
IDS182-Certification ISO 13485 : 2016 et élaboration de la documentation technique selon le règlement européen 2017/745 dans une startup d'innovation en néphrologie
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteure

Contacts
- Fatoumata Samb DIOP : fatoumatasambdiop97@gmail.com
Citation
A rappeler pour tout usage : Fatoumata Samb DIOP, « Certification ISO 13485 : 2016 et élaboration de la documentation technique selon le règlement européen 2017/745 dans une startup d'innovation en néphrologie », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet (ou de Stage), https://travaux.master.utc.fr/, réf n° IDS182, (DOI s'il existe et s'il est connu), juillet 2023, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids182/
Résumé
L’entrée en vigueur du règlement européen 2017/745 relatif aux dispositifs médicaux (MDR) [1] représente un véritable changement dans l’écosystème des acteurs de santé. Depuis mai 2021, les fabricants de dispositifs médicaux (DM) nouvellement déclarés sur le marché européen doivent se conformer directement aux exigences de cette nouvelle réglementation pour pouvoir obtenir le marquage CE et commercialiser leurs produits.
L’article 10 du règlement énonce l’obligation de mettre en place et tenir à jour une documentation technique et un système de gestion de la qualité conformes aux dispositions dudit règlement [1].
L’objectif de ce projet de fin d’étude est d’accompagner l’entreprise d’accueil Home Habilis dans sa certification ISO 13485 : 2016 et la mise en application du règlement 2017/745 en vue de l’obtention du marquage CE. Ce mémoire met en évidence les différentes étapes et exigences liées à la mise en place d’un système de management de la qualité selon la NF EN ISO 13485 : 2016 [2], en soulignant l'importance des processus et systèmes de gestion de la qualité. De plus, il explore les spécificités de la documentation technique applicable aux dispositifs médicaux de classe IIa, conformément aux exigences du règlement 2017/745 .
Abstract
The entry into force of the European Medical Device Regulation (MDR) 2017/745 [1] represents a real change in the ecosystem of healthcare players. Since May 2021, manufacturers of medical devices (MD) newly declared on the European market must comply directly with the requirements of this new regulation in order to obtain CE marking and market their products.
Article 10 of the regulation sets out the obligation to set up and maintain technical documentation and a quality management system in line with the provisions of the said regulation [1].
The aim of this end-of-study project is to support the host company Home Habilis in its ISO 13485 : 2016 certification and the implementation of Regulation 2017/745 with a view to obtaining CE marking. This brief highlights the various stages and requirements involved in setting up a quality management system in accordance with NF EN ISO 13485 : 2016 [2], emphasizing the importance of quality management processes and systems. In addition, it explores the specifics of the technical documentation applicable to Class IIa medical devices, in accordance with the requirements of regulation 2017/745.
Téléchargements

Introduction
Partie intégrante du système de santé, les dispositifs médicaux innovants sont un terrain fertile pour la prise en charge des patients, que ce soit en milieu hospitalier ou à domicile. Dans ce contexte, la startup prometteuse Home Habilis se positionne comme un acteur clé de l’innovation en néphrologie en développant un dispositif médical novateur pour la télésurveillance des patients atteints d'insuffisance rénale chronique.
Cependant, avec la transition en cours entre le règlement 2017/745 et les directives 93/42/CEE [3] et 98/79/CE [4] relatives aux dispositifs médicaux et aux dispositifs médicaux in vitro, l'obtention du marquage CE sous le nouveau règlement devient de plus en plus pressante. Les startups, en particulier, sont confrontées à des défis majeurs, tels que la rareté des organismes notifiés et la complexité des exigences réglementaires. Dans ce paysage, il est essentiel pour toutes les nouvelles entreprises du domaine médical de viser la certification ISO 13485 :2016 et de se conformer au règlement européen 2017/745 afin de pouvoir commercialiser leurs dispositifs médicaux en Europe. Home Habilis n'échappe donc pas à ces exigences réglementaires pour obtenir le marquage CE de son premier produit.
Ainsi, la problématique centrale de ce mémoire se concentre sur la manière d’accompagner Home Habilis dans sa certification ISO 13485 :2016 et l’élaboration de la documentation technique dans le cadre de la mise en application du MDR.
Ce mémoire adopte une approche en plusieurs étapes. Tout d'abord, il présente en détail l'entreprise Home Habilis et son secteur d'activité spécifique. Ensuite, il examine les missions menées et les méthodologies adoptées pour, la mise en place du système de management de la qualité en vue de la certification ISO 13485 : 2016 et, l’élaboration de la documentation technique. Enfin, il expose les résultats obtenus à l’issue des missions ainsi que les apports de ce projet.
1. Présentation de la structure d'accueil et de son environnement
1.1. L’entreprise et son secteur d’activité
Home Habilis est une jeune startup française à vocation internationale, fondée en avril 2021 et basée à Beauvais, à moins d'une heure de Paris. Son siège social est situé dans la pépinière d'entreprises StarLab de Beauvais, créée en 2008, qui soutient les jeunes entreprises dans leur démarrage rapide et leur permet de s'implanter et de se développer.
Sous la présidence de M. Julien GAUTIER, qui apporte 20 ans d'expérience internationale dans l'industrie de la dialyse, cette entreprise a été créée avec le soutien de deux associés. Les trois co-fondateurs cumulent 70 années d'expérience complémentaires.
Home Habilis développe une plateforme médicale de télésurveillance à domicile pour les patients atteints d'insuffisance rénale chronique. L’insuffisance rénale est une maladie prioritaire [5] et un enjeu mondial de santé publique [6]. L'entreprise a ainsi pour mission d'améliorer le parcours de soins, de prévenir les complications, et d'améliorer la qualité de vie des patients.
1.2. Environnement de développement de la startup
Une startup est une jeune entreprise innovante qui investit fortement dans la recherche et le développement, dans le but ultime de réaliser une croissance économique exponentielle à long terme [7]. En tant que jeune startup dans l'écosystème de la Medtech, Home Habilis s'est développée dans un environnement d'incubateurs et d'acteurs régionaux de l'innovation depuis sa création. Les incubateurs ont pour mission de favoriser l'émergence et le développement des startups, en optimisant leurs chances de succès. Home Habilis est incubée en parallèle chez iTerra et chez Eurasanté, deux incubateurs labellisés par la région Hauts-de-France.
L’incubateur iTerra a accompagné l'entreprise dans sa phase de maturation et de création depuis mai 2020. Cette incubation est axé sur la construction d'un projet d'entreprise solide et viable, l'orientation stratégique, la faisabilité du projet, l'adéquation aux marchés et le modèle économique [8].
Eurasanté, incubateur et accélérateur spécialisé dans les innovations en santé, a également soutenu l'entreprise dans le développement du projet, notamment sur les aspects économiques, techniques, scientifiques, réglementaires et de réseautage.
La labellisation DeepTech par BPI France (FrenchTech Seed) et la French Tech Emergence démontrent que Home Habilis est une entreprise innovante dans l'écosystème de la MedTech.
En plus des incubateurs, l'entreprise est soutenue par des partenaires de différentes catégories.

Figure 1 : Partenaires de Home Habilis (source : CEO de Home Habilis)
1.3. Organisation de la startup
Dans un environnement de startup, les ressources sont limitées, ce qui rend nécessaire la polyvalence et l'adaptabilité des employés. Cependant, il est essentiel que chaque personne occupe un poste clairement défini dans l'organigramme afin de connaître les compétences disponibles et de favoriser une collaboration indispensable pour atteindre un objectif commun.
L'organisation générale de l'entreprise est représentée dans l'organigramme ci-dessous :
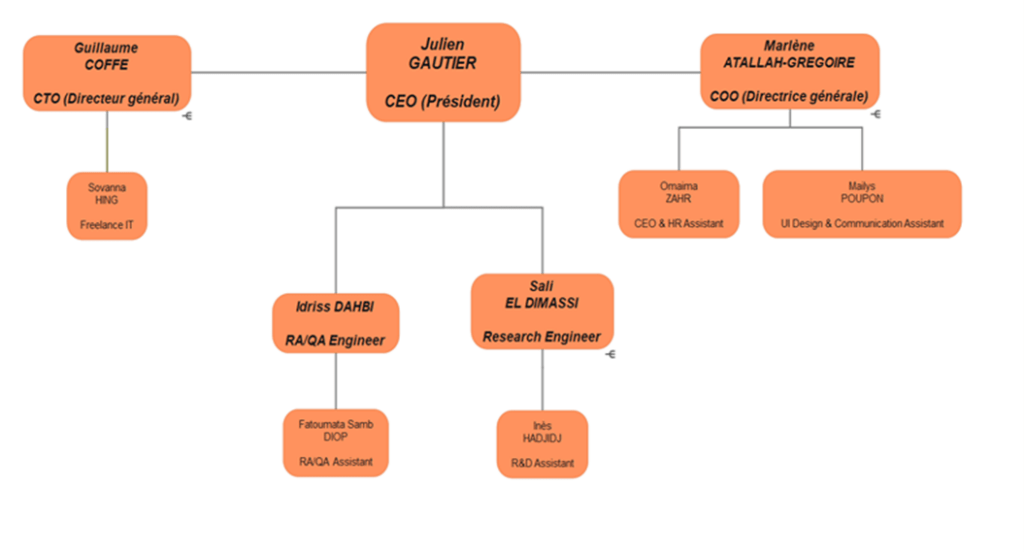
Figure 2 : Organigramme de l’entreprise (source : RH Home Habilis)
1.4. L’insuffisance rénale chronique et technologies concurrentielles
L'insuffisance rénale est une affection dans laquelle les reins ne fonctionnent pas correctement, entraînant une diminution progressive de leur capacité à filtrer et éliminer les déchets et les toxines du corps. Il existe 5 stades d'insuffisance rénale, et la maladie est considérée comme chronique à partir du stade 3, lorsque les reins cessent de fonctionner de manière irréversible, nécessitant un suivi médical pour préserver la fonction rénale [9]. L'insuffisance rénale chronique (IRC) est une maladie qui représente un enjeu mondial impactant considérablement la vie de 3 millions de patients en France [10] et 850 millions dans le monde [11]. La surcharge hydrique est une complication fréquente chez les patients atteints d'insuffisance rénale chronique. Elle entraîne une détérioration dramatique de la qualité de vie et des chances de survie de ces patients, en raison de l'accumulation d'eau qui n'est plus filtrée par les reins [12]. Cette surcharge hydrique est une complication critique et peut nécessiter une dialyse en urgence. Ces urgences représentent une perte de chance pour les patients, une pression sur les systèmes de santé parce que la prise en charge du patient n’est pas planifiée ou anticipée. La normalisation de la surcharge hydrique chez les patients en IRC est un facteur important directement associé aux chances de survie des patients[13] [14].
En France, quatre entreprises développent des DM utilisables en néphrologie et destinés aux patients atteints d’IRC :
Tableau 1 : Entreprises concurrentes de Home Habilis en néphrologie (Source : CEO de Home Habilis)

Cependant, ces dispositifs médicaux sont sous-utilisés en routine.
Parallèlement, il existe également des produits grand public capables de mesurer la composition corporelle comme le produit Bodycardio de Withings [15]. Cependant, la littérature indique que ces systèmes ne doivent pas être utilisés de manière régulière avec des patients [12].
L’objectif de Home Habilis est de rendre ces pratiques régulières et mieux adaptées à la néphrologie.
1.5. Projet en cours de développement
Le projet eKIDNEy développé par Home Habilis vise à apporter des solutions aux patients atteints d'IRC en optimisant leur parcours de soins, en améliorant leur qualité de vie et en leur permettant de mesurer régulièrement leur surcharge hydrique. Le produit en développement est un dispositif médical de classe IIa accompagné d'une plateforme médicale de télésurveillance. Pour plus de détails, se référer à l'annexe 1 (confidentielle) pour la description détaillée et la visuelle.
2. Missions, méthodes et résultats
2.1. Contexte, enjeux, problématique et objectifs
2.1.1. Contexte
En plein développement de son projet, Home Habilis a pour objectif d'obtenir simultanément le marquage CE pour son dispositif médical ainsi que la certification de son système qualité selon la norme NF EN ISO 13485:2016, intitulée "Dispositifs médicaux - Systèmes de management de la qualité – Exigences à des fins réglementaires"[2]. Étant donné que les dispositifs médicaux évoluent dans un environnement strictement réglementé, le développement de ce produit a été confronté aux réalités de la réglementation européenne dès ses premières ébauches.
En ce qui concerne l'accès au marché européen, l'entrée en vigueur du nouveau règlement 2017/745, relatif aux dispositifs médicaux en mai 2021, a entraîné une refonte totale du cycle de vie de ces derniers. Cette réglementation renforce les conditions d'obtention du marquage CE, qui est nécessaire pour commercialiser et faire circuler un dispositif médical sur le marché européen. Cette évolution représente un véritable enjeu pour tous les acteurs de la santé, notamment les fabricants. Home Habilis, en tant que nouvelle startup, est particulièrement concernée et doit se conformer directement aux nouvelles exigences du marché.
Par ailleurs, en plus de se conformer aux exigences du MDR, l'amendement A11 de la norme ISO 13485, l'ISO 13485/A11 : 2021, intègre les exigences réglementaires liées au SMQ. Les fabricants doivent donc intégrer les exigences relatives au système qualité du règlement européen dans leur processus de mise en place d'un système qualité, afin d'assurer une conformité totale pour la certification ISO 13485 [16].
À ce stade, l'obtention du marquage CE et de la certification ISO repose sur la convergence de deux aspects complémentaires :
- La conformité réglementaire selon le règlement européen 2017/745 par l’élaboration d’une documentation technique
- La conformité du système de management de la qualité (SMQ) selon la NF EN ISO 13485 :2016 et son amendement A11.
2.1.2. Enjeux
En cette période de transition entre les directives de DM [3] [4] et le règlement MDR, les acteurs de la santé, notamment les fabricants, se précipitent pour obtenir le marquage CE afin de recertifier leurs dispositifs médicaux existants ou de certifier leurs nouveaux dispositifs. L'un des impacts majeurs du règlement relatif aux dispositifs médicaux (RDM) concerne l'implication des organismes notifiés (ON), qui sont des organismes indépendants chargés d'évaluer la conformité des dispositifs médicaux aux normes de sécurité et de performance [17]. L'article 38 du règlement 2017/745 décrit les nouveaux critères de désignation des organismes notifiés aux paragraphes 1 et 2 : "Les organismes d'évaluation de la conformité introduisent une demande de désignation auprès de l'autorité responsable des organismes notifiés. La demande précise les activités d'évaluation de la conformité telles qu'elles sont définies par le présent règlement et les types de dispositifs pour lesquels l'organisme demande à être désigné, et est accompagnée des documents attestant le respect de l'annexe VII." [1]
L'article 36 et l'annexe VII du règlement RDM détaillent les exigences, considérablement renforcées, applicables aux organismes notifiés. Ces organismes d'évaluation de la conformité ont désormais plus de responsabilités et doivent respecter des exigences plus strictes pour être certifiés et désignés par les autorités responsables des organismes notifiés, contrairement à l'ancienne directive 93/42/CEE [3]. Les difficultés de mise en œuvre de ce règlement ont entraîné la perte de certification pour de nombreux organismes notifiés dès son entrée en vigueur, réduisant leur nombre de plus de 50 à seulement 38 en Europe [18] [19]. Aujourd'hui, la pénurie d'organismes notifiés constitue un enjeu majeur pour les fabricants de dispositifs médicaux dans leur processus d'obtention de marquage CE [17].
2.1.3 Problématique et objectifs
L'entreprise Home Habilis avait entamé la mise en place de son système de management de la qualité (SMQ) en 2021, et à cette fin, un audit interne a été effectué. Le SMQ était non conforme et 99 constats ont été identifiés à la suite de cet audit. Compte tenu du contexte de l'entreprise et des enjeux réglementaires, mon rôle en tant qu'alternante s'inscrit dans la problématique suivante : quelle démarche entreprendre pour accompagner Home Habilis dans sa certification ISO 13485 :2016 et la mise en application du règlement 2017/745 ?
Pour cela, mes principales missions étaient les suivantes :
- Finaliser la mise en place du SMQ conformément à la norme ISO 13485 :2016 et à son amendement A11 ;
- Élaborer un plan d'action et mettre en œuvre les mesures correctives découlant de l'audit interne de 2021 ;
- Constituer et documenter le dossier technique en vue du marquage CE ;
- Établir et gérer le dossier de gestion des risques.
Afin de remplir ces missions et de répondre à la problématique, une approche stratégique et opérationnelle a été élaborée pour agir de manière efficace et efficiente dans l'exécution des tâches.
2.2. Méthodologie
Pour avoir une vision claire de la stratégie à adopter, des opérations à effectuer et de l'environnement qualité et réglementaire de l'entreprise, une cartographie a été réalisée.
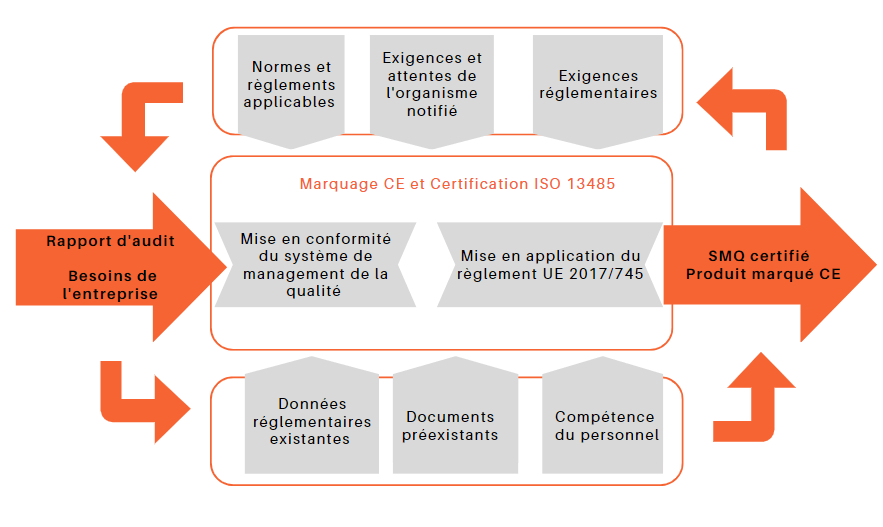
Figure 3 : Cartographie illustrant les besoins de l’entreprise (source : auteur)
Cette cartographie permet de bien définir les éléments qui alimentent le projet, tels que les données d'entrée, les données existantes et les données de sortie, et met en évidence la corrélation entre chaque partie afin d'atteindre l'objectif visé. Cela s'avère pratique pour prévisualiser mes missions par rapport aux attentes de l'entreprise et planifier les événements à réaliser pendant l'alternance. La gestion des activités sous forme de processus permet d'atteindre les objectifs de manière rationnelle et efficace.
2.2.1. Démarche de mise en conformité du SMQ dans le cadre de la certification ISO 13485
La mise en place d'un système de management de la qualité nécessite une démarche qualité et une stratégie organisationnelle appropriées. Pour les fabricants de dispositifs médicaux, cette étape est obligatoire, mais peut s'avérer être un véritable défi pour les startups de l'écosystème Medtech.
Un système de management de la qualité est un ensemble d'éléments corrélés ou en interaction utilisés pour établir des politiques, des objectifs et des processus afin d'aider un organisme à garantir la qualité de son organisation et de sa production [20].
Pour la mise en conformité du SMQ de Home Habilis, la méthode de gestion de la qualité et d'amélioration continue appelée "Roue de Deming" a été adoptée. Cette méthode, popularisée par le statisticien américain et promoteur de la qualité William Edwards Deming, se décompose en quatre phases : Check (Vérifier), Act ((Ré)Agir), Plan (Planifier) et Do (Réaliser) (CAPD). Elle permet, grâce à un cycle d'amélioration continue, de s'inscrire dans une logique de résolution de problèmes et de gestion de projet.
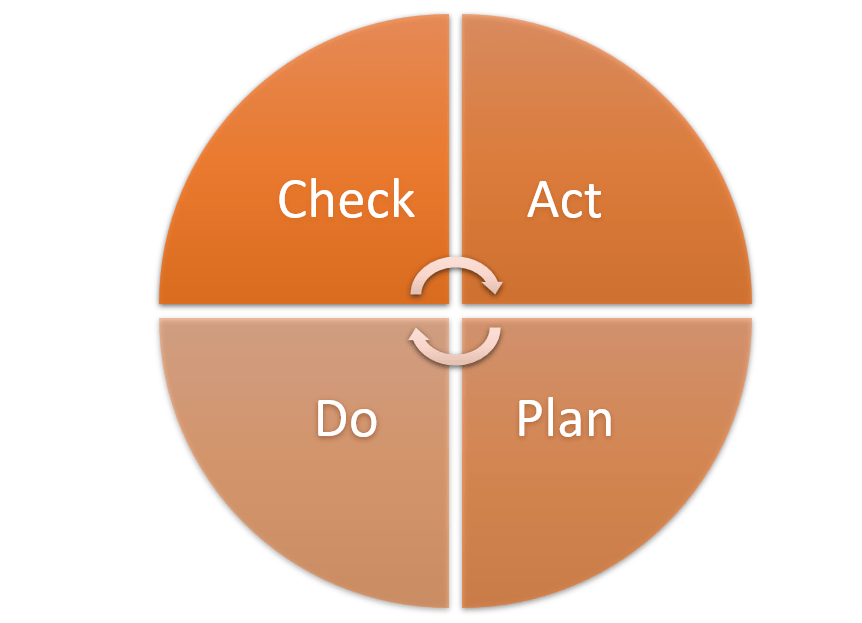
Figure 4 : Roue Deming (sens à CAPD) (source : auteur)
2.2.1.1. Check : faire l’état des lieux, connaitre ce qui est réalisé
La première étape consistait à dresser un état des lieux du système de management de la qualité afin d'analyser les documents existants ainsi que le niveau de conformité du système. Pour ce faire, une analyse du rapport d'audit a été réalisée dans un premier temps afin de détecter le nombre de constats. Au total, 99 constats ont été identifiés à la suite de l'audit. Par la suite, une étude approfondie du système qualité a été réalisée grâce à un autodiagnostic du SMQ, afin d'obtenir un aperçu du niveau de conformité du système par rapport aux exigences de la norme NF EN ISO 13485 :2016 [2].
L'outil d'autodiagnostic de la norme NF EN ISO 13485, développé par des étudiants de l'Université de Technologie de Compiègne, s'est avéré extrêmement utile pour mener à bien cette tâche [21]. Cet outil est composé de plusieurs onglets, dont l'onglet Evaluation (selon les critères de la norme) et l'onglet Résultats qui affiche graphiquement le niveau de conformité par article. Les résultats de l'évaluation du système qualité sont présentés dans le graphique suivant.
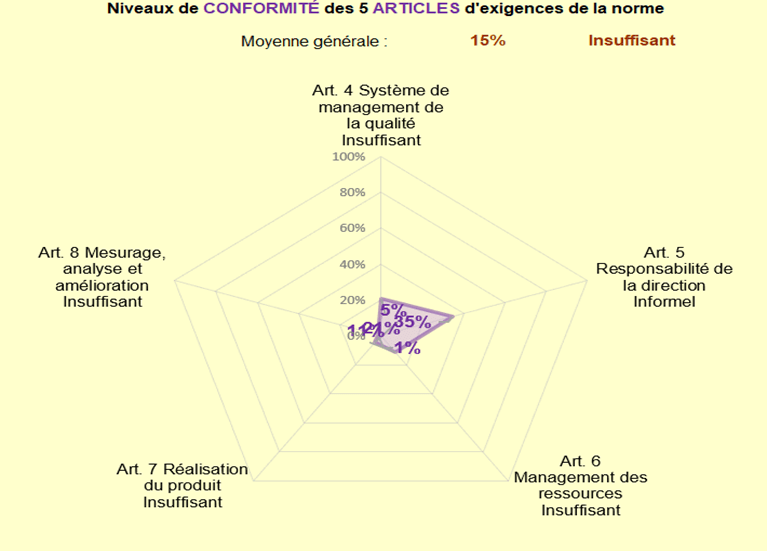
Figure 5 : Autodiagnostic initial du SMQ (source : auteur [21] )
2.2.1.2 Act : Agir, identifier les améliorations et les corrections
Après avoir identifié les écarts entre les pratiques existantes de l'entreprise et les exigences de la norme, il était important d'identifier les actions à mettre en place pour répondre aux critères de la norme et corriger les non-conformités relevées lors de l'audit. Une première priorité a été donnée à l'identification des exigences obligatoires pour obtenir la certification. Cette démarche permet de déterminer l'ensemble des documents qualité (dossiers, procédures, enregistrements, etc.) indispensables pour obtenir la certification.
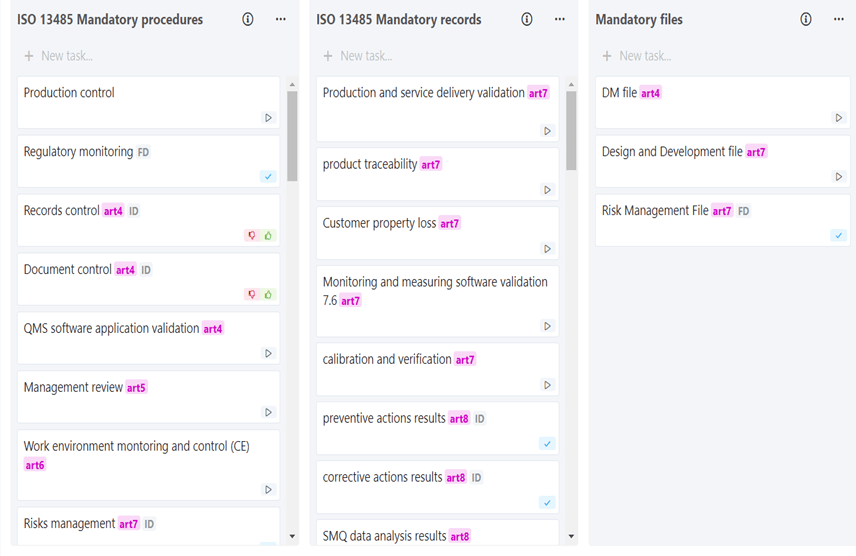
Figure 6 : Extrait d’identification des documents obligatoires selon la NF EN ISO 13485 (Source : auteur)
Ce processus d'identification de la documentation obligatoire a permis simultanément de définir les actions préventives et correctives à mettre en œuvre pour corriger les non-conformités relevées lors de l'audit. Cette étape peut également être l'occasion d'identifier les processus clés adaptés au projet et à l'organisation de l'entreprise (voir la cartographie des processus en annexe 2 confidentielle).
2.2.1.3. Plan : Planifier les actions à réaliser
Dans cette phase, l'accent est mis sur la planification des activités et des tâches à réaliser. Un registre CAPA a été développé pour planifier et enregistrer les activités et les actions nécessaires afin de corriger les non-conformités identifiées. Ce plan comprend également la description des responsabilités, des échéances et des ressources requises pour mener à bien ces activités. Le schéma suivant montre un extrait du registre des CAPA.

Figure 7 : Extrait du plan d’actions préventives et correctives (source : auteur)
Afin de garantir une alimentation efficace du système de management de la qualité (SMQ), une planification de la documentation qualité a été effectuée en utilisant une matrice de traçabilité. Cela assure la gestion des procédures et des enregistrements documentés.

Figure 8 : Extrait de la planification de l’alimentation du SMQ (source : auteur)
Ensuite, un plan de formation a été élaboré pour initier le personnel de l'entreprise à l'objectif et à l'utilité du SMQ, et les intégrer à la mise en place du système qualité.
2.2.1.4. Do : Réaliser ce qui a été prévu (alimenter, suivre, former, améliorer)
Cette étape finale consiste à mettre en œuvre toutes les activités planifiées pour assurer la conformité du SMQ et améliorer continuellement le système en vue de la certification ISO 13485. Les actions comprennent :
- La formation et la sensibilisation du personnel : Kaoru Ishikawa disait « La maîtrise de la qualité commence par la formation et se termine par la formation ». Ainsi, consciente de cela, une formation adéquate a été dispensée à tous les employés concernés par le système de gestion de la qualité. Cette formation vise à intégrer le personnel dans le système qualité, à les familiariser avec les procédures, la codification des documents qualité et les exigences spécifiques de la norme NF EN ISO 13485, et à les sensibiliser à leur rôle dans la conformité et l'amélioration continue du système.
- La mise en œuvre de la documentation : La documentation joue un rôle essentiel dans le SMQ. Il s'agit de continuellement alimenter les documents identifiés et planifiés, telles que les procédures documentées pour chaque processus clé. Ces documents doivent être conformes aux exigences de la NF EN ISO 13485 et décrire les activités à effectuer, les responsabilités et les interactions entre les processus. La documentation comprend également la création d'un manuel qualité, de formulaires, d'enregistrements et d'autres documents nécessaires au développement du SMQ.
- Le suivi, la vérification et l’amélioration continu : La dernière étape consiste à améliorer en permanence le système. Cela implique de surveiller régulièrement la documentation du SMQ à travers des revues qualité, des revues de direction et des évaluations telles que l'autodiagnostic ou l'audit interne. L'objectif est de vérifier le niveau de maturité et de conformité aux exigences de la norme NF EN ISO 13485. A cette fin, une procédure d'amélioration continue (voir annexe 3 confidentielle) a été mise en place chez Home Habilis pour décrire les activités visant à assurer et maintenir la pertinence, l'efficacité et l'amélioration continue du système qualité.
2.2.1.5. Adopter une culture qualité
« La qualité n’est pas une action – c’est une habitude. » Aristote
La qualité est une habitude qui se construit. En tant que membre du service qualité, mon objectif était d'adopter une culture qualité au sein de Home Habilis dès la mise en place du SMQ. Cela m'a conduit à interagir avec tous les départements de l'entreprise afin d'intégrer tous les employés dans l'élaboration et l'amélioration continue du SMQ. Dans ce cadre, j'ai été chargée de mener des séances de formation et des réunions régulières sur la qualité avec les employés afin d'instaurer une culture qualité et de promouvoir les bonnes pratiques pour développer le SMQ sur de solides bases. La direction de Home Habilis s'engage à placer la qualité au cœur de ses activités et à la faire appliquer par tous les employés de l'entreprise.
La mise en place du système de management de la qualité n'est pas la seule obligation du fabricant de dispositifs médicaux. En effet, comme introduit dans la partie 2.1, l'article 10 du règlement 2017/745 exige aux fabricants une documentation technique. La conformité à cette exigence sera abordée dans la section suivante.
2.2.2. Conformité réglementaire vers le marquage CE
Comme mentionné précédemment, pour commercialiser un dispositif médical en Europe, il est nécessaire d'obtenir le marquage CE. Aujourd'hui, un organisme notifié accorde le marquage CE lorsque l'entreprise et le dispositif médical à certifier satisfont aux exigences du règlement européen 2017/745. Alors que la fin de la période de transition des directives [3] [4] au règlement était initialement fixée au 26 mai 2025 selon l'article 120 du MDR [1], les difficultés de mise en œuvre du règlement ont conduit le Parlement européen à prolonger la période transitoire à travers le règlement (UE) 2023/607 du 20 mars 2023 [22]. Les périodes de prolongation sont définies en fonction de la classe de risque des dispositifs. Le tableau suivant présente le nouveau calendrier de transition en fonction des classes de DM selon le règlement (UE) 2023/607.
Tableau 2 : Périodes de transition selon le règlement 2023/607 (source : [23])
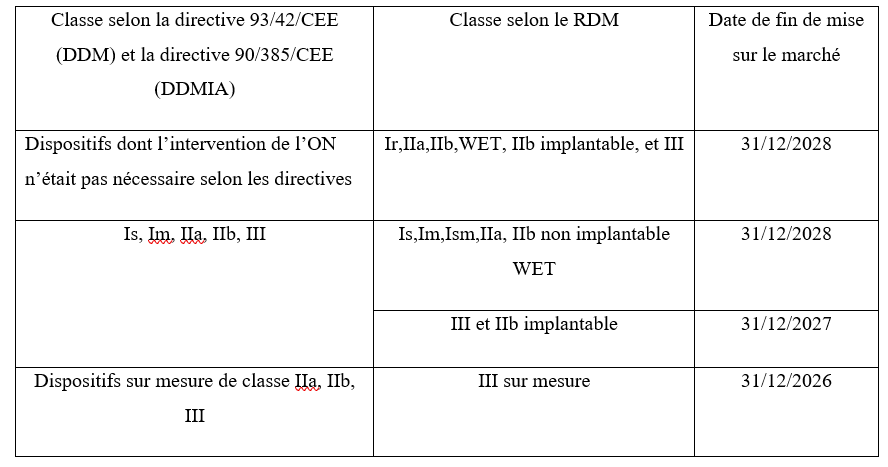
L'obtention du marquage CE est un véritable challenge. Pour réussir, les fabricants de dispositifs médicaux doivent élaborer un plan stratégique détaillant les différentes étapes nécessaires.
À mon arrivée, l'entreprise était en phase de conception d'un dispositif médical classé IIa selon le règlement 2017/745. Le planning suivant a été établi pour définir les prochaines étapes du processus d'obtention du marquage CE :
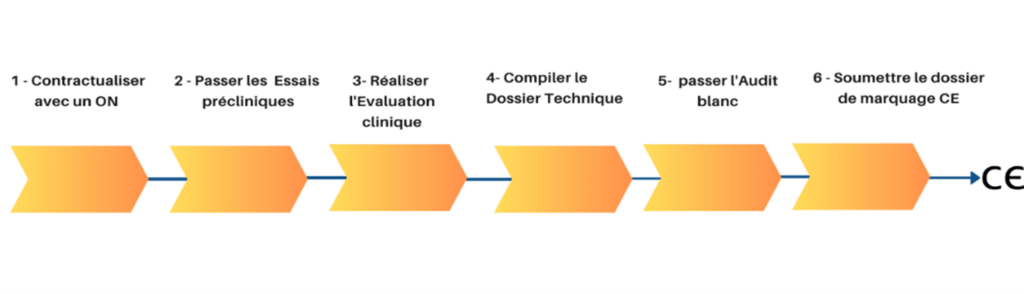
Figure 9 : Route vers le marquage CE (Source : auteur)
La toute première étape consistait à trouver un organisme notifié capable de traiter notre dossier de marquage CE dans les délais. J'ai principalement été impliqué dans la sélection des ON et la mise en place de la documentation technique.
2.2.2.1. La quête d’un organisme notifié, un véritable défi pour les startups de la Medtech
Comme mentionné précédemment, la pénurie des organismes notifiés est un problème majeur pour les fabricants, en particulier pour les nouvelles start-ups de la Medtech qui recherchent une certification CE afin de mettre leurs produits sur le marché. La recherche d'un ON a été un véritable défi chez Home Habilis, qui a été particulièrement touchée par ce phénomène. L'entreprise a été confrontée aux enjeux suivants :
- Manque d'ON disponibles : Les organismes notifiés se sont font de plus en plus rares. Ceci s’explique au durcissement des critères d'autorisation d'agrément. La plupart des ON répertoriés sur le site NANDO (base de données des organismes notifiés) ne pouvaient plus accepter de nouveaux dossiers en raison d'une quantité insuffisante de ressources humaines et matérielles.
- Ralentissement des processus d’évaluation : Avec moins d'organismes notifiés disponibles, l'entreprise s'est heurtée à des délais d'évaluation très longs (au minimum 18 mois), ce qui ne lui était pas convenable, car cela aurait eu un impact sur la planification des activités commerciales et pourrait avoir des conséquences financières significatives.
- Concurrence accrue : Le temps était compté pour trouver un ON répondant aux critères de l'entreprise. Home Habilis se trouvait en concurrence avec d'autres fabricants de dispositifs médicaux (grandes entreprises et PME) pour accéder aux services limités offerts par ces organismes de certification. En tant que nouvelle start-up disposant de ressources limitées pour faire face à cette concurrence, cela représentait une menace.
Il était donc judicieux de trouver des solutions en interne pour élargir les options de choix et accroître l'efficacité :
- Ouverture aux ON étrangers : L'entreprise a décidé de s'ouvrir à d'autres ON et d'élargir ses choix en considérant tous les ON éligibles en Europe répondant aux critères spécifiques à son type de dispositif médical.
- Mise en place d’une procédure adaptée : Une procédure de sélection des ON (voir annexe 4 confidentielle) a été établie afin d'optimiser et d'uniformiser la méthode de sélection des ON selon les critères de l'entreprise.
- Veille stratégique axée sur les ON : Un registre de veille des ON a également été créé pour suivre quotidiennement les ON certifiés sur le site NANDO, afin de rester informé des mises à jour et anticiper les changements à venir (par exemple, les ON qui doivent être certifiés prochainement).
- Priorité aux ON récemment désignés : Cette approche permettait d'éviter la saturation des ON existants et d'augmenter les chances d'acceptation de la candidature.
Grâce à cette démarche, tous les ON présents sur le site NANDO ont été suivis et contactés. Cela a permis d'obtenir des réponses plus convaincantes et de trouver un organisme notifié répondant aux critères de l'entreprise.
Le processus de conformité réglementaire est une tâche complexe qui nécessite des compétences et des ressources importantes. Les fabricants de dispositifs médicaux doivent établir une feuille de route pour pouvoir franchir les différentes étapes et alimenter le dossier technique. La partie suivante présente l’élaboration de la documentation technique.
2.2.2.2. Elaboration de la documentation technique pour le marquage CE
La mise en place de la documentation technique constitue une étape essentielle à l’obtention du marquage CE d'un dispositif médical. Selon l'article 10 du règlement, « Les fabricants de dispositifs autres que des dispositifs sur mesure établissent et tiennent à jour la documentation technique relative auxdits dispositifs. La documentation technique est de nature à permettre l'évaluation de la conformité du dispositif avec les exigences du présent règlement. Cette documentation technique contient les éléments prévus aux annexes II et III. » [1].
Ainsi, la démarche suivante a été adoptée pour l’élaboration de la documentation technique.
2.2.2.2.1 Cadre normatif et réglementaire
La mise en application du règlement peut être chose ardue pour certains fabricants, c'est pourquoi des normes d'aide à l'application du MDR sont élaborées par des organismes européens de normalisation tels que le Comité Européen de Normalisation (CEN), en collaboration avec la Commission Européenne. Ces normes, appelées normes harmonisées, sont publiées au Journal officiel de l'Union européenne et sont considérées comme des moyens présumés de conformité aux exigences du règlement. Le règlement le stipule dans le premier alinéa de l'article 8 : "Les dispositifs conformes aux normes harmonisées applicables, ou à des parties pertinentes de ces normes, dont les références ont été publiées au Journal officiel de l'Union européenne, sont présumés conformes aux exigences du présent règlement relevant de ces normes ou de parties de celles-ci" [1].
Il est donc recommandé de mettre en application ces normes dès les premières étapes de la conception et de l'élaboration de la documentation technique.
Un registre réglementaire relatant toutes les normes harmonisées applicables au dispositif de Home Habilis a été mis en place (voir annexe 5 confidentielle). Ce registre permet également d’enregistrer toutes les normes spécifiques, les directives et les règlements qui concernent le dispositif médical. Une veille réglementaire est régulièrement faite en interne pour s’assurer de la mise à jour du registre et garantir la conformité du produit avec les référentiels en vigueur.
2.2.2.2.2 Mise en place d’une feuille de gestion des preuves de conformité
Afin de répondre aux exigences des annexes II et III concernant la documentation technique, une première étape a consisté à identifier les livrables correspondant à ces annexes, suivie de l'élaboration d'un plan de collecte de ces livrables.
L'annexe 6 (confidentielle) présente la feuille de route de la documentation technique présentant les livrables à fournir à chaque phase du projet. Depuis la phase de conception jusqu'à la réalisation et l'évaluation clinique du dispositif médical final, tous les documents obligatoires pour constituer la documentation technique (par exemple, les spécifications de conception, les procédures de fabrication, les rapports d'essai) ont été planifiés dans ce tableau. La collecte et le suivi de ces documents étaient effectués à chaque phase du projet lors de points d'étape mensuels.
Différentes activités d'évaluation et d'essais doivent être réalisées par les fabricants pour alimenter la documentation technique.
2.2.2.2.3 Les principaux éléments du dossier technique
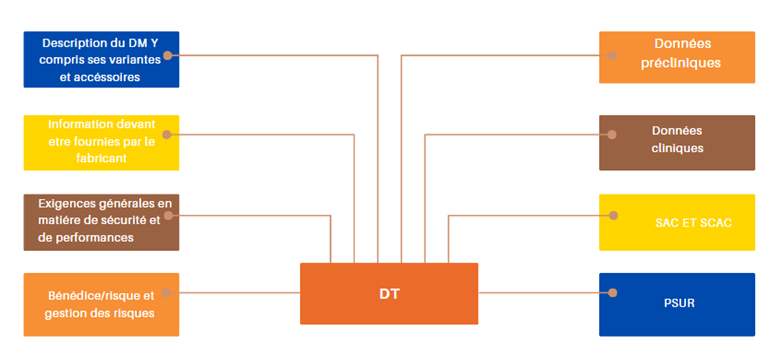
Figure 10 : Principaux éléments du dossier technique (source : auteur)
Description du dispositif, y compris les variantes et les accessoires
Les fabricants doivent fournir une description détaillée de leur produit afin que l'organisme notifié puisse l'évaluer en fonction de ses caractéristiques. A cette fin, une description complète a été réalisée, incluant le type de dispositif médical (actif ou passif), sa qualification, sa classification selon les règles de l'annexe VIII du règlement, ainsi que ses caractéristiques spécifiques et toutes les informations relatives aux accessoires contribuant à son utilisation. Cette partie contient également des indications sur les revendications, la population de patients visée, l'utilisation prévue et toutes autres informations permettant à l'organisme notifié de comprendre la fonctionnalité et les compositions techniques du dispositif médical.
Informations devant être fournies par le fabricant
Les dispositifs médicaux doivent être accompagnés des informations nécessaires pour les tracer, identifier leur fabricant et fournir toute information relative à la sécurité de l'utilisateur [1]. Deux documents ont été élaborés à cet effet :
- Des étiquettes pour le dispositif et son conditionnement, conformément à alinéas 23.2 de l'annexe I du MDR. La norme harmonisée NF EN ISO 15223 1:2021 [24] et la norme NF EN 60601-1:2007 [25] fournissent des directives sur les symboles à utiliser pour les étiquettes des dispositifs médicaux. L'application de ces normes nous a permis d'identifier les symboles applicables à notre dispositif médical.
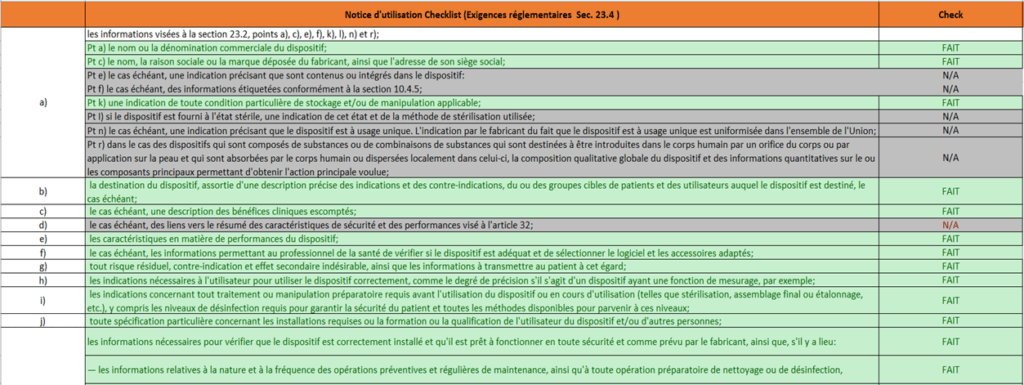
- Une notice d'utilisation conforme à la section 23.4 de l'annexe I du règlement. Des listes de vérification (checklists) ont été mises en place pour identifier toutes les exigences applicables au dispositif médical Home Habilis et structurer l'application de ces exigences (voir annexes 7 et 8).
Exigences générales en matière de sécurité et de performances
Les exigences en matière de sécurité et de performances définissent les critères essentiels que les dispositifs médicaux doivent satisfaire afin d'assurer leur sécurité et leurs performances conformément aux objectifs du règlement. Ces exigences couvrent un large éventail d'aspects tels que la conception, les matériaux, la fabrication et les informations fournies aux utilisateurs...
Dans l'annexe I du règlement, ces exigences sont réparties en trois chapitres correspondant à des catégories d'exigences [1] :
- Chapitre I : Exigences générales, qui s'appliquent à tous les dispositifs médicaux, quelle que soit leur classe.
- Chapitre II : Exigences relatives à la conception et à la fabrication, qui fournissent des orientations sur les substances, matériaux ou matières utilisés, ainsi que les critères de conception et de fabrication en fonction de la fonctionnalité du dispositif médical. L'application de ces critères dépend du type de dispositif médical et de son environnement d'utilisation.
- Chapitre III : Exigences relatives aux informations fournies avec le dispositif, qui regroupent tous les critères permettant de tracer le dispositif médical (étiquettes) et de comprendre sa fonctionnalité et son utilisation (notice d'utilisation).
J'ai été particulièrement chargée de piloter et de mettre en application cette partie du dossier technique. L'objectif était d'identifier les exigences applicables au dispositif de Home Habilis en fonction de ses caractéristiques, de déterminer les normes à appliquer pour répondre à ces critères et de définir les preuves documentaires à fournir pour justifier la conformité aux exigences.
Il s’agit de 23 exigences avec des sous sections, le schéma suivant présente un extrait des exigences répertoriées avec leurs preuves associées et les référentiels applicables :
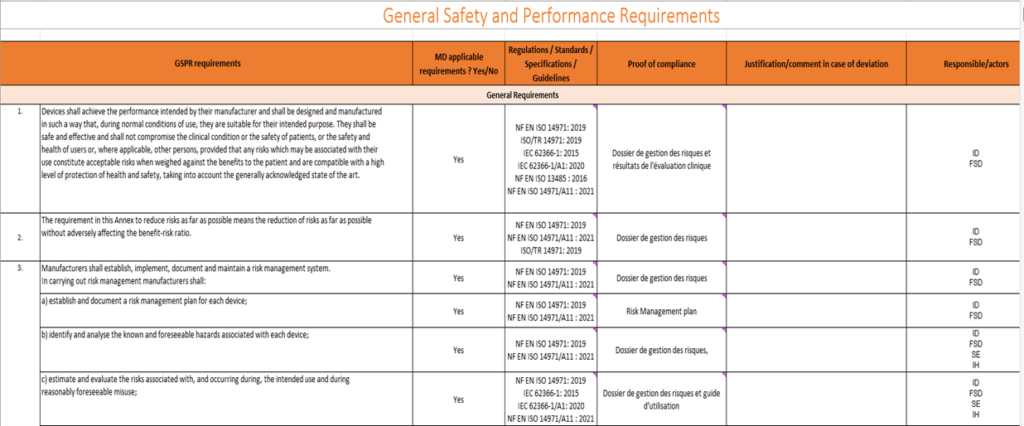
Figure 11 : Extrait des exigences répertoriées et leurs preuves associées (Source : auteur)
Les fabricants doivent prendre en compte toutes les exigences relatives à la sécurité et aux performances du dispositif médical et justifier, le cas échéant, pourquoi certaines exigences ne sont pas applicables.
Analyse bénéfice / risque et Gestion des risques
Le règlement exige que les fabricants mettent en place un système de gestion des risques selon l'alinéa 3 de l'annexe I [1].
Pour répondre à cette exigence, un dossier de gestion des risques a été élaboré conformément aux normes NF EN ISO 14971 : 2019 [26] et NF EN ISO 14971/A11 : 2021 [27], qui fournissent des lignes directrices sur la gestion des risques des dispositifs médicaux.
Le dossier de gestion des risques mis en place vise à identifier, évaluer et contrôler les risques associés à l'utilisation du dispositif médical Home Habilis afin de garantir la sécurité des patients et des utilisateurs. Il comprend :
-Un plan de gestion des risques qui permet de planifier les activités de gestion des risques du dispositif médical, d'assigner les responsabilités des employés pour ce processus, de définir les critères d'acceptabilité des risques liés au dispositif médical et les méthodes d'évaluation des risques résiduels. L’annexe 9 présente le plan de gestion des risques établi.
-Un rapport de gestion des risques qui identifie tous les risques liés à chaque fonctionnalité du dispositif médical, ainsi que les risques liés aux processus ayant un lien avec le dispositif médical (par exemple, processus de conception ou de production). Ce rapport doit analyser et évaluer ces risques afin de pouvoir mettre en place ou prévoir des actions de maîtrise permettant d'atténuer ou d'éliminer les risques identifiés. Le fabricant doit choisir les critères d’évaluation des risques liés à son dispositif en fonction de l’occurrence et de la sévérité des risques (voir annexe 10 confidentielle). L’évaluation quantitative des risques a été défini en fonction d’une estimation donné par le produit (occurrence (x) sévérité). Le niveau d’acceptabilité des risques estimés doit être fixé par le fabricant. L’annexe 11 (confidentielle) montre un extrait du rapport de gestion des risques.
La gestion des risques est un processus continu, et le dossier de gestion des risques doit être mis à jour tout au long du cycle de vie du dispositif médical, de sa conception à sa mise sur le marché et son utilisation continue.
En ce qui concerne l'analyse du rapport bénéfice/risque, l'application de la norme XP S 93 223 [28] sera intégrée au processus afin d'évaluer les avantages cliniques du dispositif médical et les risques associés à son utilisation. Ces risques seront principalement basés sur les données précliniques et cliniques du dispositif médical.
L'objectif sera d'utiliser les méthodes d'évaluation de la norme pour s'assurer que les avantages cliniques offerts par le dispositif médical l'emportent sur les risques pour les patients et les utilisateurs.
Cette étape permettra de vérifier la conformité du dispositif médical par rapport aux exigences en matière de sécurité et de performance énoncées dans l'annexe I.
Données précliniques
Les données précliniques proviennent d'essais de laboratoire ou d'essais expérimentaux appelés essais précliniques [1]. Ces essais permettent d'évaluer la sécurité, les performances et les caractéristiques du dispositif médical. Dans ce contexte, Home Habilis prévoit de réaliser des essais précliniques en collaboration avec le laboratoire LNE afin de vérifier si le dispositif médical fonctionne conformément à ses spécifications et s’il atteint les objectifs prévus.
Ces essais se concentreront sur les aspects suivants :
- Aspect technique pour vérifier la sécurité et les performances du dispositif médical par rapport aux exigences les normes NF EN 60601-1 :2007 [25], NF EN 60601-1-11 : 2015 [29] et son amendement A1 de 2021 [30], notamment en ce qui concerne les aspects électriques, mécaniques et l’utilisation à domicile.
- Aspect biologique pour évaluer les effets de biocompatibilité conformément à la norme NF EN ISO 10993-1 : 2020 [31]. Les tests à effectuer porteront surtout sur la sensibilisation, l'irritation et la cytotoxicité.
La réalisation de tests précliniques permettra d'obtenir des données précliniques justifiant la conformité du dispositif médical par rapport à ses caractéristiques spécifiques.
Données cliniques
Les données cliniques sont obtenues grâce à une évaluation clinique du dispositif médical. Selon le règlement, l'évaluation clinique est « un processus systématique et planifié visant à produire, collecter, analyser et évaluer en continu les données cliniques relatives à un dispositif afin de vérifier la sécurité et les performances, y compris les bénéfices cliniques, de celui-ci lorsqu'il est utilisé conformément à la destination prévue par le fabricant » [1].
L'article 61 du règlement stipule que les fabricants doivent justifier le niveau de preuve clinique requis afin de démontrer la conformité aux exigences générales en matière de sécurité et de performances. Dans ce contexte, Home Habilis devra réaliser une évaluation clinique conformément à l'article 61 et à l'annexe XIV, partie A du MDR.
Les fabricants peuvent obtenir des données cliniques selon trois procédures différentes :
- Une évaluation critique de la littérature scientifique : en effectuant une revue des publications scientifiques pertinentes afin de collecter des données cliniques existantes concernant la sécurité, les performances, la destination du dispositif et les caractéristiques de conception.
- Une évaluation par équivalence : en démontrant que le DM soumis à l'évaluation clinique est équivalent à un DM existant en ce qui concerne les caractéristiques techniques, biologiques et cliniques, conformément à l'annexe XIV, section 3 du règlement. Ces caractéristiques doivent être similaires de manière à ce qu'il n'y ait pas de différence en termes de performances cliniques et de sécurité du DM.
- Une évaluation des données propres au dispositif : en réalisant une investigation clinique du DM conformément à l'article 62 et à l'annexe XV du MDR afin d'obtenir des données cliniques spécifiques au DM basées sur des sujets humains.
L'entreprise a choisi de procéder à une investigation clinique pour obtenir des données cliniques propres à son DM. Cette démarche implique une demande auprès de deux comités :
- L'autorité compétente, en l'occurrence l'ANSM en France, qui évalue le dossier de demande en se basant sur la sécurité des personnes et du produit de l'étude.
- Le CPP (Comité de Protection des Personnes), qui effectue un examen d’ordre éthique.
L'investigation clinique ne peut être lancée que si le fabricant obtient l'approbation des deux parties.
SAC ET SCAC
Les fabricants doivent mettre en place un plan de surveillance après commercialisation (SAC) et un plan de suivi clinique après commercialisation (SCAC) qu'ils doivent intégrer dans le dossier technique (DT) [1].
Home Habilis devra établir un plan SAC décrivant les activités que l'entreprise doit mettre en œuvre et maintenir pour assurer une collecte systématique de données une fois que le DM est sur le marché. L'objectif de ce plan est de programmer des procédures, des méthodes ou des protocoles permettant de collecter, d'analyser et d'enregistrer les données relatives à la sécurité, à la qualité et aux performances du DM tout au long de sa durée de vie. Cela permettra de prendre des mesures préventives ou correctives appropriées et de suivre leur mise en œuvre.
Selon la section 1.1 de l'annexe III du RDM, le plan de suivi clinique après commercialisation (SCAC) fait partie intégrante du plan de surveillance après commercialisation (SAC). Le SCAC est considéré comme un processus continu d'évaluation clinique. Il décrit les méthodes et les procédures à suivre pour recueillir de nouvelles données cliniques et mettre à jour le rapport d'évaluation clinique, tout en maintenant l'objectif de sécurité et de performance du DM.
PSUR (Rapport périodique actualisé de sécurité)
En tant que fabricant d'un DM de classe IIa, Home Habilis est tenu d'intégrer un PSUR dans le DT et de le mettre à jour au moins tous les deux ans [1]. Ce rapport résume les résultats du SAC en exposant les conclusions sur les indicateurs de suivi, sur les événements importants constatés et sur la justification de toute action préventive ou corrective prise, en les décrivant. L'objectif du PSUR est de démontrer que le rapport bénéfice/risque reste favorable tout au long de la durée de vie du DM.
Le guide MDCG 2022-21 [32] constitue un référentiel approprié pour la mise en œuvre du PSUR.
Après avoir accompli toutes ces étapes, toutes les informations pertinentes ainsi que les preuves de conformité requises doivent être compilées dans le dossier technique et soumises à l'organisme notifié aux fins d'évaluation.
2.3. Résultats obtenus
-Système de management de la qualité
A mon arrivée le SMQ était non conforme avec 99 constats ont été relevés à la suite de l’audit interne.
Les schémas suivants montrent l’évolution du SMQ après 8 mois d’alternance :
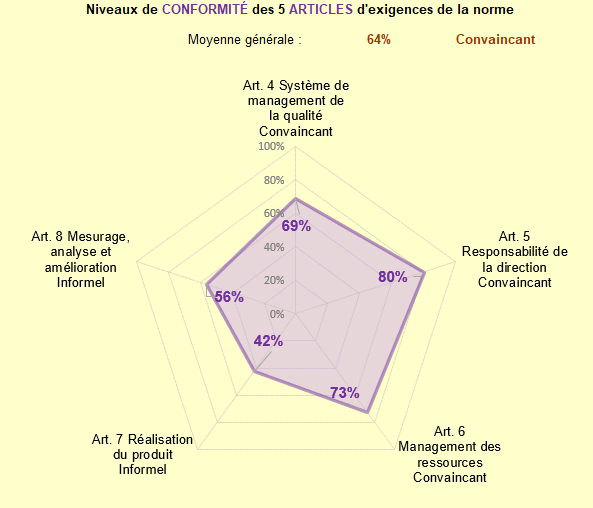
Figure 12 : Autodiagnostic du SMQ après 8 mois d’alternance (Source : auteur [21])
Les documents qualité suivants ont été élaborés conformément à la NF EN ISO 13485 : 2016 et son amendement A11 :
- Un manuel qualité
- La politique qualité de l’entreprise et l’engagement de la direction
- Une cartographie de processus décrivant les activités de l’entreprise
- Des outils de traçabilité pour le suivi de l’évolution du SMQ (voir figure 8)
- 21 procédures documentées dont 14 écrites par le service QARA
- Un registre de gestion et de suivi des CAPA (voir figure 7)
-Documentation Technique
La documentation technique de l’entreprise n’était pas encore élaborée au début de mon alternance. La réalisation de mes missions ont permis de contractualiser avec un ON et de mettre en place le dossier technique. Les documents suivants ont été élaboré dans le DT :
- Un registre de veille réglementaire
- Le registre des exigences en matière de sécurité et de performances
- Le dossier de gestion des risques du DM
- La notice d’utilisation et les étiquettes du DM
- Obtention d’un numéro d’identification unique IUD
- Collection des données précliniques et cliniques
Les schémas suivants montrent l’état d’évolution du DT avant et après 8 mois d’alternance.
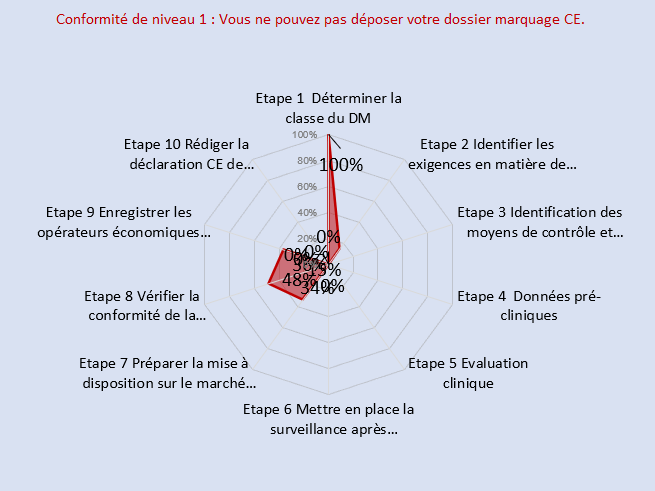
Figure 13 : Autodiagnostic initial du DT (à t = 0 ) (Source : auteur [33] )
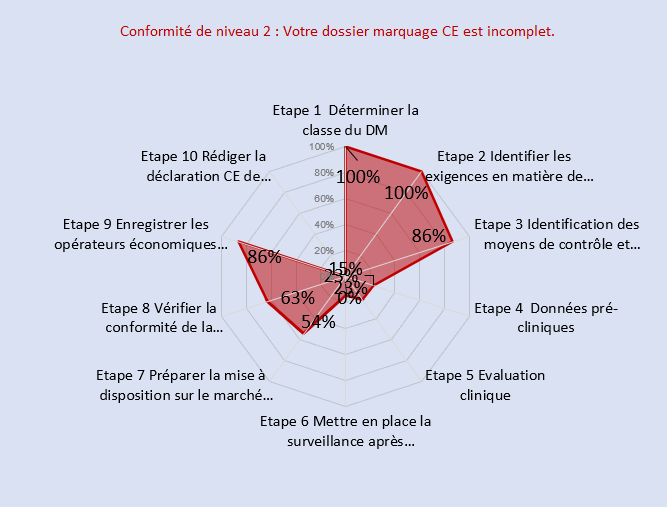
Figure 14 : Autodiagnostic du DT après 8 mois d’alternance (Source : auteur [33])
3. Apports de l'alternance
3.1. Compétences acquises
Sur le plan personnel
Mon expérience en tant qu'alternante chez Home Habilis a été enrichissante à bien des égards. Grâce à cette opportunité, j'ai pu développer plusieurs compétences professionnelles et techniques qui ont contribué à ma croissance personnelle. Cette expérience m’a permis de :
- Développer mon sens relationnel : en travaillant en équipe sur diverses missions. Dans le but de promouvoir la qualité au sein de l'entreprise et d'obtenir l'adhésion de tous les employés au système qualité, j'ai régulièrement interagi avec mes collègues. J'ai notamment organisé des sessions de formation et des réunions d'échange d'idées, encourageant ainsi la créativité et la participation de chacun dans la mise en place du SMQ. Mon approche ouverte et ma capacité d'écoute ont renforcé l'esprit d'équipe et stimulé la culture de la qualité au sein de l'entreprise. De plus, mes interactions avec les organismes notifiés et les consultants externes ont amélioré ma capacité de communication.
- Confirmer ma capacité à travailler de manière autonome : J'ai été confrontée à différentes problématiques liées à la conformité, ce qui m'a poussée à m'autoformer en suivant des webinaires et à consulter des sites réglementaires pour obtenir les informations pertinentes nécessaires à la résolution des problèmes. Cette capacité d'autonomie s'est particulièrement démontrée lors de la mise en place de la procédure de sélection des organismes notifiés, lors de l'identification des codes applicables au dispositif, lors de l'application des exigences en matière de sécurité et de performances, ainsi que lors des démarches d'investigation clinique.
Sur le plan professionnel et technique
Sur le plan professionnel et technique, mon parcours chez Home Habilis m'a permis de développer les compétences suivantes :
La réalisation de mes missions au sein de Home Habilis m’a permis de développer mes compétences professionnelles suivantes :
- Capacité d’analyse et de synthèse : mes missions étaient liées, d’une part, à l’identification et l’analyse des exigences normatives et réglementaires applicables au dispositif de l’entreprise afin de répondre aux besoins. Ces tâches m’ont permis de développer mon esprit d’analyse et de synthèse documentaire.
- Identification des besoins et résolution des problèmes : La mise en place du SMQ et l’élaboration de la documentation technique m’ont permis de confirmer ma capacité à choisir et mettre en œuvre mes connaissances en proposant des méthodes et outils de traçabilité, de gestion de projet et de maitrise documentaire.
- Développer mes capacités rédactionnelles, avec la rédaction régulière de documents qualité qui nécessite une certaine précision et rigueur.
- Identifier et exploiter les référentiels et informations pertinentes, notamment dans le cadre de la mise en place du dossier technique et la veille documentaire.
- Connaitre concrètement les démarches nécessaires pour obtenir le marquage CE.
3.2. Liens avec la formation théorique
Les connaissances théoriques acquises lors de ma formation m’ont permis d’avoir les outils et le bagage intellectuel nécessaires pour mener à bien mes missions. Les cours de l’UE IDCA (Management de la qualité et des organisations biomédicales) ont particulièrement été une source d’inspiration pendant la mise la mise en place du système qualité, notamment avec l’utilisation des outils qualité abordés dans cette UE. Les UE IDCE (Cycle de Vie d’un DM) et IDCL (Dispositifs médicaux et Affaires Réglementaires) m’ont permis de savoir les étapes du cycle de vie d’un DM ainsi que les démarches et exigences nécessaires pour mettre en place un DM sur le marché européen. Ce qui m’a permis de faire une prise en main rapide de la documentation technique de l’entreprise.
L’UE IDCK (Audit et Evaluation des organisations : normes et processus) m’a procuré les connaissances nécessaires pour mener un audit, de connaitre les normes, référentiels et les méthodes d’audit. Ceci m’a été très utile lors de la mise en place de la procédure d’audit, mais aussi à la préparation de l’audit interne de l’entreprise.
Les différents projets effectués au cours de l’année ont été d’une aide précieuse à la fois sur le plan méthodologique et théorique.
Conclusion
Au cours de notre étude, nous avons mis en évidence les différentes étapes et les exigences spécifiques associées à la certification ISO 13485 :2016, ainsi que les étapes prises par Home Habilis pour élaborer sa documentation technique et se conformer au règlement européen 2017/745.
La démarche de certification ISO 13485 :2016 et d’élaboration de la documentation technique est une étape indispensable pour le marquage CE. Il est important de souligner que ce processus est un vrai parcours du combattant et la convergence entre conformité réglementaire et celle du SMQ seront les clés pour obtenir le marquage CE.
L’entreprise devra appliquer ce processus de mise en conformité en continu, pendant toute la durée de vie de son produit pour maintenir la sécurité et la performance de son dispositif médical.
Ce travail a été une expérience enrichissante, me permettant d'approfondir mes connaissances dans le domaine de la certification ISO et de la réglementation des dispositifs médicaux. Il m'a également permis de comprendre les défis et les opportunités auxquels les startups d'innovation dans le domaine biomédical sont confrontées lorsqu'elles cherchent à se conformer aux normes et à commercialiser leurs produits sur le marché européen.
En conclusion, le parcours de Home Habilis vers la certification ISO 13485 :2016 et la mise en application du règlement européen 2017/745 démontre l'engagement de l'entreprise envers la qualité, la sécurité et l'innovation dans le domaine de la néphrologie.
Cette démarche basée sur la conformité réglementaire pourra positionner l’entreprise comme un acteur clé sur le marché européen et l’ouverture à d’autres marchés internationaux.
Références bibliographiques
[1] « RÈGLEMENT (UE) 2017/ 745 DU PARLEMENT EUROPÉEN ET DU CONSEIL - du 5 avril 2017 - relatif aux dispositifs médicaux, modifiant la directive 2001/ 83/ CE, le règlement (CE) no 178/ 2002 et le règlement (CE) no 1223/ 2009 et abrogeant les directives du Conseil 90/ 385/ CEE et 93/ 42/ CEE », Journal officiel de l’Union européenne, mai 2017, Consulté le : 27 juin 2023. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:32017R0745
[2] « Norme NF EN ISO 13485- Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires », Afnor, avril 2016. https://viewerbdc.afnor.org/pdf/viewer/diKNMBAHyBw1?proxy=true (consulté le 27 juin 2023).
[3] « DIRECTIVE 93/42/CEE DU CONSEIL du 14 juin 1993 relative aux dispositifs médicaux ». Consulté le : 27 juin 2023. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:31993L0042
[4] « DIRECTIVE 98/79/CE DU PARLEMENT EUROPÉEN ET DU CONSEIL du 27 octobre 1998 relative aux dispositifs médicaux de diagnostic in vitro ». Consulté le : 27 juin 2023. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:31998L0079
[5] HAS, « Télésurveillance médicale du patient insuffisant rénal chronique », Haute Autorité de Santé, 2022.
[6] « Maladie rénale chronique - Journée mondiale du rein », 18 septembre 2014. https://www.worldkidneyday.org/facts/chronic-kidney-disease/, https://www.worldkidneyday.org/facts/chronic-kidney-disease/ (consulté le 27 juin 2023).
[7] L. Dufour, « Qu’est-ce qu’une start up ? | Le Blog du Dirigeant », 6 juin 2023. https://www.leblogdudirigeant.com/qu-est-ce-qu-une-start-up/#la-notion-de-start-up (consulté le 27 juin 2023).
[8] « Nos Incubés », iTerra. https://iterra.fr/nos-incubes/ (consulté le 27 juin 2023).
[9] « L’insuffisance rénale », France Rein, décembre 2022. https://www.francerein.org/vivre-avec-la-maladie/maladies-et-traitements/linsuffisance-renale/ (consulté le 27 juin 2023).
[10] P. Machek, T. Jirka, U. Moissl, P. Chamney, et P. Wabel, « Guided optimization of fluid status in haemodialysis patients », Nephrol Dial Transplant, vol. 25, no 2, p. 538‑544, févr. 2010, doi : 10.1093/ndt/gfp487.
[11] C. Sommerer, P. Felten, J. Toernig, M. Zeier, et R. Dikow, « Bioimpedance analysis is not superior to clinical assessment in determining hydration status : A prospective randomized-controlled trial in a Western dialysis population », Hemodial Int, mars 2021, doi : 10.1111/hdi.12919.
[12] J. Frija-Masson, J. Mullaert, E. Vidal-Petiot, N. Pons-Kerjean, M. Flamant, et M.-P. d’Ortho, « Accuracy of Smart Scales on Weight and Body Composition : Observational Study », JMIR mHealth and uHealth, vol. 9, p. e22487, avr. 2021, doi : 10.2196/22487.
[13] louisakewell, « Prescribing Frequent Haemodialysis in Complex Patients : Highlights from the 55th ERA–EDTA Congress », EMJ, vol. 6, no 1, p. 34‑41, juill. 2018, doi : 10.33590/emjnephrol/10314381.
[14] Y.-L. Kim et W. V. Biesen, « Fluid Overload in Peritoneal Dialysis Patients », Seminars in Nephrology, vol. 37, no 1, p. 43‑53, janv. 2017, doi : 10.1016/j.semnephrol.2016.10.006.
[15] « Balance Wi-Fi avec composition corporelle et santé cardiovasculaire - Body Cardio | Withings ». https://www.withings.com/fr/fr/body-cardio (consulté le 27 juin 2023).
[16] « Amendement A11 de la norme NF EN ISO 13485 - Dispositifs médicaux — Systèmes de management de la qualité — Exigences à des fins réglementaires », Afnor, septembre 2021. https://viewerbdc.afnor.org/pdf/viewer/pDe24usNLvc1?proxy=true (consulté le 27 juin 2023).
[17] « IDS117 - Impacts des nouveaux règlements européens (2017/745 et 2017/746) sur l’ingénierie biomédicale hospitalière », Bibliothèque des travaux Master. https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids117/ (consulté le 27 juin 2023).
[18] « Communiqué : un risque réel de pénurie de dispositifs médicaux », Bulletin de l’Académie Nationale de Médecine, vol. 206, no 8, p. 921‑922, oct. 2022, doi : 10.1016/j.banm.2022.07.001.
[19] « EUROPA - Commission européenne - Croissance - Politique réglementaire - NANDO », Commission européenne. https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34 (consulté le 27 juin 2023).
[20] « Norme NF EN ISO 9000 - Systèmes de management de la qualité — Principes essentiels et vocabulaire », octobre 2015. https://viewerbdc.afnor.org/pdf/viewer/XNB_jo10kNo1?proxy=true (consulté le 27 juin 2023).
[21] L. Beuzelin, A. Desgranges, et Q. Emile, « Accompagnement à la certification ISO 13485 v2016 », Bibliothèque des travaux Master, avril 2018. https://travaux.master.utc.fr/articles-publies/2018_20_ttsap/ (consulté le 27 juin 2023).
[22] « RÈGLEMENT (UE) 2023/607 DU PARLEMENT EUROPÉEN ET DU CONSEIL du 15 mars 2023 modifiant les règlements (UE) 2017/745 et (UE) 2017/746 en ce qui concerne les dispositions transitoires relatives à certains dispositifs médicaux et à certains dispositifs médicaux de diagnostic in vitro ». Consulté le : 27 juin 2023. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:32023R0607&from=EN
[23] S. Oudjaneplan, « La prolongation de la période de transition pour les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro est adoptée », GMED Medical Device Certification, 17 février 2023. https://lne-gmed.com/fr/news/la-prolongation (consulté le 27 juin 2023).
[24] « Norme NF EN ISO 15223-1 Dispositifs médicaux — Symboles à utiliser avec les informations à fournir par le fabricant — Partie 1 : Exigences générales », septembre 2021. https://viewerbdc.afnor.org/pdf/viewer/CtJC2Qf--701?proxy=true (consulté le 27 juin 2023).
[25] « Norme NF EN 60601-1 Appareils électromédicauxPartie 1 : Exigences générales pour la sécurité de base et les performances essentielle », Afnor, janvier 2007. https://viewerbdc.afnor.org/pdf/viewer/bFIvwHSbJhE1?proxy=true (consulté le 27 juin 2023).
[26] « Norme NF EN ISO 14971 Dispositifs médicaux — Application de la gestion des risques aux dispositifs médicaux », Afnor, décembre 2019. https://viewerbdc.afnor.org/pdf/viewer/MIGJErBPUx41?proxy=true (consulté le 27 juin 2023).
[27] « Amendement A11 de la NF EN ISO 14971 Dispositifs médicaux — Application de la gestion des risques aux dispositifs médicauX », Afnor, décembre 2021. https://viewerbdc.afnor.org/pdf/viewer/_kKl2RiG2Qo1?proxy=true (consulté le 27 juin 2023).
[28] « Norme expérimentale Dispositifs médicaux — Gestion du rapport bénéfice/risque », Afnor, février 2020. https://viewerbdc.afnor.org/pdf/viewer/2kDCudDMCaM1?proxy=true (consulté le 27 juin 2023).
[29] « Norme NF EN 60601-1-11 Appareils électromédicauxPartie 1-11 : Exigences générales pour la sécurité de base et les performances essentielles –Norme Collatérale : Exigences pour les appareils électromédicaux et les systèmes électromédicaux utilisés dans l’environnement des soins à domicile », Afnor, août 2015. https://viewerbdc.afnor.org/pdf/viewer/xjFftFTmcuY1?proxy=true (consulté le 27 juin 2023).
[30] « Amendement A1 de la norme NF EN 60601-1-11 Appareils électromédicauxPartie 1-11 : exigences générales pour la sécurité de base et les performances essentielles –Norme Collatérale : exigences pour les appareils électromédicaux et les systèmes électromédicaux utilisés dans l’environnement des soins à domicile », juillet 2021. https://viewerbdc.afnor.org/pdf/viewer/a9fjxALp8ZQ1?proxy=true (consulté le 27 juin 2023).
[31] « Norme NF EN ISO 10993-1 Évaluation biologique des dispositifs médicaux — Partie 1 : Évaluation et essais au sein d’un processus de gestion du risque », Afnor, décembre 2020. https://viewerbdc.afnor.org/pdf/viewer/LXUjVh1kVKg1?proxy=true (consulté le 27 juin 2023).
[32] « mdcg_2022-21_en.pdf ». Consulté le : 27 juin 2023. [En ligne]. Disponible sur : https://health.ec.europa.eu/system/files/2023-01/mdcg_2022-21_en.pdf
[33] A. M. Aziz, P. Brochet, F. Shandy, R. Valériane, et O. Sadiqui, « IDS084 - Roadmap réglementaire pour une innovation technologique d’imagerie de haute résolution », Bibliothèque des travaux Master. https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids084/ (consulté le 27 juin 2023).
Annexes
Annexe 7 : Extrait Checklist notice d’utilisation
Annexe 8 : Extrait Checklist étiquettes

Annexe 9 : Sommaire du plan de gestion des risques
