
IDS136 - Elaboration d'un Système de Management de la Qualité : Introduction pour la conception de DMDIV
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Contact
- Yousra BOUTAROUAIT : yousra.b.21@hotmail.fr
Citation
A rappeler pour tout usage : Yousra BOUTAROUAIT , "Elaboration d'un Système de Management de la Qualité : Introduction pour la conception de DMDIV", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Stage, https://travaux.master.utc.fr/, réf n° IDS136, Juin 2022, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids136/
Résumé
Pour assurer sa pérennité, accroître sa performance et satisfaire les besoins auxquels elle veut répondre, une entreprise se doit d’instaurer un système de management de la qualité.
Pour les entreprises de dispositifs médicaux, c’est la norme EN ISO 13485 « Dispositifs médicaux – Système de management de la qualité - Exigences à des fins réglementaires » qui peut servir de référence pour le fabricant. En obtenant la certification de ce système, l’entreprise prouve à ses clients potentiels que tous les constituants mis en place respectent les exigences réglementaires applicables et que l’activité développée avec les risques associés est maîtrisée.
L’obtention du marquage CE, nécessaire pour la mise sur le marché sur le territoire européen, peut être travaillé en parallèle par le fabricant, en suivant les normes techniques spécifiques au dispositif conçu.
C’est autour de ces enjeux qu’avec l’entreprise ALGOSCOPE, startup développant des innovations dans le domaine des technologies médicales, qu’une stratégie a été mise en place pour atteindre au mieux la certification de leur système.
Mots-clés : Système Management Qualité, Règlement Européen, Conception, Dispositif Médical de Diagnostic in Vitro.
Abstract
In order to ensure its continuity, to increase its performance and to satisfy the needs it wants to meet, a company needs to set up a quality management system.
For medical device companies, EN ISO 13485 "Medical devices - Quality management system - Requirements for regulatory purposes" can serve as a reference for the manufacturer. By obtaining certification of this system, the company demonstrates to its potential customers that all the components implemented comply with the applicable regulatory requirements and that the activity developed with the associated risks is under control.
Obtaining the CE mark, which is necessary for marketing on European territory, can be worked on in parallel by the manufacturer, following the technical standards specific to the device designed.
It is around these issues that a strategy was set up with ALGOSCOPE, a start-up company developing innovations in the field of medical technologies, to best achieve the certification of their system.
Keywords : Quality Management System, European Regulation, Conception, In Vitro Diagnostic Medical Device.
Téléchargements

Liste de sigles/acronymes
- ANSM : Agence nationale de sécurité du médicament et des produits de santé ;
- DM : Dispositifs Médicaux ;
- DMDIV : Dispositif Médical de Diagnostic In Vitro ;
- EUDAMED : European Database on Medical Devices
- IA : Intelligence Artificielle ;
- IUD : Identifiant Unique du Dispositif ;
- ON : Organisme Notifié ;
- SMQ : Système Management Qualité.
Liste des figures
- Figure 1 : Organigramme fonctionnel d’ALGOSCOPE
- Figure 2 : Exemple de bannière ALGOSCOPE
- Figure 3 : Les différentes étapes de conditionnement d’un tissu dans un laboratoire de pathologie : zoom sur la phase pré-analytique
- Figure 4 : Cycle d’amélioration continue réalisé au début du stage
- Figure 5 : Composants à respecter pour la constitution du SMQ
- Figure 6 : Etapes de conception à suivre selon l’EN ISO 13485:2016 : détails sur l’étape 1
- Figure 7 : Récapitulatif du document URS, nécessaire pour l’Aptitude à l’Utilisation
- Figure 8 : Liste pour le suivi de la mise en place du SMQ : partie conception
Introduction
“L’intelligence est la capacité de s’adapter au changement” - Stephen W. Hawking, physicien théoricien et cosmologiste britannique.
Dans la société d'aujourd'hui, le prisme de l'innovation touche tous les domaines. La course au renouveau et la recherche ne laissent pas en reste le domaine médical.
Avec la crise sanitaire de ces deux dernières années, la Santé s’est encore plus placée comme sujet central. Des questions se posent : comment améliorer ce domaine en termes de nouveauté et de performance ? Quels sont les moyens pour y parvenir ? Autant d’interrogations qui nous poussent à réfléchir sur la contribution de chaque acteur du monde médical, et surtout sur l’assurance qualité des produits et services proposés par ces acteurs.
C’est dans ce contexte que des exigences réglementaires existent : elles permettent d’assurer un gage de qualité qui garde en ligne de mire le bien-être du patient. Dans le cas des dispositifs médicaux (DM), toute entreprise souhaitant être conforme se doit d’être en adéquation avec la norme ISO EN 13485 [1]. D’un point de vue réglementation, pour les Dispositifs Médicaux de Diagnostic In Vitro, il en va du Règlement (EU) 2017/746 [2], obligatoire à suivre pour tout fabricant souhaitant commercialiser son dispositif sur le marché européen.
En 2021, c’est un chiffre d'affaires de 30,7 milliards d’euros qui est recensé pour le secteur des DM en France [3]. Cet environnement pousse les jeunes structures proposant des innovations de ruptures, “qui changent radicalement les pratiques habituelles” [4], à se challenger dans un milieu évolutif.
La problématique qui peut alors se poser est la manière d’appréhender la mise en place d’un Système Management de la Qualité (SMQ) pour une startup, spécialisé dans les DMDIV dans le cas de ce rapport de stage, et les actions à mener pour assurer la garantie de bonnes performances au sein de celle-ci.
Ainsi ce rapport s’articule, dans un premier temps, par une présentation de la structure d’accueil, dont l’objectif était l’élaboration d’un SMQ adapté, puis de l’explication de certaines actions pertinentes menées pour la réalisation des missions et pour finir, d’une rétrospection sur les comportements et capacités requises lors de cette période.
I. Présentation de la structure d'accueil et de son environnement
a. Parcours de l'entreprise d'accueil
C’est en 2018 qu’est née l'idée de créer la startup ALGOSCOPE. Ce nom découle de deux termes, propres aux filières développées par l'entreprise :
- “ALGO-” pour algorithme, en lien avec les techniques utilisées ;
- “-SCOPE” pour microscope, en référence aux laboratoires.
Elle a été fondée officiellement en février 2021 par Soufiane Zakaria AZDAD, président, cofondateur et médecin anatomopathologiste, ainsi que deux associés, Samy DAHMANI, cofondateur, directeur scientifique et médecin biologiste et Moustapha EL S., cofondateur, directeur technique et ingénieur.
L’entreprise étant une startup, le modèle suivi est celui de l’adhocratie [5] : la direction générale est capable de traiter diverses fonctions en même temps telles que le service achat, le service marketing ou encore le service administratif.
Figure 1 : Organigramme fonctionnel d’ALGOSCOPE (juin 2022) (source : auteur).
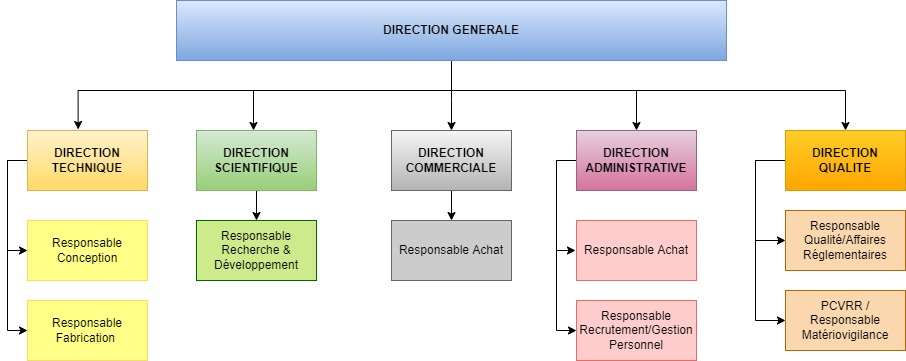
C’est lors de leurs propres travaux en laboratoire que les deux médecins ont développé l’envie de produire des solutions logicielles et matérielles afin d’améliorer la médecine de laboratoire. Ayant donc vocation à aider les soignants dans leurs tâches quotidiennes, ALGOSCOPE s’intéresse à un problème récurrent au sein des laboratoires : la sous-exploitation de la numérisation et de la valorisation des données de santé [6].
Ayant effectué, chacun dans leur domaine de prédilection une thèse sur l'Intelligence Artificielle (IA), c’est grâce à leurs techniques acquises durant leurs exercices de praticiens et les retours d'expériences accumulés au fil des années, qu’ils ont décidé d’entreprendre ce projet. Actuellement dans des locaux situés sur Compiègne, l’entreprise est également présente sur les réseaux sociaux avec une page LinkedIn et un site internet.
Figure 2 : Exemple de bannière ALGOSCOPE (source : algoscope.fr).

ALGOSCOPE a pu coopérer avec plusieurs partenaires : en 2020, c’est avec l'incubateur à projets Amiens Cluster que la startup a collaboré [7]. Cela a permis de lever les premiers fonds et de marquer le début d’autres échanges comme avec la Fondation de l’UPJV ou plus récemment avec BPI France.
D’un point de vue actualité, l’entreprise souhaite montrer son sérieux et ses performances spécifiquement dans le domaine de l’anatomopathologie : lors du “Data Challenge” en octobre 2020, coorganisé par la Société Française de Pathologie [8], l’entreprise a pu rivaliser avec d'autres sur l’analyse d’imagerie médicale concernant des biopsies du col de l’utérus. L’entreprise ALGOSCOPE est arrivée 6ème sur les 547 participants, avec un score de 92,5% (seulement dix participants à avoir obtenu plus de 90%). Ce résultat est un argument prégnant dans le milieu où la startup souhaite se démarquer.
b. Dispositifs conçus au sein de l’entreprise
n 2022, l’entreprise mène de nombreux projets dont la conception et la fabrication de DMDIV destinés à l’anatomopathologie.
L'anatomopathologie est une spécialité médicale qui étudie les tissus, les cellules et leurs anomalies, afin de contribuer au diagnostic des maladies. Il peut s’agir de tumeurs, de maladies de cause inflammatoire, dégénérative, vasculaire, métabolique ou encore infectieuse.
Cette spécialité établit une étude macroscopique (étude effectuée à l'œil nu dont l’objectif est la mise en évidence des anomalies morphologiques), ainsi que diverses méthodes de microscopie et de biologie moléculaire pour l’analyse histologique (étude des tissus vivants) et cytologique (étude des cellules vivantes) [9]. L’anatomopathologiste analyse des prélèvements pour l’examen histopathologique : ce sont par exemple des biopsies, des matériels d’expulsion spontanée, des échantillons de pièce opératoire ou des échantillons d’organes [10]. Cet examen est une partie essentielle et indispensable pour le patient dans sa prise en charge globale.
Par biopsie, il s’agit ici d’un petit fragment de tissu prélevé par voie transcutanée ou par voie endoscopique, par exemple un échantillon de tumeur. Par pièce opératoire, il est désigné ici des exérèses partielles ou complètes d’un ou plusieurs organes, prélevées au bloc opératoire lors d’une intervention chirurgicale [11]. L’examen peut se dérouler en laboratoire de pathologie, ou lors d’interventions chirurgicales.
Figure 3 : Les différentes étapes de conditionnement d’un tissu dans un laboratoire de pathologie : zoom sur la phase pré-analytique (source : auteur).

Le dispositif développé par ALGOSCOPE agit sur la phase pré-analytique (figure 3). Son rôle principal est de fournir des données images à l’opérateur. Ces données, qui sont habituellement non exploitées, vont permettre une traçabilité et un gain de temps conséquent pour le travail réalisé en macroscopie. Ce DMDIV est en cours d’obtention de brevet pour propriété intellectuelle.
II. Missions et/ou observations réalisées
a. Contexte, enjeux, problématique et objectifs
Contexte
Pour introduire le domaine de fabrication choisi par l’entreprise, voici la définition explicite d’un DMDIV d’après le règlement 2017/746 :
“ tout dispositif médical qui consiste en un réactif, un produit réactif, un matériau d'étalonnage, un matériau de contrôle, une trousse, un instrument, un appareil, un équipement, un logiciel ou un système, utilisé seul ou en association, destiné par le fabricant à être utilisé in vitro dans l'examen d'échantillons provenant du corps humain, y compris les dons de sang et de tissus, uniquement ou principalement dans le but de fournir des informations sur un ou plusieurs des éléments suivants :
a) concernant un processus ou état physiologique ou pathologique ;
b) concernant des déficiences congénitales physiques ou mentales ;
c) concernant la prédisposition à une affection ou à une maladie ;
d) permettant de déterminer si un traitement donné est sûr pour des receveurs potentiels et compatible avec eux ;
e) permettant de prévoir la réponse ou les réactions à un traitement ;
f) permettant de définir ou de contrôler des mesures thérapeutiques. Les récipients pour échantillons sont également réputés être des dispositifs médicaux de diagnostic in vitro ”[1].
Afin d’assurer et garantir la sécurité du patient et des utilisateurs dans ce secteur de grande envergure, différents textes sont à suivre :
- Le règlement européen 2017/746 pour les DMDIV. Ce texte est obligatoire pour tout fabricant souhaitant commercialiser un dispositif médical (DM) sur le sol de l’Union Européenne. Il précise différentes parties telles que la classification de risque du dispositif, la procédure à suivre pour l’Organisme Notifié (ON), qui permet l’obtention du marquage CE (Conformité Européenne) ou encore la traçabilité des dispositifs. Le respect de ce règlement permet d’obtenir le marquage CE, obligatoire pour les DM mis sur le marché de l’Union Européenne [2] ;
- La norme NF EN ISO 13485:2016 « Dispositifs médicaux - Systèmes de management de la qualité — Exigences à des fins réglementaires » : c’est une norme dite“ à des fins réglementaires”. La certification selon cette norme n’est pas obligatoire pour commercialiser les dispositifs médicaux, notamment ceux de classe A pour les DMDIV, mais est fortement recommandée afin d’assurer la sûreté des dispositifs proposés. La dernière version effective date de mars 2016. Elle permet de traiter différents aspects tels que le dossier du DM, l’approche par les risques ou encore le cycle de vie du dispositif [1]. Le 12 mai 2022 est entrée en vigueur la liste des normes européennes harmonisées selon le Règlement 2017/746 dont fait partie la EN ISO 13485:2016 [12]
Ces textes sont applicables dans toutes les entreprises de DM. L'enjeu pour une startup est de connaître les exigences applicables à son échelle et les atteindre. Avec des écrits officiels très concis sur les performances à assurer, toutes les exigences exclus vis-à-vis de l'entreprise font office de justifications argumentées et détaillées.
En revanche dans le cas d’une entreprise dont la création serait récente, n’ayant donc pas suivi les anciennes directives européennes, il n’est pas nécessaire de parler d’harmonisation car la seule réglementation suivie est celle du nouveau règlement, en l'occurrence ici le 2017/746 qui est applicable depuis le 26 mai 2022.
Enjeux
Pouvoir répondre aux attentes du marché et tenir face à la concurrence existante est un point majeur pour toute organisation qui se lance.
C’est tout autant le cas pour le domaine des DMDIV et encore plus pour l’anatomopathologie, spécialité peu connue du grand public et peu convoitée par les fabricants de dispositifs (argument obtenu par les données confidentielles de l’entreprise). En 2020, il y a 1 672 anatomopathologistes qui exercent sur le territoire français [13] : c’est un secteur non négligeable et sur lequel certaines entreprises ont décidé d’investir. Ainsi le fabricant se doit d'avoir une vision sur le long terme et d’apporter un réel bénéfice pour prendre position dans ce secteur.
Plusieurs stratégies sont à mener notamment celle de la visibilité : pour promulguer des avancées technologiques, la participation à des évènements tels que des congrès ou encore des assises est importante : c’est le cas pour le secteur de la pathologie [14] eti le secteur du DM en général [15].
Un autre enjeu qui est aussi à prendre en compte est l’enjeu sociétal : en octobre 2021, le Président de la République Emmanuel Macron a présenté le plan “France 2030” [16]. Ce plan, qui est défini comme un “grand plan d’investissement d’avenir”, s’établit sur 5 années avec 30 milliards d'euros déployés. Il présente les différents moyens de répondre aux problématiques à l’horizon de 2030, en s’intéressant entre autres à la croissance des technologies d’avenir en France. Il y est traité le sujet des startups notamment avec le Programme d’Investissement d’Avenir (PIA) : conscient des difficultés formalistes auxquelles les startups doivent faire face, ce programme se veut un moyen de soutien pour passer de l’innovation à l’industrialisation [17].
Sur ces faits, il semble donc normal de considérer la France comme territoire favorable aux jeunes entreprises novatrices. Néanmoins la concurrence est rude : d’après l’INSEE [18], il est recensé environ 1 million de startups en France en 2021. A cela s’ajoute le vaste secteur du DM dans lequel le fabricant doit faire sa place : toujours en France, le SNITEM [19] précise dans son “Panorama DM 2022” qu'il y a 1 440 entreprises recensées en 2021, avec une hausse du chiffre d’affaires de 30,2 milliards à 30,7 milliards d’euros grâce à l’export et grâce à la commercialisation de DMDIV.
Problématique
Face à ces enjeux, la problématique qui se pose concerne la mise en place d’une amélioration continue adaptée et adaptable à l’entreprise (en fonction de ses besoins et de ses attentes) et les moyens d’y parvenir afin d'accéder au marché de prédilection et d’y rester de façon pérenne.
Également liée à cette problématique, l’assurance de la conformité des dispositifs produits par le fabricant, gage de respect face aux réglementations existantes et de prise en compte de la sécurité des utilisateurs/patients concernés.
Objectifs
Durant ce stage, les objectifs qui ont été posés sont :
- L’élaboration d’un SMQ adéquat à ALGOSCOPE : mettre en place tous les éléments nécessaires, notamment pour la conception des DMDIV ;
- L’élaboration en parallèle du dossier technique du dispositif souhaité pour le marquage CE ;
- Sensibiliser l’ensemble du personnel pour la compréhension du SMQ ;
- Respecter les délais impartis pour la certification du SMQ.
b. Moyens et méthodes mis en œuvre
L'ensemble du SMQ ainsi qu’une partie de l’aspect technique pour le marquage CE du dispositif développé par ALGOSCOPE a été traité.
Cependant dans un souci de confidentialité, de respect du nombre de pages et de pertinence pour les explications, il a été choisi de traiter une seule partie qu’est la conception avec une introduction générale sur le fonctionnement d’un SMQ.
Afin d’aborder les actions menées de manière fluide et claire au sein de la startup, plusieurs facteurs doivent être établis au préalable sous forme de stratégie méthodologique.
Cela a pour objectif de préparer l’ensemble des collaborateurs à un audit blanc qui précèdera l’audit de certification du SMQ. Il a ainsi été défini un planning de Gantt avec les dates butoires et les jalons à respecter (revues d’avancements durant lesquels les étapes passées et celles à suivre sont présentées) .C’est pourquoi il est important de spécifier les outils pertinents qui seront utiles, utilisables et utilisés lors de cette élaboration.
Tout d’abord, il est primordial d’effectuer un état des lieux de l’entreprise.
Avec la norme EN ISO 13485, l'approche processus a son importance pour la mise en œuvre d’un SMQ. Le premier état des lieux s’est fait à l’aide d’un premier outil Excel, formé par d’anciens étudiants de l’UTC [23] : c’est à partir de celui qu’a pu être établi une liste provisoire des documents existants ou absents selon la norme. L'application de la méthodologie Plan-Do-Check-Act (figure 4) permet de structurer cette approche avec une gestion adaptée de l'amélioration continue pour la startup.
L’utilisation du cycle PDCA peut se décliner sous différentes formes selon la problématique sur laquelle il faut travailler et il est donc possible de commencer par une phase de cycle plutôt qu’une autre.
Figure 4 : Cycle d’amélioration continue réalisé au début du stage (source : auteur).
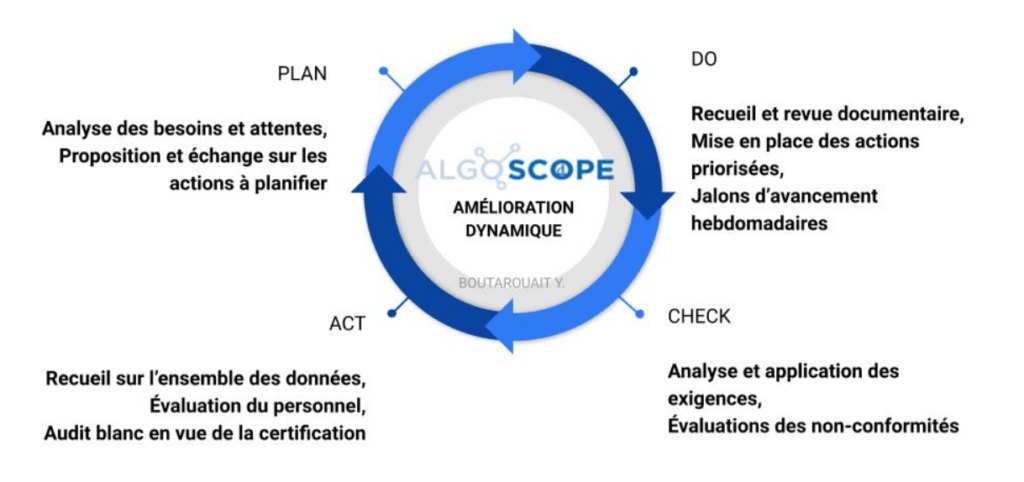
Pendant ce stage, le cycle PDCA a été adopté (figure 4) car au commencement il n’y avait pas de réelle structure pour le SMQ.
Chacune des phases comprend des éléments distincts :
- PLAN : prévision et planification sur ce qui doit être fait, autant d’un point de vue documentation qu’organisation ;
- DO : réalisation de ce qui est planifié : travail de rédaction, mise en place de jalons d’avancement, respect d’un planning selon des dates définies ;
- CHECK : vérification des résultats sur la base de la norme EN ISO 13485 : 2016 notamment ;
- ACT : amélioration et évolution : évaluation des non-conformités relevées, retour sur la sensibilisation du personnel et élaboration d’un audit blanc.
Avec la phase PLAN, il faut s’intéresser à la récolte de données d’entrée. Une acquisition et analyse des documents préexistants dans l’entreprise est à faire pour jauger ce qu’il reste pour le SMQ. Le système documentaire est défini par 5 types de documents chez ALGOSCOPE, comme précisé dans la figure 5 qui schématise la hiérarchisation de la documentation.
Figure 5 : Composants à respecter pour la constitution du SMQ (source : auteur).
Dans un premier temps, il est nécessaire pour toute structure d’entamer la rédaction de son Manuel qualité : celui-ci est la description de la gestion de la qualité au sein de l’entreprise. Il intervient notamment pour communiquer clairement, aussi bien en interne qu’avec les potentiels clients, sur la stratégie choisie (avec les objectifs et la politique qualité) et va servir pour répondre aux exigences du système qualité et faire part de l'engagement de la direction.
Il y a également les procédures qui définissent l’organisation et l’ensemble des principes à suivre selon chaque activité (méthodes de travail, les acteurs concernés par chaque tâche selon les types de processus).
Les instructions correspondent aux modes opératoires et autres documents pertinents qui viennent préciser les détails des opérations à mener.
Les formulaires correspondent à la standardisation des procédures, c’est-à-dire à valider la conformité ou la déviation du processus. ,
Les enregistrements sont des preuves formelles des résultats obtenus lors d’une activité.
De cette observation, il a pu être établi les différents domaines d’application.
En se basant sur les articles de la norme EN ISO 13485 : 2016, l’article 7 “ Réalisation du produit ” et plus précisément la partie 7.3. qui reprend les éléments en termes de conception est applicable au sein d’ALGOSCOPE.
Constitution de la partie “Conception & Développement” :
Au sein du SMQ, une procédure décrit les dispositions préétablies permettant de maîtriser les phases de conception et de développement du dispositif conçu de façon à :
- Mettre sur le marché les DM (En Europe, identifiés par un IUD dans la Base EUDAMED, base de données européenne des dispositifs médicaux) sûrs et efficaces avec une balance Bénéfice/ Risques satisfaisante, en adéquation avec l’état de l’art :
- CER : Clinical Evaluation Report : rapport du bénéfice clinique par rapport aux performances cliniques démontrés. Il permet d’évaluer si les données cliniques sont suffisantes afin d’appuyer les revendications du fabricant telles que les bénéfices, les effets secondaires ou encore les indications ;
- RMR : Risk Management Report : rapport des risques connus et acceptables compte-tenu :
- Du patient : bénéfice clinique induit par les performances cliniques du fait du principe de fonctionnement utilisé et,
- Pour les différents utilisateurs, les biens et l’environnement : de l’état de l’art pour la sécurité (généralement les normes (harmonisées en Europe))
- Être en conformité avec les exigences réglementaires applicables, notamment en Europe : le Règlement (UE) 2017/746 pour un Fabricant de DMDIV (en Europe, identifié par son SRN : Single Regulation Number de la Base EUDAMED).
Pour aborder au mieux cette partie, il est nécessaire de la segmenter : pour cela, des étapes doivent être planifiées du démarrage de la conception (figure 6) jusqu'à la mise sur le marché dans les pays « autorisés ». Selon chaque projet, ces étapes permettent d’appréhender au mieux l’organisation.
De façon générale, chaque étape de conception se conclut par une revue de conception : c’est une réunion permettant de mettre les différents partis impliqués d’accord soit sur la clôture de l’étape et le passage à la suivante, soit de revenir sur les éléments manquants avant de continuer (article 7.3.5).
Figure 6 : Etapes de conception à suivre selon l’EN ISO 13485:2016 : détails sur l’étape 1 (source : auteur)
Détails de la méthode à suivre pour l’étape 1 de la conception :
La première étape de la conception (figure 6) concerne les données d’entrées. Sans cette partie cruciale, le projet ne peut commencer correctement.
- Démarrage du DHF :
Il s’agit du Dossier Conception (7.3.10) selon la EN ISO 13485:2016. Pour chaque type de dispositif, le DHF contient ou réfère aux documents et aux enregistrements qui démontrent que la conception et le développement ont été mis en œuvre selon le plan de conception approuvé et les exigences réglementaires.
- Plan de développement du projet de conception établi :
Ce plan reprend les revues nécessaires à chaque étape de la conception et du développement, les activités de vérification, de validation et de transfert de conception selon chaque étape, les responsabilités et autorités liés, les méthodes permettant d’assurer la traçabilité des éléments de sortie par rapport aux éléments d’entrée et les ressources nécessaires, y compris la compétence nécessaire du personnel (7.3.1, 7.3.2, 7.3.6, 7.3.7 et 7.3.8).
Il doit aussi être documenté avec les normes harmonisées à la date du document, les normes non harmonisées mais qui pourraient être utiles et les autres réglementations applicables.