-device-regulation-requirements/ (consulté le déc. 19, 2020).
[27] « Marquage CE selon le Règlement (UE) 2017/745 », Qualitiso, déc. 04, 2017. https://www.qualitiso.com/marquage-ce-reglement-dispositifs-medicaux/ (consulté le déc. 01, 2020).
[28] « Norme NF EN 60601-1 Appareils électromédicaux - Partie 1 : exigences générales pour la sécurité de base et les performances essentielles (Tirage 5 (2014-09-01)) », Ed. Afnor, Paris, www.afnor.org, janv. 01, 2007.
[29] « NF EN 62366-1 Dispositifs médicaux - Partie 1 : application de l’ingénierie de l’aptitude à l’utilisation aux dispositifs médicaux (Tirage 2 (2016-01-01)) », Ed. Afnor, Paris, www.afnor.org, déc. 18, 2015.
[30] « NF EN 60601-2-22 : Appareils électromédicaux - Partie 2-22 : règles particulières pour la sécurité de base et les performances essentielles des appareils chirurgicaux, esthétiques, thérapeutiques et de diagnostic à laser », Ed. Afnor, Paris, www.afnor.org, mai 17, 2013.
[31] « IEC/TR 60825-8:2006 : Safety of laser products - Part 8 : guidelines for the safe use of laser beams on humans », Ed. Afnor, Paris, www.afnor.org, déc. 01, 2006.
[32] « NF EN ISO 10993-1 Évaluation biologique des dispositifs médicaux - Partie 1 : évaluation et essais au sein d’un processus de gestion du risque (Tirage 2 (2010-09-01)) », Ed. Afnor, Paris, www.afnor.org, juill. 01, 2010.
[33] « PR NF EN ISO 10993-1 Évaluation biologique des dispositifs médicaux - Partie 1 : Évaluation et essais au sein d’un processus de gestion du risque », Ed. Afnor, Paris, www.afnor.org, avr. 21, 2017.
[34] Isabel Scuntaro, « Clinical evaluation : a guide for manufacturers and notified bodies under directives 93/42 and 90/385 », The European Commission, Ed. European Union, MEDDEV 2.7/1 rev 4, juin 2016. Consulté le : nov. 13, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/17522/attachments/1/translations/.
[35] « Guidance on qualification and classification of software in Regulation (EU) 2017/745 - MDR and regulation (EU) 2017/746 - IVDR », Ed. the Medical Device Coordination Group, Guide MDCG 2019-11, oct. 2019. Consulté le : nov. 22, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/37581.
[36] G. CARDOEN, « MDCG 2020-13 Clinical evaluation assessment report template », Ed. the Medical Device Coordination Group, Text MDCG 2020-13, juill. 2020. Consulté le : nov. 22, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/health/md_sector/new_regulations/guidance_en.
[37] « Guide de l’utilisateur EUDAMED - Module Enregistrement des acteurs pour les opérateurs économiques. », Ed. European Union, Guide, nov. 2020. Consulté le : déc. 17, 2020. [En ligne]. Disponible sur : https://www.ansm.sante.fr/S-informer/Actualite/Base-de-donnees-EUDAMED-pour-les-dispositifs-medicaux-lancement-du-module-destine-a-l-enregistrement-des-operateurs-Point-d-Information-actualise-le-10-12-2020.
[38] DG santé, « User’s rights and obligations », The European Commission, avr. 2020. Consulté le : déc. 18, 2020. [En ligne]. Disponible sur : https://webgate.ec.europa.eu/eudamed/landing-page#/.
[39] DG santé, « Déclaration sur les responsabilités en matière de sécurité et l’information », The European Commission, 2020 805. Consulté le : déc. 18, 2020. [En ligne]. Disponible sur : https://webgate.ec.europa.eu/eudamed/landing-page#/.
[40] « Norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », Ed. Afnor, Paris, www.afnor.org, avr. 30, 2016.
[41] « NF EN ISO 14971 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux », Ed. Afnor, Paris, www.afnor.org, déc. 18, 2019.
[42] « IEC 62304/A1:2015 : Logiciels de santé - Processus du cycle de vie du logiciel », Ed. Afnor, Paris, www.afnor.org, juin 01, 2015.
[43] « NF EN 60825-1- Sécurité des appareils à laser - Partie 1 : classification des matériels et exigences (Tirage 2 (2017-07-01)) », Ed. Afnor, Paris, www.afnor.org, oct. 10, 2014.
[44] « NF EN ISO 11252 : Lasers et équipements associés aux lasers - Source laser - Exigences minimales pour la documentation », Ed. Afnor, Paris, www.afnor.org, oct. 11, 2013.
[45] The Therapeutic Goods Administration, « About IMDRF », 11 2020. http://www.imdrf.org/about/about.asp (consulté le nov. 10, 2020).
[46] « GHTF Study Group 1 - Pre-market Evaluation », Ed. The Therapeutic Goods Administration, GHTF/SG1/N78:2012, nov. 2012. Consulté le : nov. 10, 2020. [En ligne]. Disponible sur : http://www.imdrf.org/documents/doc-ghtf-sg1.asp.
[47] Essabiri Sara et Groell Agathe, « Facilitation de l’accès aux marchés biomédicaux internationaux : le STED », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Technologies Biomédicales et Territoires de Santé (TBTS) et Dispositif Médical et Affaires Règlementaires (DMAR), Mémoire de projet, réf n°IDS006, janv. 2019. Consulté le : oct. 01, 2020. [En ligne]. Disponible sur : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids006/.
[48] International Medical Device Regulators Forum, « Assembly and Technical Guide for IMDRF Table of Contents Submissions », Ed. The Therapeutic Goods Administration, Guide IMDRF/RPS WG/N27 FINAL:2019, mars 2019. Consulté le : nov. 11, 2020. [En ligne]. Disponible sur : https://www.dm-experts.fr/2019/04/imdrf-guide-pour-soumettre-un-dossier-au-format-table-des-matieres/.
[49] « NF EN ISO 14155 Investigation clinique des dispositifs médicaux pour sujets humains - Bonne pratique clinique », Ed. Afnor, Paris, www.afnor.org, août 19, 2020.
[50] EUROPEAN COMMISSION, DG Health and Consumers (SANCO), Directorate B-Consumer Affairs, et Unit B2- Health Technology and Cosmetics, « Guidance document - Market surveillance - Guidelines on a Medical Devices Vigilance System - MEDDEV 2.12/1 rev.8 », Ed. European Union, Guide MEDDEV 2.12/1 rev.8, janv. 2013. Consulté le : nov. 14, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/32305/attachments/1/translations.
[51] EUROPEAN COMMISSION, Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs, Unit GROW D.4 – Health Technology and Cosmetics, et GROW.DDG1.D.4, « Additional Guidance Regarding the Vigilance System as outlined in MEDDEV 2.12-1 rev. 8 », Ed. European Union, Guide MEDDEV 2.12-1 rev. 8, juill. 2019. Consulté le : oct. 23, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/36292.
[52] « Guidance document Medical devices - Market surveillance - Post Market Clinical Follow-up studies. », Ed. European Union, Guide MEDDEV.12/2 rev2, janv. 2012. Consulté le : nov. 17, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/10334/attachments/1/translations.
[53] « Manufacturer incident report 2020 », Ed. European Union, juin 2020. Consulté le : déc. 24, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/41681.
[54] « MDCG 2020-8 Post-market clinical follow-up (PMCF) Evaluation Report Template. A guide for manufacturers and notified bodies », Ed. European Union, avr. 2020. Consulté le : déc. 27, 2020. [En ligne]. Disponible sur : https://ec.europa.eu/docsroom/documents/40906.
Annexes
Annexe 1 : Tableau identifiant les chapitres et annexes pour classe IIa
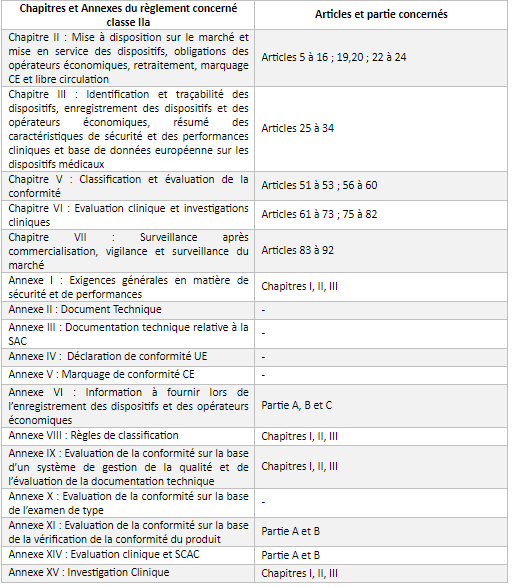
Annexe 2 : Etapes Evaluation clinique
- « Definition of the scope of the clinical evaluation » : il s’agit d’une étape permettant de cerner le domaine d’application. Elle détermine les bases des étapes suivantes. Cette étape définit le plan d’évaluation clinique en y détaillant les informations à intégrer en fonction de s’il s’agit d’un dossier réalisé avant le marquage CE ou simplement d’une mise à jour post-marquage CE. Des informations complémentaires sur le plan d’évaluation clinique sont décrites dans la partie 2.6.2.
- « Identification of pertinent data » : cette étape met en relief que l’indentification de toutes données générées, de surveillances, d’investigations cliniques, de retours utilisateurs, obtenues de la littérature scientifique doit être faite par le fabricant du dispositif médical. L’ensemble des données obtenues qu’elles soient favorables ou défavorables doivent émaner d’une méthodologie de recherche (décrite dans l’annexe A5 du MEDDEV) objective. L’annexe A1 du MEDDEV est un guide pour la démonstration de l’équivalence (cf partie 2.6.2.1). L’annexe A4 du MEDDEV, quant à elle, relève les sources de littérature scientifique les plus fiables à utiliser notamment Pubmed ou Medline et aussi décrit des pistes pour des recherches internet pertinentes sur les normes harmonisées, la matériovigilance ou encore les registres.
- « Appraisal of pertinent data » : cette étape est l’évaluation des données. Ici, les objectifs sont définis comme tels : « évaluer les données du fabricant dans chaque étude, la qualité méthodologique de l’étude, la pertinence de l’étude et pondérer la contribution de chaque étude par rapport aux autres données retenues ». Les données pouvant être prises en considération pour une évaluation de la pertinence de l’étude sont détaillées dans un tableau fournit dans le paragraphe 9 du MEDDEV. Dans ce paragraphe, chaque objectif est explicité et une aide pour les accomplir y est intégrée.
- « Analysis of the clinical data » : Le but de cette étape est de démontrer grâce à l’évaluation des données disponibles la conformité aux exigences essentielles de la performance clinique et sécurité du patient dans l’utilisation du dispositif médical. L’annexe A7 évoque l’évaluation du rapport bénéfice/risque et l’annexe A2 établit quant à elle les cas où les investigations cliniques doivent être réalisées. Cependant, le MEDDEV n’étant pas encore révisé selon le règlement pour cette partie notamment détaillée dans la partie 2.6.4.
- « The clinical evaluation report » : ce document doit rassembler toutes les étapes précédemment énoncées. Un contenu précis doit être présent. L’annexe A10 référence sous forme de checklist les éléments indispensables à avoir dans le rapport d’évaluation clinique. Une conclusion du rapport d’évaluation clinique doit être réalisé sur les points suivantes :
- Conformité aux exigences essentielles ;
- Evaluation de l’acceptabilité du rapport bénéfice/risque par rapport aux solutions alternatives existantes ;
- Relevance des informations fournies ;
- Aptitude à l’utilisation du DM ;
- Concordance des revendications du fabricant ;
- Identification des risques résiduels, incertitudes et impacts sur l’obtention du marquage CE avec les descriptions des actions préventives inclues dans le SCAC ;
- Documentation du DM, dossier de gestion des risques (dossier des gestions du bénéfice/risque demandé dans le nouveau règlement) et les données cliniques en concordance.
Annexe 3 : Tableau identifiant les guides utiles pour l’évaluation clinique
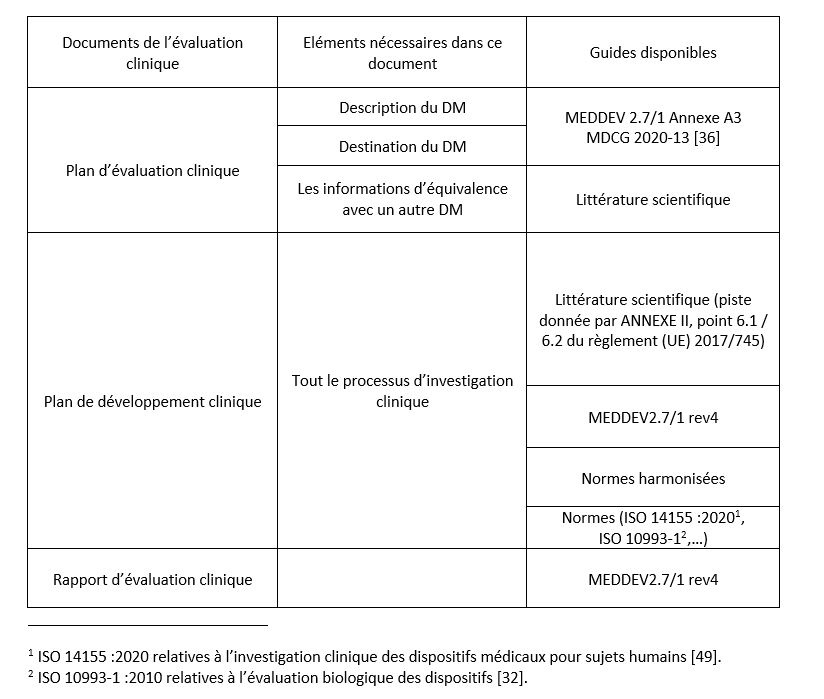
Annexe 4 : Tableau identifiant les guides utiles pour la Surveillance Après Commercialisation (SAC)
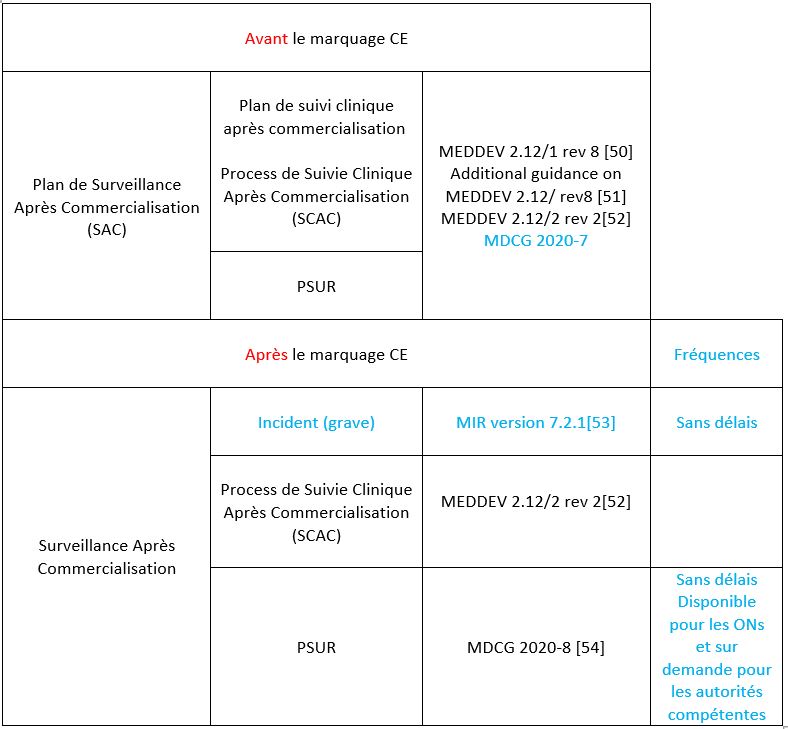
Annexe 5 : Exemple de déclaration de conformité UE
