IDS224 - Maitriser les marchés européens et américains : Documentation Technique
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Contacts
- EL MANSOURI Hamza : Hamza.elmansouri79@gmail.com
Citation
A rappeler pour tout usage : EL MANSOURI Hamza, « Maitriser les marchés européen et américain : Documentation Technique », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire d'apprentissage, https://travaux.master.utc.fr/, réf n° IDS224, juillet 2024, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids224/
Résumé
Ce mémoire traite de l'élaboration et de la gestion de la documentation technique pour les dispositifs médicaux au sein de l'entreprise FineHeart. L'objectif principal est d'assurer la conformité aux réglementations européennes (Règlement européen 2017/745) et américaines (FDA, PMA). L'étude présente les diverses étapes de préparation de la documentation, incluant la création de formulaires, la validation interne, les mises à jour continues, et la gestion documentaire. Des outils de suivi et de veille réglementaire ont été mis en place pour garantir le respect des exigences de sécurité et de performance. En plus de la documentation technique, des missions réglementaires connexes, telles que la veille réglementaire, la mise en place d'un plan de mise sur le marché, et la formation des équipes sur les sujets réglementaires, ont été effectuées. Ce travail a permis d'acquérir une compréhension approfondie des normes et des processus nécessaires à la conformité des dispositifs médicaux, tout en contribuant à l'amélioration des processus internes de FineHeart.
Abstract
This thesis focuses on the development and management of technical documentation for medical devices at FineHeart. The main objective is to ensure compliance with European regulations (European Regulation 2017/745) and American regulations (FDA, Premarket Approval). The study outlines the various steps in preparing documentation, including template creation, internal validation, continuous updates, and document management. Tracking and regulatory monitoring tools were implemented to ensure adherence to safety and performance requirements. In addition to technical documentation, the intern also undertook related regulatory tasks such as regulatory monitoring, setting up a market entry plan, and training teams on regulatory topics. This work provided a deep understanding of the standards and processes required for medical device compliance while contributing to the improvement of FineHeart’s internal processes.
Téléchargement

Mémoire Complet :
Maîtriser les marchés européen et américain : Documentation Technique
Remerciements
Je souhaite exprimer ma profonde gratitude à toutes les personnes qui ont contribué à la réalisation de ce mémoire.
Tout d'abord, je tiens à remercier chaleureusement Mme Virginie Rivet, ma maître d'apprentissage, pour son soutien, ses conseils précieux tout au long de cette expérience. Sa vision et son expertise ont été essentielles pour mener à bien ce projet.
Je remercie également Mme Isabelle Claude, ma tutrice à l'UTC, pour son encadrement académique et ses orientations éclairées. Sa rigueur et ses encouragements ont joué un rôle crucial dans la structuration et la qualité de ce travail.
Un remerciement tout particulier à Mme Shahrzad Hoseiny, qui m'a embauché en premier lieu et a été ma tutrice initiale avant son départ. Son soutien et sa confiance en mes capacités ont été déterminants pour mon intégration et mon épanouissement au sein de FineHeart.
Je suis également particulièrement reconnaissant envers toute l'équipe de FineHeart pour leur accueil chaleureux et leur collaboration. Merci à chaque membre du service Qualité et Affaires Réglementaires pour leur patience, leurs partages de connaissances et leur aide précieuse. Travailler au sein de cette équipe dynamique et compétente a été une expérience extrêmement enrichissante.
Je tiens aussi à exprimer ma reconnaissance à mes collègues à l’UTC pour les moments de partage qui ont rendu cette période plus agréable et motivante.
Enfin, je remercie ma famille et mes amis pour leur soutien constant, leur compréhension et leurs encouragements tout au long de mon parcours académique. Leur présence à mes côtés m'a permis de surmonter les défis et de rester concentré sur mes objectifs.
À tous, un grand merci.
Liste des Abréviations
| CE | Conformité Européenne |
| DM | Dispositif Médical |
| DMAR | Dispositifs Médicaux et Affaires Réglementaires |
| QARA | Quality Assurance and Regulatory Affairs (Assurance Qualité et Affaires Réglementaires) |
| IC | Insuffisance Cardiaque |
| HF | Heart Failure (Insuffisance Cardiaque) |
| AHA | American Heart Association (Association Américaine du Cœur) |
| LVAD | Left Ventricular Assist Device (Dispositif d'Assistance Ventriculaire Gauche) |
| DT | Documentation Technique |
| GSPR | General Safety and Performance Requirements (Exigences Générales de Sécurité et de Performance) |
| FDA | Food and Drug Administration (Administration des Aliments et Médicaments) |
| PMA | Premarket Approval (Approbation de Mise sur le Marché) |
| 510k | Section 510(k) du Food, Drug, and Cosmetic Act (Procédure de Notification Préalable à la Mise sur le Marché) |
| MDR | Medical Device Regulation (Règlement sur les Dispositifs Médicaux) |
| RDM | Règlement sur les Dispositifs Médicaux |
| PMS | PostMarket Surveillance (Surveillance Postcommercialisation) |
| PMCF | PostMarket Clinical Followup (Suivi Clinique Postcommercialisation) |
| UDI | Unique Device Identification (Identification Unique du Dispositif) |
| MDCG | Medical Device Coordination Group (Groupe de Coordination des Dispositifs Médicaux) |
| ISO | International Organization for Standardization (Organisation Internationale de Normalisation) |
| AAMI | Association for the Advancement of Medical Instrumentation (Association pour l'Avancement de l'Instrumentation Médicale) |
| TIR | Technical Information Report (Rapport d'Information Technique) |
| EUDAMED | European Database on Medical Devices (Base de Données Européenne des Dispositifs Médicaux) |
| DHF | Design History File (Dossier de l’Historique de la Conception) |
| DMR | Device Master Record (Dossier Maître du Dispositif) |
| DHR | Device History Record (Dossier de l’Historique du Dispositif) |
| SMQ | Système de Management de la Qualité / QMS : Quality Management System (Système de Management de la Qualité) |
| GED | Gestion Électronique des Documents |
| EMC | Electrical Safety and Electromagnetic Compatibility (Sécurité Électrique et Compatibilité Électromagnétique) |
| IFU | Instructions for Use (Instructions d'Utilisation) |
| CFR | Code of Federal Regulations (Code des Règlements Fédéraux) |
| USA | United States of America (ÉtatsUnis d'Amérique) |
| UTC | Université de Technologie de Compiègne |
Glossaire
| Marquage CE | un marquage au travers duquel un fabricant indique que dispositif médical qu’il fabrique est conforme aux exigences essentielles du règlement européen et aux autres actes législatifs harmonisés de l'Union Européenne qui en prévoient l'apposition |
| Cycle de vie du dispositif médical | ensemble des phases de la vie d’un dispositif médical de la conception initiale à la réforme finale. |
| Dispositif médical | Selon le règlement (UE) 2017/745, un dispositif médical est tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes - diagnostic, prévention, surveillance, prédiction, pronostic, traitement ou atténuation d’une maladie, - diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci, - investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique, - communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus, Et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. |
| Dossier technique de marquage CE | document qui permet à un fabriquant d’attester la conformité de son produit aux exigences applicables du règlement (UE) 2017/745. |
| Enregistrement | Processus de documentation des informations clés sur le développement, la fabrication et la gestion des dispositifs médicaux pour assurer la traçabilité et la conformité réglementaire. |
| Règlement sur les Dispositifs Médicaux (RDM) | Règlement Européen 2017/745, établissant les exigences pour la mise sur le marché, la mise en service et la surveillance des dispositifs médicaux dans l'Union Européenne. |
| Food and Drug Administration (FDA) | Agence américaine responsable de la protection de la santé publique en assurant la sécurité, l'efficacité et la sûreté des médicaments, des dispositifs médicaux, des aliments et des cosmétiques. |
| Premarket Approval (PMA) | Processus de soumission rigoureux de la FDA pour les dispositifs médicaux de classe III sans équivalence sur le marché, exigeant des preuves scientifiques de sécurité et d'efficacité avant la mise sur le marché. |
| Section 510(k) | Procédure de notification préalable à la mise sur le marché d'un dispositif médical, où le fabricant doit démontrer que le dispositif est substantiellement équivalent à un dispositif déjà commercialisé légalement. |
| Code of Federal Regulations (CFR) | Code des Règlements Fédéraux, recueil des règlements généraux et permanents émis par les départements et les agences du gouvernement fédéral des États-Unis. |
Introduction
L'insuffisance cardiaque (IC) survient lorsque le cœur ne pompe pas suffisamment de sang comme il le devrait, ce qui met en péril le bon fonctionnement de l'organisme. En d'autres termes, le cœur est incapable de fournir un débit sanguin adéquat pour répondre aux besoins du corps. Cela peut entraîner une accumulation de liquide dans les poumons, les jambes et d'autres parties du corps, provoquant des symptômes tels que l'essoufflement, la fatigue et le gonflement.
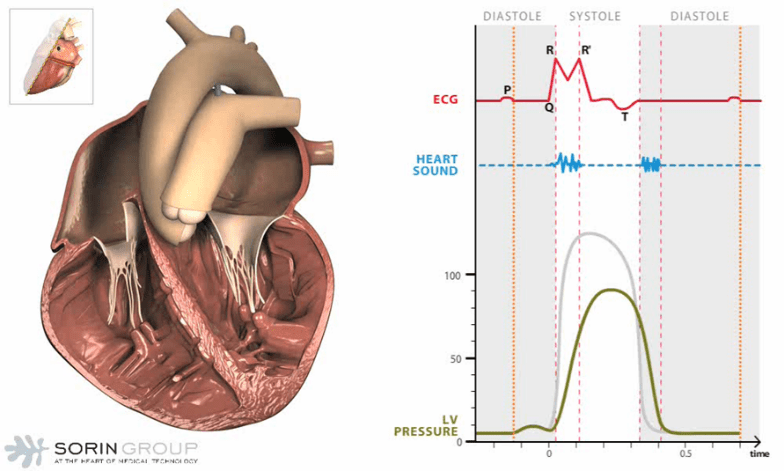
La figure 1 présente deux illustrations du cœur humain en action, accompagnées de graphiques représentant les activités électrocardiographiques (ECG), les sons cardiaques et la pression dans le ventricule gauche (Left Ventricular Pressure). Ces graphiques permettent de visualiser le fonctionnement du cœur au cours d'un cycle cardiaque complet, comprenant les phases de diastole et de systole.
Figure 1 : Fonctionnement du cœur (Contraction-Remplissage vs hémodynamique… cœur altéré) [Source – FineHeart-interne]
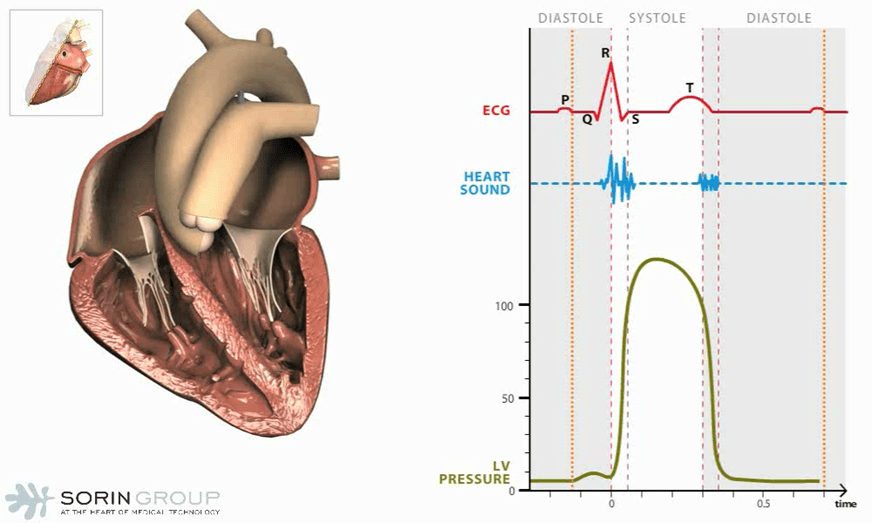
La partie gauche de la figure 1 montre une coupe transversale du cœur, permettant de voir les structures internes et le flux sanguin. Le tracé électrocardiographique en rouge montre les différentes ondes associées à l'activité électrique du cœur, notamment les ondes P, QRS et T. Les sons cardiaques, en bleu, représentent les bruits produits par les battements du cœur, généralement causés par la fermeture des valves cardiaques. La courbe en vert montre la pression dans le ventricule gauche, augmentant pendant la systole (contraction) et diminuant pendant la diastole (relaxation).
La partie droite de la figure 1 présente une autre coupe transversale du cœur, similaire à celle de gauche, mais avec des détails supplémentaires. Le tracé ECG en rouge montre encore une fois les ondes P, QRS et T, indiquant l'activité électrique du cœur. Les sons cardiaques en bleu sont synchronisés avec les battements du cœur et les phases du cycle cardiaque. La courbe en vert de la pression ventriculaire gauche montre les changements de pression pendant les phases de systole et de diastole.
La diastole est la phase de relaxation du cœur où les ventricules se remplissent de sang. La pression ventriculaire gauche est basse pendant cette phase. En cas d'insuffisance cardiaque, le cœur peut ne pas se remplir adéquatement, réduisant ainsi le volume de sang éjecté lors de la systole. La systole est la phase de contraction du cœur où le sang est éjecté des ventricules vers les artères. La pression ventriculaire gauche augmente pendant cette phase. En cas d'insuffisance cardiaque, le cœur peut ne pas se contracter efficacement, diminuant ainsi la quantité de sang éjecté et entraînant une accumulation de sang dans le cœur.
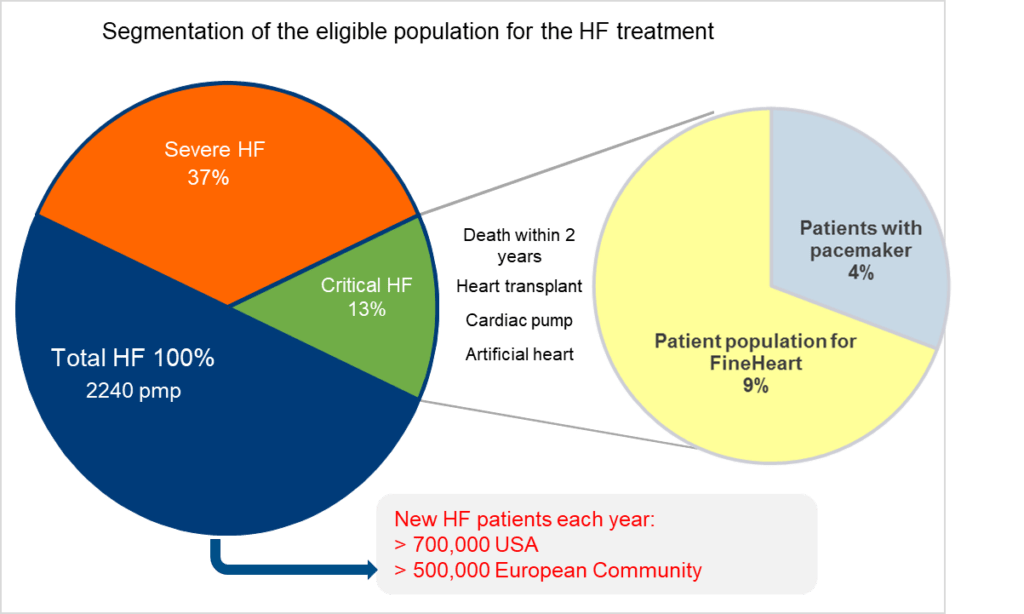
L'insuffisance cardiaque (IC) représente un défi significatif pour les systèmes de santé mondiaux. Selon les dernières statistiques de l'AHA (Task Force on Heart Disease and Stroke Statistics, Circulation 2012)[3], la segmentation de la population éligible pour le traitement de l'IC est la suivante : 37% des patients souffrent d'une IC sévère, tandis que 13% sont dans un état critique figure 2. La prévalence totale de l'IC est de 2240 patients par million de population (pmp). Parmi ces patients, 4% sont équipés d'un pacemaker et 9% constituent la population cible pour les traitements innovants de FineHeart. Chaque année, plus de 700 000 nouveaux cas d'IC sont diagnostiqués aux États-Unis, tandis que la Communauté Européenne enregistre plus de 500 000 nouveaux cas.
Figure 2 : Epidémiologie de l'Insuffisance cardiaque [Source : Auteur]
L'insuffisance cardiaque critique (IC) touche 13 % de la population souffrant d'IC en Europe et aux États-Unis, soit environ 302 patients par million d'habitants par an (pmy). Parmi ces patients, 135 pmy sont éligibles pour des stimulateurs cardiaques, représentant 100 000 patients par an, avec un marché évalué à 3 milliards de dollars aux États-Unis. Cependant, 45 pmy ne répondent pas aux stimulateurs cardiaques. Par ailleurs, 212 pmy sont des candidats pour une pompe cardiaque, soit 220 000 patients par an, mais seulement 6 pmy, soit 7 000 patients par an, se font effectivement implanter une pompe cardiaque en raison de diverses limitations technologiques et cliniques, avec un marché estimé à 1 milliard de dollars. D'autre part, 167 pmy ne sont pas éligibles aux stimulateurs cardiaques. De ces patients, 6 pmy, soit 5 000 patients par an, reçoivent une greffe de cœur. Une solution possible pour ceux qui ne peuvent pas bénéficier de ces options pourrait être un cœur artificiel.
Présentation de l’environnement professionnel
Présentation de l’entreprise

FineHeart est une entreprise française innovante dans le domaine des dispositifs médicaux, spécifiquement axée sur le développement de technologies de pointe en cardiologie. Fondée en 2010, cette société est basée à Pessac, près de Bordeaux, et possède également des installations technologiques à Cestas ainsi qu'un bureau à Tours. Depuis 2020, FineHeart a établi son propre siège, à Pessac (33600), comprenant des bureaux et des laboratoires, renforçant ainsi sa présence et ses capacités opérationnelles [4].
L'invention brevetée de FineHeart, l'ICOMS FLOWMAKER® [5], est au cœur de ses innovations. Il s'agit d'un dispositif de soutien circulatoire mécanique, entièrement implantable et alimenté sans fil, conçu pour optimiser le débit cardiaque tout en préservant la contractilité naturelle du cœur. Cette technologie représente une avancée majeure, offrant une solution thérapeutique durable pour l'assistance circulatoire chez les patients souffrant d'insuffisance cardiaque sévère.
L'équipe fondatrice de FineHeart comprend des chirurgiens cardiaques et des cardiologues de renommée internationale, notamment le Dr Stéphane Garrigue, inventeur du FLOWMAKER®, et le Dr Philippe Ritter, co-inventeur de la thérapie de resynchronisation cardiaque. Sous la direction d'Arnaud Mascarell, le PDG de FineHeart, l'équipe intègre également des experts en gestion issus du domaine des dispositifs médicaux, notamment de chez Medtronic, renforçant ainsi l'expertise technique et commerciale de l'entreprise.
La vision de ces co-fondateurs était de créer un système de soutien circulatoire pulsatile, entièrement implantable et miniaturisé, qui surmonte de nombreux défis associés aux LVAD actuellement sur le marché. L'objectif principal de FineHeart est de restaurer un débit cardiaque normal et de permettre aux patients atteints d'insuffisance cardiaque de retrouver une qualité de vie normale.
Implantée au sein de Coeur Bersol et soutenue par Bordeaux Métropole et la ville de Pessac, FineHeart fait partie de l'opération Bordeaux Inno Campus, un nouveau centre d'excellence. Avec plus de 45 employés, FineHeart se positionne comme un acteur clé dans le domaine des dispositifs médicaux pour le traitement des maladies cardiovasculaires, combinant expertise médicale de pointe et innovation technologique.
Organigramme de FineHeart
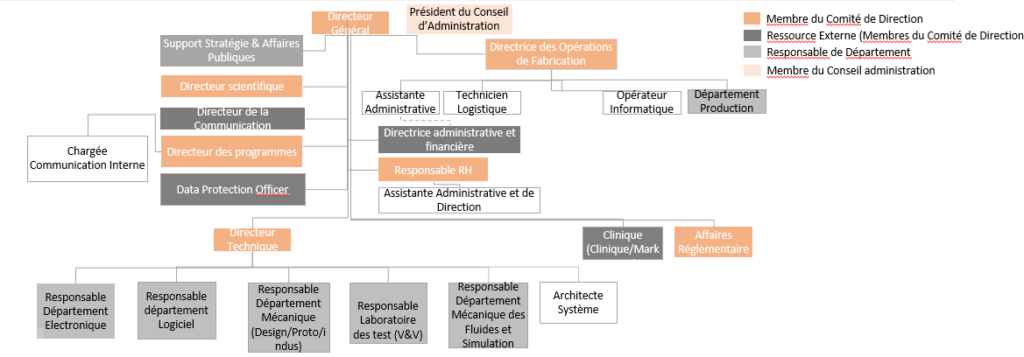
Pour une compréhension plus approfondie de la structure et de la dynamique interne de FineHeart, il est essentiel de considérer son organigramme. L'organigramme reflète non seulement la hiérarchie et les relations de reporting au sein de l'entreprise, mais il met également en lumière les différents départements et leurs rôles clés dans la réalisation des objectifs de FineHeart (figure 3).
Figure 3 : Organigramme de FineHeart [Source : FineHeart-Interne]
Présentation du service Qualité et Affaires Réglementaires
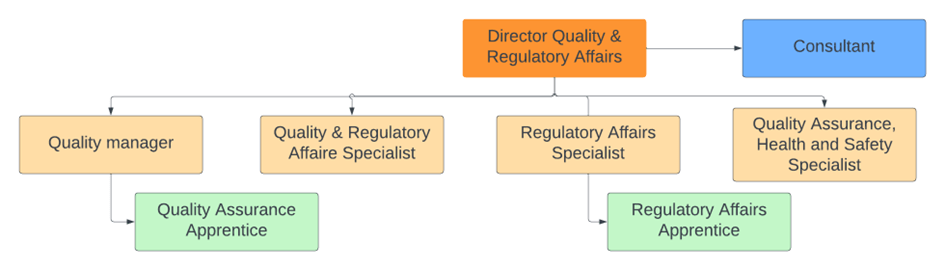
L’équipe Qualité et Affaires Réglementaires est le pilier central garantissant l'excellence et la conformité de nos produits et services. Chaque membre joue un rôle crucial, collaborant étroitement pour s'assurer que toutes les normes et régulations sont non seulement respectées, mais aussi intégrées dans l'ADN du travail quotidien. La structure de l’équipe reflète l’engagement envers une gestion rigoureuse et une amélioration continue. En figure 4, une présentation de l’organigramme qui illustre l’organisation interne du service, mettant en lumière les responsabilités et la synergie entre les différents rôles.
Figure 4 : Organigramme service Qualité et Affaires Réglementaires [Source : FineHeart-Interne]
Solution de l’entreprise : FLOWMAKER®
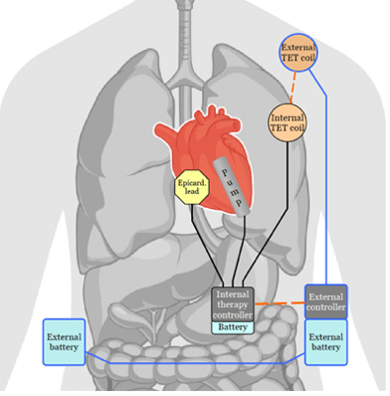
Le FlowMaker® est un dispositif d'assistance cardiaque entièrement implantable, conçu pour les patients souffrant d'insuffisance cardiaque sévère (stades intermédiaires à avancés). Le FlowMaker® ajoute un flux sanguin supplémentaire au flux sanguin natif pendant chaque systole (figure 5). Le cœur continue à contribuer au flux sanguin global, mais un flux sanguin plus satisfaisant est rétabli grâce à l'action complémentaire du FlowMaker®.
Figure 5 : ICOMS FLOWMAKER [Source : FineHeart-Interne]
Le dispositif comprend une pompe sanguine implantable, fixée à l'aide d'un système de pose, placée en position intraventriculaire via une approche trans-apicale minimalement invasive (figure 6). Cette pompe dirige un flux vers la valve aortique, soutenant ainsi le ventricule gauche défaillant en ajoutant un flux sanguin supplémentaire à chaque systole, tandis que le cœur continue de contribuer au flux sanguin global. Un conducteur épicardique permet à la pompe de se synchroniser avec la systole cardiaque en captant le signal de dépolarisation du ventricule gauche, servant ainsi de capteur physiologique du début de la systole.
Figure 6 : DM Implantable ICOMS FLOWMAKER [Source : FinHeart-Interne]

Le système est alimenté par des batteries externes qui rechargent continuellement la batterie interne par induction, via un transfert d'énergie transcutané.
Missions de l'alternance
En tant qu'Alternant Affaires Réglementaires, ma mission est centrée sur la compréhension approfondie et l'application des normes réglementaires qui gouvernent le secteur des dispositifs médicaux. L'objectif est de garantir que les produits de FineHeart non seulement respectent ces normes, mais aussi maintiennent les standards les plus élevés de qualité et de sécurité pour les utilisateurs finaux.
Veille réglementaire
Maintenir à jour la liste des normes applicables, qu'elles soient harmonisées ou reconnues par des organismes tels que la FDA [6].
J’ai mis en place un outil de veille réglementaire. Cet outil, structuré autour des volets Normes, Réglementations et Guides.

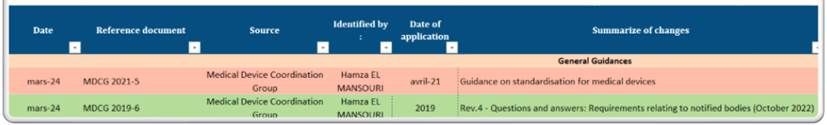
Il nous permet de surveiller, documenter et analyser les évolutions pertinentes pour notre secteur. Chaque changement est minutieusement documenté, incluant la source, la date d'application et un résumé des modifications (figure 7).
Figure 7 : Outil de veille réglementaire - partie description [Source : Auteur]
Les parties prenantes sont informées et un suivi rigoureux est mis en place pour la mise en œuvre des actions correctives nécessaires (figure 8).
Figure 8 : Outil veille réglementaire - Partie Suivi [Source : Auteur]

Plan Réglementaire de mise sur le marché
La mise en place d'un plan réglementaire, tel que détaillé dans la table des matières (figure 9), comporte plusieurs étapes concrètes. Tout d'abord, décrire le dispositif médical en précisant son usage prévu, ses groupes cibles et son indication médicale. Ensuite, établir un calendrier pour l'accès au marché et identifier les normes principales du produit.
Pour la stratégie réglementaire, une analyse détaillée des exigences pour les marchés clés est nécessaire. En Europe, par exemple, cela comprend le scope de cette stratégie règlementaire, la classification, les marchés ciblés, les exigences réglementaires applicables, la procédure d'évaluation de la conformité et la date de soumission prévue. Pour les États-Unis, cela inclut aussi, la classification spécifique à l’FDA, les exigences réglementaires et le type de soumission (PMA[2], 510k[7]).
Figure 9 : Table des matières d'un plan réglementaire de mise sur le marché [Source : FineHeart-Interne]

Conformité au Règlement des Dispositifs Médicaux 2017/745 (MDR)
L'analyse des écarts évalue la conformité aux exigences réglementaires et identifie les domaines nécessitant des améliorations. Cette analyse couvre plusieurs catégories essentielles, telles que l'évaluation générale, le SMQ [18], la base de données EUDAMED, la PMS et le PMCF, l'évaluation clinique et UDI.

Pour chaque article réglementaire, les éléments suivants sont analysés et documentés (figure 10):
- Exigence : Par exemple, l'article 10.1 du MDR 2017/745 stipule que les dispositifs doivent être conçus et fabriqués conformément aux exigences réglementaires.
- Applicabilité : Détermination de la pertinence de l'exigence pour l'organisation.
- Rationnel : Justification de la couverture partielle ou totale de l'exigence.
- Référence dans ISO 13485[18] : Indication de la manière dont la norme ISO 13485 couvre cette exigence.
- Résumé : Description de l'obligation réglementaire et de son intégration dans le système de gestion de la qualité (QMS).
- Processus : Identification des processus internes affectés par cette exigence.
- Procédures/Documents : Documentation nécessaire pour assurer la conformité.
- Conformité : Évaluation du statut de conformité actuel.
- Actions : Propositions de mesures correctives ou préventives pour combler les écarts identifiés.
Figure 10 : Formulaire de l'analyse des écarts du RDM [Source : Auteur]

Formation sur les sujets règlementaires
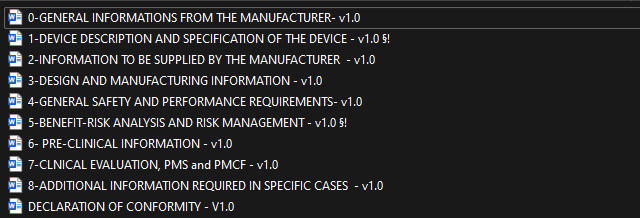
J'ai eu l'opportunité de travailler avec l'équipe QARA. Nous avons organisé des sessions de formation pour approfondir la compréhension des documents réglementaires essentiels (figure 11). Ces documents comprennent le QMS [8], DHF [8] DMR [9], DHR [10] et la DT [11] (annexe 1).
Figure 11 : Formulaire de l'analyse des écarts du RDM [Source : Auteur]

J'ai également mené des sessions de formation sur la veille réglementaire (figure 12). Ces sessions visaient à former les collègues sur la stratégie interne de veille réglementaire, en leur montrant comment surveiller les mises à jour réglementaires pertinentes pour l'entreprise.
Figure 12 : Formation des collaborateurs [Source : FineHeart-Interne]

Autres
Support aux Missions du Département QARA
J'ai apporté mon aide au département Qualité dans la gestion des documents DHF en interne. Cela incluait la préparation des documents nécessaires pour les révisions du design. En outre, j'ai contribué à la préparation de la mise en place de la GED. Mon rôle a consisté à préparer les documents DHF pour leur hébergement dans le nouveau système GED.
Maintien de la mise a jours des Procédures
Contribuer à la mise à jour de procédures et formulaires internes pour optimiser les processus réglementaires.
Préparation de la documentation technique
Cartographie du Processus
Cette méthodologie présente un cadre structuré pour la création d'un dossier technique, essentiel pour accéder aux marchés européens et américains. En s'appuyant sur des données de conception, de test, et en respectant les exigences réglementaires telles que MDR 2017/745 et exigences du FDA pour PMA, le processus vise à garantir la conformité et la sécurité des dispositifs médicaux (figure 13).
Les ressources documentaires et humaines démontrent la capacité à répondre efficacement aux exigences réglementaires. Le développement des formulaires et la collaboration interservices sont au cœur de cette approche, assurant une documentation cohérente et complète.
Figure 13 : Cartographie du processus de rédaction de la documentation Technique [Source : Auteur]
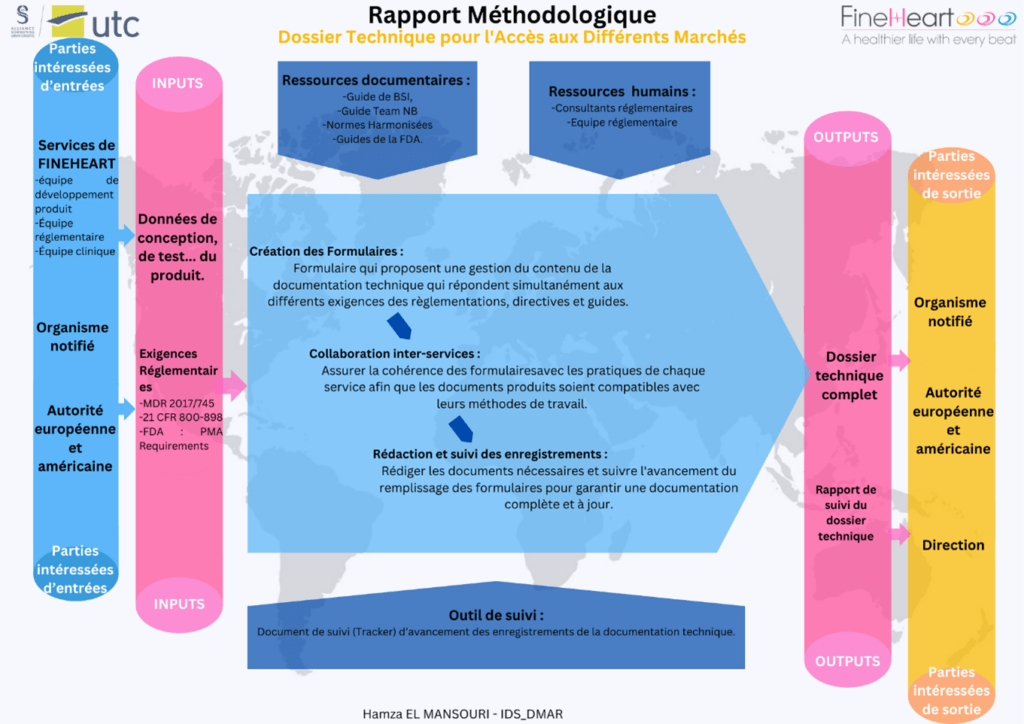
Dans les sections suivantes, les étapes spécifiques de ce processus, en expliquant comment chaque composant contribue à la constitution d'un dossier technique robuste et conforme aux normes réglementaires.
Contexte Règlementaire
En Europe et aux États-Unis, la réglementation des DMs définit des critères précis pour la documentation technique, qui est cruciale pour garantir la conformité et permettre la commercialisation de ces produits. En Europe, le MDR, également appelé Règlement (UE) 2017/745[1], exige que la documentation contienne des informations détaillées sur la conception, la production, les performances et la sécurité des dispositifs. Avant la mise sur le marché, un organisme notifié vérifie la conformité à ces aspects, ce qui est indispensable pour obtenir le marquage CE. Selon l’Annexe II du MDR, cette documentation doit inclure :
- Une description générale du produit
- Des spécifications, y compris les variantes et les accessoires
- Des informations sur les matières et substances utilisées
- Les résultats des évaluations précliniques et cliniques
- Autres
Aux États-Unis, la réglementation des DMs impose des critères précis pour la documentation technique, essentielle pour garantir la conformité et permettre la commercialisation de ces produits. La FDA, par le biais du processus de PMA, exige que la documentation contienne des informations détaillées sur la conception, la production, les performances et la sécurité des dispositifs. Avant la mise sur le marché, la FDA vérifie la conformité de ces aspects, ce qui est indispensable pour obtenir l'approbation PMA. Selon les exigences de la PMA, détaillées dans le Code of Federal Regulations [12], cette documentation doit inclure :
- Une description générale du produit
- Des spécifications, y compris les variantes et les accessoires
- Des informations sur les matériaux et substances utilisés
- Les résultats des évaluations précliniques et cliniques
- Une analyse des risques
- Les procédures de fabrication et de contrôle de la qualité
- Des preuves de conformité aux normes applicables
- Autres informations pertinentes
Les procédures de contrôle de tous les documents et enregistrements, y compris les dossiers de conception et de fabrication, sont requises par la sous-partie G du 21 CFR Part 820[8], qui est cruciale pour garantir la qualité et la conformité réglementaire tout au long du cycle de vie du produit. Il est essentiel de rendre ces documents accessibles et révisés régulièrement afin de s'assurer qu'ils correspondent aux pratiques actuelles et aux données à jour.
La documentation technique revêt une grande importance : elle permet non seulement de démontrer le respect des exigences réglementaires rigoureuses, mais elle joue également un rôle essentiel dans la gestion des risques, la qualité et la sécurité des dispositifs. Si le produit ne respecte pas les normes ou présente des dysfonctionnements, une documentation bien tenue peut faciliter la détection rapide des problèmes et la mise en place d'actions correctives. Cela garantit la protection des fabricants face aux éventuels litiges et renforce la confiance des utilisateurs et des autorités réglementaires dans la sécurité des dispositifs médicaux commercialisés.
Ressources Utilisées
Ressources documentaires
Pour élaborer une documentation technique conforme aux réglementations des dispositifs médicaux en Europe et aux États-Unis, il est essentiel de s'appuyer sur un éventail de ressources documentaires diversifiées.
Données internes
Une multitude de ressources internes sont essentielles pour la création de la documentation technique du dispositif médical. Ces ressources comprennent principalement les données internes issues des processus documentaires clés tels que le Système de Management de la Qualité[18], le DHF[8], le DMR[9] et le DHR[10] (figure 14).
Figure 14 : Données de la documentation technique suivant la stratégie de l’entreprise [Source : FineHeart-Interne]
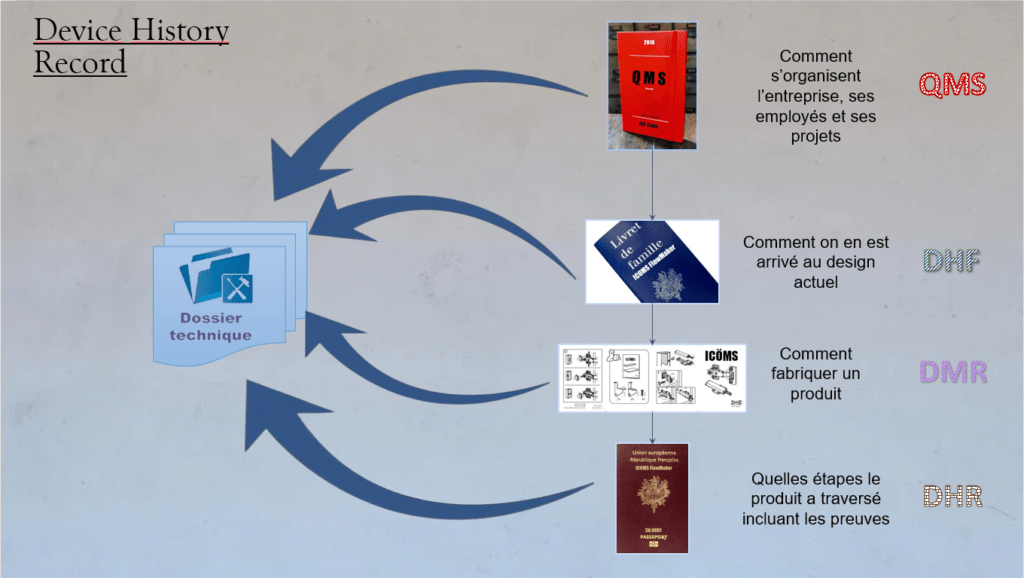
Ces documents, qui constituent l'épine dorsale de notre approche qualité, renferment des informations cruciales sur la conception, la fabrication et le développement de nos produits. Notre défi majeur est de consolider ces données en vue de composer une documentation technique conforme aux exigences rigoureuses du Règlement sur les Dispositifs Médicaux 2017/745.
Il est impératif que ces données soient soigneusement collectées, vérifiées et intégrées dans les dossiers de conformité requis, afin de garantir leur exactitude et leur pertinence. Cette approche nous permet de démontrer la conformité de nos dispositifs médicaux à toutes les phases du processus de production, et de répondre efficacement aux attentes des autorités réglementaires.
Guides européens
En Europe, il est crucial de se référer aux guides de l'organisme notifié (BSI [19]) qui est chargé de l'évaluation des dispositifs médicaux, ce guide a fourni des directives précieuses pour structurer et préparer les documents nécessaires à la conformité réglementaire, ainsi qu'aux guides du MDCG [13] qui fournissent des clarifications et des recommandations sur l'application du Règlement (UE) 2017/745. Les guides du Team NB[14] sont également essentiels, car ils offrent des perspectives spécifiques des organismes notifiés sur des questions réglementaires clés, permettant ainsi une meilleure compréhension des attentes réglementaires.
Guidance documents de la FDA
Aux États-Unis, les fabricants doivent se conformer aux Guidance documents de la FDA, qui expliquent les exigences spécifiques, y compris celles liées à la préparation de la PMA [2]— un processus de soumission très rigoureux pour les dispositifs médicaux de classe III qui assurent une évaluation approfondie de la sécurité et de l'efficacité du produit avant sa mise sur le marché.
Documents de l'IMDRF
En outre, les documents de l’IMDRF [15], qui visent à harmoniser les pratiques réglementaires au niveau international, constituent une ressource précieuse pour comprendre les standards globaux et faciliter la conformité dans différents marchés.
Normes spécifiques
En ce qui concerne les normes spécifiques,
- ISO 14708-5 :2020 spécifie les exigences relatives à la sécurité et aux performances des dispositifs d'assistance circulatoire implantables actifs, y compris les exigences des essais de type, des études sur les animaux et des évaluations cliniques.
- La norme ISO 14155 est incontournable pour la gestion des investigations cliniques des dispositifs médicaux, fournissant un cadre pour assurer la protection des droits, la sécurité et le bien-être des sujets humains, ainsi que la crédibilité des données cliniques recueillies.
- AAMI TIR17 :2017(R2023) Ce rapport d’information technique fournit des conseils aux fabricants de produits de soins de santé pour la qualification des matériaux polymères, des céramiques et des métaux destinés à être utilisés dans les produits de soins de santé stérilisés.
Comme nous visons le marché européen et américain, nous nous efforçons toujours de nous conformer aux normes applicables et reconnues par la FDA. Cette reconnaissance est vérifiée dans les documents du registre fédéral [6].
Ressources Humaines
Au cœur de la gestion de notre documentation technique, l'équipe réglementaire, composée du spécialiste des affaires réglementaires, de la consultante en affaires réglementaires, et de la responsable de validation, joue un rôle pivot.
Le spécialiste des affaires réglementaires s'assure que tous les processus au sein de l’entreprise soient conformes aux normes européennes et américaines, mettant à jour les dossiers techniques en fonction des évolutions réglementaires. La consultante externe apporte une expertise supplémentaire, vérifiant l'exactitude de notre documentation et conseillant sur les meilleures pratiques réglementaires.
La responsable de validation joue un rôle essentiel en apportant ses connaissances approfondies sur les besoins spécifiques et les spécifications requises par chaque service de l'entreprise. Sa compréhension détaillée des processus de validation et des exigences réglementaires lui permet de contribuer de manière significative à la conception de modèles de documents qui répondent précisément aux attentes des autorités réglementaires et aux besoins internes de chaque département. Grâce à son expertise, elle aide à garantir que les formulaires de documentation facilitent la cohérence, la précision et la complétude des informations, ce qui est crucial pour une mise en conformité efficace et la réussite des audits réglementaires.
Elaboration de la Documentation Technique
Création des Templates
La création des formulaires pour la documentation technique de nos dispositifs médicaux est un élément clé pour assurer la conformité réglementaire et faciliter la révision des dossiers par les organismes notifiés. Chaque Template a été méticuleusement conçu pour couvrir intégralement toutes les exigences des réglementations européennes et américaines, ainsi que pour répondre aux besoins internes de suivi et de validation de produit.
Processus de Développement des formulaires
Analyse des deux règlementations
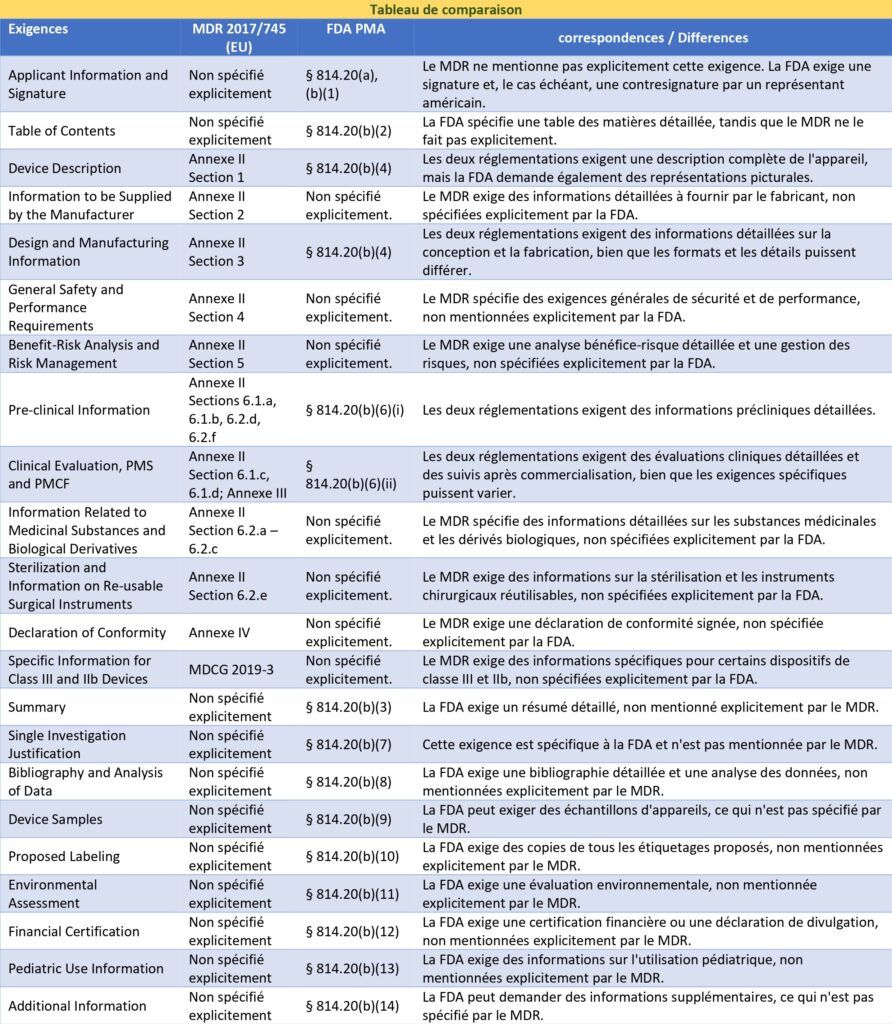
Dans le cadre de cette étude, une comparaison des exigences de la documentation technique des dispositifs médicaux selon deux cadres réglementaires majeurs a été réalisée : le MDR et la PMA de la FDA aux États-Unis. Cette comparaison vise à identifier les similitudes et les différences entre les deux réglementations (annexe 2), facilitant ainsi la préparation de la documentation pour les dispositifs médicaux destinés à être commercialisés sur les deux marchés.
Le tableau 1 présente une vue d'ensemble des exigences des deux cadres réglementaires, en mettant en évidence les sections correspondantes et les principales différences.
Tableau 1 : Comparaison des deux réglementations, européenne et américaine [Source : Auteur]
Définition des Sections Principales
Chaque section du formulaire correspond à un aspect spécifique de la réglementation ou à une exigence interne. La structure a été pensée pour guider l'utilisateur à travers toutes les étapes nécessaires à la documentation exhaustive du dispositif, depuis la conception jusqu'à la mise sur le marché (figure 15).
Figure 15 : Sections Principales de la documentation technique [Source : Auteur]
Incorporation des Exigences Réglementaires
Une comparaison entre les deux réglementations a été effectuée. Cependant, en réponse à un besoin et à une stratégie interne visant à commencer par le marquage CE, l'accent a été mis en premier temps sur les exigences du MDR pour la création des formulaires.
« General Information from the Manufacturer » : Comprend le nom, l'adresse, les coordonnées du fabricant, une description des installations et toutes certifications telles que l'ISO 13485. Cela peut également inclure des informations sur le système de gestion de la qualité du fabricant et la structure organisationnelle.
<
- Legal manufacturer
- Commercial designation of the medical device
- EU Technical Documentation Assessment Certificate
- QMS Certification
>
« Device Description and Specification » : Description détaillée de l'appareil incluant ses caractéristiques physiques, composants, fonctionnalités, utilisation prévue et les principes de fonctionnement. Les spécifications peuvent inclure les dimensions, les matériaux, les algorithmes logiciels et tout critère de performance critique.
<
Device Description
Intended Purpose and Intended Users
Classification
Basic UDI-DI / Unique Device Identifier (UDI-DI) / EMDN code
Devices covered by Technical Documentation
Market History (If Applicable)
>
« Information to be Supplied by the Manufacturer » : Détails sur ce qui sera inclus dans l'emballage, tels que les manuels d'utilisation, les étiquettes, les spécifications du produit, les informations de sécurité et réglementaires, et toutes les exigences de formation nécessaires pour garantir l'utilisation sûre de l'appareil.
<
User Information – Labelling
User Information – Device Operating Manual & Instruction for use
User Information – Electronic IFU
User Information – Patient/Physicians handbook (If applicable)
User Information – Implant card information (If applicable)
User Information – URL of the website
- Other
>
« Design and Manufacturing Information » : Comprend les enregistrements du développement de la conception, les entrées et sorties de conception, les activités de vérification et de validation, les points de contrôle critiques dans la fabrication, et les descriptions de l'environnement et des processus de fabrication.
<
Design History File
Product/design specifications of the device
User requirements
Manufacturing Information
Sites involved in design and manufacturing activities.
>
« General Safety and Performance Requirements » : Évaluation de toutes les exigences de sécurité et de performance générales applicables telles que définies par le cadre réglementaire (par exemple, annexe I du RDM UE). Cela inclut la preuve de conformité avec les normes, la gestion des risques tout au long du cycle de vie du produit, et la justification globale de la classification de l'appareil.
<
GSPR checklist of MDR Annex II section 4
Standards applied (21 CFR 814.20(b)(5))
Other applicable Regulations & Directives (PPE, Machinery, e-IFU regulation, etc)
>
Afin de mettre en œuvre cette section, nous avons mis en place un système de suivi des GSPR (GSPR Tracker) ainsi qu'une procédure de veille normative et réglementaire. Ce système comprend :
GSPR Tracker
Outil dynamique pour enregistrer (figure 16) et suivre la conformité (figure 17) de chaque dispositif aux exigences spécifiques des GSPR. Cela assure que toutes les exigences sont couvertes, et que la documentation est prête pour les audits réglementaires et les revues internes.
Figure 16 : Enregistrement des exigences GSPR [Source : Auteur]
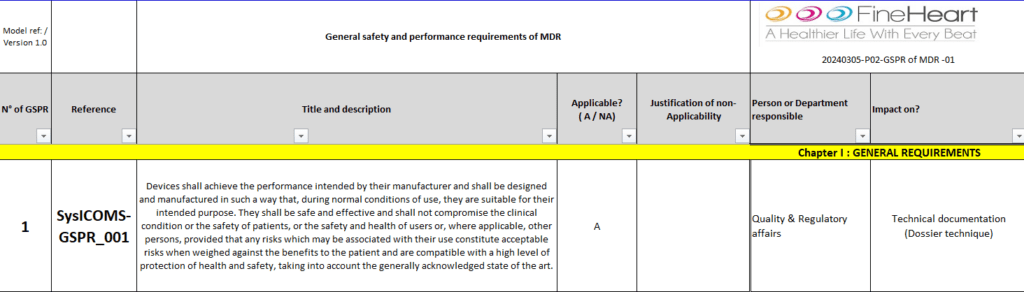
Figure 17 : Suivi de la conformité par rapport à l'exigence GSPR [Source : Auteur]
Veille Normative et Réglementaire
Pour rester à jour avec les normes en constante évolution et les directives réglementaires, nous avons établi un processus de veille réglementaire. Ce processus comprend la surveillance systématique des publications et des mises à jour réglementaires liées aux dispositifs médicaux, y compris les normes ISO pertinentes, les modifications dans le RDM UE, ainsi que d'autres régulations internationales et guides soit de l’IMDRF []15)ou du MDCG [13]
« Benefit-risk Analysis and Risk Management » : Comprend un processus systématique d'identification, d'évaluation et de contrôle des risques, ainsi qu'un résumé du processus d'analyse des bénéfices-risques. Documentation de toutes les activités de gestion des risques selon l'ISO 14971, et mises à jour continues basées sur les données de surveillance post-commercialisation.
<
Benefit-risk analysis.
Risk Management
o Risk management procedure/plan
o Risk scoring system
o Design risk assessment
o Production/process risk assessment
o Clinical/Application/Product risk assessment
o Risk management report
>
« Pre-clinical information » : Pour documenter toutes les études de laboratoire et sur les animaux menés pour évaluer la sécurité et l'efficacité de l'appareil médical avant qu'il ne subisse des tests cliniques sur les humains. Comprend des descriptions des méthodologies utilisées, des plans d'étude, des résultats et des analyses de données complètes. Cela couvre également toutes les études de biocompatibilité pertinentes, essentielles pour démontrer que l'appareil ne produit pas d'effets indésirables lorsqu'il est en contact avec les tissus humains. Assure la conformité avec l'ISO 10993-1 et autres normes applicables, cruciales pour obtenir les approbations réglementaires.
<
Biocompatibility
Electrical safety and electromagnetic compatibility (EMC)
Software Verification and Validation
Stability, including shelf life.
Performance and Safety – Design Verification and Validations
Packaging and Transit (Transport) testing
Sterilization of sterile product
Sterilization of reusable surgical instruments
Devices with a measuring or diagnostic function
Devices intended to be connected to other devices to operate as intended.
>
« Clinical evaluation, PMS and PMCF » : Pour établir et maintenir un processus systématique et planifié visant à évaluer et mettre à jour en continu l'évaluation clinique et surveiller la performance et la sécurité cliniques de l'appareil après sa mise sur le marché. Contient :
Clinical Evaluation : Résumé des données cliniques, revues de la littérature et analyses soutenant l'utilisation prévue de l'appareil.
<
Clinical development strategy/plan
Clinical evaluation plan/report
Personnel associated with the Clinical evaluation report.
Clinical investigation protocols/results
Statistical analysis plans
Copies of literature articles
Summary of Safety and Clinical Performance
>
Post-Market Surveillance (PMS) : Procédures et résultats du suivi continu de la performance de l'appareil dans le cadre clinique après sa commercialisation.
Post-Market Clinical Follow-up (PMCF) : Un sous-ensemble de la PMS, qui implique des études supplémentaires ou la collecte de données nécessaire lorsque les données existantes ne répondent pas suffisamment aux risques émergents ou à la performance à long terme.
<
Post Market Surveillance data/plan.
Periodic Safety Update Reports
Post market clinical follow-up plan & protocols/reports.
>
Répond aux exigences de l'annexe XIV du RDM pour maintenir la sécurité et la performance de l'appareil grâce à une surveillance proactive.
« Declaration of Conformity » : Une déclaration que l'appareil est conforme à toutes les législations pertinentes, listant les normes et réglementations applicables, telles que le RDM UE ou les parties 800-900 du CFR 21 de la FDA[16], et les détails de l'Organisme Notifié ayant examiné l'appareil, le cas échéant.
« Additional information required in specific cases » : Informations sur toutes déviations par rapport aux réglementations standard, mesures de sécurité supplémentaires pour des populations spécifiques, ou données supplémentaires demandées par les organismes réglementaires.
Validation Interne
Chaque template a été revu par les membres de chaque équipe concernée du projet, y compris la responsable de validation, pour garantir leur pertinence et leur complétude.
Révisions et Mises à Jour Continues
Les formulaires sont prévus d’être revus régulièrement pour intégrer les changements dans les réglementations et les retours des utilisateurs.
Rédaction des Enregistrements
La rédaction des enregistrements est assurée par les équipes techniques et est rigoureusement supervisée par l'équipe QARA pour garantir la conformité avec les exigences réglementaires. Les équipes utilisent des formulaires qui facilitent la précision et la cohérence des documents.
Les formulaires guident les équipes pour assurer une documentation complète et bien structurée, conforme au Règlement sur les Dispositifs Médicaux (RDM 2017/745). L’Équipe QARA veille à ce que tous les enregistrements soient correctement rédigés et conformes aux normes réglementaires.
Suivi d’avancement
Le suivi de l'avancement de la documentation technique est effectué via un tracker dédié (figure 18). Ce système permet de suivre chaque section de la documentation, de répertorier les documents exigés, et d'identifier leur localisation dans notre base documentaire. Le tracker inclut également les informations concernant les responsables de chaque tâche.
Fonctionnalités Clés du Tracker :
- Description Détaillée : Chaque partie de la documentation est clairement définie dans le tracker, facilitant la référence rapide et l'accès aux documents nécessaires et la désignation des parties est définie en se basant sur l’analyse des données de comparaison des deux règlementations, européenne et américaine.
- Localisation des Documents : Le tracker indique précisément où chaque document est stocké dans notre système de gestion documentaire, ce qui simplifie la récupération et la gestion des documents.
- Attribution des Responsabilités : Les responsables de la rédaction, de la révision et de l'approbation de chaque document sont identifiés, assurant la clarté des rôles et la responsabilité.
- Suivi de l'Avancement : Le tracker est régulièrement mis à jour pour refléter l'état d'avancement actuel des documents, permettant ainsi une gestion efficace du temps et des ressources.
Figure 18 : Suiveur (Tracker) d'avancement de la documentation technique [Source : Auteur]
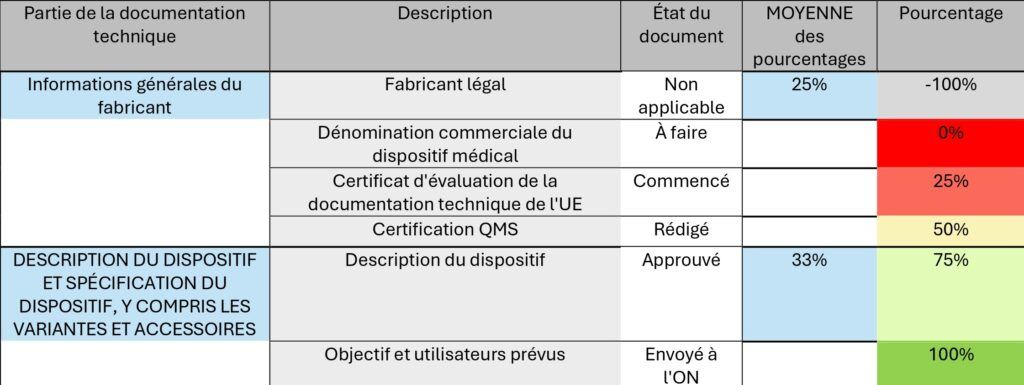
Les pourcentages reflètent le niveau de progression pour chaque exigence ou section spécifique de la documentation.
- Non applicable (N/A), avec un pourcentage de -100%, mais n’est pas pris en compte dans le calcul de la valeur moyenne des pourcentages.
- À faire, avec un pourcentage de 0%
- Commencé, avec un pourcentage de 25%
- Rédigé, avec un pourcentage de 50%
- Approuvé, avec un pourcentage de 75%
- Envoyé à l'ON (Organisme Notifié), avec un pourcentage de 100%
Bilan personnel
Durant mon alternance chez FineHeart en tant qu'Alternant Affaires Réglementaires, j'ai eu l'opportunité de m'immerger dans un environnement professionnel exigeant et stimulant. Ce bilan personnel vise à résumer les compétences acquises, les défis rencontrés, ainsi que les réussites et les leçons tirées de cette expérience enrichissante.
Compétences Techniques et Réglementaires
Au cours de mon alternance, j'ai développé une compréhension approfondie des normes réglementaires qui régissent le secteur des dispositifs médicaux, tant en Europe qu'aux États-Unis. J'ai notamment acquis des compétences dans les domaines suivants :
- Veille Réglementaire : Mise en place et gestion d'un outil de veille réglementaire structuré, permettant de surveiller, documenter et analyser les évolutions pertinentes pour le secteur.
- Conformité Réglementaire : Réalisation d'analyses des écarts pour évaluer la conformité aux exigences du Règlement des Dispositifs Médicaux 2017/745 (MDR) et du titre 21 CFR Part 820 de la FDA.
- Documentation Technique : Participation à l'élaboration et à la mise à jour de la documentation technique, incluant la création de formulaires conformes aux exigences réglementaires, ainsi que la gestion des enregistrements et des contrôles documentaires.
Défis et Résolutions
Un des principaux défis rencontrés fut la nécessité de rester constamment à jour avec les modifications réglementaires et les nouvelles normes. Pour surmonter cela, j'ai :
- Développé une méthodologie rigoureuse pour la veille réglementaire.
- Collaboré étroitement avec l'équipe QARA pour assurer une mise en conformité continue et efficace.
- Organisé des sessions de formation pour les collègues afin de partager les connaissances acquises et assurer une compréhension collective des exigences réglementaires.
- S’aligner avec la stratégie interne de l’entreprise en termes d’accès aux marchés et élaboration de la documentation technique dans la période d’alternance.
Réalisations et Contributions
Je suis fier des contributions suivantes :
- Mise en Œuvre d'outils de suivi : Développement d'outils de suivi pour les GSPR (General Safety and Performance Requirements) et la documentation technique, assurant une traçabilité et une conformité optimales.
- La mise en place du plan réglementaire de mise sur le marché européen et américain du produit de FineHeart.
- Préparation de la stratégie et des formulaires pour la mise en place de la documentation technique de l’ICOMS® FLOWMAKER®.
- Amélioration des Processus Documentaires : Participation active à l'amélioration des processus de gestion documentaire, incluant la préparation et l'intégration des documents dans un nouveau système de gestion électronique des documents (GED).
Compétences Interpersonnelles et Professionnelles
Cette expérience m'a également permis de développer des compétences interpersonnelles et professionnelles essentielles, telles que :
- Communication : Capacité à communiquer efficacement avec des équipes multidisciplinaires.
- Gestion du Temps et des Priorités : Apprentissage de la gestion du temps et des priorités pour répondre aux exigences strictes des projets réglementaires.
- Travail d'Équipe : Renforcement de mes compétences en travail d'équipe, en collaborant étroitement avec les membres de l'équipe QARA et d'autres départements pour atteindre les objectifs communs.
- Autoformation : Développement de la capacité à apprendre de manière autonome en recherchant activement des informations et des ressources pertinentes. Cela inclut la maîtrise de nouvelles réglementations, la compréhension des processus internes, et l'acquisition de compétences techniques supplémentaires nécessaires à la gestion efficace de la documentation et de la conformité réglementaire.
Points d’amélioration
Pour les prochains mois, plusieurs axes d'amélioration peuvent être envisagés :
- Traitement du Guide IMDRF "Non-In Vitro Diagnostic Device Market Authorization Table of Contents (nIVD MA ToC)" [17]: analyser l’impact de ce guide pour aider à structurer et compléter la documentation technique, en s'assurant qu'elle répond aux différentes réglementations.
- Schéma Radar : Élaborer un schéma radar pour visualiser l'avancement de l'élaboration de la documentation technique.
Conclusion
L'élaboration et la gestion de la documentation technique pour les dispositifs médicaux sont des éléments cruciaux pour garantir la conformité aux réglementations internationales, telles que le Règlement Européen 2017/745 et les exigences de la FDA. Ces processus permettent non seulement de démontrer la sécurité et l'efficacité des dispositifs, mais aussi de faciliter leur mise sur le marché dans différents pays.
Les initiatives actuelles montrent des progrès significatifs dans l'harmonisation des exigences réglementaires, ouvrant ainsi de nouvelles perspectives pour les fabricants de dispositifs médicaux. Cette harmonisation permet de simplifier les processus de conformité et de réduire les délais d'approbation, tout en maintenant des standards élevés de qualité et de sécurité.
Travailler sur la documentation technique signifie également aborder tous les autres aspects réglementaires essentiels tels que les exigences générales de sécurité et de performance (GSPR), les aspects cliniques, les logiciels, et les validations. Cela implique une compréhension approfondie de chaque composant du dispositif médical, assurant ainsi une conformité complète et intégrée.
Pour améliorer encore ces processus, l'utilisation de ressources documentaires spécifiques peut fournir des recommandations précieuses pour structurer la documentation technique. De plus, l'élaboration de schémas visuels pour suivre l'avancement de la documentation technique offre une méthode efficace pour suivre les progrès et identifier les domaines nécessitant encore des efforts.
En conclusion, l'expérience acquise chez FineHeart a non seulement enrichi mes compétences en matière de réglementation, mais a également ouvert des perspectives prometteuses pour mon avenir professionnel. En tant que spécialiste des affaires réglementaires, je suis désormais mieux préparé à relever les défis de conformité et à contribuer à l'innovation dans le domaine des dispositifs médicaux. Pour FineHeart, cette expérience a renforcé notre capacité à naviguer dans un paysage réglementaire complexe et en évolution constante, et a posé les bases pour une amélioration continue de nos processus de documentation technique.
Références bibliographiques
[1] Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ), vol. 117. 2017. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra
[2] C. for D. and R. Health, « Premarket Approval (PMA) », FDA. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-approval-pma
[3] S. S. Martin et al., « 2024 Heart Disease and Stroke Statistics : A Report of US and Global Data From the American Heart Association », Circulation, vol. 149, no 8, févr. 2024, doi : 10.1161/CIR.0000000000001209.
[4] « ABOUT US », FineHeart. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://fineheart.fr/about-us/
[5] « The Technologie », FineHeart. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://fineheart.fr/the-technologie/
[6] « Recognized Consensus Standards : Medical Devices ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfstandards/search.cfm
[7] C. for D. and R. Health, « Premarket Notification 510(k) », FDA. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k
[8] « 21 CFR Part 820 -- Quality System Regulation ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.ecfr.gov/current/title-21/part-820
[9] « 21 CFR 820.181 -- Device master record. » Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.ecfr.gov/current/title-21/part-820/section-820.181
[10] « 21 CFR 820.184 -- Device history record. » Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.ecfr.gov/current/title-21/part-820/section-820.184
[11] « Differences between DHF, DMR, and DHR | Scilife ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.scilife.io/blog/differences-dhr-dmr-dhr
[12] « 21 CFR Part 814 -- Premarket Approval of Medical Devices ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-814?toc=1
[13] « Guidance - MDCG endorsed documents and other guidance - European Commission ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en
[14] PascalePerspectives, « Team-nb Documents », Welcome to Team NB | Team NB. Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.team-nb.org/team-nb-documents/
[15] « Documents | International Medical Device Regulators Forum ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.imdrf.org/documents
[16] « Title 21 of the CFR -- Food and Drugs ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.ecfr.gov/current/title-21
[17] R. Wg, « Non-In Vitro Diagnostic Device Regulatory Submission Table of Contents (nIVD ToC) ».
[18] « NF EN ISO 13485 - Avril 2016 - Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/viewer-docs?urn=FA161550&type=norme&cleID=18158618000000
[19] « MDR Technical Documentation submission guidance ». Consulté le : 27 juin 2024. [En ligne]. Disponible sur : https://www.bsigroup.com/fr-FR/Dispositifs-Medicaux/Espace-Presse/Mises-a-jour-en-ligne/2022-news/mdr-documentation-guide/
[20] « Compatibility of materials subject to sterilization », AAMI TIR17:2017/(R)2023 ; Compatibility of materials subject to sterilization, mai 2022, doi : 10.2345/9781570207006.ch1.
Annexes
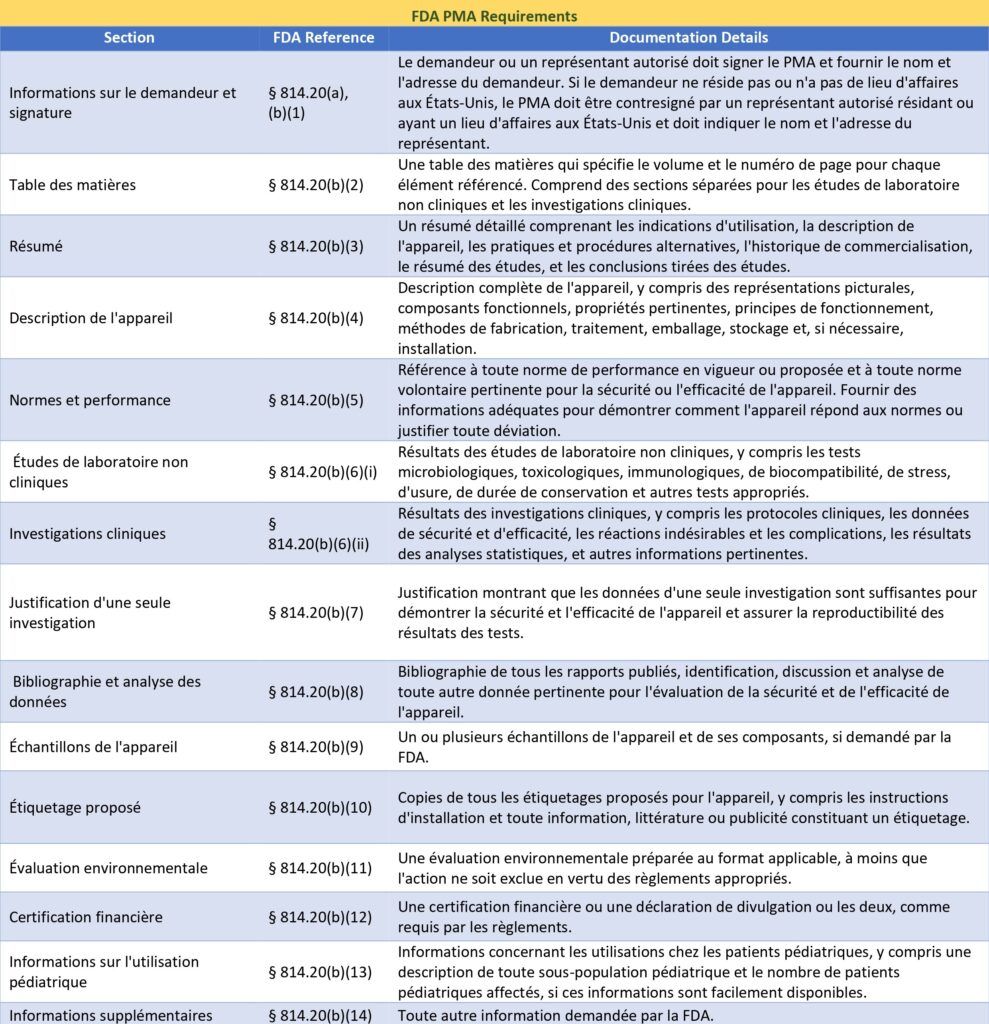
Annexe 1 : Tableaux des exigences du MDR pour la documentation technique [Source : Auteur]
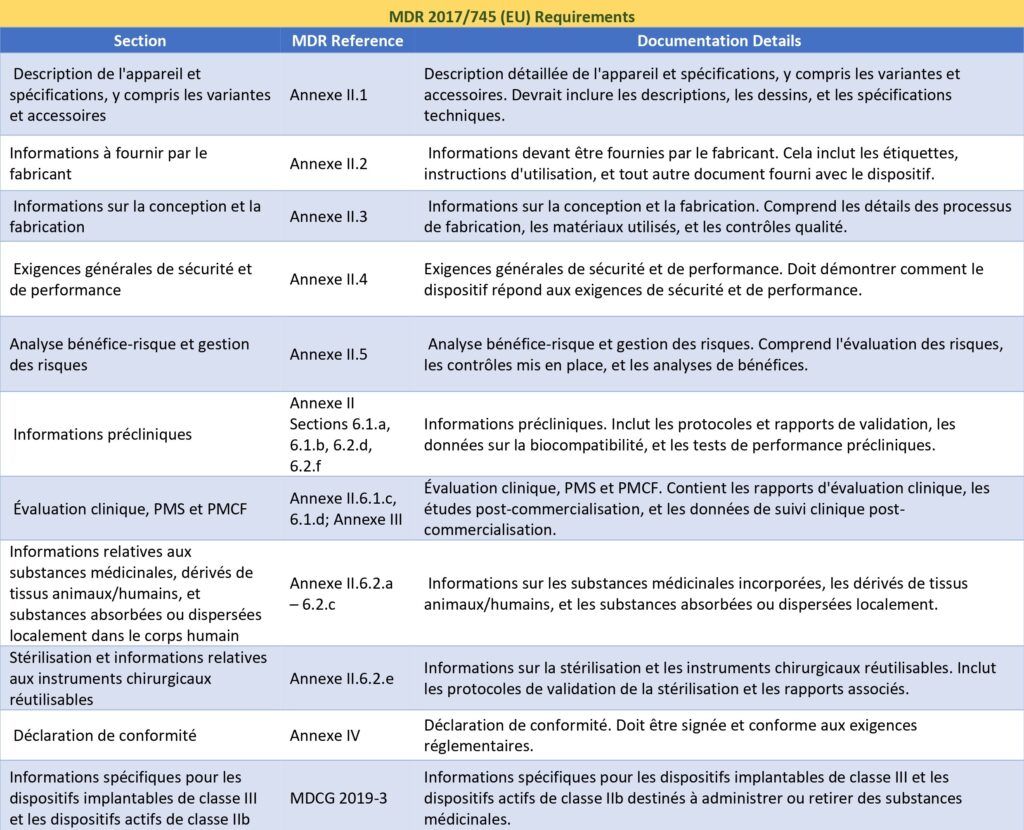
Annexe 2 : Exigences de l’FDA pour la PMA [Source : Auteur]