
IDS203 - Bilan du nouveau Règlement Européen sur les Dispositifs Médicaux 2017/745 : De l’entrée en vigueur à l’application
DOI mémoire
https://doi.org/10.34746/ids203Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- Aissetou BATHILY : agbathily@gmail.com
- Oumaima ELKOUACH : elkouachoumaima@gmail.com
- Ouissal HAKIK : ouissal.hakik20@gmail.com
- Catiana RAKOTOZAFY : fcatiana@hotmail.fr
- Anaïs SIMO : chloekouajip@gmail.com
Citation
Aissetou BATHILY, Oumaima ELKOUACH, Ouissal HAKIK, Catiana RAKOTOZAFY et Anaïs SIMO, " Bilan du nouveau Règlement Européen sur les Dispositifs Médicaux 2017/745 : De l’entrée en vigueur à l’application", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, parcours Dispositif Médical et Affaires Règlementaires, Mémoire de Projet, https://travaux.master.utc.fr/ids203,DOI : https://doi.org/10.34746/ids203, Janvier 2024, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids203
Résumé
Après six années d'application du règlement européen sur les dispositifs médicaux, il devient essentiel de réaliser une enquête approfondie auprès des acteurs clés de l'écosystème du dispositif médical, afin d’établir un état des lieux de la mise en place du Règlement relatif aux Dispositifs médicaux 2017/745, tant au niveau européen que français. Cette enquête a été menée à différentes échelles afin d’enrichir au maximum les données collectées et être rigoureux dans nos constatations. Nos sources d’informations comprennent la participation lors d'un colloque organisé par le SNITEM, des entretiens individuels avec des acteurs nationaux et européens, la diffusion d’un questionnaire sur LinkedIn, ainsi que des données provenant d’un syndicat européen. Les ressentis et les répercussions diffèrent en fonction du milieu local, dans lequel l’acteur opère mais plusieurs tendances majeures ont émergé.
Cette étude sur le terrain a permis d'explorer en détail les raisons sous-jacentes des périodes de transitions et quantifier les impacts durant les périodes de transition, offrant ainsi une perspective claire sur les défis auxquels font face les différentes parties prenantes dans la mise en application de cette réglementation.
En réponse à ces constatations, nos conclusions suggèrent que certains de ces défis pourraient être relevés grâce à une plus ample acquisition d'informations. C’est la raison pour laquelle nous avons décidé de contribuer à cet écosystème en élaborant un guide d'accompagnement destiné aux fabricants dans le but d'obtention du marquage CE et ainsi être compétitifs et innovants face à la concurrence internationale dans le secteur des dispositifs médicaux.
Abstract
After six years of application of the European Medical Device Regulation, it is becoming essential to carry out an in-depth survey of key players in the medical device ecosystem, in order to establish the current state of implementation of the Medical Device Regulation 2017/745, both at European and French level. This survey was conducted at different scales in order to enrich the data collected as much as possible and be rigorous in our findings. Our sources of information include participation at a symposium organized by SNITEM, individual interviews with national and European players, the distribution of a questionnaire on LinkedIn, as well as data from a European trade union. Feelings and repercussions differ according to the local environment in which the player operates, but several major trends have emerged.
This field study explored in detail the underlying reasons for transition periods and quantified impacts during transition periods, providing a clear perspective on the challenges faced by different stakeholders in implementing these regulations.
In response to these findings, our conclusions suggest that some of these challenges could be addressed through further information acquisition. For this reason, we have decided to contribute to this ecosystem by developing a guide for manufacturers, with the aim of obtaining CE marking, and thus being competitive and innovative in the face of international competition in the medical devices sector.
Téléchargements



Remerciements
Avant de développer ce rapport, nous souhaitons exprimer notre gratitude envers toutes les personnes ayant contribué à la réalisation de ce projet.
En premier lieu, nous tenons à remercier chaleureusement nos trois superviseurs de l'Université de technologie de Compiègne, Madame Isabelle CLAUDE, Madame Julie FOLLET et Monsieur Jean-Matthieu PROT, pour leurs conseils précieux à chaque étape et leur soutien, qui ont été inestimables, et qui nous ont guidé tout au long de ce semestre.
Nous exprimons également notre reconnaissance envers Monsieur Jean-Matthieu PROT pour sa perspective critique, ses conseils avisés et sa disponibilité. En sa qualité de suiveur de projet, il a partagé son expérience et nous a orienté vers les professionnels appropriés, facilitant ainsi l'obtention des informations nécessaires.
Ensuite, nous remercions sincèrement Madame Béatrice KONIG pour son examen attentif de notre bibliographie et ses remarques constructives.
Enfin, nous exprimons notre gratitude envers les entreprises qui ont participé à nos sondages et nos entretiens. Leur contribution précieuse a enrichi notre étude et nous les remercions sincèrement pour avoir pris le temps de partager leurs connaissances et expériences.
Abréviations
AFSSAPS : Agence Française de Sécurité Sanitaire et des Produits de Santé
ANSM : Agence nationale de sécurité du médicament et des produits de santé
CE : Conformité Européenne
CEE : Communauté Economique Européenne
DM : Dispositif Médical
DMDIV : Dispositifs Médicaux In Vitro
DMIA : Dispositifs Médicaux Implantables Actifs
DMS : Dispositifs médicaux stériles
DT : Dossier technique
IVDR : In Vitro Diagnostic Regulation
MDD : Medical Device Directives
MDCG : Groupe de Comité d’experts des dispositifs médicaux
MDR : Medical Device Regulation
NANDO : New Approach Notified and Designated Organizations
ON : Organisme Notifié
PIP : Poly Implant Prothèse
RDM : Règlement relatif aux Dispositifs Médicaux
SMQ : Système de Management de la Qualité
UE : Union Européenne
QARA : Quality Assurance and Regulatory Affairs
Liste des figures
Figure 1 : Présentation chronologique des principales directives
Figure 2 : Calendrier initial de la mise en application du règlement 2017/745 et 2017/746
Figure 3 : Synthèse de la mise sur le marché européen d’un DM
Figure 4 : Procédure de candidature d’un futur organisme notifié
Figure 5 : Répartition géographique d'organismes notifiés dans l’Union européen
Figure 6 : Répartition des organismes notifiés selon la procédure d'évaluation de conformité
Figure 7 : Répartition des dispositifs médicaux en fonction des certificats MDR/MDD
Figure 9 : Représentation des ressources utilisées
Figure 10 : Treemap des types de DM susceptibles de quitter le marché
Figure 11 : Taille des entreprises
Figure 13 : Localisation privilégiée des entreprises pour une approbation réglementaire
Figure 16 : Nouvelles échéances de la deuxième période transitoire du règlement 2017/745
Liste des tableaux
Tableau 1 : Comparaison du règlement et des directives
Tableau 4 : Tarifs des organismes notifiés pour le règlement des dispositifs médicaux.
Liste des annexes
Mémoire Complet :
Bilan du nouveau Règlement Européen sur les Dispositifs Médicaux 2017/745 : De l’entrée en vigueur à l’application
Introduction
Les dispositifs médicaux occupent une place centrale dans le système de santé et sont en constante évolution. En effet, il est important de souligner que le marché mondial des dispositifs médicaux a généré un chiffre d'affaires de 447 milliards de dollars en 2022 [1]. En Europe, ce marché était évalué à 150 milliards d'euros en 202, représentant 27,3 % [1] du marché mondial. En ce qui concerne la France, elle se positionne deuxième derrière l'Allemagne, comptant 1502 entreprises en 2019 et 1440 en 2021. De plus, à elle seule, la France a généré un chiffre d'affaires estimé à 30,7 milliards d'euros en 2021 [2].
Ces dispositifs médicaux, au-delà de leur impact sur la santé, ont également un rôle capital dans l'économie. Dès lors, la réglementation était impérative, marquée par des directives élaborées dès 1990 et modifiées à plusieurs reprises, culminant en 2007 avec la directive 2007/47/CE. (figure1)
Une transformation majeure a été entamée par l'avènement du Règlement relatif aux Dispositifs Médicaux 2017/745, couramment appelé le Règlement sur les Dispositifs Médicaux (RDM). En vigueur depuis mai 2017, ce règlement a instauré un cadre réglementaire plus rigoureux et complet pour les dispositifs médicaux, dans le but d'améliorer la qualité et les performances de ces produits. Son objectif est également d'assurer un niveau élevé de protection de la santé des patients et des utilisateurs, tout en harmonisant le marché intérieur de l'Union européenne.
En effet, face aux enjeux de sécurité et d'efficacité des dispositifs médicaux, le RDM répond aux lacunes exposées par des scandales, tels que celui des prothèses mammaires PIP ou encore l'affaire Implant files. Ces crises ont précipité le remplacement des directives par les règlements 2017/745 et 2017/746, apportant une traçabilité renforcée pour la sécurité des dispositifs médicaux et des patients.
Après six années d'application du règlement, il est pertinent de s'interroger sur les impacts observés chez les parties concernées : opérateurs économiques, autorités compétentes, les organismes notifiés, personnels hospitaliers… De plus, il semble important d’étudier les raisons sous-jacentes aux retards dans la mise en œuvre du nouveau règlement européen sur les dispositifs médicaux.
Afin de répondre à ces questions, ce mémoire d'intelligence méthodologique plonge dans une évaluation approfondie de l'impact de ce règlement sur l'écosystème des dispositifs médicaux. Il analyse les influences exercées sur les autorités compétentes, les organismes notifiés, les fabricants, les professionnels de la santé et les patients, en interrogeant ces différents acteurs pour recueillir leurs impressions au cours des six dernières années.(Figure1)
I. Contexte de la réglementation des dispositifs médicaux
A. Les dispositifs médicaux sous les directives européennes
1. Historique de la réglementation sur les dispositifs médicaux
Les dispositifs médicaux jouent un rôle essentiel dans l'amélioration de la qualité des soins prodigués aux patients. Pour cela, il est essentiel de veiller à ce que ces dispositifs médicaux, qu'ils soient implantables, actifs ou in vitro, répondent aux normes strictes en matière de sécurité, de performance, d'efficacité et de qualité. C’est pourquoi il était important que l’Union européenne réglemente leur introduction sur le marché européen par la mise en place d’une législation harmonisée et respectée par tous les pays membres [3] . Cette législation harmonisée a pour objectif de définir les exigences réglementaires que les fabricants doivent respecter tout au long du cycle de vie du DM (dès la conception jusqu’à la réforme du DM par l’utilisateur). Cette réglementation repose sur quatre directives essentielles ( voir la Figure 1):
Figure 1 : Présentation chronologique des principales directives
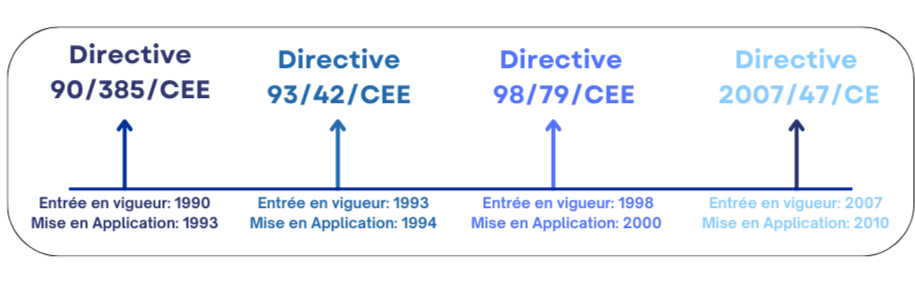
- Directive 90/385/CEE : Cette directive remonte aux années 1990, elle décrit le cadre réglementaire relatif aux DM implantables actifs qui est entré en vigueur le 1er janvier 1993 et mise en application 3 ans plus tard. Cette directive européenne vise à établir des normes de sécurité et d'efficacité pour les dispositifs médicaux implantables actifs, tels que les stimulateurs cardiaques et les défibrillateurs implantables, dans le but de protéger la santé des patients [4].
- Directive 93/42/CEE : Cette directive succède la 90/385/CEE en 1993 et s'applique aux dispositifs médicaux et à leurs accessoires [5]. Son objectif principal était l’obligation du marquage CE des dispositifs médicaux, ce qui permettait aux produits de circuler librement sur le marché européen. Demandant ainsi aux fabricants de respecter les normes harmonisées lorsqu'il s'agit de concevoir, fabriquer, et de suivre les produits après leur mise sur le marché. Tout cela dans le but de de garantir leur sécurité et leur efficacité. Cette directive est très détaillée, elle évoque les points suivants[6] :
- Définitions utiles telles que “dispositif médical”, “accessoire”, “données cliniques”, “
- Les exigences essentielles pour les dispositifs médicaux
- Les normes harmonisées comme moyen de réponse aux exigences
- Les règles de classification des DM
- Les différentes procédures du marquage CE
- Des détails sur le rôle et la nature des organismes notifiés, des évaluations cliniques…
- Directive 98/79/CEE : Cette directive adoptée en 1998, est relative aux Dispositifs Médicaux de Diagnostic in Vitro (DMDIV), qui fixe les exigences réglementaires pour leur mise sur le marché. Elle oblige les fabricants de DMDIV d’obtenir le marquage CE pour démontrer la conformité aux exigences en termes de sécurité et de performance [5].
En moins de vingt ans, la réglementation des DM a connu six modifications majeures. Ces changements ont été nécessaires pour que les fabricants s’adaptent aux avancées technologiques, à la sécurité et aux besoins des patients. L’un de ces changements a été mis en place par la directive 2007/47/CE.
2. Présentation de la directive 2007/47/CE
La directive 2007/47/CE est un amendement des deux directives 90/385/CEE et 93/42/CE, faite par l’Union Européenne en septembre 2007. Cette directive est entrée en application le 21 mars 2010. Les modifications significatives introduites par la directive 2007/47/CE sont les suivantes [3]:
La classification des dispositifs médicaux : La directive 2007/47/CE a apporté des modifications concernant la classification des dispositifs médicaux, par l’introduction d’une nouvelle annexe, l'Annexe IX, qui établit des critères précis pour classer les dispositifs médicaux en fonction de leur niveau de risque, allant de classe I (risque faible) à la classe III (risque élevé) .
Les exigences essentielles : La présente directive a apporté des améliorations aux exigences essentielles appliquées aux dispositifs médicaux. En effet, elle a introduit la notion de risque de mésusage lié aux caractéristiques ergonomiques du dispositif médical dans les exigences générales. En outre, elle a ajouté l’obligation de prendre en compte la notion de l’aptitude à l’utilisation lors de développement et la conception du DM en accord avec la norme 62366:2008 « Dispositifs médicaux - Application de l'ingénierie de l'aptitude à l'utilisation aux dispositifs médicaux ».
Les informations fournies par le fabricant : Différentes modifications sont faites, notamment l’obligation à désigner un mandataire si les fabricants ne sont pas établis dans la communauté européenne.
Notion de l’usage unique des DM : La directive a clarifié la notion de l’usage unique du DM, tout en demandant aux fabricants de fournir des informations sur les risques liés à la réutilisation des dispositifs médicaux à usage unique.
Le renforcement de l’évaluation clinique : La directive 2007/47/CE a renforcé la notion d'évaluation clinique. En effet, les fabricants sont tenus de mener des évaluations cliniques approfondies pour démontrer l’efficacité et la sécurité de leurs dispositifs médicaux. Cela implique la collecte de données cliniques pertinentes et, le cas échéant, la réalisation d’essais cliniques. L’objectif est de garantir que les DM fournissent des bénéfices cliniques substantiels tout en minimisant les risques pour les patients et les utilisateurs.
La surveillance après commercialisation : La directive 2007/47/CE a imposé des obligations aux fabricants liée à la surveillance après la commercialisation, notamment établir une procédure de surveillance post-commercialisation. Ensuite la mise à jour de l’évaluation clinique conformément aux données collectées après la mise sur le marché. Enfin la justification d'absence de suivi clinique dans le plan de surveillance post-commercialisation.
Ainsi, l’objectif principal de cet amendement a été de renforcer la sécurité et le niveau de protection des patients. Cependant, étant donné que ces textes sont des directives, leur transposition varie de manière disparate parmi les différents pays membres de l'Union européenne, laissant libre interprétation des textes.
3. Scandales sanitaires liés aux dispositifs médicaux sous l'application des directives
Comme mentionné précédemment, la réglementation liée aux dispositifs a remarquablement évolué au cours des trente dernières années. Les différentes directives applicables à l’ensemble des États membres de l’Union européenne ont été transposées dans les législations de chaque pays. Ainsi, chaque État membre interprète librement les obligations relatives à la mise sur le marché des dispositifs médicaux ce qui crée des hétérogénéités au sein de l’UE.
En 2010, l’affaire PIP a suscité un grand émoi médiatique. Le fabricant français Poly Implant Prothèse, une société fondée en 1991, spécialisée dans la production de prothèses mammaires internes, a été impliqué dans un scandale. Cette société a trompé pendant plusieurs années le marché mondial en commercialisant des prothèses remplies d’un gel différent de celui déclaré dans le dossier de conception et de fabrication des implants. Un rapport complet [7] de la Direction générale de la Santé à la demande du ministère de la Santé a dressé un état des lieux de cette affaire en 2012. Il ressort de ce rapport le manque de réactivité de l'autorité compétente AFSSAPS, devenue l’ANSM. En 2016, le fondateur de la société a été condamné à quatre ans de prison ferme et à une amende de 75 000 euros pour escroquerie et tromperie aggravée [8]. L'organisme notifié a également été condamné en 2013 pour avoir "manqué à ses devoirs de contrôle et de vigilance" et de nouveau en 2021 pour manquement aux obligations de vigilance et de contrôle par la cour d’appel de Paris [9]. Au total, près d'un million de prothèses défectueuses ont été vendues entre 2001 et 2010, et 400 000 victimes ont été recensées dans le monde [9].
En 2018, à la suite de l’affaire PIP, un nouveau scandale éclate : l’affaire des “Implant Files”. Une enquête internationale regroupant 252 journalistes de plus de 36 pays [10] a fait référence à une enquête journalistique internationale réalisée par le Consortium international des journalistes d'investigation. Des dispositifs médicaux tels que des pacemakers défaillants, des prothèses de hanche, des stents, des valves et des prothèses vaginales ont été déployés sur le marché alors qu'ils présentaient des défaillances. Aux États-Unis, environ 5 477 285 incidents ont été recensés, 82 000 décès et 1,7 million de blessés [10] ont été liés aux dispositifs médicaux. Grâce à la création d'une méga base de données, l’International Medical Devices Database [11], recensant plus de 120 000 rappels, alertes et avis de sécurité sur les dispositifs médicaux défectueux voire potentiellement mortels à travers le monde ont été recensés. Les Implant Files ont permis de révéler que moins de 20 % des pays [12] partageaient des informations de vigilance liées aux dispositifs médicaux. Des faux dossiers volontairement erronés ou contenant des erreurs ont été validés par des organismes notifiés [13]. Cette enquête a mis en lumière les pratiques de l'industrie des dispositifs médicaux implantables, mettant en évidence des lacunes réglementaires, des problèmes de sécurité et des préoccupations concernant la transparence et la supervision de ces dispositifs.
Finalement, cela a davantage suscité des interrogations quant à l'efficacité des directives censées garantir la sécurité des produits et la protection des patients.
B. Création du Règlement (UE) 2017/745 et 2017/746
1. Présentation du règlement et analyse comparative avec les directives
Les scandales à répétition et les problèmes d'interprétation des textes ont mis en lumière les limites des directives. Cela a accéléré l’introduction du Règlement UE 2017/745 et 2017/746 en mai 2017. Il est essentiel de rappeler qu’un règlement est une mesure législative adoptée soit conjointement par le Conseil de l'Union européenne et le Parlement, soit individuellement dans certains cas. À la différence de la directive, il est appliqué directement dans tous les pays de l'UE sans nécessiter une transposition dans le droit national. Ainsi le Règlement (UE) 2017/745 concerne les dispositifs médicaux et le Règlement (UE) 2017/746 concerne les dispositifs médicaux de diagnostic in vitro.
Lors de la comparaison des deux réglementations (RDM vs MDD), il ressort en premier lieu que le nouveau règlement présente des améliorations tant quantitatives que qualitatives (Voir tableau 1) . [14]
Tableau 1 : Comparatif du règlement et des directives, Source [14]

Le nouveau règlement conserve les fondamentaux de la "nouvelle approche" dite de marquage CE. Cependant, il apporte des axes d’améliorations par rapport aux directives et renforce de nombreuses exigences [15] [16]:
Redéfinition des dispositifs médicaux : Les avancées technologiques, notamment dans le domaine de l'informatique et de l'électronique, ont considérablement élargi la portée des dispositifs médicaux. Les nouveaux règlements visent à adapter la définition de ces dispositifs pour englober ces avancées, garantissant ainsi que les dispositifs tels que les applications médicales, les dispositifs connectés, et les logiciels médicaux soient couverts par la réglementation.
Classification des DM : Une nouvelle classification des dispositifs médicaux en classe I (Is : pour stérile, Im:pour ceux avec fonction de mesurage, Ir : pour chirurgical réutilisable), IIa,IIb et III est faite par le nouveau règlement dans son article 51 compte tenu de leurs objectifs et de leurs risques inhérents. Le niveau de risque augmentant de la classe I à la classe III ayant ainsi un impact sur le marquage CE et les obligations réglementaires. En conséquence, plusieurs dispositifs médicaux pourraient voir leur classe modifiée en fonction de leur niveau de risque [17].
Contrôle des organismes notifiés : Ces derniers sont placés sous contrôle de l’autorité compétente de leur pays. De plus, la Commission Européenne assure la coordination et la coopération entre les ONG via un groupe de coordination des organismes notifiés (NBCG). Ce groupe se réunit régulièrement, au minimum une fois par an.
Amélioration de la traçabilité et de la transparence des certifications CE : Le marquage CE indique que le dispositif médical est conforme aux normes de sécurité et de performance européennes. Les nouveaux règlements renforcent la traçabilité des dispositifs en exigeant des données plus complètes sur leur fabrication, leur historique, et leurs performances. Cela permet aux autorités de réglementation et aux professionnels de santé de mieux surveiller la conformité.
Renforcement de la sécurité sanitaire au sein de l’UE : L'harmonisation des règles applicables aux dispositifs médicaux dans toute l'Union européenne garantit un niveau de sécurité et de qualité cohérent pour les patients. Cela évite également les divergences réglementaires entre les États membres, favorisant ainsi un accès équitable à des dispositifs médicaux de haute qualité dans toute l'UE.
Facilitation de la gouvernance européenne du secteur des dispositifs médicaux : La création de groupes de coordination (MDCG) permet une véritable régulation du secteur à l'échelle européenne. Cela permet une coordination plus efficace entre les autorités nationales, une meilleure harmonisation des procédures d'autorisation, en somme une surveillance du marché coordonnée réduisant les disparités entre les États membres.
Désignation d’un responsable du SMQ et de la documentation technique : L'article 15 du règlement énonce les obligations qui incombent aux fabricants. Désormais, il leur est requis de désigner une personne responsable de garantir la conformité réglementaire tant au sein de l'entreprise du fabricant que chez son mandataire.
Surveillance après commercialisation proactive : Le RDM oblige le fabricant à collecter et évaluer les données cliniques de l’utilisation pour garantir le rapport bénéfice/risque et détecter les risques ou les dommages et mettre en place des plans d’actions correctives ou préventives ou curatives [18].
EUDAMED : Le dispositif de vigilance est amélioré avec la mise en place d’une base européenne couramment appelée EUDAMED. Les fabricants, sous contrôle des organismes notifiés, sont tenus de fournir des résumés périodiques de sécurité, permettant aux patients de s'informer sur les dispositifs.
Essais cliniques renforcés : L’encadrement des investigations cliniques converge avec l'encadrement applicable au médicament pour les essais cliniques. De plus, pour les nouveaux dispositifs implantables de classe III, le recours aux investigations cliniques pour l'évaluation devient incontournable. En effet, l'organisme notifié devra consulter un panel d’experts européens sur le dossier clinique.
Amélioration de l'évaluation préalable : Renforcement des processus d'évaluation de la conformité des dispositifs médicaux avant leur mise sur le marché.
- De nouvelles exigences essentielles sont introduites, telles que la nécessité de justifier l’utilisation de substances dangereuses (CMR, "Cancérogène, Mutagène, Reprotoxique" et le Polyéthylène) ou encore pour la cybersécurité ;
- Pour les DM à base de substances absorbées : de nouvelles procédures de consultation sont instaurées pour obtenir la certification CE auprès d’une autorité compétente en matière de médicament.
Ainsi ce règlement cherchent à moderniser la réglementation des dispositifs médicaux pour refléter l'évolution des technologies, à harmoniser les pratiques au sein de l'UE, à renforcer les preuves cliniques, la surveillance préalable et post-commercialisation, et à favoriser une meilleure gouvernance du secteur, le tout dans l'intérêt de la santé et de la sécurité des patients en Europe.
2. Présentation de la période de transition (Mai 2021 - Mai 2024)
Le règlement entre en vigueur depuis mai 2017 et applicable à partir du mai 2020 selon les dispositions de l'article 123, paragraphe 2, qui prévoyait l'abrogation des directives précédentes à cette date. Cependant, cette prévision s'est confrontée à la réalité, décalant à 2024 l’application du règlement (Voir figure 2).
Figure 2 : Calendrier initial de la mise en application du règlement 2017/745 et 2017/746. Source [19]
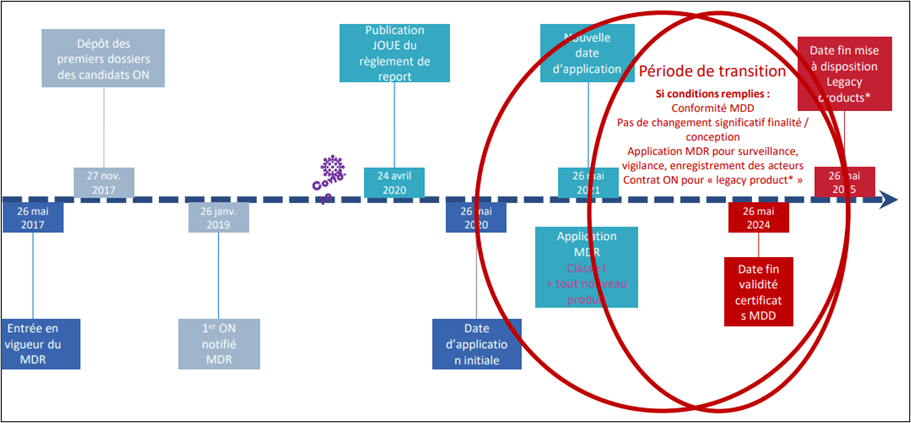
Tout d’abord, la crise de COVID-19 a eu un impact significatif dans plusieurs domaines. En France une enquête a été menée en 2021 [20] par le Syndicat National de l'Industrie des Technologies Médicales (SNITEM) illustrant les impacts liés au Covid. En R&D, les projets ont subi en moyenne une perte de 2 à 4 mois, avec des essais cliniques ralentis voire stoppés. Les restrictions comme les confinements et les protocoles hospitaliers ont limité toute visite des commerciaux d'entreprises dans les hôpitaux pendant une année. Dans le secteur financier, les négociations avec les investisseurs ont été interrompues, entraînant une baisse du chiffre d'affaires jusqu'à -20 % pour la moitié des entreprises. Concernant la production, pour 50 % des répondants, les lignes de production ont été stoppées pendant 2 semaines à 1 mois. De plus, les coûts des matières premières ont augmenté et les délais d'approvisionnement ont triplé, particulièrement dans le secteur de l’électronique.
Ensuite, le manque d’organismes notifiés MDR, qui sont des organisations indépendantes autorisées par les autorités compétentes pour évaluer la conformité des dispositifs médicaux aux exigences réglementaires. En 2019, les fabricants sont face à un seul organisme accrédité RDM. Cela a provoqué un ralentissement du processus de mise en œuvre de ce règlement, initialement prévu pour la première date d'application en 2020. Le nombre de certificats émis représente un nombre très faible en 2018, soit 4 992 [21]. La crainte de pénurie de DM, les difficultés rencontrées par les fabricants à déposer les dossiers et les organismes notifiés à répondre aux demandes de certification à été l'initiative d’une lettre ouverte [22] rédigée en avril 2019 par Medtech Europe accompagné d’une centaine de parties prenantes. Ce syndicat a joué un rôle crucial en permettant aux différents acteurs de faire valoir leurs voix auprès de la Commission européenne. Cela leur a été profitable, car la date d’application a été reportée d’un an, à 26 mai 2021, par le règlement (UE) 2020/561.
Cependant les efforts menés par les fabricants et organismes notifiés n’ont pas permis d’atteindre les objectifs de recertification. Un deuxième appel à l’aide est lancé par l’Association européenne des organismes notifiés, Team NB, auprès des instances européennes. C’est l’une des raisons pour laquelle une période de grâce est accordée jusqu’au 26 mai 2024 [23] permettant la libre circulation sur le marché de certains dispositifs, nommés “Legacy Devices”, qui sont encore conformes aux directives 90/385/CEE, 93/42/CEE et 98/79/CE.
3. Nouvelles procédures de mises sur le marché des dispositifs médicaux
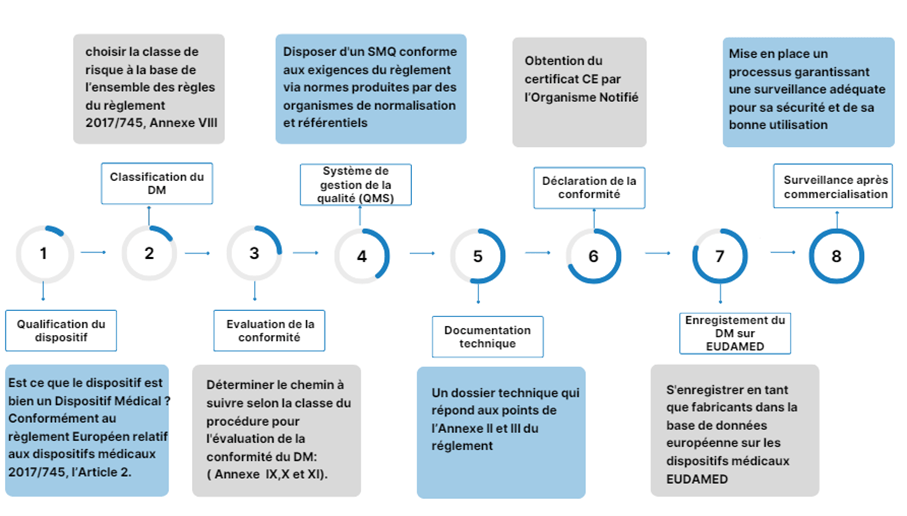
Avec l'adoption du nouveau règlement sur les dispositifs médicaux en Europe, régissant la mise sur le marché des produits, il est essentiel de décrire minutieusement les étapes de ce processus. La figure 3 ci-dessous illustre les étapes pour accéder au marché des dispositifs médicaux.
Figure 3 : Synthèse de la mise sur le marché européen d’un DM, Source Auteur.
II. État des lieux depuis 6 ans d’entrée en vigueur du Règlement (UE) 2017/745
A. Enquêtes auprès d’organismes notifiés et autorités compétentes
1. Implication des autorités compétentes : Exemple de l’ANSM en France
Les impacts liés à la mise en œuvre du Règlement (UE) 2017/745 sur les dispositifs médicaux peuvent varier d'un pays à un autre et d'une autorité compétente à une autre. Nous avons eu le privilège d’interviewer un membre de l’autorité compétente française qui est l’Agence nationale de sécurité du médicament et des produits de santé. Au travers de son regard éclairé, un témoignage édifiant émerge, soulignant l’importance cruciale de l’ANSM dans cette période de transition. Cet expert offre une perspective sur l’implication de l’ANSM dans l’accompagnement des fabricants de dispositifs médicaux. L’implication de l’ANSM s’illustre dans :
L’accompagnement stratégique : Ce professionnel confirme la démarche proactive de l’agence dans l’accompagnement des fabricants, notamment en accordant des dérogations réglementaires à des fabricants, à travers l’article 59, puis en interprétant judicieusement l'article 97. Cet article 97 autorise les Etats membres de l’UE à considérer qu’un DM qui était légalement mis sur le marché avant la date d’application du RDM peut continuer à être mis sur le marché et à être utilisé dans cet État membre, même en l’absence de certificat conforme au nouveau règlement, atténuant ainsi les potentielles ruptures.
Le rôle pédagogique : L’ANSM s'investit dans l’accompagnement des fabricants, en s’impliquant activement dans l’éducation et la sensibilisation de l’industrie. Des webinaires publics sont organisés dans le but de partager des connaissances clés concernant le RDM. Cette initiative va au-delà du simple rôle de régulateur, montrant son engagement à être un véritable partenaire dans le succès des fabricants.
Le soutien actif à l’innovation : L'expert témoigne de l'implication soutenue de l'ANSM dans la promotion de l'innovation en accompagnant les starts-up novatrices. Il évoque le contraste avec une situation antérieure, se souvenant d'un laboratoire de recherche, « 10 ans en arrière j’ai des souvenirs d’un laboratoire de recherche qui avait développé dans son petit coin, son produit et qui n’avait pas du tout tenu compte de l’aspect réglementaire et sans même connaître le contexte réglementaire, ce qui n’est plus le cas maintenant pour les starts-up qui nous contacte » témoigne-t-il. Il souligne donc que cette réalité a changé, notamment pour les start-ups qui les contactent aujourd'hui.
La surveillance rigoureuse du marché : Le professionnel souligne la posture vigilante de l’ANSM, dans la surveillance du marché pour garantir la conformité réglementaire. Les contrôles proactifs et les demandes de données auprès des fabricants, éclairent la responsabilité renforcée de l’ANSM dans la supervision active du paysage des dispositifs médicaux.
Cependant, il souligne qu’il n’y a pas d'interaction significative avec le GMED qui est le seul organisme notifié en France. Leur seul interlocuteur concernant le cycle de vie d’un DM reste le fabricant. Ainsi, l’ANSM se positionne non seulement comme une autorité de régulation, mais aussi comme un acteur proactif, collaboratif et éducatif, prêt à soutenir et à guider l’industrie des dispositifs médicaux vers l’avenir réglementaire.
2. Réorganisation des organismes notifiés
Le recrutement des organismes notifiés dans le cadre du nouveau règlement européen sur les dispositifs médicaux Règlement (UE) 2017/745 est soumis à des exigences strictes pour garantir leur compétence, indépendance et transparence. Ces organismes jouent un rôle fondamental dans l'évaluation de la conformité des dispositifs médicaux avant et après leur mise sur le marché.
Rôle des organismes notifiés : Un organisme notifié est une entité désignée par un État membre de l'Union européenne ou par d'autres nations dans le cadre d'accords spécifiques, chargée d'évaluer la conformité de produits spécifiques avant leur commercialisation. La Commission européenne publie une liste de ces organismes notifiés dans le système d’information NANDO [24].
Procédure pour être un organisme notifié : Depuis l’introduction du RDM, les organismes notifiés sont invités à de nouveau être certifiés. Pour ce faire, ils doivent postuler pour devenir à nouveau un organisme notifié dans un État membre de l'Union européenne. Ces derniers doivent démontrer qu'ils remplissent les critères spécifiques et les exigences du chapitre IV et l’annexe VII du règlement. Des évaluations sont portées sur la capacité technique, l'indépendance, les ressources humaines, les processus internes, etc. Si ces organismes répondent aux critères exigés, ils sont alors accrédités pour opérer en tant qu’organismes notifiés.
La figure 4 présente une timeline simplifiée résumant la procédure de candidature lors du dépôt d’un dossier d’un nouveau candidat :
Figure 4 : Procédure de candidature d’un futur organisme notifié. Source [25]
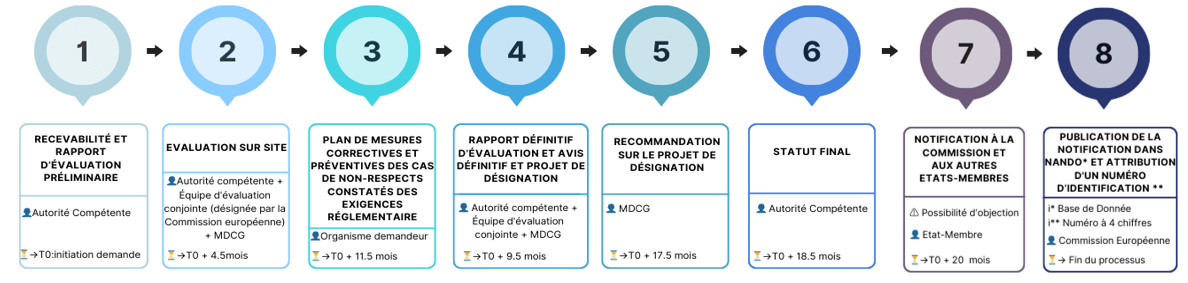
Tout ce processus est détaillé dans le guide rédigé par le MDCG. La procédure de recertification des ON est différente de celle-ci à quelques activités près. Selon le guide, le temps estimé pour avoir une réponse finale suite au dépôt du dossier de candidature est de 20 mois en moyenne [26]. Nous avons eu l'occasion d’interviewer un directeur d’un organisme notifié, il nous a confirmé que la désignation de son organisme notifié au titre de RDM a pris plus de 29 mois, indiquant que le processus est devenu plus long qu'auparavant.
Surveillance renforcée : Une fois accrédités, les organismes notifiés sont soumis à une surveillance régulière pour garantir le maintien de leurs standards de qualité, de compétence et d'indépendance. Des évaluations périodiques sont menées pour s'assurer que ces organismes continuent de respecter les exigences. En effet, ces évaluations sont réalisées en collaboration avec l'autorité nationale compétente, telle que l’ANSM en France, conjointement avec une équipe d'évaluation européenne et se poursuivent de manière continue à travers des mesures de surveillance assurées par ladite autorité nationale [27]. Les organismes notifiés doivent désormais respecter des critères de compétence plus stricts et se conformer à de nouvelles procédures obligatoires, telles que des inspections inopinées chez les fabricants et des contrôles des produits, définis dans des cahiers des charges renforcés.
🔊 Témoignage 1 : Interview d’un organisme notifié européen, Novembre 2023
Nous avons eu l’occasion d'interviewer un membre de l’ON Belge qui a souligné l’importance des ressources humaines dans cette étape de la surveillance renforcée, Ainsi, pour son organisme notifié, l’effectif a été augmenté par rapport au MDD, indiquant que avant MDR , l’équipe comptait 45 professionnels tandis qu'après la transition vers le MDR, elle compte désormais plus de 150 professionnels internes et plus de 250 auditeurs externes inclus. Cependant, le processus de recrutement a été confronté à des difficultés pour trouver des experts dans l’industrie des DM pour répondre aux exigences réglementaires, ce qui entraîne de la compétitivité entre les ON pour maintenir leurs ressources humaines.
Toutefois, il souligne l'absence d'une collaboration étroite avec d'autres organismes notifiés. Cependant, ils sont membres de l'association Team NB, axée sur les aspects réglementaires, où ils discutent et cherchent à harmoniser leurs services.
En ce qui concerne les avantages de la réorganisation des organismes notifiés, selon son point de vue aucune amélioration n'est identifiée jusqu'à présent. L'examen des objectifs émis par la Commission européenne révèle des préoccupations, notamment en ce qui concerne la stimulation de l'innovation en Europe, où ils estiment que l'innovation est entravée par ce changement. De plus, l'environnement innovant et compétitif entre les entreprises et les PME semble être impactés négativement, avec les PME et les PE étant les plus touchées. Quant au point de système de soins efficace et sécurisé pour les patients, l'élimination prévue de 25% des dispositifs médicaux suscite des doutes quant à l'atteinte de cet objectif.
🔊 Témoignage 2 : Novembre 2023
Colloque SNITEM, Septembre 2023 : Nous avons participé au Colloque où le Président du GMED a partagé les informations suivantes : Le GMED a dû ajuster son Système de Management de la Qualité (SMQ) pour répondre aux exigences du Règlement sur les Dispositifs Médicaux (RDM). Cette adaptation a nécessité le recrutement de plus d'une centaine de personnes ainsi que la création de nouveaux services. Par exemple, des chefs de projets en certification sont désormais disponibles pour accompagner les fabricants dans leurs projets. Actuellement, le processus de mise sur le marché d'un nouveau produit conforme au RDM prend 2 ans, comparé à 1 an sous les directives précédentes.
Interview Auditeur , Novembre 2023 : Nos échanges avec un auditeur ont mis en évidence que l'introduction du RDM a profondément perturbé la routine quotidienne tant des auditeurs que des examinateurs. Ces derniers se voient désormais contraints d'anticiper et de planifier l'intégralité de leurs rendez-vous. Pour certains, cela implique de connaître jusqu'à un an à l'avance les visites et les rencontres qu'ils auront à effectuer. De plus, il y a une évaluation continue des compétences des auditeurs grâce à la mise en place d'une cellule de qualification dédiée, qui évalue régulièrement leurs capacités.
3. L’évolution du nombre d’organismes notifiés, certificats et coût
a) L’évolution du nombre d’organismes notifiés
L'une des conséquences majeures de l'application du nouveau règlement RDM (2017/745) est l'évolution du nombre d'organismes notifiés. Avant le RDM, en 2012, le nombre d'organismes notifiés s'élevait à 86 dans l’UE. Cependant, après l'adoption du règlement d'exécution 2013/920 relatif à la désignation et au contrôle des organismes notifiés au titre de la directive 90/385/CEE et de la directive 93/42/CEE en 2013, ce nombre a diminué, passant de 86 à 59 en 2016. Ensuite, après la mise en application de la RDM, le nombre d'organismes notifiés actifs a encore diminué pour atteindre 20 en 2020.
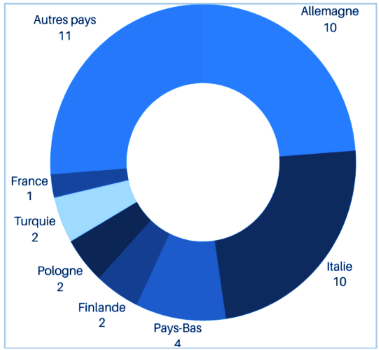
À l'heure actuelle, 42 organismes notifiés ont été désignés dans le cadre du RDM. Parmi ces 42 organismes notifiés, 31 le sont en vertu du RDM, 10 le sont à la fois en vertu du RDM et de l’IVDR, et un seul organisme notifié en vertu de l’IVDR. Il convient de noter qu’il existe une forte disparité géographique, comme le montre Figure 5, avec un seul ON en France et deux autres en cours d’évaluation [28]. Malgré l’augmentation du nombre d’organismes notifiés par rapport à 2020, le chiffre reste toujours insuffisant pour satisfaire aux demandes de certifications de l'ensemble des fabricants de dispositifs médicaux en Europe, ce qui représente plus de 33 000 entreprises [29].
Figure 5 : Répartition géographique d'organismes notifiés dans l’Union européen. Source [28].
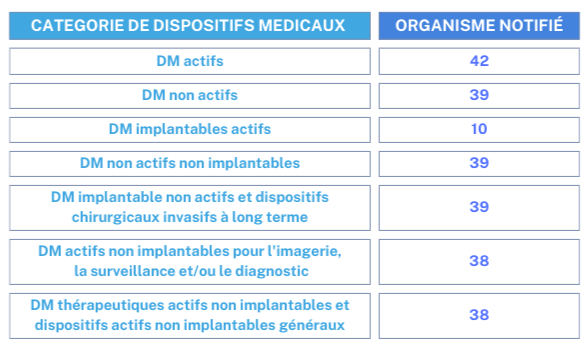
Les organismes notifiés (ON) conformes aux exigences du règlement sont capables de certifier différentes catégories de dispositifs médicaux (tableau 2). Il est important que les fabricants de dispositifs médicaux choisissent un ON compétent et habilité à évaluer la conformité de leur dispositif en fonction de sa catégorie spécifique. La base de données NANDO permet de visualiser ces informations spécifiques.
Tableau 2 : Répartition des organismes notifiés selon la catégorie des dispositifs médicaux. Source [28]
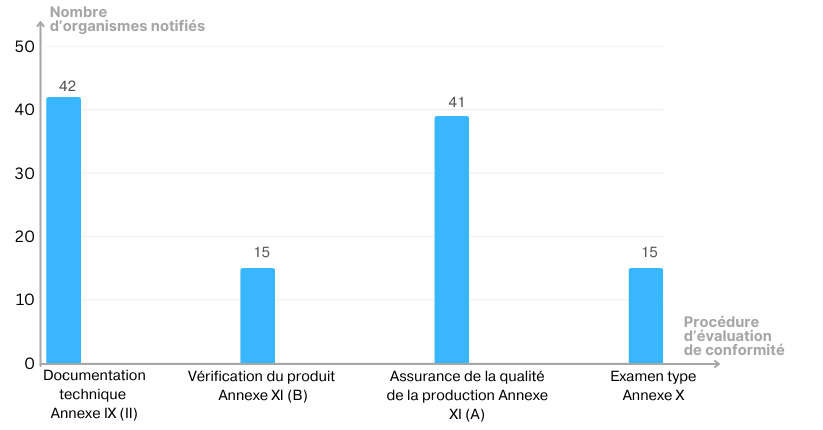
Enfin une représentation des organismes notifiés selon la procédure d’évaluation de conformité (Voir Figure 6).
Figure 6 : Répartition des organismes notifiés selon la procédure d'évaluation de conformité[28]
b) Évolution des demandes de certification en fonction des classes de DM
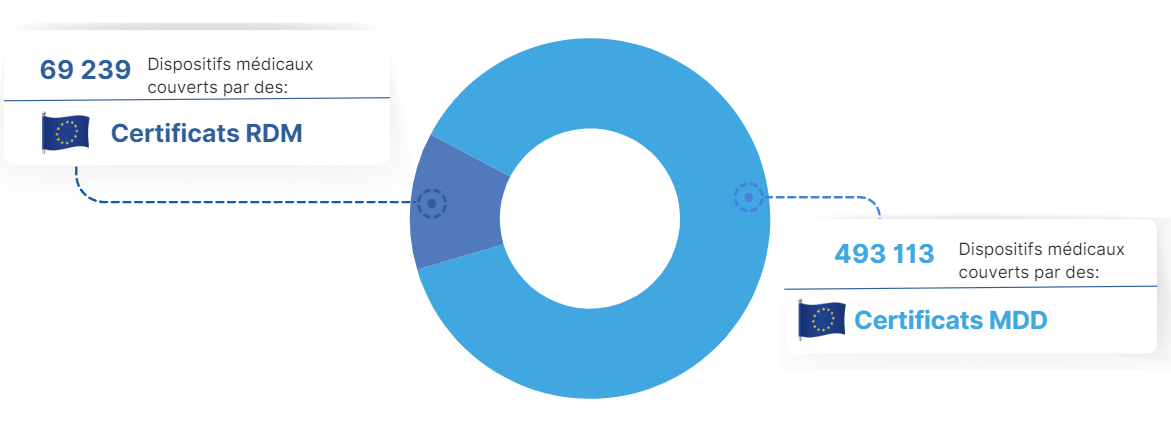
L'enquête de Medtech Europe publiée en 2022 permet d’avoir une vision globale des impacts. Selon les chiffres, parmi les plus de 500 000 dispositifs médicaux disponibles sur le marché, une grande partie reste couverte par des certificats MDD (figure 7). [30]
Figure 7:Répartition des dispositifs médicaux en fonction des certificats MDR/MDD , Source [30]
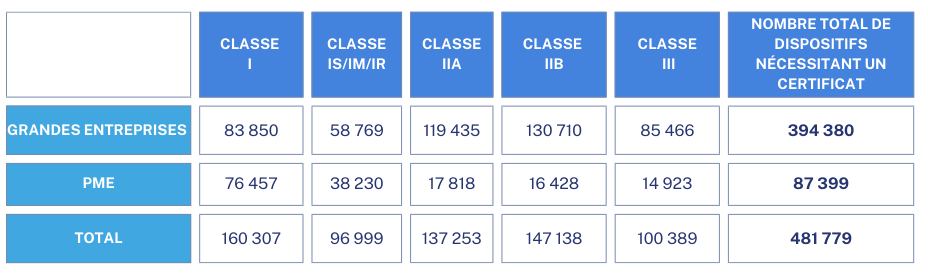
Ensuite le tableau 3 présente la répartition du nombre prévu de dispositifs sous le Règlement sur les dispositifs médicaux (RDM) classés par classe de dispositif (Classe I, Classe Is/Im/Ir, Classe IIa, Classe IIb et Classe III) et fait la distinction entre les grandes entreprises et les PME .[30]
Tableau 3:Répartition des dispositifs médicaux en fonction de la classe et taille des entreprises, Source [30]
c) L’évolution du nombre des demandes de certifications et les certificats délivrés en 2023
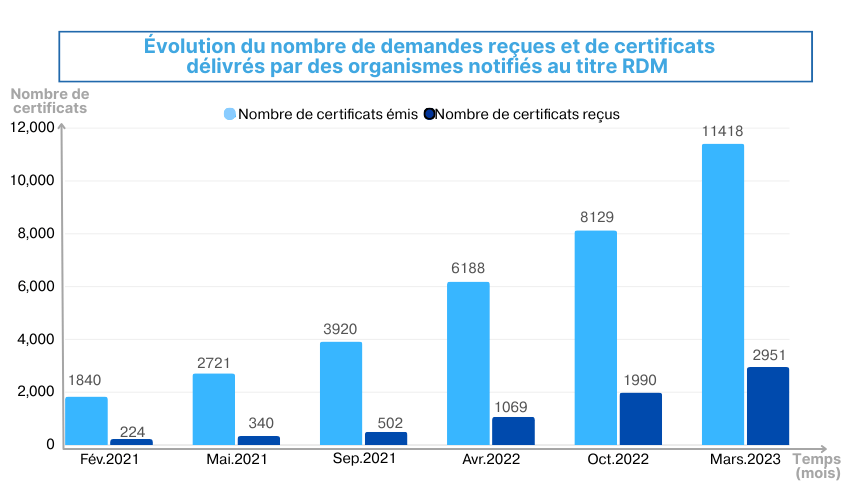
D’après la dernière enquête réalisée par la Commission européenne, le 23 mars 2023, les organismes notifiés actifs ont reçu 11 418 demandes de certification de la part des fabricants, réparties en 7 551 demandes pour la certification SMQ et 3 867 pour la certification du produit. Cependant, seulement 2 951 certificats ont été délivrés au titre de RDM, comprenant 1 954 certificats pour SMQ et 997 certificats pour le produit, ce qui représente un nombre moyen de 95 certificats délivrés par l'organisme notifié actif. En revanche, 22 793 certificats délivrés au titre du MDD restent encore actifs, et sur ces 22 793, 4 311 certificats vont expirer en fin 2023 et 17 095 certificats au cours des cinq premiers mois de 2024 [31] (Voir figure 8).
Figure 8 : Évolution du nombre des demandes reçues et de certificats délivrés par les ONs au titre du RDM. Source [31].
Pour mieux appréhender la réalité du processus de certification au sein de l'industrie des dispositifs médicaux, voici quelques chiffres extraits de la dernière enquête réalisée par la Commission européenne auprès des organismes notifiés [31]:
- Délai moyen entre une demande déposée et un accord signé : 2-3 mois.
- Délai moyen entre une demande déposée et la réception de la certification :
- Certificat système qualité et produit : 13 à 18 mois
- Certificat système qualité : 6 à 12 mois
- Pourcentage des demandes satisfaisantes en termes de documentation fournie :
- 55% des ONs déclarent que moins de 25% des demandes sont satisfaisantes
- 32% des ONs déclarent qu’entre 25% et 50% des demandes sont satisfaisantes
- 8% des ONs déclarent que plus de 75% des demandes sont satisfaisantes
- 5% des ONs déclarent qu’entre 21% et 75% des demandes sont satisfaisantes.
- Les principales raisons de refus des dossiers (mars 2023) :
- Demande hors champ de désignation de l’organisme notifié. Le témoignage du directeur de l'organisme notifié belge confirme que pour eux la principale cause de rejet des dossiers réside dans le fait que la demande est en dehors du champ de désignation de l'ON.
- Dossier incomplet
- Mauvaise qualification du produit / classification du DM incorrecte
- Ressources humaines insuffisantes des organismes notifiés car le RDM impose aux ON une charge de responsabilités plus importante, notamment la rédaction des rapports SSCP (Résumé des caractéristiques de sécurité et des performances cliniques) et PSUR (Rapport de sécurité périodique actualisé)
- Mauvais choix de procédure d'évaluation de la conformité
Les écarts entre les demandes de certification et les certifications délivrées, ainsi que les retards et les possibles refus, suscitent de vives inquiétudes chez les fabricants de dispositifs médicaux. Ces derniers craignent que ces retards et refus potentiels compromettent leur capacité à fournir des dispositifs médicaux essentiels, mettant en danger la santé des patients.
d) Tarification des organismes notifiés
Depuis l’entrée en vigueur du règlement 2017/45, les exigences réglementaires pour avoir la certification sont devenues plus strictes en ce qui concerne l’évaluation de la conformité et la surveillance post-commercialisation. Tout cela génère des coûts supplémentaires pour les fabricants. De plus, rappelons que le règlement 2017/45 des dispositifs médicaux a établi une obligation plus stricte pour les organismes notifiés. Cette obligation vise à garder la transparence et l’équité dans le processus de certification des DM. En outre, cela signifie que les organismes notifiés sont tenus de rendre les tarifs de leurs services accessibles aux publics.
Le coût de certifications varie en fonction de nombreux facteurs, notamment la taille de l’entreprise, la complexité du processus et de la structure organisationnelle de chaque entreprise, et de la classe de risque du DM, etc. Le coût global d’une certification regroupe l’ensemble des coûts suivants :
- Les charges administratives, frais du transport.
- Audit inopiné, Audit SMQ, certification.
- Revue de la documentation technique, revue de la documentation clinique.
Pour donner une visibilité concrète sur la tarification des organismes notifiés, nous avons réalisé un tablea (Tableau 4) comparatif des tarifs de quelques organismes notifiés selon le RDM à partir des tarifs publiés sur les sites des ONs :
Tableau 4 : Tarifs des organismes notifiés pour le règlement des dispositifs médicaux.
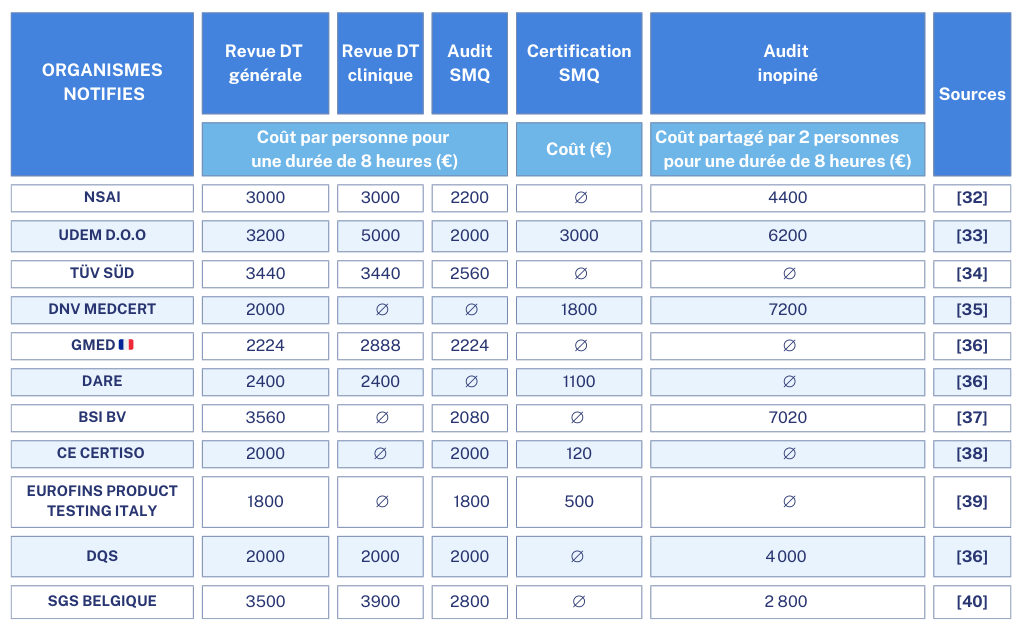
Selon l'analyse comparative des tarifs (tableau 4), il apparaît que parmi les organismes notifiés, seuls l’UDEM et SGS se conforment au règlement en fournissant des informations détaillées sur leurs tarifs et en prenant en considération la taille des entreprises.
B. Enquêtes auprès des fabricants
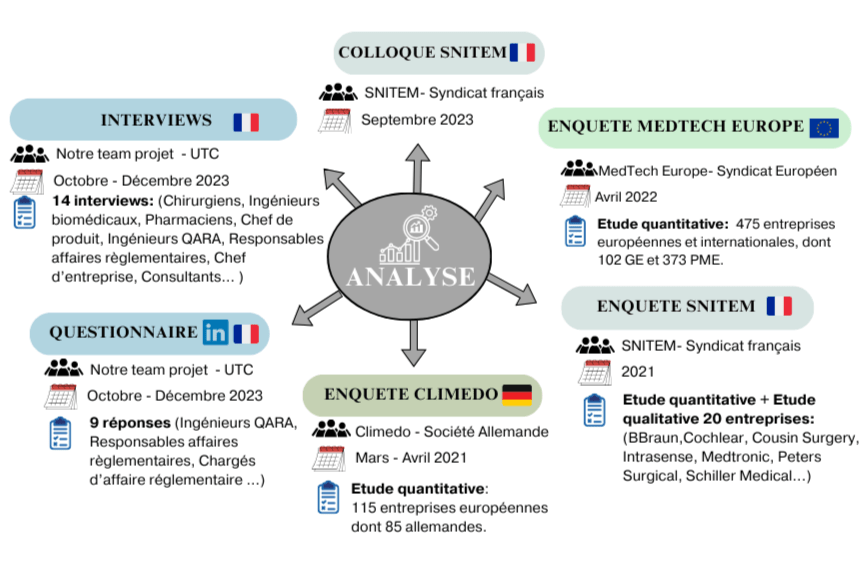
Dans cette section, il est pertinent d'examiner les défis auxquels sont confrontés les fabricants pendant cette période transitoire. Pour ce faire, des enquêtes sur le terrain ont été entreprises afin de recueillir des informations auprès des fabricants et des professionnels de la santé. Nous avons également pris part à un colloque organisé par le SNITEM [19] et diffusé un questionnaire sur LinkedIn (Annexe 1). De plus, pour obtenir davantage de données, nous avons analysé trois enquêtes dressant un bilan depuis l'introduction du dispositif médical réglementaire : Snitem [20], Medtech Europe [30] et Climedo [41] (Voir figure 9)
Figure 9 : Représentation des ressources utilisées, Source Auteur.
1. Principales difficultés rencontrées pendant la phase de transition
La compréhension et interprétation des nouvelles règles : La nouvelle réglementation est souvent complexe et nécessite une compréhension approfondie pour assurer la conformité. Plus de la moitié des répondants, soit 55 %, consacrent désormais plus de 5 heures par semaine pour se conformer aux demandes spécifiques du RDM (Climedo, 2021). Ce surcroît d'investissement en temps souligne l'impact considérable que cette réglementation a sur la charge de travail des entreprises concernées. Les activités principales auxquelles ce temps supplémentaire est consacré sont principalement axées sur la "compréhension des nouvelles exigences", identifiée par 68% des participants comme étant la priorité. Ensuite, viennent les "évaluations cliniques et les études cliniques", mentionnées par 63 % des entreprises, soulignant l'importance de ces aspects pour la conformité au RDM (Climedo, 2021).
Les coûts accrus : Le RDM a été facteur de l'augmentation des coûts et des ressources pour 70% des sondés. Une grande partie des ressources sont allouées à la conformité, aux tests, à la documentation ainsi qu’aux évaluations cliniques et études cliniques. Les activités de surveillance et suivi clinique post-commercialisation absorbent 52 % des ressources financières pour certaines entreprises. 31 % des entreprises estiment que le RDM entraînera dans les années à venir des coûts en plus compris entre 5 % et 10% de leur chiffre d'affaires annuel. Ces chiffres soulignent l'impact financier croissant que cette réglementation aura sur ces entreprises, pouvant mettre en péril leur santé financière. (Climedo, 2021)
La mise en place de systèmes de qualité et de suivi post-commercialisation renforcée : Les exigences en matière de gestion de la qualité et de surveillance après mise sur le marché sont renforcées, ce qui nécessite des mises à jour importantes des processus internes. Une interview (Annexe 2) avec un gestionnaire de réclamation en matériovigilance témoigne de la difficulté d’ajouter ces aspects de vigilance car les ressources documentaires sont limitées et la méthodologie reste propre à chaque entreprise. (Interview PME, 2023)
Interaction avec les organismes notifiés : La nécessité d'une interaction avec les organismes notifiés pour la certification et l'approbation des produits est un défi. Particulièrement pendant la période de transition où ces organismes sont eux-mêmes en train de s'adapter aux nouvelles réglementations. Pour certains fabricants, il s’avère que c’est très difficile de trouver un ON au titre de RDM, soit 40% des participants de l'enquête Climedo. (Climedo, 2021)
Les délais : L'enquête de Medtech Europe met en lumière que les délais de certification peuvent varier en fonction de la classe du dispositif médical et des spécificités du processus de certification. En effet les nouveaux DM et les DM de classe III sont sujets à des délais de traitement plus longs allant jusqu’à plus de 24 mois. Ce qui ralentit le déploiement des produits sur le marché, entraînant des pertes de marché pour certaines entreprises. (Medtech Europe, 2022)
La réévaluation et reclassement des produits existants : Certains dispositifs médicaux sont reclassés dans une catégorie de risque plus élevée, ce qui exige une réévaluation et une conformité accrues. Cela implique des tests supplémentaires, la collecte de nouvelles données cliniques, et une documentation plus détaillée. Depuis l'implémentation du RDM, 54 820 dispositifs médicaux de classe I, dont 78 % appartiennent à de grandes entreprises, doivent être reclassés selon les exigences du RDM. Ce chiffre représente 8 % du total des legacy devices en circulation sur le marché. Ces dispositifs étaient auto-déclarés et doivent désormais obtenir une certification selon le RDM (Snitem, 2022). Un directeur scientifique témoigne sur la transition d'un défibrillateur de classe IIb à la classe III. Ce changement de classe a suscité de nombreuses questions et difficultés, les exigences supplémentaires pour la classe III, notamment en ce qui concerne la documentation clinique, ont suscité des ressources significatives et une expertise accrue (Interview PME, 2023).
Les risques de pénuries de produits sur le marché : Les entreprises ne parviennent pas à se conformer dans les délais, ce qui entraîne des pénuries de certains dispositifs médicaux sur le marché. En 2022, 66% à 80% des industriels français sont favorables pour renoncer à une partie de leurs produits, ce qui représente environ 150 000 dispositifs médicaux [42]. A l'échelle européenne, 54% des répondants ont déclaré qu'ils n'ont pas l'intention de faire passer une partie de leur portefeuille sous le RDM. Globalement, les portefeuilles des fabricants seront réduits en moyenne de 20%, ce qui est significatif. (Medtech Europe, 2022). Le graphique ci-dessous représente les types de DM les plus susceptibles d'être rationalisés ou cessés d'être mis sur le marché à long terme. La taille des carrés est proportionnelle au nombre de réponses reçues. (figure 10)
Figure 10 : Treemap des types de DM susceptibles de quitter le marché. Source [30]

La gestion des stocks et du cycle de vie des produits : Les entreprises doivent gérer efficacement les stocks des produits existants tout en assurant la transition vers les nouvelles spécifications pour les produits futurs.
Compétitivité et Innovation : La transition des Legacy Devices vers le RDM a généré du retard dans la mise sur le marché de nouveaux produits. Tout cela entraîne une perte de compétitivité chez les fabricants.
2. Impacts selon la taille de l’entreprise
Après avoir analysé les impacts à une échelle macroscopique, il est intéressant de voir comment ils se répercutent en fonction de la taille des entreprises. Tout d’abord il est essentiel de rappeler que les entreprises peuvent se présenter sous différents types selon leurs activités. On distingue la GE (Grande Entreprise), l’ETI (Entreprise de Taille Intermédiaire), la PME (Petite et Moyenne Entreprise), la TPE (Très Petite Entreprise). (Voir figure 11)
Figure 11 : Taille des entreprises. Source [43]

- Start-ups et TPE
Les petites entreprises sont les premières à ressentir les répercussions du Règlement sur les Dispositifs Médicaux (RDM) en raison de leurs ressources et de leur capacité limitées à se conformer aux exigences réglementaires strictes. Elles rencontrent des défis pour mener à bien les tests cliniques et financer les coûts associés à la mise en conformité. Lors du colloque du SNITEM [19], il a été mentionné le destin tragique des petites entreprises. Ces dernières, incapables de réunir les fonds nécessaires pour obtenir leur marquage CE, se retrouvent malheureusement contraintes à mettre la clé sous le tapis.
De plus, lors des interviews, les dirigeants de start-up innovantes se retrouvent seuls et peu accompagnés. Le coût d’une certification peut atteindre 450 000€ [44], ce qui représente un coût important pour des startup qui ont très peu de moyens. Cela nécessite de trouver et convaincre de plus en plus d’investisseurs, ce qui n’est pas facile, nous a confié la CEO d’une start up d’un dispositif médical numérique (Interview, 2023).
- PME
Les petites et moyennes entreprises (PME) opérant dans le domaine des dispositifs médicaux représentent la plus grande part du marché des DM. Les PME sont fortement impactées, car elles sont confrontées à des coûts additionnels pour se conformer. En 2021, le président de Vygon a exprimé son inquiétude concernant les coûts engendrés pour se conformer, mentionnant que l'entreprise familiale a dépensé plus de 12 millions d'euros [45] à cet effet.
La complexité du RDM a nécessité une révision approfondie des documentations techniques, des méthodes de travail, ainsi que l'acquisition de nouvelles compétences pour répondre à des exigences plus strictes. En terme de coût, il ressort de notre questionnaire (Annexe 1) que les PME consacrent un budget assez important allant de 200 000€ à plus d’1 000 000€ pour être conforme au RDM.
L'enquête de Medtech Europe révèle qu’au moins 15% à 30% PME interrogées n’ont pas d’organisme notifié ou ont recours à un organisme notifié qui n'est pas encore désigné pour être conforme au RDM [30].
De plus, certains fabricants ont réduit leur portfolio, en retirant certaines gammes sur le marché comme vu dans l'enquête de Medtech Europe. Selon les informations de notre questionnaire, plus de la moitié des répondants ont dû modifier leur portfolio. Par exemple, une PME d’un dispositif de classe IIa, a répondu au questionnaire qu’elle se limite désormais à un seul produit. (Annexe 1)
- ETI
Les entreprises à taille intermédiaire pourraient être mieux positionnées pour se conformer aux exigences du RDM par rapport aux PME et TPE. Elles disposent souvent de plus de ressources financières et de capacités techniques leur permettant de répondre et s'adapter aux exigences élevées du règlement. En effet c’est le cas du groupe Menix, spécialisé en orthopédie, en implantologie dentaire et chirurgie cranio-maxillo-faciale ; qui a partagé son expérience lors du colloque [19]. Le président du groupe affirme qu’ils ont déposé 100% de leur dossier auprès de leurs organismes notifiés, mais jusqu’à présent ils n’ont pas eu de retour sur plusieurs dossiers. En terme de coût sous la MDD le renouvellement d’un legacy device leur coûtait 150 000€ aujourd’hui c’est moyennement 500 000€.
Ces défis nécessitent une planification stratégique, des ressources adéquates, et une collaboration étroite entre toutes les parties prenantes pour assurer une transition réussie vers les nouvelles normes de classification des dispositifs médicaux.
- GE
Les grandes entreprises se démarquent par leur meilleure adaptation aux changements réglementaires. Environ 95% de ces entreprises disposent déjà d'organismes notifiés conformes au règlement. En effet, un grand nombre d'entre elles ont anticipé, facilitant ainsi la transition de leurs anciens dispositifs vers les nouvelles réglementations. En raison de leurs ressources financières importantes, les grandes entreprises ont plus de facilité à obtenir la certification nécessaire, contrairement aux PME, comme le montre clairement Figure 12 ; Ces chiffres démontrent que le passage au MDR est moins fréquent pour les petites entreprises que pour les grandes.
Figure 12 : Comparaison des dispositifs médicaux sous MDR / MDD en fonction de la taille des entreprises. Source [30]

3. Stratégies adoptées par les fabricants pour être en conformité avec le règlement
Pour faire face aux impacts du RDM, différentes stratégies ont été adoptées par les entreprises du secteur. Tout d'abord, l'une des approches privilégiées est le recrutement de personnel supplémentaire. D'après l'enquête menée par Climedo Health en 2021, environ 67 % des entreprises interrogées [41] ont déjà embauché ou envisagent de recruter au moins un nouvel employé. Ce renforcement des effectifs vise à mieux gérer et répondre aux exigences imposées par le RDM. Lors du colloque du SNITEM [19] en 2023, le groupe Menix a adopté la même stratégie, en ajoutant 30 nouveaux employés (chargés d'affaires réglementaires, des spécialistes en affaires cliniques et des employés de bureau d'études…) aux 132 employés, au cours des cinq dernières années.
Pour continuer, parmi les réponses obtenues dans le questionnaire que nous avons diffusé, certaines entreprises ont choisi de faire appel à des consultants spécialisés dans les affaires réglementaires, pour un coût moyen de 60 000€ (Annexe 1). Lors de nos interviews, 3 consultants ont pu partager leur retour d'expérience notamment en accompagnant et formant leur clients sur les exigences du RDM (Annexe 3).
Ensuite d'autres entreprises ont pris la décision de changer d'organisme notifié pour des raisons diverses. Un sondé a indiqué que leur ancien organisme notifié ne satisfaisait pas aux exigences du RDM. Un autre a fait le choix de changer d’ON pour des raisons économiques, de délais plus avantageux proposant des revues de dossier plus efficaces et rapides.
Enfin, une autre stratégie mentionnée dans le questionnaire (Annexe 1), le colloque du SNITEM, et dans l'enquête de Medtech Europe, est de vendre les nouveaux DM hors Europe. En effet depuis mai 2021, environ 4306 nouveaux dispositifs ont été déployés sur le marché américain [30]. Comme représenté dans la figure 13, les États-Unis occupent la première place devant l'Europe pour le déploiement des DM. Cette solution est choisie car la procédure de certification est “plus simple, moins coûteuse et rapide”, nous confie ingénieure QARA pendant une interview (Annexe 4).
Figure 13 : Localisation privilégiée des entreprises pour une approbation réglementaire, Source [30]

C. Impact du Règlement (UE) 2017/745 dans le domaine hospitalier
1. Impact sur l’approvisionnement en dispositifs médicaux
Après avoir analysé les impacts chez les ON, les autorités compétentes et les fabricants, détaillons comment le secteur hospitalier est aujourd'hui confronté aux défis du RDM notamment dans l'approvisionnement en dispositifs médicaux. L'entrée en vigueur du nouveau RDM a considérablement modifié la donne, suscitant des préoccupations majeures au sein des établissements de santé. Pour mieux comprendre l'impact de ces changements sur le terrain, dans un premier temps nous avons recueilli le témoignage d'une pharmacienne (Annexe 5) spécialisée en hospitalisation à domicile, qui œuvre directement au contact des patients et des équipes de soins. Dans un second temps, nous avons relevé une étude menée au sein d’un centre hospitalo-universitaire français entre novembre 2019 et septembre 2020 [46]. Suite à ces ressources nous avons pu identifier divers impacts :
Pénuries de DM et causes : Depuis l'implémentation du nouveau RDM, l'approvisionnement en DM à l'hôpital a été touché par des pénuries. “Les fabricants évoquent plusieurs raisons. Il n’y a pas vraiment de transparence. Certains évoquent des pénuries de matières premières, d'autres pointent du doigt des composants manquants, mais dans l'ensemble, les explications demeurent rares” déplore la pharmacienne.
Impact budgétaire : En ce qui concerne l'impact budgétaire, elle confirme une hausse des budgets alloués aux DM en raison du nouveau RDM, mais elle ne dispose pas de chiffres précis.
Détérioration de la disponibilité : La pharmacienne souligne également les retraits de DM, tels que les tampons obturateurs anaux, sans explications valables de la part des fabricants, ce qui a contribué à une détérioration de la disponibilité de ces dispositifs médicaux. La situation devient particulièrement problématique car il n'existe pas d'alternatives viables pour ces produits. Il arrive même que la pharmacienne se trouve dans la difficile position d'informer les patients de la rupture de stock, les privant ainsi de soins appropriés. Cette réalité souligne l'urgence de trouver des solutions pour atténuer l'impact direct sur les patients.
Réponses aux appels d'offres : Concernant les réponses aux appels d'offres, la pharmacienne souligne que son service n'est pas directement impliqué dans ces procédures.
Catégories de DM les plus touchées : Elle ajoute que les DM de la catégorie des injectables rencontrent le plus de problèmes de disponibilité. Cependant, le DM qui fait face à le plus de problème en termes d’indisponibilité sont les sondes urinaires, leurs disponibilités sont compromises malgré un changement de fournisseur.
🔊 Témoignage : AP-HP/ CHU Amiens/ CH Compiègne, 2023 À la suite de nos échanges avec divers professionnels de la santé, notamment des neurochirurgiens, des pharmaciens hospitaliers et des ingénieurs biomédicaux, il est évident que les hôpitaux sont principalement impactés par le Règlement sur les Dispositifs Médicaux (RDM). En effet, dans certains services, des dispositifs tels que des vis, des seringues auto-injectables, des pompes à perfusion, etc., se font rares, car les fabricants n'ont pas nécessairement souhaité renouveler leurs certifications. Par conséquent, les établissements hospitaliers sont contraints de trouver de nouveaux marchés, souvent à des coûts plus élevés.
Une étude menée au sein du centre hospitalo-universitaire français a quantifié ces problèmes à une plus grande échelle. Entre novembre 2019 et septembre 2020, 96 désignations de produits de DM stériles ont été signalées en rupture ou arrêt de commercialisation, concernant un total de 402 références. Cette situation a eu un impact considérable, touchant principalement les DM de classe IIa, IIb et III, couvrant diverses catégories de DM.
Les pénuries de DM et les retraits de produits entraînent des retards dans les traitements médicaux, affectant ainsi la qualité des soins. La figure 5 illustre les pics de ces ruptures d’approvisionnements.
Figure 14 : Distribution mensuelle des déclarations de rupture d’approvisionnement ou d’arrêt de commercialisation. Source [46]
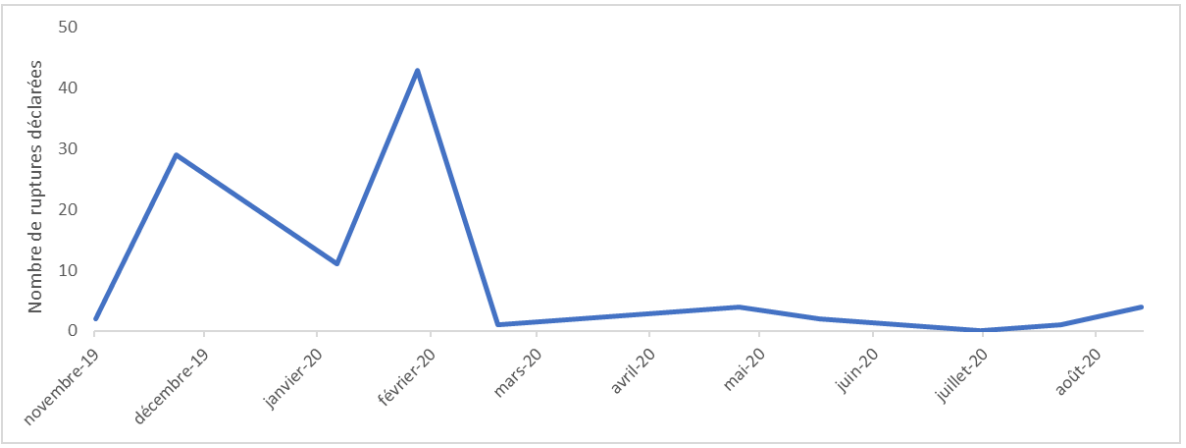
82.4% des déclarations étant survenues entre novembre 2019 et février 2020. Le niveau de déclaration a diminué considérablement à partir de mars 2020, parallèlement avec l’évolution de la crise sanitaire pandémique de COVID-19.
L'étude a évalué les conséquences économiques de ces ruptures, révélant un coût mensuel estimé à 3052 euros aux Hospices Civils de Lyon pour la gestion de ces 96 désignations produits de dispositifs médicaux stériles déclarées en rupture ou arrêt.
En conclusion, l'impact sur l'approvisionnement en DM est un défi majeur auquel le secteur hospitalier est confronté, avec des implications à la fois cliniques et économiques. La combinaison du témoignage de la pharmacienne en hospitalisation à domicile et des résultats de l'étude met en évidence la nécessité cruciale de trouver des solutions pour atténuer ces perturbations, garantir la continuité des soins de qualité pour les patients et maintenir une gestion budgétaire efficace au sein du secteur hospitalier.
2. Stratégies adoptées par le domaine hospitalier
Pour faire face à aux difficultés énumérées précédemment et garantir la continuité des soins aux patients, professionnels de la santé ont dû mettre en place des approches stratégiques. Nous pouvons porter notre regard sur les stratégies adoptées par les Hôpitaux Universitaires de Strasbourg [47]. Ces stratégies comprennent :
L’anticipation des retards de livraison : Une surveillance quotidienne des délais de livraison est effectuée au moyen de requêtes informatiques. Cette démarche permet d'entrer rapidement en contact avec les fournisseurs en cas de retard, de recueillir des informations sur les raisons du retard et d'estimer le délai nécessaire pour rétablir l'approvisionnement. Les commandes de dispositifs médicaux en stock sont planifiées en fonction de critères spécifiques, tels que le seuil de sécurité, la fréquence des commandes, les quantités prévisionnelles annuelles et les coûts unitaires.
La priorisation des ruptures : L'évaluation des délais de livraison annoncés et de l'état des stocks au sein de la pharmacie à usage intérieur et des services de soins permet de déterminer quelles ruptures nécessitent une intervention immédiate. Cette analyse vise à identifier les ruptures qui doivent être traitées en priorité et celles qui peuvent attendre.
La validation des substitutions : Afin d'assurer la continuité des soins, les pharmaciens valident les solutions de remplacement en collaboration avec les unités de soins concernées et les services spécialisés tels que le service d'hygiène ou la médecine du travail. Cette validation fait suite à une phase d'anticipation et de priorisation, qui permet d'envoyer des échantillons des dispositifs de substitution aux services pour évaluation. L'objectif est de s'assurer que les dispositifs de substitution répondent aux besoins du service.
L’accompagnement des équipes soignantes et médicales : Lorsqu'une substitution est mise en place, les pharmaciens communiquent avec les équipes soignantes et médicales en envoyant des notes d'information aux services concernés. Ces notes expliquent les différences entre les dispositifs médicaux stériles en rupture et les dispositifs de substitution. Les chefs de service et les cadres de santé sont responsables de diffuser ces informations aux professionnels de l'unité de soins. Il est important de noter que certaines ruptures appartenant à la même gamme ont été regroupées en une seule note d'information pour réduire leur nombre.
La mobilisation des ressources : Pour mettre en œuvre efficacement ces stratégies, il est essentiel de mobiliser les ressources informatiques et techniques nécessaires pour faire face aux ruptures d'approvisionnement en dispositifs médicaux.
Ces approches stratégiques globales reflètent l'engagement du secteur hospitalier à maintenir la continuité des soins malgré les défis posés par les ruptures d'approvisionnement en dispositifs médicaux stériles.
3. Expérimentation des dispositifs médicaux
L'article 61.1 du règlement sur les dispositifs médicaux stipule que lorsqu'un fabricant demande une évaluation de conformité de son dispositif médical à un organisme notifié, il doit inclure les résultats d'une évaluation clinique dans le dossier technique. Cette évaluation clinique est définie comme un processus planifié et systématique visant à collecter, analyser et évaluer en continu des données cliniques liées au dispositif, dans le but de confirmer sa sécurité, sa performance et ses avantages cliniques. Elle doit fournir des preuves cliniques adéquates pour étayer la conformité du dispositif avec les exigences réglementaires. (article 2.44 du RDM)
Pour les dispositifs médicaux implantables et de classe III, le règlement sur les dispositifs médicaux exige la réalisation d'investigations cliniques, conformément à l'article 61.4. Ces études cliniques doivent démontrer un bénéfice clinique défini comme l'impact positif d'un dispositif sur la santé d'un individu, mesurable par des résultats cliniques significatifs et pertinents pour le patient, comme stipulé à l'article 2.53 du RDM. Par conséquent, bien que la nouvelle réglementation n'aborde pas explicitement l'efficacité clinique, elle renforce la nécessité de disposer de preuves cliniques solides. Les nouvelles exigences impliquent que les fabricants devront conduire un nombre accru d'études cliniques avant de commercialiser leurs dispositifs.
Désormais, tout fabricant qui souhaite obtenir l'approbation de son dispositif en se basant sur l'équivalence avec un dispositif déjà sur le marché doit établir un contrat formel avec le fabricant de ce dernier. Ce contrat doit garantir un accès complet à la documentation et aux données cliniques du dispositif de référence, conformément à l'article 61.5 du RDM. Les critères qui doivent être satisfaits avant de pouvoir envisager l'équivalence englobent des aspects techniques, biologiques et cliniques, comme précisé dans l'Annexe XIV du RDM, section 3. Par exemple, le nouveau dispositif doit présenter une conception similaire, être fabriqué à partir des mêmes matériaux en contact avec les tissus humains et être destiné au traitement de la même condition clinique que le dispositif de référence. Ces mesures pourraient potentiellement entraîner une réduction du nombre d'approbations de dispositifs à haut risque basées sur l'équivalence.
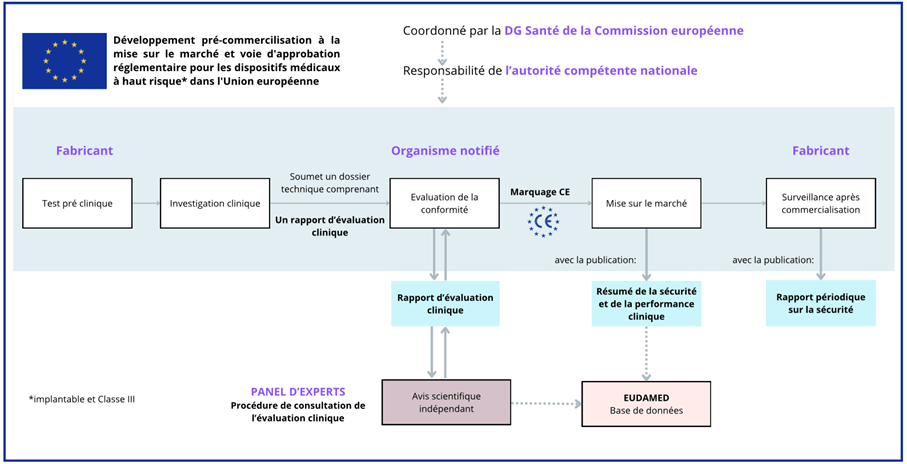
Figure 15 : Représentation schématique du processus d'approbation et de la procédure de consultation pour l'évaluation clinique des dispositifs médicaux à haut risque. Source [48].
III. Limites et perspectives
A. Comparaison des objectifs initiaux du Règlement (UE) 2017/745 vs la réalité du terrain
Les objectifs initiaux du RDM visent à renforcer à la fois la transparence, la traçabilité et la sécurité des patients comme énoncé dans le chapitre 1. Toutefois, la réalité du terrain démontre que ce n’est pas encore le cas (Voir Tableau05).
Tableau 5 : Comparaison des objectifs initiaux du Règlement (UE) 2017/745 vs la réalité du terrain
| OBJECTIFS DU RDM | RÉALITÉ DU TERRAIN |
| Améliorer la qualité, la sécurité et la fiabilité des dispositifs médicaux | Les fabricants sont confrontés à des défis pour se conformer aux nouvelles exigences en matière d'évaluation des dispositifs médicaux. Cela a entraîné des retards dans la mise sur le marché de certains produits ou des pénuries des DM, pouvant mettre en danger la vie des patients. Le nombre des organismes notifiés est insuffisant pour répondre aux demandes des fabricants, ce qui entraîne des délais d'évaluation plus longs. |
| Assurer la transparence des informations : EUDAMED | La base de données EUDAMED a eu du retard pour sa mise à disposition et jusqu’à présent, il n’y a pas de visibilité sur la date à laquelle cette base sera opérationnelle. |
| Favoriser l’innovation tout en garantissant la sécurité des patients | La réalité du terrain, telle qu'elle ressort du projet « Coordinating Research and Evidence for Medical Devices (CORE-MD), financé par l'UE dans le cadre du programme Horizon 2020, révèle un paysage beaucoup plus complexe et nuancé. Les principales difficultés résident dans le recrutement de patients, en particulier les nourrissons et les jeunes enfants, pour les essais cliniques. La faible prévalence de certaines maladies pédiatriques et les défis éthiques entourant la participation des enfants à des essais cliniques sont autant de facteurs qui limitent la réalisation d'investigations cliniques rigoureuses. [49] |
| Améliorer la surveillance après commercialisation | Lors d’une interview avec un chargé de matériovigilance, deux défis sont mis en évidence : des difficultés à interpréter les critères de signalement d'incidents à l’ANSM et l’absence de ressources pour une meilleure compréhension de la méthodologie de rapport des tendances. (Annexe 2) |
B. Aménagement décidé par la Commission Européenne : Le deuxième report (2024 à 2028)
Le chapitre précédent a démontré que l’application du RDM demeure fragile. La période de grâce octroyée jusqu’en 2024 permettant la libre circulation des "legacy devices" n'est pas suffisante pour accompagner les acteurs du secteur. Par exemple, les résultats d'une enquête menée par le SNITEM en 2023 démontrent que parmi les 50 réponses, seulement 15% des entreprises n'ont pas encore soumis de demande, tandis que 40% ont des demandes en attente de traitement. Quant aux organismes notifiés, ils sont tellement submergés par les demandes de certification, qu'ils auraient besoin en moyenne de 18 mois pour effectuer les évaluations [50].
Tout cela a contribué de nouveau à des discussions au sein de la communauté européenne du dispositif médical. En réponse à la volonté commune du Parlement européen, des États membres et des parties prenantes, une proposition a été publiée au Journal officiel de l'Union européenne le 15 mars 2023 (Voir figure16).
Figure 16 : Nouvelles échéances de la deuxième période transitoire du règlement 2017/745. Source [19]
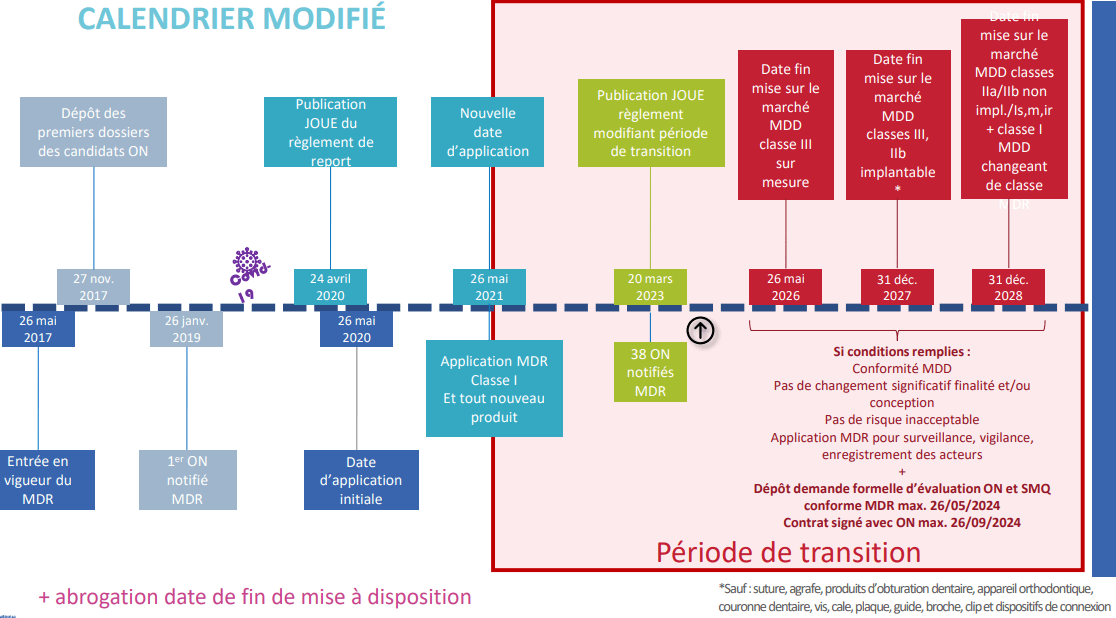
Cet amendement modifie l'article 120 du RDM qui concerne les dispositions transitoires. D’une part, il prévoit que la validité des certificats émis conformément aux anciennes directives est maintenue, et d’autre part que les dispositifs correspondants peuvent être mis sur le marché sous certaines conditions (figure 16).
Par ailleurs il est important de rappeler que pour bénéficier de cette “dernière” période de transition, les dispositifs doivent respecter certains critères [51] :
- Les dispositifs ne doivent pas présenter de risques inacceptables pour la santé et la sécurité ;
- Les dispositifs ne doivent pas faire l’objet de modifications significatives de conception ou de destination ;
- Les fabricants doivent engager au plus tard le 26 mai 2024 un processus de certification au titre du règlement tels que l’adaptation de son système de management de la qualité et la soumission et/ou l’acceptation d’une demande d’évaluation de la conformité par un ON.
C. Perspectives pour la bonne évolution du cadre réglementaire
Quant bien même la deuxième période de transition octroyée, les acteurs du secteur portent toujours leur voix et sensibilisent sur l'avenir de la réglementation en matière de dispositif médical en Europe. C’est pourquoi une seconde lettre ouverte à été rédigée par Medtech Europe et 34 autres syndicats européens en Septembre 2023 [52]. Cette lettre à destination de la présidente de la Commission Européenne, Mme Stella Kyriakides, alerte sur trois points :
- Faciliter l’obtention du marquage CE : Les auteurs de la lettre soutiennent l'établissement d'un système de marquage CE plus efficace et efficient, dans le but d'améliorer la prévisibilité et de réduire la charge administrative. À long terme, l'idée serait de développer un système similaire au modèle américain, qui, avant l'introduction du règlement, était considéré comme difficile d'accès, mais qui est désormais plus accessible et moins coûteux grâce au Règlement sur les Dispositifs Médicaux (RDM). En effet, le cycle quinquennal des certifications CE est remis en question en raison de sa durée, contrairement au modèle de la FDA (Food and Drug Administration) américaine.
- Soutien à l'innovation : Leur proposition vise à instaurer un processus favorisant la mise sur le marché des innovations, permettant ainsi un accès rapide des dernières avancées technologiques médicales aux patients et aux systèmes de santé européens. Cela se ferait par le biais de voies d'évaluation spécialisées et d'échanges précoces avec les concepteurs. Depuis l'introduction du Règlement sur les Dispositifs Médicaux (RDM), la priorité est accordée aux dispositifs existants. Actuellement, de nombreuses PME et grandes entreprises ciblent principalement le marché américain. Cette fuite d'innovation profite davantage aux patients américains, compromettant ainsi l'accessibilité des innovations aux patients européens.
- Structure de gouvernance responsable : Leur recommandation porte sur la mise en place d'une entité dédiée à la supervision, à la gestion et à la surveillance des organismes notifiés. Des différences notables se sont révélées entre les organismes notifiés européens. Il est impératif d'assurer la transparence du processus de certification en utilisant des indicateurs fiables. De nombreux fabricants soulignent des problèmes de favoritisme dans le traitement des dossiers. Cette structure de gouvernance des organismes notifiés serait l'occasion de centraliser et d'harmoniser l'ensemble des pratiques à l'échelle européenne. Un modèle qui se rapprocherait de l’Agence européenne des médicaments (EMA).
D. Nos solutions proposées à travers la création d’un guide
À l'issue de ce travail, les principales difficultés auxquelles font face les opérateurs économiques ont été relevées. À travers les différents témoignages, entretiens et retours d’expérience, un guide a été élaboré dans le but de répondre au mieux aux problèmes mis en évidence dans ce rapport. Bien que ce guide ne soit pas exhaustif, des solutions pertinentes ont été sélectionnées. Cela n'exempte pas les lecteurs de conduire d'autres recherches pour acquérir des informations complémentaires. Ce guide cible différents objectifs énoncés ci dessous :
Clarifier la procédure de mise sur le marché d’un DM :
Les éléments de ce rapport révèlent que les organismes notifiés rejettent des dossiers en raison d'erreurs de classification DM, manque d’informations de la part des fabricants, entraînant des coûts supplémentaires. Le guide fournit une procédure détaillée ainsi que des outils d'aide, à prendre en compte pour la mise sur le marché d'un dispositif médical.
- Le guide MDCG qui explique les règles de classification des dispositifs médicaux pour une manipulation plus aisée et sûre se basant sur le nouveau règlement.
- Un caneva de choix de la procédure d’évaluation de la conformité qui présente les différentes options de procédures d'évaluation de la conformité en fonction de la classe du DM.
- Un descriptif de la documentation technique qui détaille le contenu et les documents nécessaires d’un dispositif médical en vue de l’obtention du marquage CE.
- Un template de la déclaration de conformité pour les dispositifs médicaux permettant de donner un contenu selon le règlement.
Éclairer le choix de sélection d’un organisme notifié accrédité selon le règlement :
Les différentes enquêtes témoignent qu’un grand nombre de fabricants, principalement les PME et TPE, n'ont pas encore sélectionné d'organisme notifié en raison d'un manque d'information et de moyen. Le guide suggère des recommandation pour sélectionner un ON :
- Une procédure pour choisir un organisme notifié.
- Une grille tarifaire comparant les tarifs des différents organismes notifiés.
- Une cartographie interactive permettant de localiser les ON en fonction de leur champ de compétence.
Répertorier des aides financières et plans d'accompagnement pour les fabricants :
Ce rapport met en lumière les difficultés financières rencontrées par de nombreuses entreprises depuis l'instauration de cette réglementation. En France, diverses aides sont disponibles pour soutenir principalement les startups et les PME dans leur développement.
- Solutions concernant les investigations cliniques : (F-CRIN, CIC-IT, Base de données ….)
- Aides pour l’innovations des DM (Guichet diagnostic BPIFrance, Guichet innovation ANSM, Plan innovation 2030..)
- Supports et offres de formations (SNITEM, Tech4Health, EIT Health…)
- Accompagnement dans l'acquisition de DM au sein de structures de soins (Aide Région Haut de france)
- Et d'autres aides consultables dans le guide.
Conclusion
À six ans de l'entrée en vigueur du règlement européen sur les dispositifs médicaux, il est essentiel de noter que les exigences sont élevées, mais les ressources disponibles ne permettent pas une mise en œuvre complète des exigences réglementaires. L’un des enjeux du règlement sur les dispositifs médicaux (RDM) était l'homogénéisation des textes. Cependant, à ce jour, certains acteurs soulignent la difficulté de compréhension du RDM.
Du côté des organismes notifiés, le règlement a renforcé les règles pour une meilleure transparence, entraînant la nécessité d'une revalidation des organismes qui, jusqu'à présent, est insuffisante pour répondre à la demande croissante de certifications.
Quant aux fabricants, ils font face à de nombreuses difficultés, qui les conduisent à revoir leur stratégie notamment en réduisant leur portefeuille de produits ou en mettant fin à leurs activités pour des raisons financières.
Les hôpitaux et les patients rencontrent également des obstacles, notamment en ce qui concerne la gestion des approvisionnements en dispositifs médicaux, en particulier les produits stériles, dans les pharmacies. Cela se traduit par une diminution de la qualité des soins, mettant indirectement en danger la sécurité des patients.
Malgré les initiatives prises par la Commission Européenne suite aux diverses actions des syndicats, fabricants, médecins…, l'écosystème réglementaire demeure instable. Cette situation suscite une diminution de la confiance parmi les acteurs du secteur, car ces derniers se sentent incompris et mal accompagnés. C'est pourquoi la prédictibilité devrait être un critère sur lequel les instances européennes devraient travailler pour anticiper et mieux contrôler le système réglementaire.
Enfin, il est important de souligner que l'Europe représente le deuxième marché mondial des dispositifs médicaux, et des préoccupations émergent quant à l'innovation. De nombreux fabricants ont été contraints de mettre fin à leurs activités de recherche et développement au profit de la mise en conformité des legacy devices. Cela met en danger la réputation de l'Europe en termes d'innovation et un risque de perte de compétitivité face à la concurrence internationale dans le secteur des dispositifs médicaux.
Références Bibliographies
[1] Statista, « Medical Devices - Worldwide », juin 2023. Consulté le : 24 septembre 2023. [En ligne]. Disponible sur : https://www.statista.com/outlook/hmo/medical-technology/medical-devices/worldwide
[2] SNITEM, « Le marché du DM », octobre 2023. Consulté le : 24 septembre 2023. [En ligne]. Disponible sur : https://www.snitem.fr/le-dispositif-medical-dm/lessentiel-sur-le-dm/dm-et-economie-de-la-sante/
[3] M. Foglietti, « Entrée en vigueur de la directive 2007/47/CE : impacts des exigences cliniques pour un fabricant de dispositifs médicaux », Thèse de doctorat, Faculté de pharmacie de Grenoble, janvier 2023. Consulté le : 2 octobre 2023. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-03925422/document/
[4] J. Mimouni, « Dispositifs médicaux : réglementation et approche par les risques. Application à une industrie de la santé », Thèse de doctorat, Faculté de pharmacie d’Aix-Marseille université, mars 2018. Consulté le : 2 octobre 2023. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-01740135/document/
[5] G. Promé, « Directive 93/42/CEE relative aux Dispositifs Médicaux », Qualitiso, mai 2015. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.qualitiso.com/directive-93-42-cee-dispositifs-medicaux
[6] G. Promé, « Principaux textes réglementaires européens pour les dispositifs médicaux et de santé », Qualitiso, septembre 2017. Consulté le : 22 octobre 2023. [En ligne]. Disponible sur : https://www.qualitiso.com/directives-europeennes/
[7] Direction générale de la santé, « Rapport complet PIP », Ministère de la santé et de la prévention, février 2012. Consulté le : 22 septembre 2023. [En ligne]. Disponible sur : https://sante.gouv.fr/IMG/pdf/Rapport_complet_PIP_def_01_02_12.pdf
[8] Médiapart, « Le fondateur de PIP condamné à 4 ans ferme et 75 000 euros d’amende », décembre 2013. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.mediapart.fr/journal/france/101213/le-fondateur-de-pip-condamne-4-ans-ferme-et-75-000-euros-damende
[9] V. Cantié, « Scandale des prothèses PIP : 11 ans après, la cour d’appel de Paris condamne l’organisme certificateur TÜV », France Inter, mai 2021. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.radiofrance.fr/franceinter/scandale-des-protheses-pip-11-ans-apres-la-cour-d-appel-de-paris-condamne-l-organisme-certificateur-tuev-8374588
[10] J. Monin, « Ce que révèlent les “Implant Files” sur les failles du système de certification des dispositifs médicaux », Franceinfo, novembre 2018. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.francetvinfo.fr/sante/implant-files/ce-que-revelent-les-implant-files-sur-les-failles-du-systeme-de-certification-des-dispositifs-medicaux_3051203.html
[11] International Medical Devices Database. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://fr.medicaldevices.icij.org/
[12] Le Monde, « ‘Implant files ’ : l’ICIJ a créé une base de données recensant les dispositifs médicaux défaillants », décembre 2018. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.lemonde.fr/implant-files/article/2018/12/01/implant-files-l-icij-a-cree-une-base-de-donnees-recensant-les-dispositifs-medicaux-defaillants_5391242_5385406.html
[13] L. Cohen, « Scandale sanitaire des « Implant files », novembre 2018. Consulté le : 12 novembre 2023. [En ligne]. Disponible sur : https://www.senat.fr/questions/base/2018/qSEQ181107966.html
[14] « RÈGLEMENT (UE) 2017/ 745 DU PARLEMENT EUROPÉEN ET DU CONSEIL - du 5 avril 2017 - relatif aux dispositifs médicaux, modifiant la directive 2001/ 83/ CE, le règlement (CE) no 178/ 2002 et le règlement (CE) no 1223/ 2009 et abrogeant les directives du Conseil 90/ 385/ CEE et 93/ 42/ CEE ». Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/PDF/?uri=CELEX:32017R0745
[15] V. Boissart, « Impacts du nouveau règlement 2017/745 sur la gestion biomédicale des dispositifs médicaux », IRBM News, vol. 42, no 2, p.100300, avril 2021, Disponible sur : https://doi.org/10.1016/j.irbmnw.2021.100300
[16] ANSM, « Réglementation relative aux dispositifs médicaux (DM) et aux dispositifs médicaux de diagnostic in vitro (DMDIV) », janvier 2021. Consulté le : 8 novembre 2023. [En ligne].Disponible sur :https://ansm.sante.fr/documents/reference/reglementation-relative-aux-dispositifs-medicaux-dm-et-aux-dispositifs-medicaux-de-diagnostic-in-vitro-dmdiv
[17] Universal Medica, « Points clés du nouveau règlement européen sur les dispositifs médicaux (UE) 2017/745. », novembre 2021. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.universalmedica.com/fr/points-cles-du-nouveau-reglement-europeen-sur-les-dispositifs-medicaux-ue-2017-745/
[18] J-F. Lataste,« Le point sur le Suivi Clinique après Commercialisation (SCAC) », Medalps | Medical Device Experts, septembre 2021. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.medalps.com/le_point_sur_le_suivi_clinique_apres_commercialisation_scac/
[19] SNITEM, « Nouveau Règlement DM : premier bilan de la nouvelle période de transition et perspectives », Colloque, septembre 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.snitem.fr/wp-content/uploads/2023/09/Colloque-MDR-3-Presentation.pdf
[20] SNITEM, « Panorama et analyse quantitative de la filière industrielle des dispositifs médicaux en France en 2021 ». 2021, Consulté le : 5 décembre 2023. [En ligne]. Disponible sur : https://www.snitem.fr/wp-content/uploads/2022/02/Snitem-Panorama-DM-2022.pdf
[21] M. Mezher, « CE certificates up nearly 50% in 2019 in anticipation of MDR », RAPS, mai 2020. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.raps.org/news-and-articles/news-articles/2020/5/team-nb-ce-certificates-up-nearly-50-in-2019-in-an
[22] MedTech Europe, « Open letter on the implementation and readiness status of the new Medical Device Regulation 745/2017 (MDR)», avril 2019. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.medtecheurope.org/wp-content/uploads/2019/04/MedTech-Europe_VP-Katainen_MDR-implementation-status_15-April-2019.pdf
[23] Business France, « Union européenne - Réglementation des dispositifs médicaux “période de grâce” », mars 2020. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.businessfrance.fr/union-europeenne-reglementation-des-dispositifs-medicaux-periode-de-grace
[24] Commission européenne, « Organismes notifiés », Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://health.ec.europa.eu/medical-devices-topics-interest/notified-bodies_fr
[25] ANSM, « Évaluation, désignation et notification des organismes d’évaluation de la conformité en France », octobre 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://ansm.sante.fr/documents/reference/evaluation-designation-et-notification-des-organismes-devaluation-de-la-conformite-en-france
[26]Commission européenne, « MDCG 2022-13 Désignation, re-assessment and notification of conformity assessment bodies and notified bodies », août 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://health.ec.europa.eu/system/files/2022-08/mdcg_2022-13_en.pdf
[27] ANSM, « Actualité - Certification des dispositifs médicaux : le GMED est désigné comme organisme notifié au titre du nouveau règlement européen », août 2021. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://ansm.sante.fr/actualites/certification-des-dispositifs-medicaux-le-gmed-est-designe-comme-organisme-notifie-au-titre-du-nouveau-reglement-europeen
[28] Commission européenne, « EUROPA – European Commission – Growth – Regulatory policy - SMCS », Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://webgate.ec.europa.eu/single-market-compliance-space/#/notified-bodies/free-search
[29] MedTech Europe, « The European Medical Technology Industry in figures », 2021. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.medtecheurope.org/wp-content/uploads/2021/06/medtech-europe-facts-and-figures-2021.pdf
[30] MedTech Europe, « MedTech Europe Survey Report analysing the availability of Medical Devices in 2022 in connection to the Medical Device Regulation (MDR) implementation », avril 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.medtecheurope.org/wp-content/uploads/2022/07/medtech-europe-survey-report-analysing-the-availability-of-medical-devices-in-2022-in-connection-to-the-medical-device-regulation-mdr-implementation.pdf
[31] Directeur général de la santé et de la sécurité alimentaire, « Notified Bodies Survey on certifications and applications(MDR/IVDR) », Commission européenne, octobre 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://health.ec.europa.eu/latest-updates/updated-document-notified-bodies-survey-certifications-and-applications-mdrivdr-survey-results-data-2023-07-25_en
[32] NSAI, « NSAI Fees for Conformity Assessment Activities (EUR) ». Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.nsai.ie/images/uploads/medical-devices/Client_Communication_cdg.pdf
[33] UDEM, « List of Standard Fees for Conformity Assessment Activities under the MDR (2017/745)», juin 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.udemadriatic.com/lib_dokuman/24.pdf
[34] TÜV SÜD, « MDR conformity assessment procedures », Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.tuvsud.com/en/industries/healthcare-and-medical-devices/medical-devices-and-ivd/medical-device-market-approval-and-certification/medical-device-regulation/mdr-conformity-assessment-procedures
[35] Medcert, « Prices/Fees », septembre 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.med-cert.com/en_prices-fees/
[36] G. Promé, « Comparatif : tarifs des Organismes Notifiés 2017/745 », Qualitiso, septembre 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.qualitiso.com/comparatif-des-tarifs-des-organismes-notifies-2017-745/
[37] BSI, « Fees for Conformity Assessment Activities (EUR) », novembre 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.bsigroup.com/globalassets/meddev/localfiles/en-gb/documents/bsi-md-conformity-assessment-services-and-fees-eur-uk-en.pdf
[38] CE Certiso, « MDR price list », Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://cecertiso.hu/?page_id=14941
[39] Eurofins, « Price list - Medical devices - Regulation (EU) 2017/745 (MDR)», Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://cdnmedia.eurofins.com/european-west/media/12154507/price-list-medical-devices-mdr.pdf
[40] SGS, « List of Standard Fees for Conformity Assessment Activities Under the MDR (2017/745), Notified Body NB1639 ». Consulté le : 5 décembre 2023. [En ligne]. Disponible sur : https://www.sgs.com/en-be/-/media/sgscorp/documents/corporate/technical-documents/sgs-hn-mdr-cost-sheet-en.cdn.en-BE.pdf
[41] Climedo, « Survey : One Year after the EU MDR Delay – Lack of Clarity, Manual Processes and High Costs for Manufacturers Persist », avril 2021. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://climedo.de/en/press/eumdr-survey-results-2021/
[42] R. L. Quéré, « Perte du marquage CE : les enjeux critiques des nouvelles réglementations européennes sur les dispositifs médicaux », Med it up, avril 2022. Consulté le : 5 décembre 2023. [En ligne]. Disponible sur : https://www.meditup.fr/2022/04/27/150-000-dispositifs-medicaux-pourraient-disparaitre-en-europe/
[43] Officéo, « Taille des entreprises : classification générale », Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://officeopro.com/creation-gestion-entreprise/ge-eti-pme-tpe/
[44] P. Renarda, « Les dangers liés au MDR : la profession tire la sonnette d’alarme ! », DeviceMed, février 2022. Consulté le : 5 décembre 2023. [En ligne]. Disponible sur : https://www.devicemed.fr/dossiers/reglementation/les-dangers-lies-au-mdr-la-profession-tire-la-sonnette-dalarme/29735
[45] SNITEM, « Start-up du DM la force de l’innovation ». 2022. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.snitem.fr/wp-content/uploads/2022/07/Snitem-info-226-web.pdf
[46] H. Sayin, et al., « Impact du nouveau règlement européen 2017/745 relatif aux dispositifs médicaux sur l’activité des pharmacies hospitalières : exemple de la fonction d’approvisionnement pharmaceutique au sein d’un centre hospitalo-universitaire français », vol. 80, no 5, p. 730‑737, sept. 2022, doi : https://doi.org/10.1016/j.pharma.2021.12.007
[47] A. Ressault, et al., « Ruptures en dispositifs médicaux stériles : un défi quotidien », Euro-Pharmat, 2022, [En ligne]. Disponible sur : https://www.euro-pharmat.com/les-journees-annuelles/archives/download/5764/4651/160
[48] A. G. Fraser et al., « Implementing the new European Regulations on medical devices—clinical responsibilities for evidence-based practice : a report from the Regulatory Affairs Committee of the European Society of Cardiology », Eur. Heart J., vol. 41, no 27, p. 2589‑2596, juillet 2020, doi : https://doi.org/10.1093/eurheartj/ehaa382
[49] K. Guerlich et al., « European expert recommendations on clinical investigation and evaluation of high-risk medical devices for children », Acta Paediatr., vol. 112, no 11, p. 2440‑2448, 2023, doi : https://onlinelibrary.wiley.com/doi/full/10.1111/apa.16919
[50] R. Fournier, « Une période transitoire étendue jusqu’en 2028, sous conditions », DeviceMed, mai 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.devicemed.fr/dossiers/reglementation/une-periode-transitoire-etendue-jusquen-2028-sous-conditions/34416
[51] S. Oudjaneplan, « La prolongation de la période de transition pour les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro est adoptée », GMED, février 2023. Consulté le : 31 octobre 2023. [En ligne]. Disponible sur : https://lne-gmed.com/fr/news/la-prolongation
[52] MedTech Europe, « Open letter to Commissioner for Health, Stella Kyriakides : Need for comprehensive structural reforms of the medical technology regulatory frameworks », septembre 2023. Consulté le : 13 novembre 2023. [En ligne]. Disponible sur : https://www.medtecheurope.org/resource-library/open-letter-to-commissioner-for-health-stella-kyriakides-need-for-comprehensive-structural-reforms-of-the-medical-technology-regulatory-frameworks/
Annexes
Annexe 1 : Résultats de notre questionnaire



Annexe 2 : Interview d’un gestionnaire de réclamation en matériovigilance, PME (Octobre, 2023)

Annexe 3 : Interview de 3 consultants spécialisés dans l' assurance qualité et les affaires réglementaires des dispositifs médicaux (QARA), Cabinet parisien, (Novembre 2023).

Annexe 4 : Interview d’une ingénieure QARA pour un logiciel de santé, TPE (Décembre 2023)

Annexe 5 : Interview d’une pharmacienne (Novembre, 2023)
