IDS199 - La place centrale du dossier de gestion des risques dans le dossier technique de marquage CE
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

Contacts
- SOH KOUDJOU Marie Josée : mariejoks@gmail.com
Citation
A rappeler pour tout usage : SOH KOUDJOU Marie Josée, « Place centrale du dossier de gestion des risques dans le dossier technique de marquage CE », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs médicaux et affaires réglementaires (DMAR), Mémoire d'apprentissage, https://travaux.master.utc.fr/, réf n° IDS199, Juillet 2023, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids199
Résumé
Le règlement (UE) 2017/745 impose aux fabricants de dispositifs médicaux des exigences relatives à la sécurité des patients. Le fabricant doit de ce fait s’assurer que les risques associés aux dispositifs qu’il fabrique est toujours réduit au plus bas niveau que possible. Lors la rédaction du dossier technique de marquage CE, les données issues de la gestion des risques sont utilisées et elles permettent de justifier que le dispositif médical respecte les exigences de sécurité.
L’importance du dossier de gestion des risques dans le dossier technique entraine un échange permanent de données entre les deux dossiers. Afin comprendre les interactions entre les éléments du dossier de gestion des risques et les documents du dossier technique de marquage CE, un schéma interactif a été élaboré.
Pour les fabricants de dispositifs médicaux, ce schéma permet de visualiser les liens entre les différents documents de chaque dossier. Il répond au besoin d’identification des preuves pour justifier la sécurité du dispositif médical. Le schéma élaboré ainsi que ses fonctionnalités d’utilisation sont revus par la société de conseil KAPSIKUM pour permettre des améliorations et des mises à jour continues.
Mots clés : dossier technique, gestion des risques, marquage CE.
Abstract
Regulation (EU) 2017/745 requires manufacturers of medical devices to comply with patient safety requirements. Manufacturer must therefore ensure that the risks associated with the devices it manufactures are always reduced to the lowest possible level. Risk management data is used in drawing up technical file for CE marking, and to justify that medical device complies with safety requirements.
Importance of the risk management file within technical file leads to a turnover of data between the files. To understand interactions between documents in the risk management file and those in CE marking technical file, an interactive diagram has been drawn up.
For medical device manufacturers, this diagram shows how between the different documents in each file are interrelated. It answers manufacturers’ need to identify evidence to justify the safety of the medical device. The diagram design and functionality are reviewed by KAPSIKUM, a medical device consulting firm, to enable continuous improvements and updates.
Keys word : CE marking, risk management, technical file.
Remerciements
Mes remerciements s’adressent :
A l’équipe pédagogique du Master Ingénierie de la Santé l’Université de Technologie de Compiègne.
En particulier aux responsables de formation du master, Isabelle Claude et Jean-Matthieu Prot pour leur soutien durant mon parcours, à Gilbert Farges pour ses enseignements, et à ma tutrice d’apprentissage Julie Follet pour toutes les connaissances et compétences professionnelles acquises.
A l’équipe de la société KAPSIKUM. En particulier à Cynthia Cottereau et Jean-Philippe Christoph pour l’attention particulière apportée à mon égard afin d’assurer la réussite de mon apprentissage. A Ilias Kraimi pour sa disponibilité et à tous les consultants qui ont contribué à ma formation. Merci d’avoir répondu à mes préoccupations et de m’avoir aidée à clarifier mon projet professionnel.
A Jean-Marc Picard, enseignant du management de la qualité à l’UTC pour ses encouragements.
A mes camarades de promotion pour les deux années de master passées ensemble.
A ma mère Suzanne Soh, source intarissable d’espoir.
Téléchargements

Liste des abréviations
| BPI | Banque Publique d’Investissement |
| BSER | Biological Safety Evaluation Report (rapport d’évaluation de la sécurité biologique) |
| CE | Conformité Européenne |
| CEE | Communauté Economique Européenne |
| CER | Clinical Evaluation Report (rapport d’évaluation clinique) |
| DCAP | Do Check Act Plan |
| DM | Dispositif Médical |
| DMAR | Dispositif Médicaux et Affaires Réglementaires |
| DT | Dossier Technique |
| EGSP | Exigences Générales de Sécurité et de Performance |
| EN | European Norm (Norme Européenne) |
| EUDAMED | European Database on Medical Devices (Base de données européenne des dispositifs médicaux) |
| GDR | Gestion des Risques |
| IAU | Ingénierie de l’Aptitude à l’Utilisation |
| IEC | International Electrotechnical Commission |
| ISO | International Organization for Standardization |
| IUD | Identifiant Unique des Dispositifs |
| MDCG | Medical Device Coordination Group |
| MEDDEV | MEDical DEVices documents |
| MDSAP | Medical Device Single Audit Program |
| NF | Norme Française |
| SAC | Suivi Après Commercialisation |
| SCAC | Suivi Clinique Après Commercialisation |
| SMQ | Système de Management de la Qualité |
| USA | United States of America |
| UTC | Université de Technologie de Compiègne |
Glossaire
Analyse des risques : utilisation des informations disponibles sur un dispositif médical pour déterminer les dangers afin d’estimer le risque [NF EN ISO 14971:2019].
Appréciation du risque : processus qui prend en compte l’analyse des risques et l’évaluation du risques [NF EN ISO 14971:2019].
Cycle de vie du dispositif médical : ensemble des phases de la vie d’un dispositif médical de la conception initiale à la réforme finale.
Dispositif médical : Selon le règlement (UE) 2017/745 [1], un dispositif médical est tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
- diagnostic, prévention, surveillance, prédiction, pronostic, traitement ou atténuation d’une maladie,
- diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
- investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
- communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus,
et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens
Dossier technique de marquage CE : document qui permet à un fabriquant d’attester la conformité de son produit aux exigences applicables du règlement (UE) 2017/745.
Dossier de gestion des risques : dossier constitué des enregistrements et des documents issues de la gestion des risques [NF EN ISO 14971:2019].
Estimation du risque : processus permettant d’attribuer des valeurs à la probabilité d’apparition d’un dommage et la gravité du dommage.
Evaluation du risque : processus permettant de déterminer l’acceptabilité du risque par comparaison des risques estimés à des critères de risques définis [NF EN ISO 14971:2019].
Gestion des risques : processus permettant l’application des activités d’analyse, d’évaluation, et de maîtrise des risques [NF EN ISO 14971:2019].
Maîtrise des risques : processus durant lequel les décisions concernant le risque sont prises et les mesures raisonnables de réduction des risques sont mises en place [NF EN ISO 14971:2019].
Marquage CE : un marquage au travers duquel un fabricant indique que dispositif médical qu’il fabrique est conforme aux exigences essentielles du règlement européen et aux autres actes législatifs harmonisés de l'Union Européenne qui en prévoient l'apposition [2].
Postproduction : étape du cycle de vie du dispositif médical après les phases de conception et de fabrication : stockage, installation, utilisation, maintenance, réforme [NF EN ISO 14971:2019].
Risque : association de la probabilité de d’apparition d’un dommage et de sa gravité.
Risque résiduel : risque restant à la suite de l’implémentation des mesures de maîtrises de risques [NF EN ISO 14971:2019].
Sécurité : absence de risques inadmissible [NF EN ISO 14971:2019].
Mémoire - Place centrale du dossier de gestion des risques dans le dossier technique de marquage CE
Introduction
Les dispositifs médicaux sont des équipements à visée médicale dont la fabrication et la commercialisation sont soumises au règlement (UE) 2017/745. En effet, à la suite des différents scandales sur les dispositifs médicaux tels que le scandale sur les prothèses mammaires Poly Implant Prothèses[3], il était important de renforcer le niveau de sécurité des dispositifs mis à disposition des patients.
Afin de répondre aux exigences du règlement (UE) 2017/745, les fabricants doivent constituer le dossier technique des dispositifs médicaux qu’ils fabriquent. Les exigences du règlement (UE) 2017/745 font appellent à des normes harmonisées dont la prise en compte vaut présomption de conformité.
La norme NF EN ISO 14971:2019 est harmonisée au règlement (UE) 2017/745 et permet d’assurer la gestion des risques relative aux dispositifs médicaux. La gestion des risques a pour but de s’assurer que le dispositif médical fabriqué présente plus de bénéfices pour l’utilisateur que de risques. En d’autres termes, la gestion des risques contribue à assurer la sécurité du patient et de l’utilisateur tout au long du cycle de vie du dispositif médical.
La gestion des risques fait l’objet d’un processus continu c’est-à-dire qu’elle oblige à constituer et mettre à jour un dossier de gestion des risques. Le dossier technique regroupe de ce fait, l’ensemble des preuves concernant la sécurité et les performances du dispositif médical et, interagit avec le dossier de gestion des risques.
Ce mémoire présentera la place du dossier de gestion des risques dans le dossier technique de marquage CE.
Dans la première partie, il sera présenté comment la gestion des risques s’applique aux dispositifs médicaux. La deuxième partie concernera la mise à jour d’un dossier technique de marquage CE sous le règlement (UE) 2017/745 relatif aux dispositifs médicaux. La troisième partie permettra de présenter la relation entre le dossier technique de marquage CE et le dossier de gestion des risques. Dans la dernière partie, il s’agira du bilan effectué à la suite de la formation en apprentissage.
1 L'application de la gestion des risques inhérent aux dispositifs médicaux
1.1 Définition
La gestion des risques est un processus regroupant plusieurs activités dont l’exécution nécessite une planification et un suivi régulier.
Selon la norme ISO 14971 relative à l’application de la gestion des risques aux dispositifs médicaux, gérer un risque c’est appliquer une politique de gestion à ce risque, c’est-à-dire, l’analyser, l’évaluer, le contrôler, et le maîtriser.
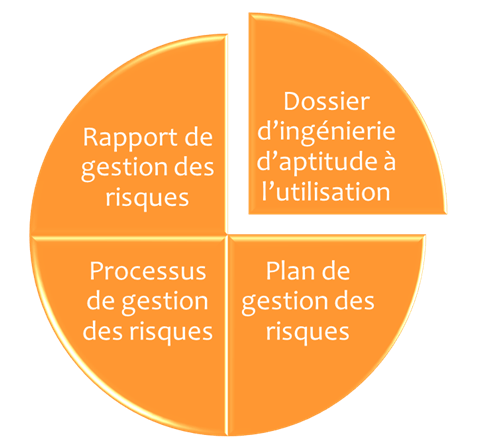
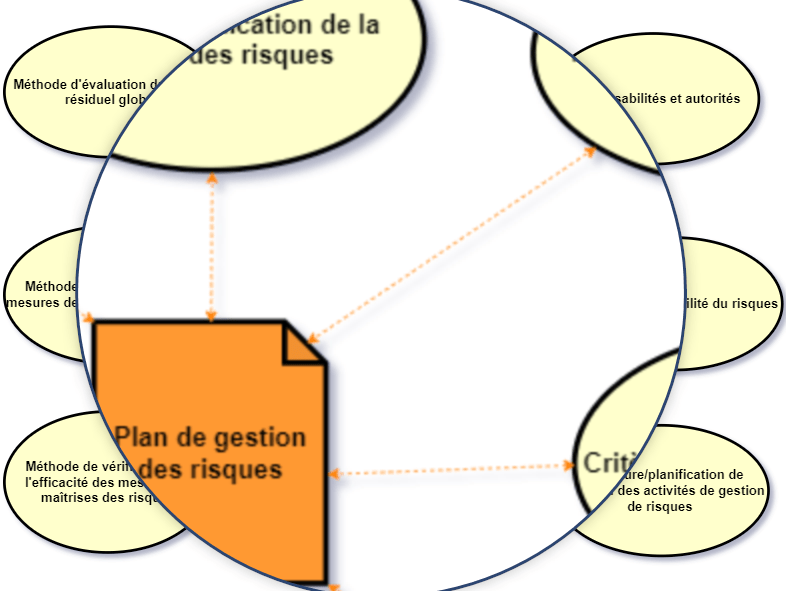
Le schéma ci-dessous (Figure 1) présente les parties principales du dossier de gestion des risques :
1.2 Mise en œuvre opérationnelle de la gestion des risques
Avant tout il est important de comprendre quel est le processus de gestion des risques.
Tous les fabricants sont tenus de mettre en place et de documenter le processus de gestion de risques de leur dispositif dans leur système de management de la qualité. Ils s’assurent que ce processus est continuellement à jour, de la phase de conception à la phase de mise au rebut.
Dans ce contexte, il est nécessaire d’établir un plan de gestion des risques.
1.2.1 Plan de gestion des risques
Le plan de gestion des risques définit le champ d’application des actions de la gestion des risques. Il permet d’identifier « qui fait quoi » dans la mise en œuvre de la gestion des risques. C’est-à-dire le niveau d'implication des personnes et les autorités responsables. Il permet également de définir le produit et le(s) étape(s) du cycle de vie concernés.
Le plan de gestion des risques énonce aussi les critères d’appréciation du risque, définit les données à réunir en production et en postproduction et planifie les actions à réaliser pour gérer les risques tout au long du cycle de vie du dispositif médical (voir annexe).
Au cours de la vie du dispositif, le plan de la gestion des risques est amené à évoluer pour tenir compte des risques identifiés. En effet, en fonction de la disponibilité des informations et de l’analyse de celles-ci, le plan de gestion des risques est appelé à évoluer et à être mis à jour durant tout le cycle de vie du dispositif médical.
Enfin, le plan de gestion des risques encadre le processus de gestion des risques.
1.2.2 Mécanisme de fonctionnement de la gestion des risques
Le processus de gestion des risques décrit comment le risque sera géré sur la base du plan préalablement défini : tout commence donc par l’établissement d’un plan de gestion des risques (§1.2.1).
Le schéma ci-dessous (Figure 2) présente les activités du processus de gestion des risques :
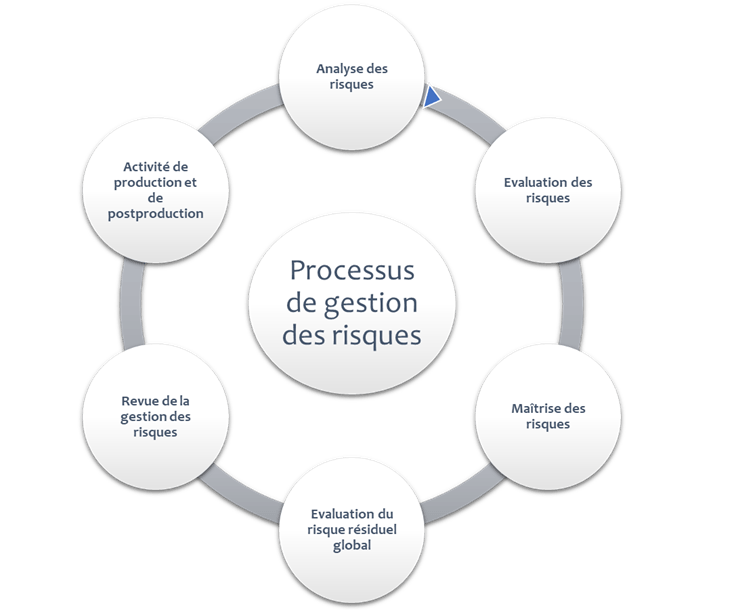
a. L'analyse du risque
- La détermination et la définition de toutes les utilisations prévues et les mauvaises utilisations raisonnablement prévisibles du dispositif médical.
Il s’agit :
- De décrire l’usage médical prévu (s’il s’agit d’un diagnostic, d’un traitement, d’une surveillance, etc.) en précisant l’environnement d’utilisation ;
- De préciser la population de patients à qui le produit est destiné ;
- D’indiquer les parties du corps cibles qui seront en interaction avec le dispositif ;
- D’expliquer le principe de fonctionnement du dispositif.
Les mauvaises utilisations du dispositif médical peuvent être identifiées au travers d’une erreur d’utilisation, de l’usage volontaire du dispositif pour une application différente de celle prévue par le fabricant ou encore une mauvaise utilisation volontaire.
L’identification des mauvaises utilisations se fait continuellement durant le cycle de vie du dispositif (par exemple par des simulations en phase de conception ou de développement, en postproduction par observation d’une réelle utilisation, etc.)
Les mauvaises utilisations peuvent provenir d’un principe de fonctionnement du dispositif non intuitif, d’une idée inadéquate ou erronée des risques ou encore d’une perception limitée des conséquences. Quoi qu’il en soit, l’identification de l’utilisation prévue et de la mauvaise utilisation raisonnablement prévisible du dispositif médical sont issues du dossier d’ingénierie de l’aptitude à l’utilisation (IAU). Les exigences relatives à l’IAU sont détaillées dans la norme IEC 62366-1 :2015.
- La détermination des caractéristiques relatives à la sécurité
Il s’agit de tous les paramètres du dispositif médical qui peuvent compromettre la sécurité du patient ou des utilisateurs. Le but est de pouvoir identifier l’ensemble des dangers et situations dangereuses relatifs au dispositif médical. De même, l’identification des performances du dispositif peut être utile pour identifier les situations dangereuses pouvant survenir à cause d’un dysfonctionnement.
Par ailleurs, les informations issues de l’état de l’art et du rapport des évènements indésirables peuvent être utiles à l’identification des caractéristiques relatives à la sécurité. En outre, les caractéristiques de sécurité identifiées serviront à enrichir les recherches de la littérature, y compris le rapport d’incidents.
- La détermination des dangers et des situations dangereuses
La gestion des risques repose sur le principe qu’un danger est une source potentielle de dommage [NF EN ISO 14971:2019]. Le danger est caractérisé par une ou plusieurs séquences d’évènements conduisant à une situation dangereuse dans laquelle le dommage est susceptible de se produire.
Commencer l’identification des dangers et situations dangereuses par un inventaire des retours d’expérience sur les dispositifs déjà existants, les incidents déclarés, ou par les données de la littérature scientifique, peut être un point de départ.
Ensuite, il est nécessaire de tenir compte des séquences d’évènements qui pourraient, en association au danger, conduire à une situation dangereuse et entraîner un dommage. L’identification des séquences d’évènements combinées au danger, se fait tout au long du cycle de vie du dispositif et dès que possible en phase de conception et de développement.
En pratique, une situation dangereuse peut être :
- la conséquence d'une ou plusieurs séquences d'évènements combinés ;
- due à une défaillance associé ou non à une séquence d'évènements.
Il est important de souligner qu’une situation dangereuse peut se produire dans le cadre d’une utilisation normale, en absence de défaillance.
- Estimation du risque
Estimer le risque revient à s’intéresser d’abord aux évènements autour d’un danger présent et à la séquence d’évènements pouvant entraîner une situation dangereuse. Il est nécessaire d’établir la probabilité que la séquence d’évènements se produisent et entraine la situation dangereuse. Ensuite, il faut estimer la probabilité que le dommage se produise en présence de la situation dangereuse. Enfin, pour que l’estimation du risque soit complète, il faut estimer la gravité du dommage résultant.
Le risque est alors estimé par cotation des deux probabilités et de la gravité à l’aide d’échelles de cotation. Ces échelles doivent être adaptées au dispositif médical et sont définies dans le plan de gestion des risques.
Le choix du niveau de cotation d’un risque doit toujours être justifié et il doit être cohérent dans l’ensemble de l’analyse des risques.
b. Evaluation des risques
L’évaluation des risques se fait par rapport à la politique du fabricant sur la détermination des risques acceptables qui est inclue dans le plan de gestion des risques. Cette politique permet d’établir les critères d’acceptabilité du risque. Lors de l’évaluation des risques, ces critères sont comparés aux risques estimés afin de savoir s’ils sont remplis.
- La politique définissant les critères d’acceptabilité du risque.
La politique est documentée et traite des parties telles que :
- La finalité : pour décrire le but de la politique (pourquoi la politique de mise en place des critères d’appréciation est établie) ;
- L’étendue d’application qui précise les personnes concernées par la politique, où elles interviennent et à quel moment ;
- Les éléments à considérer pour identifier les risques acceptables. Par exemple, au travers des mesures techniquement possibles, la réduction du risque sans nuire sur l’utilisation prévue ou sur le rapport bénéfice/risques ;
- La démarche de maîtrise des risques en se basant soit sur la faisabilité, soit sur l’ampleur ou l’étendue du risque ;
- Les spécifications de validation en identifiant la personne responsable en charge de l’approbation documentaire, les révisions apportées, etc. ;
Ce document fait partie du dossier de gestion des risques.
- Les critères d’appréciations (définis dans le plan de gestion de risques)
Ces critères sont définis à partir de la politique du fabricant préalablement établie. Il s’agit de l’appréciation de tous les risques, même dans le cas où il n’est pas possible d’estimer la probabilité d’apparition d’un dommage. Cela concerne aussi l’appréciation du risque résiduel global.
A noter que les critères établis sont fonction des caractéristiques du dispositif, de son utilisation prévue et des performances attendues.
c. Maîtrise des risques
La maîtrise des risques se réalise en respect avec la politique de gestion des risques, en se basant, soit sur la faisabilité c’est-à-dire, tout ce qu’il est possible de mettre en œuvre techniquement ou économiquement, soit sur l’ampleur du risque.
- Analyse de maîtrise des risques
Afin de maîtriser les risques, il est nécessaire de mettre en place des actions dans un ordre défini par la norme :
- La sécurité inhérente à la conception et à la fabrication ;
- Les mesures de protection dans le dispositif médical lui-même ou au sein du processus de fabrication ;
- L’information pour la sécurité et, le cas échéant, la formation des utilisateurs.
La maîtrise du risque doit être assurée dans toutes les étapes du cycle de vie du dispositif y compris sur les phases de postproduction comme le stockage, la mise au rebut, etc.
- Mesures de maîtrises des risques
Une fois chaque mesure de maîtrise des risques définie, elle doit être documentée, mise en œuvre et vérifiée. Le plan de gestion de risques doit inclure les modalités de vérification de la mise en œuvre des activités de maîtrise de risques et la vérification de l’efficacité de ces mesures.
- Evaluation du risque résiduel
Le risque résiduel est le risque persistant après la mise en œuvre des mesures de maîtrise du risque. L’évaluation du risque résiduel est faite avec les critères d’acceptabilité définis dans la politique de gestion des risques. Chaque risque résiduel est acceptable ou inacceptable. Dans le cas où le risque résiduel n’est pas acceptable, il convient d’analyser les mesures de maîtrise du risque ou de faire une analyse du rapport bénéfice/risque spécifique si aucune autre maîtrise n’est possible.
L’évaluation du risque résiduel est évolutive durant le cycle de vie du dispositif. Spécifiquement, elle est revue en cas de changement en production et en postproduction et à la lumière des données recueillies durant le suivi après la mise sur le marché.
- Analyse bénéfice/risque
L’analyse du bénéfice/risque est faite pour les risques jugés inacceptables selon les critères d’acceptabilité définis dans le plan de gestion des risques et dont une maîtrise approfondie du risque ne peut se faire. Le but de l’analyse est de voir si le bénéfice lié à l’utilisation prévue du dispositif est supérieur au risque résiduel.
L’estimation du bénéfice est relative à l’amélioration attendue de la santé du patient à la suite de l’utilisation du dispositif médical. L’estimation du bénéfice se réalise à partir de la performance clinique et des résultats de l’évaluation clinique. La nature du bénéfice attendu pour la population visée, le degré et la durée du bénéfice attendu et l’efficacité du dispositif pour répondre aux attentes, sont des aspects à prendre en compte pour estimer le bénéfice/risque.
- Risques issus des mesures de maîtrise des risques
Les dispositions prises pour maîtriser le risque peut entraîner de nouveaux risques ou augmenter les risques qui étaient déjà identifiés comme acceptables. Ces risques doivent être ajoutés s’ils n’étaient pas présents et suivre tout le processus de gestion des risques. Les risques modifiés doivent être réévalués pour s’assurer qu’ils restent acceptables.
- Maîtrise complète des risques
Maîtriser complètement les risques revient à s’assurer que tous les risques et les situations dangereuses identifiés sont bien pris en compte sans omettre un cas. Pour cela il est possible d’inscrire dans une liste de vérification toutes les situations dangereuses associées aux risques et tous les dangers. Cette liste doit bien évidemment être tenue à jour.
d. Evaluation du risque résiduel global
A ce stade, le risque résiduel est étudié de façon générale. Le risque résiduel global doit être évalué en rapport avec l’utilisation prévue du dispositif. Pour ce faire, le plan de gestion des risques doit également inclure la méthode d’évaluation et les critères d’acceptabilité du risque résiduel global.
Le risque résiduel global doit être acceptable par rapport au bénéfice global du dispositif pour que celui-ci puisse être mis sur le marché. L’acceptation du risque résiduel global doit être justifiée et documentée, et les résultats issus de l’évaluation des risques résiduels globaux sont intégrés dans le dossier de gestion des risques.
e. Revue de la gestion des risques
Il s’agit ici d’analyser les résultats finaux du processus de gestion des risques dans le but est de s’assurer :
- De la bonne exécution du plan de gestion des risques ;
- Que le risque résiduel global est acceptable ;
- De la mise en place des actions pour obtenir et analyser les données en production et en postproduction.
La revue de la gestion des risques est faite avant la mise sur le marché du dispositif médical. Elle est décrite dans le rapport de gestion des risques.
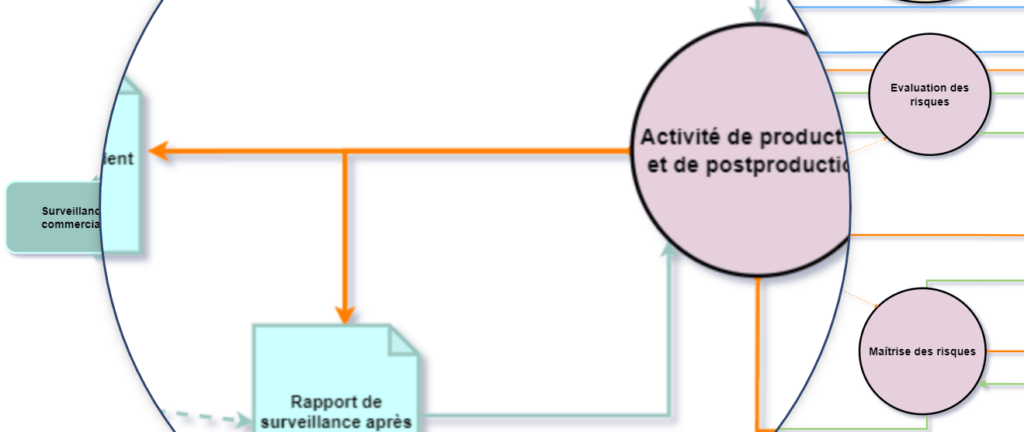
f. Activité de production et de postproduction
La vérification des activités de production et de postproduction fait de la gestion des risques un processus continu durant le cycle de vie du dispositif médical.
Les données collectées en production et postproduction sont examinées afin d'évaluer leur pertinence en terme de sécurité. Dans le cas où ces données ne sont pas pertinentes en matière de sécurité, elles sont renvoyées aux premières étapes du processus de gestion des risques.
A noter que les activités de production et de postproduction peuvent être incluses dans la surveillance après commercialisation (SAC) ainsi que dans le suivi clinique après commercialisation (SCAC).
Les informations collectées en production et postproduction peuvent provenir :
- Des retours utilisateurs, des enquêtes clients, des distributeurs, des personnes chargées de la maintenance et de l’installation, des formateurs ;
- Des données de la littérature scientifique, des rapports d’incidents ;
- Et plus généralement de tout enregistrement dans le système de management de la qualité.
2 La mise à jour d'un dossier technique de marquage CE sous le règlement 2017/745 relatif aux dispositifs médicaux
2.1 Cas concret : Accompagnement d'un fabricant de dispositif médicaux par une société de conseil
2.1.1 KAPSIKUM société de conseil des dispositifs médicaux
KAPSIKUM est cocréée en 2022 par Cynthia Cottereau et Jean-Philippe Christoph.
La société KAPSIKUM est une société de conseil dans le secteur des dispositifs médicaux de tous types et de toutes classes de risques. Elle travaille pour le compte d’entreprises du secteur de la santé de toutes tailles qui souhaitent bénéficier de son expertise.
KAPSIKUM est une entreprise récente qui regroupe une équipe jeune. Voir Figure 3.
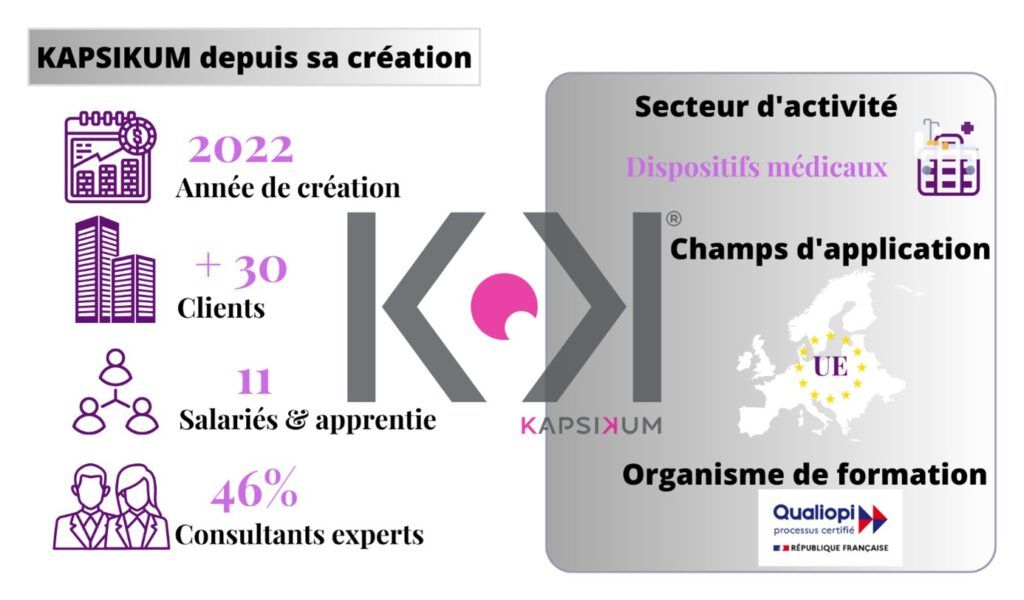
La taille de l’entreprise offre une agilité certaine pour la gestion de ses activités, et ce, tant à l’égard des clients qu’en interne, par exemple la planification des réunions, l’organisation des suivis réguliers pour la gestion des projets, etc.
Les salariés sont localisés dans différentes régions en France.
Les déplacements chez les clients sont établis et planifiés à l’avance en fonction des offres, en prenant également en compte les facteurs de distance, de temps de travail, etc.
Cependant, l’entreprise dispose d’un siège légal basé dans la ville de Saclay (91400) au 4 Rue René Razel. Ce siège :
- A un nom qui lui a été donné par l’équipe : « Kapsule » ;
- Renferme des locaux aménagées, des postes de travails, une salle de réunion, une cuisine ;
- Accueille l’équipe pour les réunions périodique organisées par la direction ;
- Est le site principal des jours de travail pour les salariés non consultants localisés dans les villes environnantes. Soit 3 jours sur site et 2 jours en télétravail ;
KAPSIKUM est aussi organisme de formation certifiée Qualiopi. Elle se prépare également à un audit de certification ISO 9001 prévu en fin d’année 2023.
Toutefois, l’activité principale revendiquée par KAPSIKUM est le conseil, c’est-à-dire inspirer, suggérer, et guider les entreprises qui n’ont pas de ressources suffisantes en internes pour atteindre leurs objectifs ou alors, des entreprises qui n’ont tout simplement pas de connaissances dans le secteur des dispositifs médicaux et qui souhaitent se développer dans ce sens.
Les clients de KAPSIKUM sont principalement des entreprises basées en France :
- Qui souhaitent mettre ou qui ont déjà mis à disposition leur produit sur le marché français et/ou européen (conformément au règlement (EU) 2017/745 relatif aux dispositifs médicaux).
- Qui souhaitent mettre ou qui ont déjà mis à disposition leur produit Australie, au Canada, au Japon et aux USA conformément au programme MDSAP (Medical Device Single Audit Program).
Les clients ont la possibilité de bénéficier de l’offre de la Banque Publique d’Investissement (BPI) pour leurs prestations car la société dispose des consultants reconnus par la BPI.
Les clients sont accompagnés par :
- La direction ;
- Des consultants du pôle d’expertise des dispositifs médicaux, une apprentie au poste de chargé d’affaires réglementaires des dispositifs médicaux, une rédactrice médicale ;
- Une rédactrice médicale (voir évolution de l’entreprise de mai 2022 à mai 2023).
Toutefois, KAPSIKUM a aussi une chargée de veille et une animatrice qualité. Ces deux fonctions ne sont pas en contact avec les clients.
N’étant pas l’unique entreprise de conseil en France, KAPSIKUM est entourée de plusieurs concurrents qui sont également des sociétés de conseil et des organismes de formations à l’instar de : CADUCEUM, NEXIALIST, ISOCELE, CEISO, etc. Ces entreprises sont des grands groupes ou des entreprises indépendantes dont les activités s’étendent en France, en Europe, et/ou hors Europe.
2.1.2 La mission de KAPSIKUM face à ID-TANDEM fabricant des dispositifs médicaux
Dans le cadre de la réalisation des prestations pour son client ID-TANDEM, KAPSIKUM a pour mission la mise en conformité des dossiers techniques de marquages CE et du système de management de la qualité dans le cadre de la transition vers le règlement (UE) 2017/745 relatif aux dispositifs médicaux.
Cette mission est également celle initialement définie en août 2022 pour le déroulement de l’apprentissage.
2.1.3 Les enjeux de ID-TANDEM face au changement réglementaire
ID-TANDEM est une entreprise qui fabrique les chambres d’inhalation (voir Figure 4) permettant l’administration de médicament sous forme d’aérosol aux personnes souffrant de pathologies respiratoires.
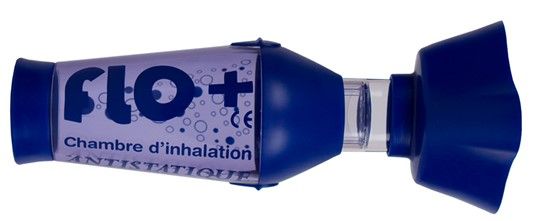
Les chambres d’inhalation fabriquées par ID-TANDEM sont marquées CE selon la directive 93/42/CEE. À la suite de la publication du règlement (UE) 2017/745, ID-TANDEM doit se mettre en conformité avec ce règlement pour continuer la commercialisation de ses chambres d’inhalation en Europe.
Le règlement (UE) 2017/745 permet une période de transition de la directive 93/42/CEE vers le règlement (UE) 2017/745 dans son article 120. Le règlement (EU) 2023/607 du 15 mars 2023 a allongé cette période de transition jusqu’en décembre 2028 pour permettre à tout fabricant de dispositifs médicaux qui souhaite continuer de commercialiser son produit en Europe d’être conforme aux exigences dudit règlement.
Pour que la période transitoire soit applicable à ID-TANDEM, celle-ci doit introduire une demande de conformité avant mai 2024 qui aboutit à un contrat signé avec l’organisme notifié avant septembre 2024. En d’autres termes, le dossier technique des chambres d’inhalation devra être prêt pour évaluation au plus tard en fin d’année 2023.
Du fait des nouvelles exigences réglementaires, les chambres d’inhalation marquées CE en classe I sous directive, passent en classe IIa sous règlement, compte tenu de la règle 20 de l’annexe VIII du règlement (UE) 2017/745. Le changement de classe de risque implique l’intervention d’un organisme notifié, ce qui n’était pas le cas jusqu’ici. Le changement de texte réglementaire implique d’identifier et d’introduire dans le dossier technique, les nouvelles exigences du règlement (UE) 2017/745 qui sont applicables au dispositif et de mettre en conformité le système de gestion des risques avec ces nouvelles exigences.
Plusieurs tâches issues de la mission principale énoncée ci-dessus, sont à effectuer pour assurer la conformité des chambres d’inhalation au règlement. Il s’agit :
- D’analyser les exigences générales de sécurité et de performance (EGSP) et de déterminer les essais manquants nécessaires à réaliser pour compléter le dossier technique ;
- De mettre à jour la documentation technique réalisée sous la directive vers le plan du règlement ;
- D’intégrer les exigences supplémentaires et les essais supplémentaires ;
- De mettre à jour les dossiers d’ingénierie de l’aptitude à l’utilisation (IAU) et de gestion des risques (GDR) ;
- De mettre à jour l’évaluation clinique et l’évaluation de la sécurité biologique avec les nouvelles exigences réglementaires et normatives ;
- De préparer les plans de suivi après commercialisation (SAC) et suivi clinique après commercialisation (SCAC) ;
- De mettre à jour la documentation du système de management de la qualité en conformité avec le règlement.
Ces tâches ont été définies dans le but d’avoir un schéma de travail pour réaliser la mise en conformité documentaire des chambres d’inhalation fabriquées par ID-TANDEM.
Ce passage au règlement est très important pour la société ID-TANDEM car il conditionne le maintien du produit sur le marché et donc la survie de la société. Les délais imposés par le règlement sont contraignants et doivent être obligatoirement respectés.
2.2 Méthode utilisée pour la mise en conformité du dossier technique de marquage CE : le DCAP (Do Check Act Plan)
Le marquage CE des chambres d’inhalation fabriquées par ID-TANDEM n’est pas une activité nouvelle car les produits sont déjà sur le marché conformément à la directive. Il existe donc déjà un dossier technique, des documents à dispositions, des processus en place. Compte tenu de l’existant, le DCAP (Figure 5) est l’outil utilisé pour présenter la méthode de travail.
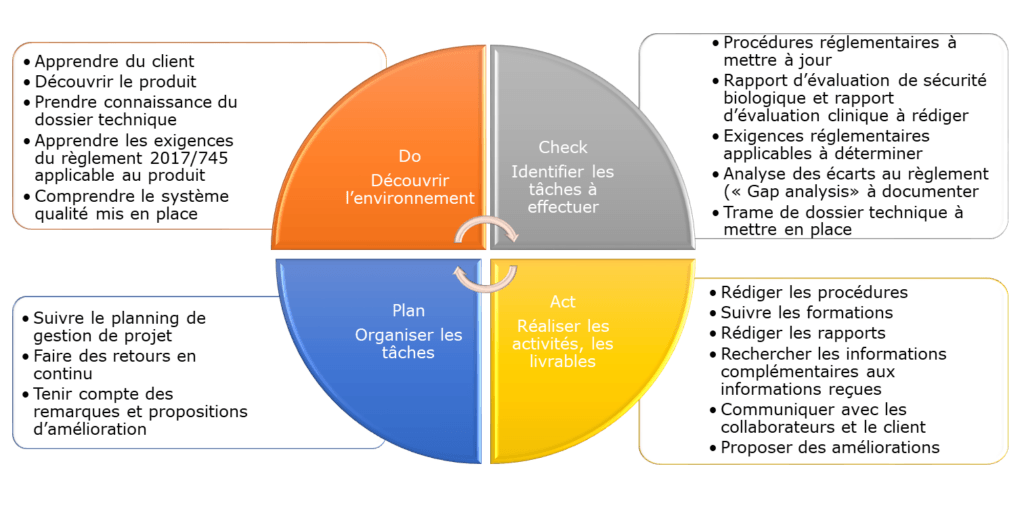
Figure 5 : Méthode utilisé pour la mise à jour du dossier technique de marquage CE par le DCAP (Do Check Act Plan) (source : auteur)
La réalisation de la mission a été progressive (voir le Tableau 1) et a commencé par une phase d’acquisition (ou imprégnation). Le but était de :
- Comprendre le règlement (UE) 2017/745 (savoir l’interpréter) pour pouvoir l’exploiter aisément par la suite ;
- Prendre connaissance du dispositif médical (la chambre d’inhalation), son utilisation, son fonctionnement, … ;
- Prendre connaissance de la documentation existante et comprendre le déroulement des activités effectuées par le fabricant.
Après avoir pris connaissance du produit, de la documentation existante et des exigences du règlement (UE) 2017/745, la seconde phase fut une phase d’adaptation : réalisation des tâches en parallèle, rédaction des différents documents nécessaires. Le but était de :
- Exploiter les informations identifiées dans le règlement (UE) 2017/745
- Concrétiser les connaissances acquises lors des formations ;
- Gérer le temps de travail ;
- Utiliser simplement et efficacement les ressources ;
- Améliorer les compétences acquises.
La dernière phase, la phase d’appropriation (ou consolidation) avait pour but de :
- Gagner en temps dans l’exécution des tâches ;
- Acquérir de nouvelles compétences ;
- Mettre en avant la productivité dans le travail ;
- Développer de nouvelles idées et proposer des améliorations.
Tableau 1 : Plan d'action de la mise à jour du dossier technique
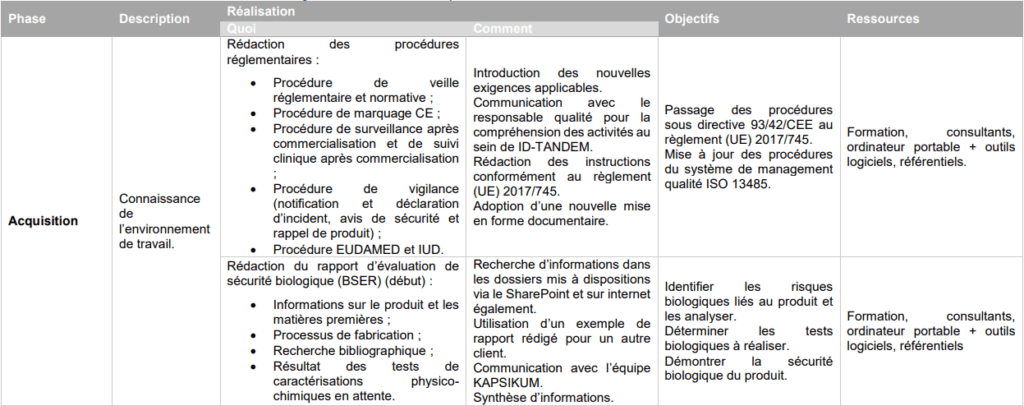
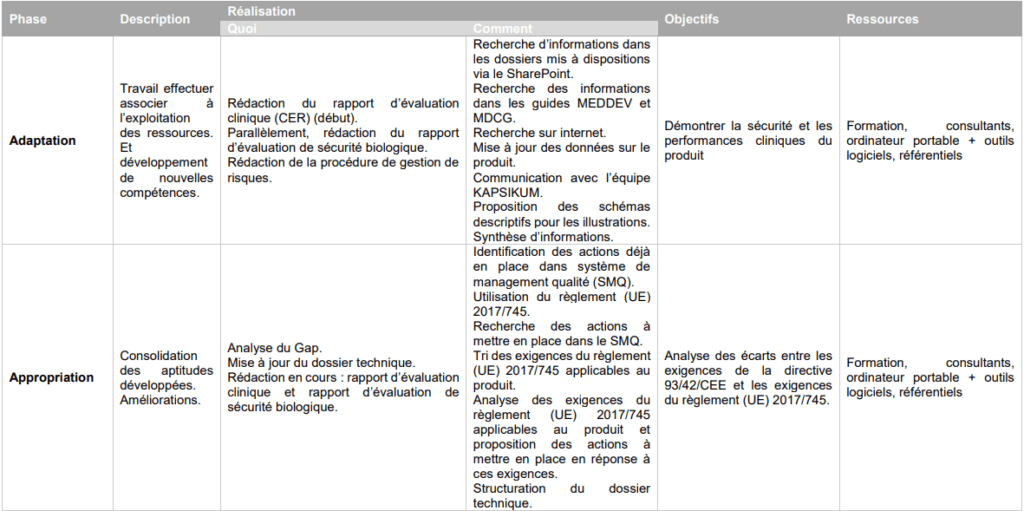
Il est à noter que toutes les ressources fournies par KAPSIKUM ont bien été exploitées
3 La relation entre le dossier technique de marquage CE et le dossier de gestion des risques
3.1 Importance du dossier de gestion des risques dans le dossier technique de marquage CE
Le processus de gestion des risques fait partie du système de management de la qualité. Le dossier de gestion des risques qui en est issu permet de rédiger la partie 5 du dossier technique de marquage CE selon l’annexe II du règlement (UE) 2017/745. Ce dossier est constitué en conformité à la norme ISO 14971 : 2019 sur l’application de la gestion des risques aux dispositifs médicaux.
Le dossier technique est un dossier constitué en réponse aux exigences de sécurité et de performance de l’annexe I et en conformité avec le plan défini aux annexes II et III du règlement (EU) 2017/745 relatifs aux dispositifs médicaux. Il prouve la conformité du dispositif médical aux exigences de sécurité et de performance du dispositif médical.
Le dossier technique comprend :
- Les informations du fabricant ;
- La description générale du dispositif médical ;
- Les informations fournies par le fabricant ;
- Les informations concernant la conception et la fabrication du dispositif médical ;
- L’analyse et la réponse aux exigences de sécurité et de performance applicable au dispositif médical ;
- L’analyse du bénéfice risque et le management de risque ;
- Les preuves de vérification et de validation du dispositif.
Chaque partie du dossier technique (voir la Figure 6) contient des informations utiles à l’application du processus de gestion des risques. Il est à noter que la norme EN ISO 14971 : 2019 portant sur l’application de la gestion des risques est une norme harmonisée au règlement (UE) 2017/745 relatif aux dispositifs médicaux. Ainsi, le respect de cette norme et la mise à disposition des résultats du processus de gestion des risques dans le dossier technique permet la réponse aux exigences de l’annexe I dudit règlement et donc de garantir la sécurité des utilisateurs du dispositif médical.
3.2 Les parties du dossier technique dont les informations servent à la gestion des risques
Les éléments du dossier technique sont rédigés en parallèle avec le dossier de gestion des risques. La mise à jour d’une information dans le dossier de gestion des risques entraîne la mise à jour des informations contenues dans le dossier technique, et réciproquement.
Les parties du dossier technique qui interagissent avec le dossier de gestion des risques sont les suivants :
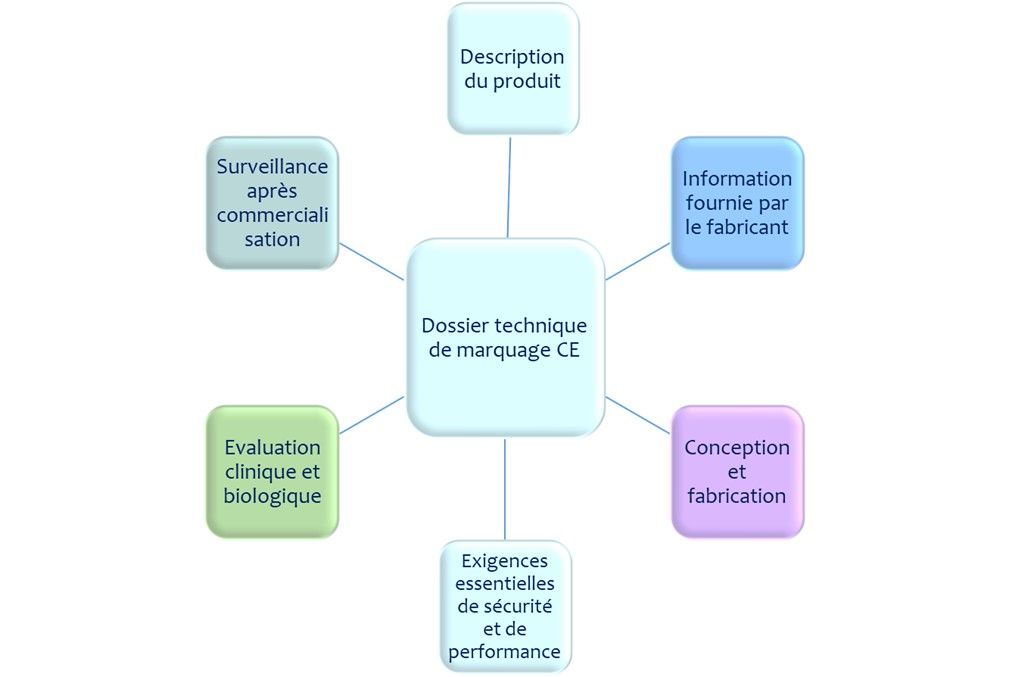
3.2.1 La description générale du dispositif médical y compris ses variantes et ses accessoires
Décrire le dispositif médical revient à identifier le type de dispositif, expliquer l’utilisation à laquelle le dispositif est destinée, expliciter le principe de fonctionnement du dispositif et déterminer sa classe de risque selon l’annexe VIII du règlement.
Par la description de l’utilisation prévue du dispositif, le fabricant peut identifier des critères d’acceptabilité du risque. Ces critères font partie de la politique à établir par la direction pour la gestion des risques.
De l’utilisation prévue par le fabricant doivent être déterminées les mauvaises utilisations raisonnablement prévisibles qui doivent être documentées dans la gestion des risques.
Par ailleurs, la description de l’utilisation prévue est le point de départ du processus d’analyse des risques. Le principe de fonctionnement du dispositif médical et la population cible font également partie des éléments à prendre en compte lors de la description de l’utilisation du dispositif médical.
Enfin, le dossier de gestion des risques contient une partie en lien avec le dossier d’ingénierie de l’aptitude à l’utilisation (IAU). Au travers de l’IAU, il est possible d’évaluer les risques liés à l’utilisation normale du dispositif.
3.2.2 Les informations fournies par le fabricant
Les informations fournies par le fabricant sont composées principalement de la notice et de l’étiquetage. Ils décrivent l’utilisation correcte du dispositif médical. La défaillance de ces informations peut engendrer une ou plusieurs situations dangereuses. Par exemple, une défaillance pendant l’étiquetage du dispositif peut causer une erreur d’utilisation.
Une analyse des risques par l’identification des dangers liées aux informations fournies par le fabricant est nécessaire ; d’autant plus que les risques résiduels doivent être mentionnés sur ces mêmes supports sous forme de précautions d’emploi, d’avertissements… La défaillance peut donc se produire sur les informations de bonne utilisation ou sur les informations de sécurité issues de la gestion des risques.
3.2.3 Les informations relatives à la conception et à la fabrication du dispositif médical
Toutes les activités effectuées en conception et en fabrication sont suivies par le processus de gestion des risques :
- La description des principales fonctionnalités du dispositif médical permet de déterminer les mauvaises utilisations raisonnablement prévisibles.
- Le planning de conception inclut la revue de conception pour l’examen des activités de gestion des risques. Il spécifie aussi à quel moment les mesures de maîtrise de risques mises en œuvre sont vérifiées. Enfin, le planning de conception est pris en compte dans le plan de gestion des risques.
- Les décisions prises durant la conception servent à la maîtrise des risques. Par exemple, le choix des tests et essais à réaliser. De ces décisions prises, ressortent des solutions possibles ou des mesures de protection à étudier pour maîtriser les risques. Le résultat des décisions prises durant la phase de conception peut influencer les performances cliniques et donc l'estimation du bénéfice.
- La maîtrise de la production et de l’environnement de production ainsi que les contrôles en cours de production permettent de limiter les risques de non-conformité pendant la production et sont donc des mesures de maîtrise du risque qui sont réfléchies durant le transfert de conception.
- Le contrôle du produit fini est une étape qui décrit les contrôles à réaliser pour la libération des lots. Les règles définies dans cette partie doivent être prises en compte dans la politique d’établissement des critères d’acceptabilité du risque.
- La définition de la chaîne de traçabilité sert à la maîtrise des risques tout au long du cycle de vie du dispositif médical.
Il est à noter que tout écart lors des phases de conception et de fabrication peut nuire à la sécurité et/ou aux performances du dispositif médical.
3.2.4 Les exigences essentielles de sécurités et de performances
Vingt-trois exigences générales en matière de sécurité et de performance sont identifiées dans l’annexe I du règlement (UE) 2017/745. Ces exigences permettent de définir les performances à atteindre et les risques à lever. Elles sont prises en compte lors de l’analyse du bénéfice/risque du dispositif médical. La détermination des exigences de sécurité et de performances applicables dépend du dispositif médical. Il en est de même pour l’analyse du bénéfice/risque.
3.2.5 L'analyse du bénéfice risque et le management des risques
Cette partie sur l’analyse du bénéfice/risque et le management des risques est entièrement basée sur les résultants du dossier de gestion des risques. Voir Maîtrise des risques à la partie 1.2.2.
3.2.6 La vérification et la validation du produit
La vérification et la validation du produit comprend les données précliniques et cliniques. Les données précliniques comprennent les essais de vérifications et de validation, y compris l’évaluation des risques biologiques, la validation des procédés spéciaux et l’ingénierie de l’aptitude à l’utilisation. Les données cliniques comprennent l’évaluation clinique et le suivi clinique après commercialisation.
Les résultats cliniques issus des performances obtenues lors de l’utilisation clinique du dispositif médical permettent de déterminer les bénéfices estimés dans le processus de gestion des risques.
De même, les données issues de l’investigation clinique (dans le cas où une investigation clinique est menée), permettent de confirmer que le ratio bénéfice/risque est conforme. Elles peuvent aussi justifier l’exposition des patients à un risque résiduel.
A titre d’illustration, des recherches dans la littérature scientifique sont effectuées lors de la rédaction du rapport d’évaluation de sécurité biologique et du rapport d’évaluation clinique. Il s'agit, respectivement, de recherches bibliographiques sur la sécurité biologique des matières premières utilisées pour la fabrication du dispositif et d’un état de l’art en vue de recueillir les informations scientifiques pertinentes sur la sécurité et la performance clinique du dispositif.
Les informations issues de la littérature scientifique sont utilisées dans le dossier de gestion des risques comme méthode d’évaluation du risque résiduel global. Ces informations peuvent également être exploitées dans les activités de production et de postproduction du dossier de gestion des risques afin de comparer les informations recueillies avec celles de l’état de l’art.
Les informations issues du suivi clinique après commercialisation permettent de confirmer le rapport bénéfice/risque et peuvent servir à comprendre les problèmes qui sont susceptibles de se produire lors de l’utilisation du dispositif médical.
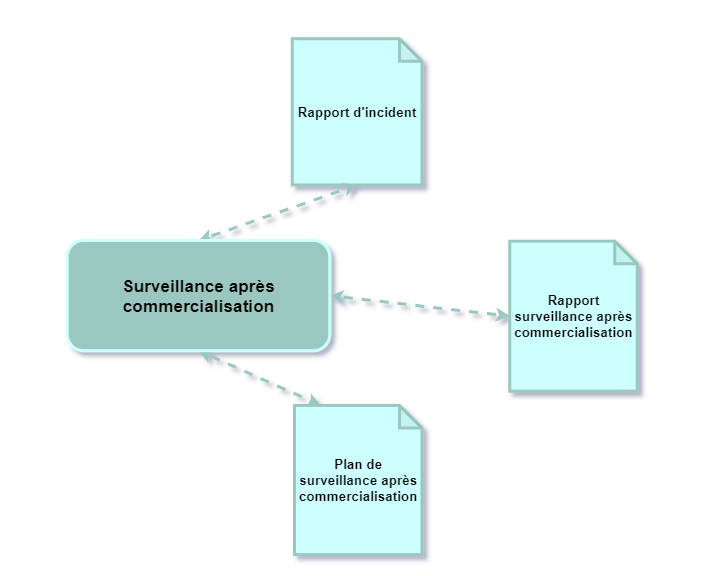
3.2.7 La surveillance après commercialisation
Le processus de surveillance après commercialisation commence par la rédaction d’un plan de surveillance après commercialisation. Ce plan fait référence aux procédures de surveillance mises en place par le fabricant, à la gestion des réclamations et des incidents. Il permet de définir le suivi spécifique mis en place pour un dispositif médical.
La mise en œuvre de ce plan, permet de rédiger le rapport de surveillance après commercialisation ou le rapport périodique actualisé de sécurité (en fonction de la classe du produit) dans lequel l’analyse des résultats permettra de déterminer les actions correctives et préventives à mettre en place.
Le rapport de surveillance après commercialisation contient les informations collectées lors des activités de production et de postproduction à intégrer dans le processus de gestion des risques.
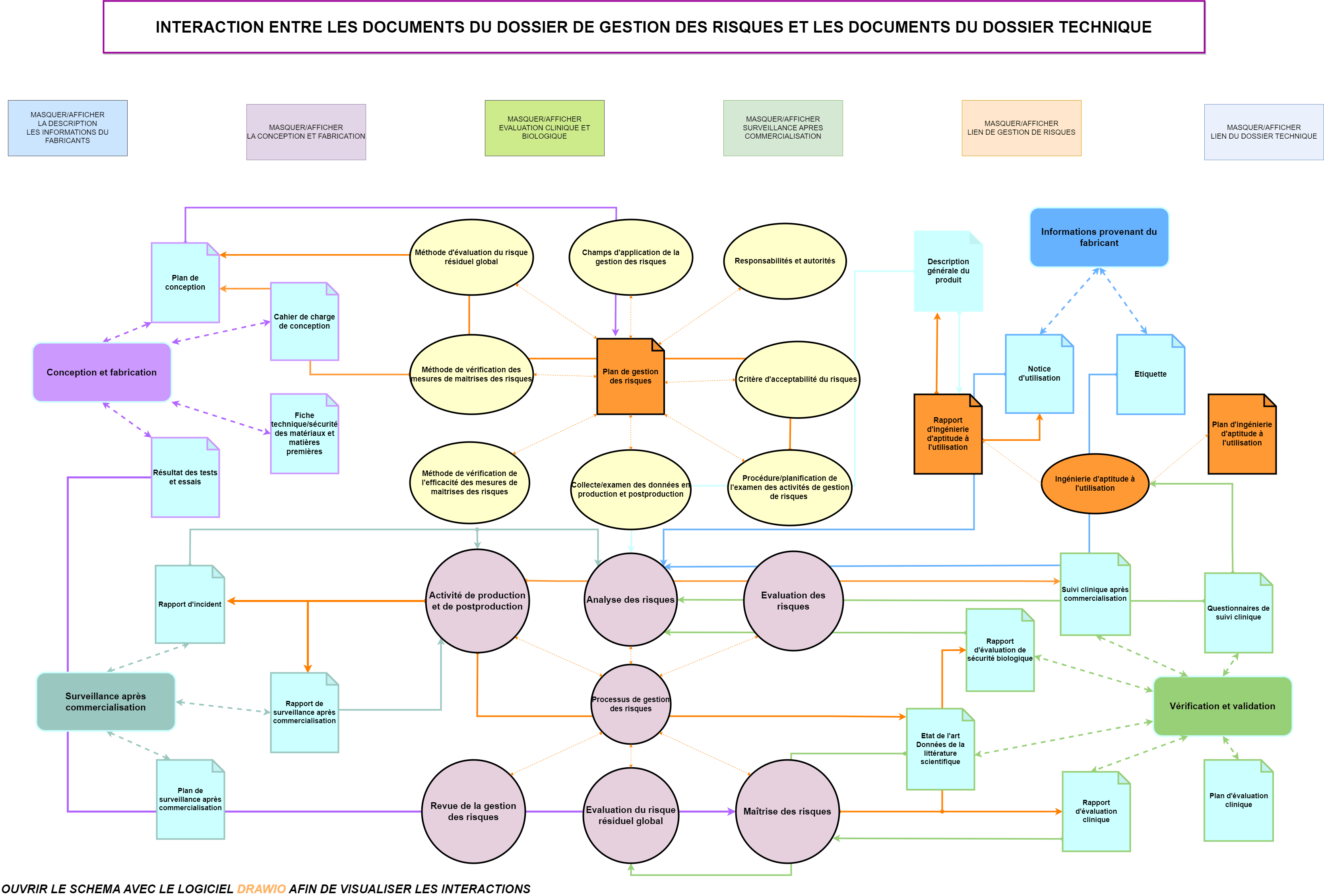
3.3 Schématisation des interactions entre le documents du dossier technique et du dossier de gestion des risques
3.3.1 Problématique rencontrée suite à la mise à jour du dossier technique de marquage CE
La mission principale de mise en conformité du dossier technique de marquage CE vers le règlement (UE) 2017/745 initialement définie, a été confrontée lors de sa réalisation, à des orientations diverses sur la notion du risque. En effet, dans chaque document, une relation différente aux risques étaient relevée. Ces orientations ont conduit à la question suivante : « En termes de gestion des risques, quelles sont les interactions qui se produisent entre le dossier technique et le dossier de gestion des risques ? » Figure 7. Il a été trouvé que plus de 90% des documents contenus dans le dossier technique interagissent avec le dossier de gestion des risques, à la fois en tant que donnée d’entrée et en tant que donnée de sortie.
3.3.2 Solution proposée en accord avec le règlement 2017/745
La schématisation des interactions est une solution qui permet d’avoir la visibilité sur les liens entres les documents afin de mieux organiser la rédaction de ceux-ci.
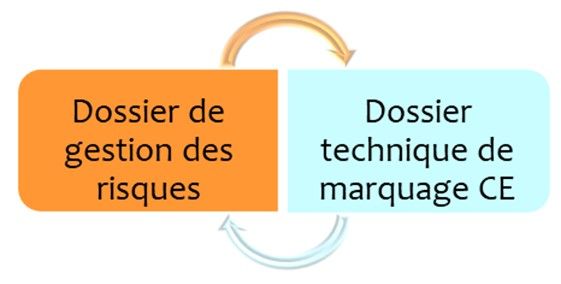
Le schéma élaboré (document à télécharger) est une représentation des relations entre les parties du dossier technique et le dossier de gestion des risques.
En effet, concernant la gestion des risques, le plan de gestion des risques est un document de plusieurs parties (voir le schéma ci-dessous). Il peut faire référence (ou pas) à d’autre plan comme, le plan d’évaluation clinique, le plan de conception, etc. Le processus de gestion des risques quant à lui est un ensemble de plusieurs activités de gestion des risques corrélées.
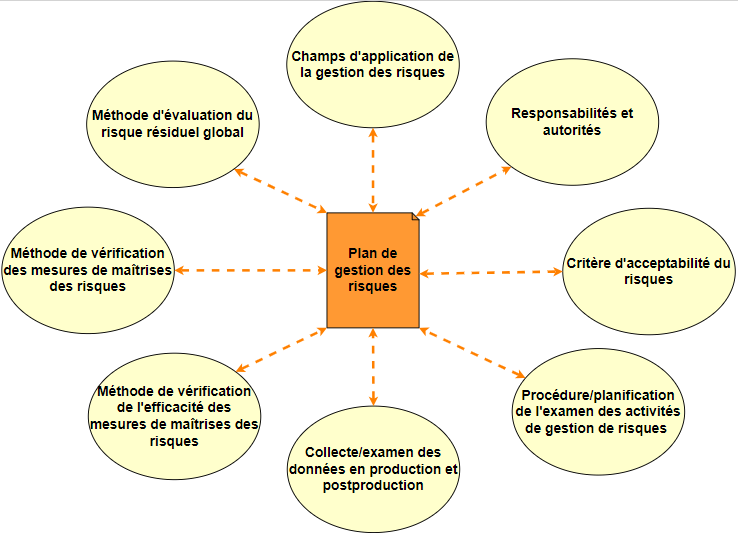
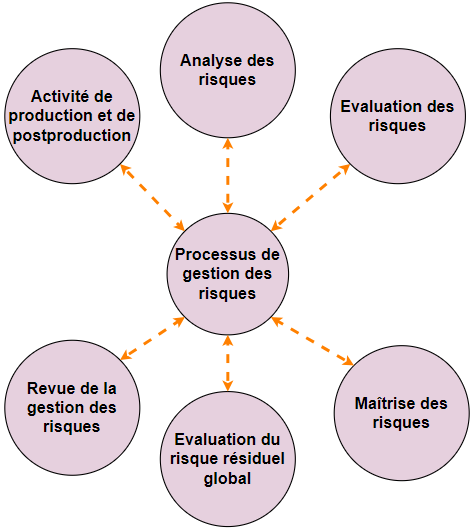
Figure 8 : Les parties du plan de gestion des risques et les activités du processus de gestion des risques. (Source : Auteur)
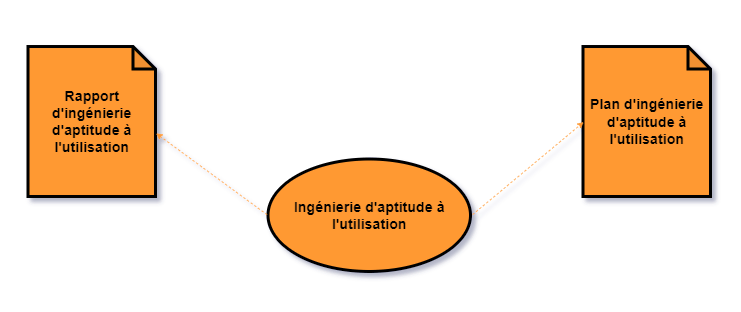
L’ingénierie d’aptitude à l’utilisation (Figure 9) est une partie qui peut faire partir du dossier de gestion des risques ou être constituer dans un dossier séparé. Néanmoins, les informations issues de l’ingénierie d’aptitude à l’utilisation sont prises en compte par les éléments du dossier de gestion des risques.
En ce qui concerne le dossier technique, chacune des parties fait référence à un ou plusieurs documents (voir l’exemple ci-dessous, Figure 10).
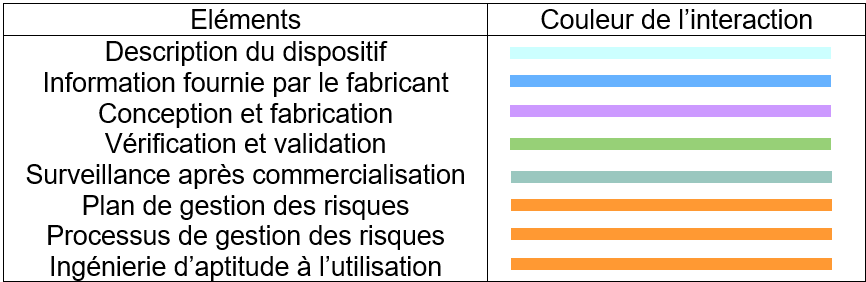
Sur le schéma (Figure 13) une couleur est attribuée pour chaque partie afin de différencier les interactions qu’elles émettent.
Il est important de préciser que dans chaque partie du dossier technique les documents interagissent entre eux. C’est-à-dire que les informations d’un document peuvent servir dans un autre document du dossier. Il en est de même pour les éléments du dossier de gestion des risques
Ces interactions en interne sont matérialisées dans le schéma pas des flèches en à double sens (voir la Figure 11 ), suivant le code couleur du tableau ci-dessus.
Un élément du dossier de gestion des risques peut fournir des interactions à un ou plusieurs documents du dossier technique et réciproquement (Voir la Figure 12).
Afin de visualiser le schéma avec toutes ses fonctionnalités (notamment en utilisant les fonctions « MASQUER / AFFICHER » pour ne voir que les interactions voulues), vous pouvez cliquer sur le lien.
4 Bilan personnel
4.1 Apport durant l'apprentissage
L’apprentissage chez KAPSIKUM, une société de conseil de petite taille (10 salariés) est un bon moyen d’insertion professionnelle du fait de la possibilité de travailler réellement sur des dossiers utiles aux clients.
La société possède un catalogue de prestations variées. Les prestations proposées sont compatibles aux métiers de responsable affaires réglementaires des dispositifs médicaux ainsi qu’au métier de responsable du système de management de la qualité.
Les activités apprises et mises en pratique sont les suivantes :
- La rédaction des procédures ;
- La rédaction des rapports : évaluation clinique, évaluation de la sécurité biologique ;
- La recherche d’information dans la littérature scientifique : l’état de l’art ;
- L’analyse des écarts à la réglementation (ou GAP analysis) ;
- La rédaction des dossiers techniques.
Il a été possible de travailler pour d’autres clients que ID-TANDEM ainsi que de travailler sur des sujets différents de la mise à jour du dossier technique. Par exemple, la préparation et la participation à un audit interne MDSAP pour un client ou encore la rédaction de quelques dossiers techniques pour d’autres clients.
Le travail effectué a permis à KAPSIKUM d’optimiser ses ressources (de temps et de moyens) et aussi d’avoir un outil présentant les interactions entre les documents du dossier technique et les documents du dossier de gestion des risques à présenter à ses clients.
4.2 Les compétences développées au sein de KAPSIKUM
Les compétences qui ont été acquises sont les suivantes :
- L’organisation des idées et des notes : notamment à l’aide du mind mapping ;
- La gestion des priorités : dans la pratique, pour rédiger un document plusieurs paramètres sont à considérer en amont avant la rédaction complète du document. Matérialiser ce qu’il faut faire avant et après a été un exercice pas simple au début mais bien acquis par la suite ;
- La gestion du temps : qui s’est améliorée durant la formation ;
- La communication : avec un client, la direction, les collaborateurs.
4.3 Le rapport entre la formation institutionnelle à l'UTC et la formation professionnelle chez KAPSIKUM
Le contenu de la formation en DMAR (Dispositif Médicaux et Affaires Réglementaires) est adapté aux attentes professionnelles. En effet, les unités enseignées sont des contenus directement liés aux métiers avec quelques notions générales en plus.
Quelques enseignements liés au métier de responsable affaires réglementaires, et utile durant l’apprentissage :
- Les affaires réglementaires de dispositifs médicaux :
Un enseignement qui parcourt l’ensemble des textes normatifs et réglementaires du domaine, les fonctionnalités des personnes impliquées, la gestion des risques et l’ingénierie de l’aptitude à l’utilisation, etc. C’est en effet une base du métier. Les informations reçues de cette unité d’enseignement ont été utilisées dans la mise en œuvre de toutes les tâches techniques des différentes activités réalisées.
- L’audit :
Un enseignement pratique sur l’audit. L’exploitation de cet enseignement a été possible lors de l’audit MDSAP pour un client.
- Le management de la qualité :
Une unité d’enseignement sur l’ensemble du management qualité, l’utilisation des outils qualités, les normes de management de la qualité. Le plus important de cet enseignement a été le travail de synthèse fait en équipe, à chaque étape de l’avancement des enseignements. Ceci a été appliqué durant l’apprentissage lors des points projets en équipe.
- La veille bibliographique :
Par la mise en application des recherches dans la littérature scientifique, pour la rédaction de l’état de l’art. Les outils connus comme Zotero ou les sites de recherches fiables sont exploités lors de la rédaction des différents rapports (évaluation clinique ou évaluation de la sécurité biologique).
Orientation professionnelle
Au travers des enseignements reçus et des compétences acquises durant dans le master dispositif médicaux et affaires réglementaires, plusieurs orientations professionnelles se présentent. Une profession en tant que chargée d’affaires réglementaires des dispositifs médicaux pour commencer, ensuite un poste de responsable affaires réglementaires experts, avec des qualifications d’auditeurs et de formateurs, et dans une suite évolutive, une extension vers d’autre pôle d’expertise (pharmaceutique, cosmétique, alimentaire, ...).
Conclusion
Le dossier de gestion des risques contient des informations utiles à la rédaction du dossier technique. La mise à jour du dossier de gestion des risques entraine une revue du dossier technique afin de s’assurer de la cohérence entre les dossiers.
La question sur la sécurité des dispositifs médicaux est à se poser lors de la rédaction de chaque partie du dossier technique. Chaque partie du dossier technique contient des informations provenant du dossier de gestion des risques ou alors exploitables par celui-ci.
Il est aussi important de préciser que le rapport de gestion des risques est le document central du dossier de gestion des risques. Il contient tous les résultats issus des activités de la gestion des risques ainsi que le résumé issu de l’examen de ces résultats. Sa mise à jour est aussi fonction de la mise à jour des documents du dossier technique et réciproquement.
Le schéma proposé est une aide, tant pour les fabricants de dispositif médicaux que pour les chargés affaires réglementaires et qualité, ou tout simplement pour toutes personnes concernées par la rédaction des dossiers techniques de marquage CE des dispositifs médicaux. Le schéma donne une visibilité sur les interactions entre les documents du dossier de gestion des risques et les documents du dossier technique. Ce schéma est bien évidemment inscrit dans une amélioration continue afin d’apporter des solutions plus optimales entre les liens du dossier technique et du dossier de gestion des risques.
Références bibliographiques
[1] Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) no 178/2002 et le règlement (CE) no 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE)Texte présentant de l’intérêt pour l’EEE. 2023. Disponible sur : http://data.europa.eu/eli/reg/2017/745/2023-03-11/fra. Consulté le : 12 juin 2023. [En ligne].
[2] « Marquage CE - Dispositifs Médicaux - AFNOR Certification ». https://certification.afnor.org/qualite/marquage-ce-des-dispositifs-medicaux (consulté le 15 juin 2023).
[3] C. Tur, « Mise en place du Règlement (UE) 2017/745 et impacts pour les fabricants et les distributeurs » Page17. https://dumas.ccsd.cnrs.fr/dumas-03513281 (Consulté le 19 juin 2023).
Annexe
Exemple de cycle de vie du dispositif médical (Source : Auteur)