
IDS248 - Gestion de la transition des dispositifs médicaux de diagnostic in vitro "legacy devices" vers le Règlement européen 2017/746
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteure

Contact
- Charline VIGUIER : charline.viguier01@gmail.com
Citation
A rappeler pour tout usage : C.VIGUIER, « Gestion de la transition des dispositifs médicaux de diagnostic in vitro "legacy devices" vers le Règlement européen 2017/746 », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire d'apprentissage, https://travaux.master.utc.fr/, réf n° IDS248, juillet 2024, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids248/
Résumé
Le 26 mai 2022 a marqué un changement important dans le secteur des Dispositifs Médicaux de Diagnostic In Vitro puisque à cette date, le texte encadrant leur cycle de vie, la directive 98/79/CE, a été officiellement abrogé ; remplacé par le Règlement européen 2017/746.
Vis-à-vis de ce nouveau contexte réglementaire, les DM-DIV peuvent avoir différents statuts possibles : parmi ceux-ci, il y le cas des « Legacy devices », un terme désignant les dispositifs, couverts par un certificat UE valide délivré selon la directive, qui continuent à être mis à disposition sur le marché ou mis en service après la date d’application du Règlement. Pour ces dispositifs, la période de transition vers le Règlement, qui a débuté à son entrée en vigueur, c’est-à-dire le 26 mai 2017, n’est pas encore terminée.
Durant cette période, il y a tout de même certaines exigences réglementaires à appliquer par les fabricants afin que leurs dispositifs puissent continuer à être mis sur le marché de l’UE ou mis en service : par exemple, en matière de Surveillance Après Commercialisation et de Vigilance (cf. article 110 du Règlement européen 2017/746).
Ce mémoire de fin d’études s’attache donc à décrire une stratégie qui a été employée afin de les mettre en place. Le choix a été pris ici de coupler leur implémentation à la mise à jour du Système de Management de la Qualité de la start-up conformément à la norme EN ISO 13485 : 2016, ainsi qu'aux exigences réglementaires en matière de qualité (article 10, annexes IX et XI) grâce à son amendement A11.
Abstract
May 26th 2022 marked an important change in the In Vitro Diagnostic Medical Devices sector, as on this date, the text framing their life cycle, Directive 98/79/EC, was officially repealed ; replaced by European Regulation 2017/746.
With regard to this new regulatory context, IVDs can have different possible statuses : among these, there is the case of « Legacy devices », a term designating devices, covered by a valid EU certificate issued under the Directive, which continue to be made available on the market or put into service after the date of application of the Regulation. For these devices, the transition period to the Regulation, which began on May 26th, 2017, is not yet over.
During this period, there are still certain regulatory requirements for manufacturers to apply so that their devices can continue to be placed on the EU market or put into service : for example, with regard to Post-Market Surveillance and Vigilance (cf. Article 110 of Regulation (EU) 2017/746).
This master’s thesis therefore describes the strategy used to implement them. The choice was made here to couple their implementation with the updating of the start-up's Quality Management System in accordance with EN ISO 13485 : 2016, as well as with regulatory quality requirements (article 10, annexes IX and XI) thanks to its amendment A11.
Téléchargements


Remerciements
En premier lieu, j’adresse ma gratitude à toute l’équipe enseignante du Master Ingénierie de la Santé.
Je tiens à remercier les responsables de formation, Mme CLAUDE et M. PROT, pour leur implication et leur investissement dans la réussite de leurs étudiants.
Je remercie en particulier ma tutrice durant cette année d’apprentissage, Mme FOLLET, pour avoir toujours été très réactive à mes demandes de relecture de mes rapports, pour m’avoir continuellement encouragée à aller chercher plus loin afin de produire un travail construit et crédible ainsi que pour m’avoir motivée à prendre confiance en mes compétences.
Je souhaite ensuite remercier mon maître de stage, M. RAUX, pour m’avoir offert l’opportunité d’intégrer sa structure et m’avoir fait confiance dans mes missions ; cela a transformé cet apprentissage en une expérience particulièrement enrichissante à tous les niveaux.
Je voudrais également témoigner toute ma reconnaissance à Mme HARRIRI, ma collègue chez GENOTROPY et incroyable binôme de travail durant cette année.
J’adresse également tous mes remerciements à l’ensemble des enseignants de la Licence Professionnelle Maintenance et technologie : technologie médicale et biomédicale de l’Université de Paris qui m’ont tous permis d’intégrer la formation à l’UTC : notamment Mme MONDOLINI qui m’a donné envie de me lancer dans les Affaires Réglementaires et la Qualité des Dispositifs Médicaux, Mme LENAT et Mme BAUDOT.
Enfin, mes dernières pensées s’adressent à mes frères et ma sœur pour leur incroyable soutien. Merci à mes parents qui ont toujours cru en moi et sont le moteur de ma réussite. À ma maman, le pilier de ma vie.
Table des abréviations
Mémoire complet
Gestion de la transition des dispositifs médicaux de diagnostic in vitro "legacy devices" vers le règlement européen 2017/746
Introduction
Les tests effectués en cas de suspicion de maladie à coronavirus 2019 (COVID-19), les tests de grossesse et les dispositifs d’autosurveillance glycémique pour le diabète sont tous des produits de santé avec un point commun particulier.
Ils font tous en effet partie de la catégorie des Dispositifs Médicaux de Diagnostic In Vitro (DM-DIV) ; les « collègues » moins connus des Dispositifs Médicaux (DM) alors que jusqu’à 70% des décisions médicales reposent sur leurs résultats et qu’ils représentent moins de 1% de la dépense totale de santé en Europe en 2022 (Figure 1) [1]:
Figure 1 : Répartition des dépenses totales de santé en Europe en 2022 (Source : auteure d'après MedTech Europe 2022):

Les DM et les DM-DIV ont tous deux une finalité médicale mais les DM-DIV, contrairement aux DM, ne sont jamais utilisés ni à l’intérieur ni sur le corps humain de par leur définition réglementaire. En effet, les DM-DIV fournissent une information sur la base d’un échantillon prélevé sur le corps humain : des liquides biologiques comme le sang et l’urine ou des échantillons de tissu humain. Les DM-DIV sont principalement utilisés par le corps médical au sein des laboratoires privés de biologie médicale et des laboratoires des centres hospitaliers ; parfois directement par des utilisateurs non formés dits « profanes » , on parle alors d’autotests [2].
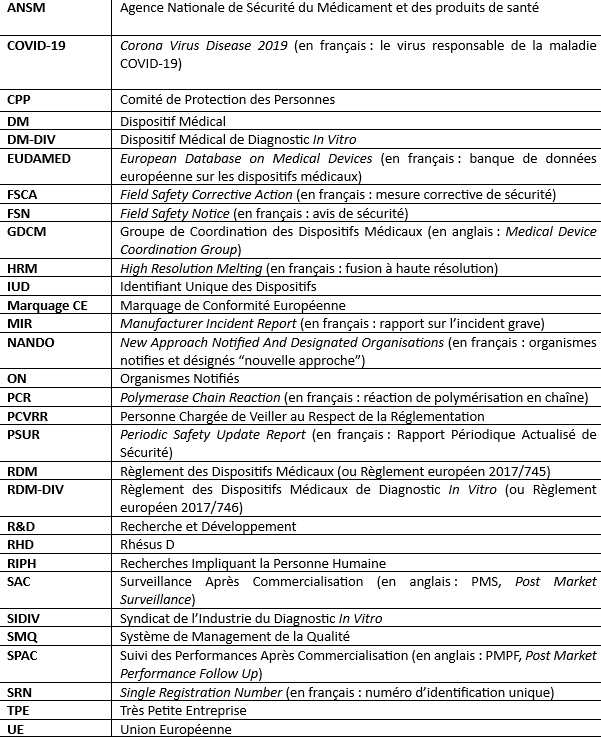
Dans le monde, le marché des DM-DIV était estimé à plus de 52.622 milliards de dollars en 2017 avec une perspective de croissance de 6.1% jusqu’en 2024 [3]. Cette croissance peut être expliquée par l’augmentation de l’incidence des maladies chroniques et infectieuses, de la population gériatrique et par les nombreuses innovations dans le domaine grâce aux multiples investissements dans les activités de Recherche & Développement (R&D). Dans le monde, ce marché est dominé, à près de 60%, par cinq entreprises : Roche, Abbott Laboratories, Danaher, Siemens Healthineers et Thermo Fisher Scientific (Figure 2) [3]:
Figure 2 : Vue d'ensemble du marché mondial du DM-DIV en 2018 (Parts de marché estimées pour 2024 en fonction de la croissance des ventes sur la période 2017-2024 (Source : EvaluateMedTech, Septembre 2018):
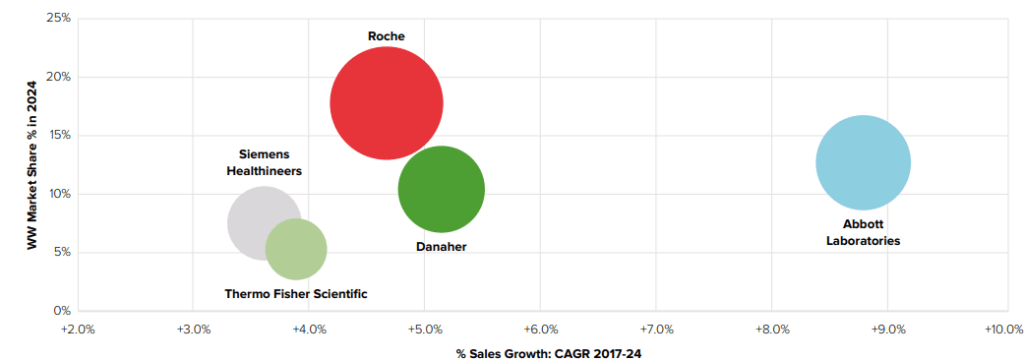
En Europe, la croissance du marché des DM-DIV enregistrée en 2021 a été sans précédent principalement à cause de la pandémie de COVID-19 : 41,2% selon les chiffres de MedTech Europe publiés en 2022 (Figure 3) [4]:
Figure 3 : Évolution du marché européen du DM-DIV sur deux axes : croissance (en blanc) et revenus en millions d'euros (en rouge) (Source : MedTech Europe, 2022):
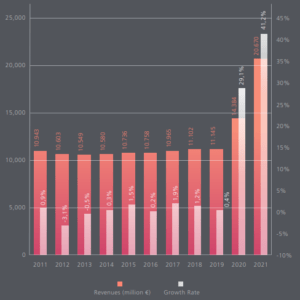
Au sein de ce marché européen, la France a atteint la seconde place en 2021, derrière l’Allemagne et devant l’Italie, avec un marché estimé à 2,610 milliards d’euros, en croissance de 22% par rapport à 2020 (Figure 4) [4]:
Figure 4 : Classement des pays par taille de marché pour les DM-DIV en 2021 (en millions d'euros) (Source : MedTech Europe, 2022):
D’après le Syndicat de l’Industrie du Diagnostic In Vitro (SIDIV) créé en 1977 qui fédère une centaine d’entreprises responsables d’environ 90% du chiffres d’affaires total du secteur en France, le marché français du DM-DIV reste dominé par 80% de Petites et Moyennes Entreprises (PME) et Très Petites Entreprises (TPE) en 2023. Dans un contexte post pandémie, son chiffre d’affaires s’est maintenu à 2 milliards d’euros (hors autosurveillance glycémique) en 2023 [5].
De plus, le marché a généré 15 100 emplois répartis dans plus de 200 métiers [5] ; une catégorie que j’ai rejoint dans le cadre de mon année d’apprentissage en intégrant en septembre 2023, la start-up GENOTROPY, TPE qui commercialise un DM-DIV utilisé pour le diagnostic génétique du rhésus fœtal dans le cadre du suivi de grossesse.
Comme le secteur des DM, celui des DM-DIV a subi un bouleversement majeur avec l’entrée en vigueur le 26 mai 2017 du règlement européen 2017/746 en remplacement de la directive 98/79/CE qui encadrait leur cycle de vie.
Pour les DM-DIV possédant un marquage de Conformité Européenne (CE) à la directive comme celui de GENOTROPY, cette date a aussi marqué le début d’une période de transition dont la fin ne cesse d’être repoussée. Bien que le règlement soit entré en application le 26 mai 2022, les fabricants concernés disposent en effet d’un temps plus long avant d’être contraints à l’appliquer dans son entièreté : jusqu’au 26 mai 2025 pour les dispositifs de classe D, 26 mai 2026 pour les classes C, 26 mai 2027 pour les classes B et 26 mai 2028 pour les classes A à l’état stérile.
Pour bénéficier de cette prolongation de la période transitoire, les fabricants doivent appliquer certaines exigences réglementaires.
C’est dans ce cadre que j’ai pris mes fonctions au sein de l’entreprise GENOTROPY, en tant qu’apprentie chargée des Affaires Réglementaires et de la Qualité.
Le travail d’apprentissage s’est concentré sur la résolution de la problématique suivante :
Quelle démarche opérationnelle les fabricants de dispositifs médicaux de diagnostic in vitro « legacy devices » doivent-ils suivre pour bénéficier de la prolongation de la période de transition de la directive 98/79/CE vers le règlement européen 2017/746 ?
La première partie de ce mémoire pose le contexte réglementaire lié aujourd’hui aux DM-DIV.
La seconde partie présente un cas d’usage : le produit commercialisé par GENOTROPY.
La troisième partie pose les exigences du règlement 2017/746 applicables aux « legacy devices » en période de transition en juillet 2024 :
- En matière de Surveillance Après Commercialisation,
- De vigilance,
- D’enregistrements sur EUDAMED.
Dans cette dernière partie, une attention particulière est donnée à l’identification des liens entre les exigences réglementaires présentées et le domaine qualité.
I) Contexte réglementaire des dispositifs médicaux de diagnostic in vitro
A) Un nouveau cadre réglementaire en Europe : le Règlement 2017/746
1) Le principe de la "Nouvelle Approche"
Les DM-DIV mis sur le marché en Europe sont encadrés par le règlement relatif aux dispositifs médicaux de diagnostic in vitro 2017/746 (RDM-DIV) [6]. Celui-ci est entré en vigueur le 26 mai 2017 mais son entrée en application n’a été promulguée que cinq ans plus tard le 26 mai 2022 afin de laisser un temps suffisant aux acteurs impliqués dans le cycle de vie des DM-DIV pour effectuer la transition par rapport au précédent texte, la directive 98/79/CE [7].
Que ce soit sous directive ou sous règlement, tout dispositif qui est commercialisé sur le marché unique européen doit obtenir en amont un marquage CE médical car celui-ci sert de preuve que le dispositif est conforme aux exigences de sécurité et de performance qui lui sont applicables en vertu du texte en vigueur : les « exigences essentielles ».
Dans les deux textes, sont ainsi retrouvés les fondamentaux de la « Nouvelle Approche » : un principe développé en 1985 selon lequel les législations européennes se limitent à fixer les exigences essentielles que les produits doivent obligatoirement respecter ; le choix étant ensuite laissé aux entreprises des moyens techniques à appliquer pour les respecter.
C’est le « renvoi aux normes » dont certaines élaborées par les organismes européens de normalisation, le Comité Européen de Normalisation (CEN) et le Comité Européen de Normalisation en Électronique et en Électrotechnique (CENELEC) sur mandat de la Commission Européenne, sont en plus harmonisées car elles fournissent une présomption de conformité à certaines exigences établies par la directive/Règlement. Toute norme harmonisée est identifiée par une décision d’exécution de la Commission Européenne et reste d’application volontaire [2] [8].
2) Le champs d'application : les dispositifs médicaux de diagnostic in vitro
i) Sous la directive 98/79/CE
La directive 98/79/CE définissait la notion de DM-DIV à son premier article. Pour connaître la classe de son DM-DIV, le fabricant devait ensuite se reporter à son annexe II où deux classes étaient distinguées sur la base de la destination d’utilisation du DM-DIV :
- La liste A (haut risque pour la santé publique et pour le patient) reprenant les dispositifs pour la détermination des groupes sanguins et le diagnostic des maladies hautement infectieuses ;
- La liste B (risque modéré) avec les dispositifs pour le diagnostic de groupes sanguins spécifiques, la détermination du marqueur tumoral « Prostate Specific Antigen » (PSA) etc.
Sous la directive, il était possible pour le fabricant ayant un dispositif ne faisant pas partie d’une des deux listes de recourir à une auto certification.
ii) Sous le Règlement 2017/746
Le règlement 2017/746 s’applique de même au « dispositif médical de diagnostic in vitro » à usage humain et à ses accessoires. Le terme de DM-DIV est défini à son article 2.2 ; il est plus précis que sous la directive 98/79/CE avec notamment : l’inclusion de la prédisposition à une affection ou une maladie et de la prédiction de la réponse à un traitement. Son champs comprend aussi les logiciels et dispositifs compagnons ce qui n’était pas le cas sous la directive 98/79/CE [2].
Seuls les dispositifs « in house », c’est-à-dire ceux dont la fabrication et l’utilisation se font dans le même établissement de santé, sont dispensés de se conformer aux exigences réglementaires autres que la conformité aux exigences essentielles de l’Annexe I et la déclaration aux autorités avant leur mise en service s’il remplissent un certain nombre de conditions (cf. article 5.5 du RDM-DIV). Cela signifie par exemple que ces dispositifs ne portent ni de marquage CE ni d’Identifiant Unique de Dispositif (IUD) et ne font pas l’objet de certificats délivrés par des organismes notifiés (ON).
Des DM-DIV peuvent être retrouvés à toutes les étapes du parcours de soin des patients (Figure 5):
Figure 5 : Représentation de certaines des étapes du parcours de soins dans lesquelles interviennent les DM-DIV (Source : auteure d'après le site du SIDIV, 2024)
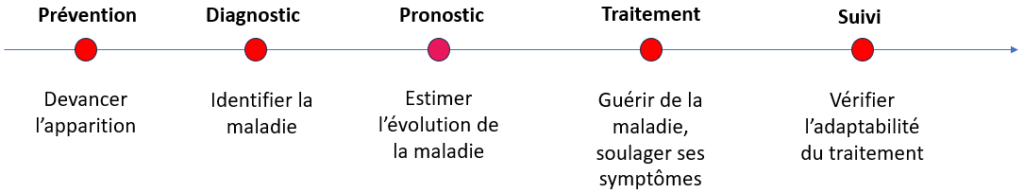
Ces dispositifs sont donc indispensables pour la santé des patients.
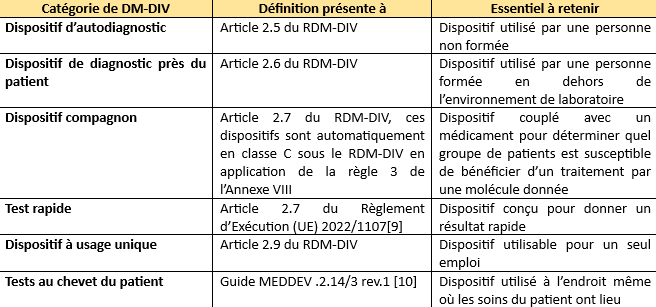
De plus, il existe une très grande diversité de DM-DIV sur le marché ; pour les désigner, plusieurs termes faisant référence à leur finalité d’usage sont ainsi fréquemment rencontrés (Tableau 1) :
Tableau 1 : Exemples de finalités d'usage de DM-DIV (Source : auteure d'après le RDM-DIV):
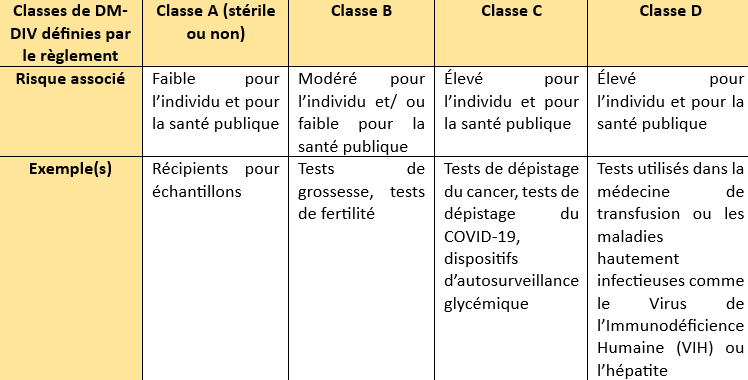
Avec le RDM-DIV, les DM-DIV sont désormais répartis en 4 classes (Tableau 2) en fonction de leur usage revendiqué et du risque qu’ils engendrent pour l’individu et pour la santé publique en cas de défaillance ; les règles les plus strictes s’appliquant aux classes de risque les plus élevées [11] :
Tableau 2 : Les quatre classes de risque des DM-DIV sous le Règlement 2017/746 (Source : auteure d'après le RDM-DIV):
Pour identifier la classe de son dispositif, le fabricant doit se référer à l’Annexe VIII du RDM-DIV, s’appuyer sur le guide MDCG 2020-16 Rev.2 [12] et le document de l’International Medical Device Regulators Forum (IMDRF) (en français : Forum international des régulateurs des dispositifs médicaux) intitulé « Principles of In Vitro Diagnostic Medical Devices Classification » [13].
De manière générale, il est essentiel que le fabricant lise, dans un premier temps, le chapitre « Règles d’application » du RDM-DIV car c’est un préalable à l’identification, dans un second temps, de la ou les règles, parmi les 7 listées, qui sont applicables afin de déterminer la classe de son DM-DIV.
La classification du DM-DIV est vérifiée par l’ON lors de la procédure d’évaluation de la conformité. Il est alors possible que l’ON soit en désaccord avec le fabricant quant à la classe retenue. Dans ce cas, l’autorité compétente de l’État membre où se trouve le fabricant est chargée de trancher sur la classe (cf. article 47 « Classification des dispositifs » du RDM-DIV).
La définition des DM-DIV et leur classification n’ont cependant pas été les seules modifications induites par le règlement 2017/746.
3) Le règlement 2017/746 : principales différences avec la directive 98/79/CE
Sur de nombreux autres aspects, le règlement 2017/746 renforce le texte de la directive 98/79/CE : il est plus long, plus précis et intègre des nouveautés (113 articles et 15 annexes contre 24 articles et 10 annexes dans la directive).
i) Le droit
De par sa nature, jusqu’au 26 mai 2022, la directive 98/79/CE était transposée dans le droit interne de chaque pays de l’UE avec certain délai accordée pour son application : en France, la transposition était ainsi faite dans le Code de la Santé Publique (livre II titre II cinquième partie) via l’ordonnance n°2001-198 du 1er mars 2001 et le décret n°2004-198 [14].
Contrairement à la directive, le règlement 2017/746 ne fait toutefois pas l’objet d’une transposition dans le droit national : il est directement applicable dans tous les États membres de l’UE ce qui permet notamment d’éviter les différences d’interprétation liées à la transposition des directives.
ii) Les exigences pour obtenir le marquage CE
Dans l’intérêt des utilisateurs des dispositifs, les exigences à respecter pour obtenir le marquage CE ont été considérablement renforcées par le RDM-DIV, notamment en matière de :
- Surveillance Après Commercialisation (SAC) ;
- Évaluation des performances : en particulier en ce qui concerne les règles relatives aux preuves cliniques à fournir ;
- Traçabilité de la chaîne d’approvisionnement : principalement avec l’IUD.
iii) Les organismes notifiés : possibilité d'intervention dans la procédure d'évaluation et évolution de leur nombre
Les ON sont des acteurs, désignés (ou « notifiés ») pour évaluer de façon indépendante la conformité de certains DM-DIV avec les exigences de sécurité applicables [11] en vertu du texte en vigueur (Directive ou Règlement) avant leur mise sur le marché sur le territoire de l’UE. Cette désignation n’est possible qu’après une réussite de la procédure dite de « joint assessment » (ou évaluation conjointe, cf. Article 38 du RDM-DIV) auxquels ils sont soumis ; procédure menée par l’autorité compétente nationale (l’Agence Nationale de Sécurité du Médicament et des produits de santé en France ou ANSM en France), la Commission Européenne et deux autres autorités compétentes européennes désignées par la Commission et le Groupe de Coordination des Dispositifs Médicaux (GDCM).
Avec la transition vers le règlement, les exigences et compétences demandées aux ON ont néanmoins été renforcées, notamment en ce qui concerne leurs ressources humaines et leurs processus internes. Les règles de compétence, d’impartialité et d’indépendance auxquelles sont soumis les ON figurent au Chapitre IV ainsi qu’en Annexe IV du RDM-DIV.
Ce renforcement des exigences peut expliquer la réduction du nombre d’ON désignés : 22 organismes désignés dans le cadre de la directive [16] contre 12 organismes au titre du RDM-DIV. Tous les ON désignés selon les directives et règlements européens sont recensés sur la plateforme NANDO (New Approach Notified And Designated Organisations, en français : organismes notifies et désignés “nouvelle approche“) ; chaque décision de désignation prenant effet un jour après sa publication sur la plateforme [2] [16].
Cette réduction a d’autant plus d’impact que leur nombre est toujours inférieur dans le secteur des DM-DIV par comparaison à celui des DM : ainsi, en juillet 2024, seuls 12 ON sont enregistrés sur NANDO dans le premier cas contre 45 dans le second.
En outre, le fabricant doit sélectionner, parmi ces 12 ON, ceux désignés pour l’évaluation de la conformité au RDM-DIV des types de DM-DIV qui le concernent (Figure 6).
Figure 6 : Nombre d'organismes notifiés auxquels un fabricant peut réellement faire appel pour évaluer sa conformité au RDM-DIV (Source : auteure):

Il y a donc moins d’ON disponibles sous le RDM-DIV par rapport à la directive 98/79/CE alors que les chiffres montrent qu’en vertu du RDM-DIV, ils devront superviser dix fois plus de DM-DIV (80% au lieu de 8%) ; la plupart pour la première fois [15] avec un niveau de participation toujours proportionné à la classe de risque du dispositif (Figure 7):
Figure 7 : Évolution de l'obligation d'intervention des organismes notifiés dans la procédure d'évaluation de la conformité des DM-DIV (Source : auteure d'après la thèse de Quentin Bugnet, 2019):
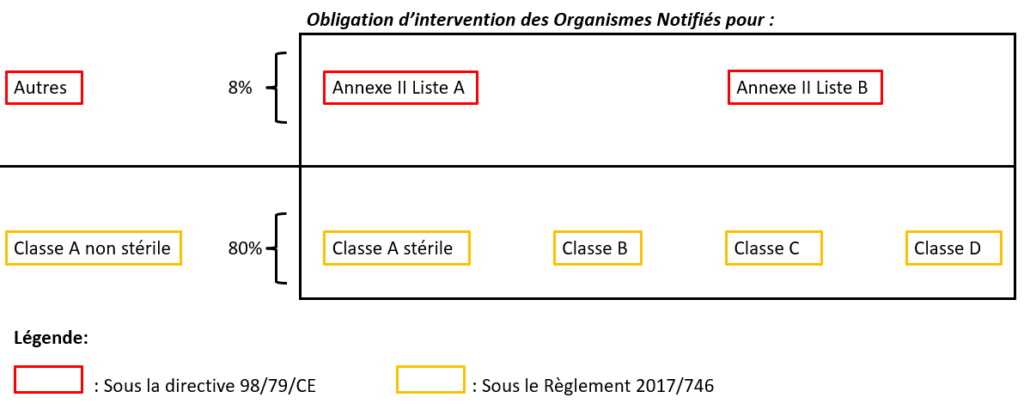
En somme, entre la Directive 98/79/CE et le règlement européen 2017/746, 10 fois plus de DM-DIV nécessitent l’intervention d’un ON pour leur procédure d’évaluation de la conformité ; avec deux fois moins d’ON disponibles et des exigences à respecter par les différents opérateurs économiques renforcées et nouvelles [17].
B) Le processus de marquage CE sous le Règlement 2017/746
Le processus de marquage CE sous le RDM-DIV obéit au raisonnement suivant (Figure 8) :
Figure 8 : Logique de raisonnement pour le marquage CE médical d'un DM-DIV (Source : auteure):
1) Vue d'ensemble sur le processus
Dans un premier temps, indépendamment de la classe de son DM-DIV, le fabricant doit connaître et comprendre les étapes pour aboutir à son marquage CE médical de son dispositif (Tableau 3) :
Tableau 3 : Les différentes étapes du processus de marquage CE d'un DM-DIV (Source : auteure):

Au sein de ce processus, l’étape la plus importante est la sixième car elle fait intervenir des organismes tiers au fabricant : les ON.
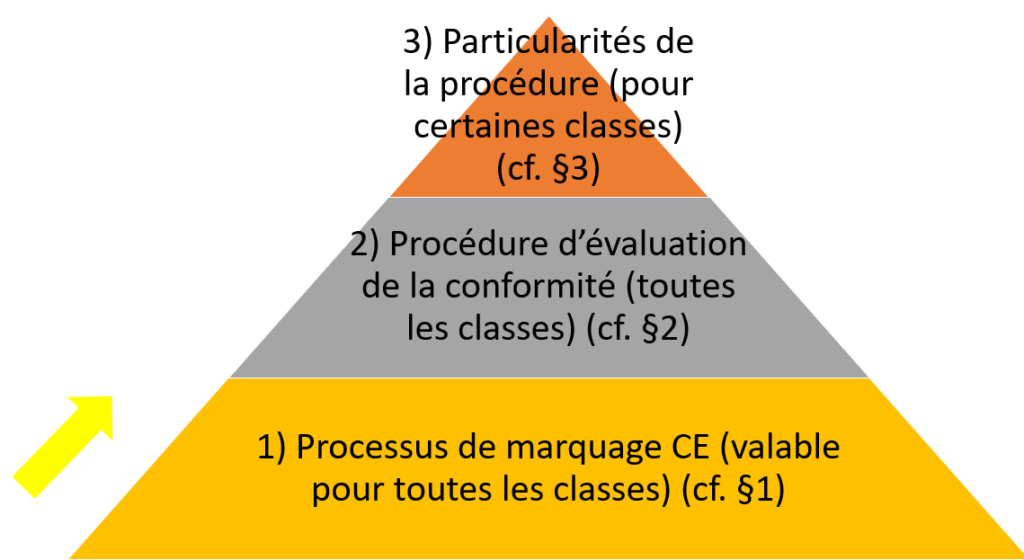
2) Zoom sur l'étape 6 : la procédure d'évaluation de la conformité d'un dispositif médical de diagnostic in vitro sous le Règlement 2017/746
Au niveau de l’étape 6, c’est seulement dans le cas où le dispositif est en classe A non stérile que le fabricant du DM-DIV peut s’auto marquer CE c’est-à-dire que la procédure d’évaluation de la conformité de son produit sera placée sous sa seule responsabilité.
Pour le reste des classes, le fabricant doit donc obligatoirement passer par un des ON désignés pour l’évaluation de la conformité de son DM-DIV selon le Règlement.
Quand ils sont impliqués, les ON réalisent des procédures d’évaluation de la conformité qui comprennent toujours deux parties (Figure 9) :
- Partie 1 : Examen de la Documentation Technique ;
- Partie 2 : Audit de certification du SMQ.
Figure 9 : Les deux parties de la procédure d'évaluation de la conformité de tous les DM-DIV (à partir de la classe A stérile) (Source : auteure d'après le RDM-DIV):
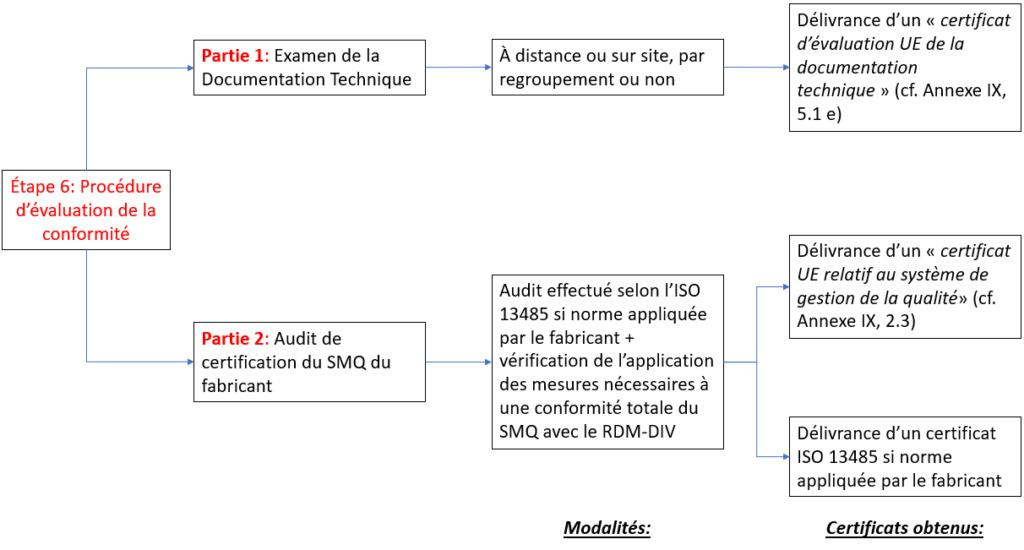
Il est à noter que les certificats EN ISO 13485 :2016 obtenus à l’année n sont valables 3 ans : un audit de suivi a lieu chacune des deux premières années suivant la certification (années n+1 et n+2) ; à la troisième année a lieu l’audit de re certification (année n+3). Les certificats UE sont quant à eux valides 5 ans sans audit intermédiaire.
3) Les particularités de la procédure pour certains types de dispositifs médicaux de diagnostic in vitro
La procédure d’évaluation de la conformité de certains DM-DIV peut inclure des particularités supplémentaires aux démarches présentées plus haut :
- Pour les dispositifs compagnons : participation d’une autorité compétente désignée par les États membres conformément à la directive 2001/83/CE [18] ou participation de l'European Medicines Agency (EMA) (conformément à l’Annexe IX section 5.2 du RDM-DIV) ;
- Pour les DM-DIV de classe D (avec ou sans spécifications communes) : participation des laboratoires de référence de l’Union Européenne (conformément à l’Annexe IX section 4.9 du RDM-DIV) ; laboratoires désignés en accord avec l’article 100 du RDM-DIV ;
- Pour les DM-DIV de classe D sans spécification communes : conformément à l’article 48.6 du RDM-DIV, l’ON fait en plus appel à un panel d’experts désignés par la Commission en concertation avec le GDCM pour les domaines d’application du règlement nécessitant une expertise de pointe (cf. article 106 du règlement 2017/745).
C’est précisément dans ce cadre réglementaire que s’inscrit mon entreprise d’apprentissage pour l’année : GENOTROPY avec son DM-DIV, l’AIO RHD Fetal DNA Kit.
II) Présentation de l'organisme d'accueil et son produit
A) Présentation de GENOTROPY
1) Les missions de l'entreprise
GENOTROPY est une start-up française établie à Rouen (Normandie) crée en novembre 2018 par M. Grégory RAUX, son président, qui reste le seul employé de l’entreprise jusqu’à aujourd’hui.
Depuis 2021, GENOTROPY commercialise en France un DM-DIV de diagnostic du Rhésus D (RHD) fœtal ; ce kit ayant été conçu puis breveté par l’entreprise à la suite de plusieurs années de travaux de R&D en interne.
Ce test est réalisable chez les femmes enceintes de RHD négatif, dès la douzième semaine d’aménorrhée, par des laboratoires d’analyse accrédités par le COFRAC selon la norme NF EN ISO 15189 : 2022 « Laboratoires de biologie médicale -Exigences concernant la qualité et la compétence » [19] pour réaliser des tests prénataux : c’est le cas de Laboratoires de Biologie Médicale (LBM) hospitaliers ainsi que de laboratoires d’analyses médicales privés [20].
Par ailleurs, les tests de génotypage du rhésus fœtal comme celui de GENOTROPY sont remboursés à 100% en France depuis juillet 2017 par l’Assurance Maladie [21]; et depuis 2022 en Allemagne [22], le premier marché du DM-DIV en Europe [4].
2) Analyse des forces et faiblesses de GENOTROPY
Une analyse SWOT a été conduite (Figure 10) : elle permet d’identifier les forces (« Strengths ») et faiblesses (« Weaknesses ») (des facteurs internes), menaces (« Threats »), et opportunités (« Opportunities ») (des facteurs externes) liées à l’organisation actuelle de GENOTROPY :
Figure 10 : Matrice SWOT de GENOTROPY (Source : auteure):

Cette analyse met en évidence une entreprise prometteuse existant déjà depuis plusieurs années et dont l’organisation future doit être développée si elle souhaite prospérer.
B) Le produit de GENOTROPY : l'AIO-RHD Fetal DNA Kit
Il s’agit ici de présenter le contexte clinique et le fonctionnement du produit commercialisé par GENOTROPY.
1) Contexte clinique de réalisation : les incompatibilités fœto-maternelles
Sur la membrane des érythrocytes, peuvent être présents ou absents un certain nombre d’antigènes : parmi ceux-ci, on retrouve l’antigène majeur RHD. Sa présence fait que l’individu est dit RH positif (RH :1), son absence RH négatif (RH : -1).
Il y a une incompatibilité fœto-maternelle de RHD quand la femme enceinte est de RH : -1 et son fœtus de RH :1.
En effet, dans ce cas, la femme enceinte peut produire des anticorps contre les antigènes RHD de son fœtus lors d’un contact sanguin se produisant principalement lors de l’accouchement ou d’une interruption de grossesse (dans les deux cas, lors de l’expulsion du fœtus, le placenta peut être perturbé ce qui augmente le risque de passage des globules rouges fœtaux). Cette production d’anticorps (ou « alloimunisation ») engendre la destruction des érythrocytes du fœtus : une anémie hémolytique sévère apparaît alors, responsable de la Maladie Hémolytique du Fœtus et du Nouveau-né [23].
Cette anémie engendre généralement la mort du fœtus en l’absence soit de traitement transfusionnel in utéro (curatif) soit d’une injection préventive d’anticorps de prophylaxie (ou Immunoglobulines anti-D) qui supprime le phénomène d’alloimunisation. Des risques et effets indésirables existent en lien avec cet injection ; ils sont les même que ceux liés à la vaccination.
On comprend de ce fait l’intérêt d’un diagnostic précoce du RHD fœtal, qui s’il est négatif, permet d’éviter l’injection inutile des Immunoglobulines anti-D chez la femme enceinte de RH :-1 [23].
2) Fonctionnement du kit de GENOTROPY
i) Extraction de l'ADN fœtal
Le produit de GENOTROPY fonctionne à partir de l’ADN fœtal : celui-ci est extrait à partir du plasma issu d’un prélèvement sanguin effectué chez la femme enceinte de RH : -1 (non invasif), grâce à un kit d’extraction de l’ADN. Ce kit d’extraction n’est pas commercialisé par GENOTROPY qui recommande simplement à ses clients d’utiliser un kit marqué CE optimisé pour l’extraction d’ADN fœtal [20].
En France, les prélèvements sanguins pour dépister les anticorps irréguliers sont obligatoires chez les femmes enceintes de RH :-1 : au premier trimestre et au cours des 6e, 8e et 9e mois de grossesse [23]. Or, seul un certain pourcentage de ces femmes ont un fœtus RH : 1. Par conséquent, la réalisation précoce du test de GENOTROPY chez ces femmes permet aussi d’éviter ces multiples dépistages inutiles.
ii) Gène RHD
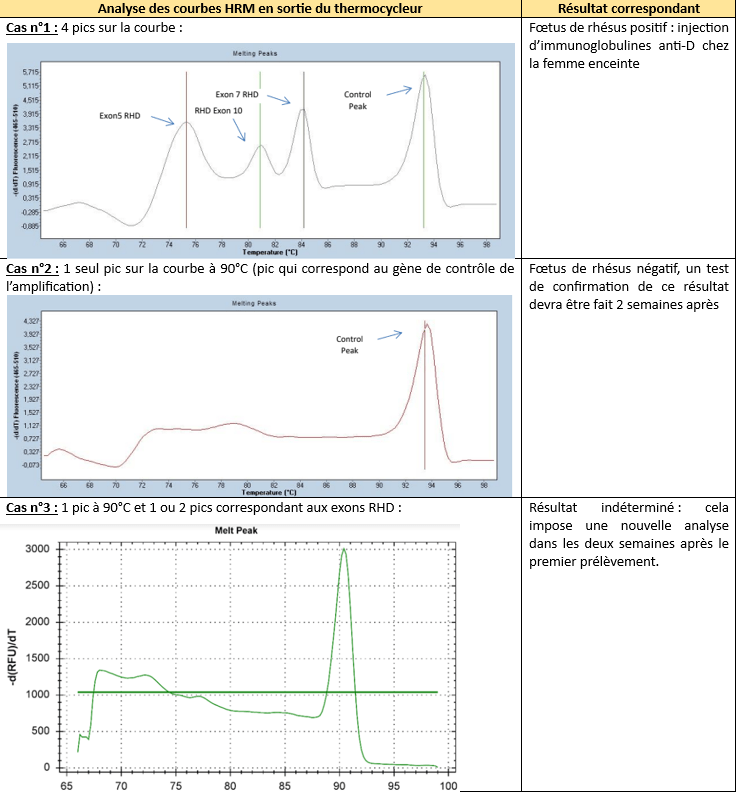
Sur l’ADN fœtal libre circulant dans le plasma maternel, le kit de GENOTROPY détecte spécifiquement les exons 5, 7 et 10 du gène RHD.
Les gènes RHD et RHCE, qui forment le locus RH, sont positionnés sur le chromosome 1. Il s’agit de deux gènes séparés par le gène SMP1 et présentant 92% de similitudes [23] dans leurs séquences respectives de 10 exons. Le gène RHD présente 2 boites rhésus en amont et en aval de sa séquence qui sont de même hautement homologues (Figure 11):
Figure 11 : Organisation génétique du locus RH (Source : thèse de doctorat de Fabien Sohet, 2008):
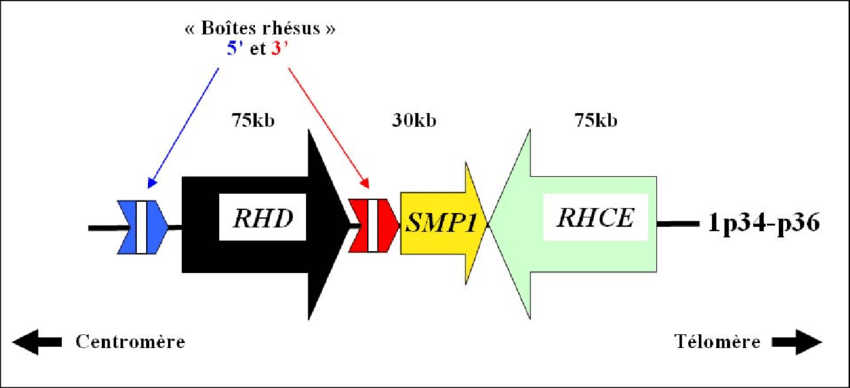
Les gènes RHD et RHCE codent respectivement l’antigène RHD et l’antigène RHCE qui traversent la membrane des érythrocytes.
Chez les individus caucasiens, le phénotype RH : -1 est présent à une fréquence de 15%, dans la population africaine à 8% et asiatique à moins de 1% [20] [24]. Le cas le plus fréquent expliquant l’absence de l’antigène RHD est celui de la délétion totale et homozygote du gène RHD.
iii) Principe de fonctionnement du kit de GENOTROPY
Grâce au kit de GENOTROPY, les exons 5, 7 et 10 du gène RHD de l’ADN fœtal, sont amplifiés par technique de « Polymerase Chain Reaction » (PCR) au sein d’un appareil programmable, un thermocycleur. Cette technique permet en effet l’amplification, grâce à l’utilisation d’une enzyme Taq polymérase thermorésistante, d’une région spécifique d’un acide nucléique donné afin d’en obtenir une quantité suffisante pour le détecter et l’étudier. Un cycle de PCR comprend trois étapes, toutes effectuées à des températures différentes afin de contrôler l’activité enzymatique :
- Dénaturation : les doubles brins d’ADN sont séparés l’un de l’autre ;
- Hybridation : des amorces spécifiques se couplent à chaque fragment d’ADN ;
- Polymérisation : les fragments d’ADN sont répliqués par l’enzyme à partir des amorces et des oligonucléotides présents dans le milieu de réaction.
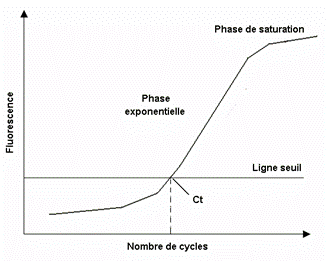
Dans le cas de GENOTROPY, le type de PCR utilisé est la PCR en temps réel ou q-PCR : cela signifie que la quantité d’ADN est mesurée à chaque cycle de PCR grâce à un « reporter » fluorescent. Le Ct est le nombre de cycles de PCR à partir duquel le signal de fluorescence qu'il émet est statistiquement différent du bruit de fond représenté par la ligne seuil sur le graphique ci-dessous (Figure 12):
Figure 12 : Courbe de suivi d'amplification de la PCR représentant l'intensité de fluorescence en fonction du nombre de cycles de PCR (Source : Thèse de Mariko Matsui, Novembre 2009):
iv) Méthode d'analyse post-PCR
En fin de la réaction de PCR, une analyse « High Resolution Melt » (HRM) (en français : fusion à haute résolution) est réalisée, également dans le thermocycleur. Cela signifie que les produits de la PCR, les amplicons, sont soumis à une augmentation de température progressive provoquant une séparation des deux brins d’ADN appelée fusion. Cette fusion est observable en temps réel grâce à des colorants intercalants ayant la propriété d’augmenter en fluorescence lorsque fixés à de l’ADN double brin. En revanche, lorsqu’il n’y a pas de double brin, les intercalants ne peuvent pas se fixer ; par conséquent, la fluorescence est faible.
La courbe représentant l’intensité de fluorescence en fonction de la température est appelée courbe de fusion (Figure 13).
Figure 13 : Courbe de fusion obtenue avec la technique de HRM (Source : Wikipedia, page "High Resolution Melt"):
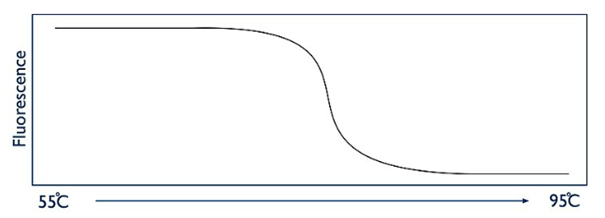
Le Tableau 4 ci-dessous montre plusieurs dérivées de cette courbe de fusion : ces dérivées sont appelées courbes d’analyse. Elles permettent de mettre en évidence les pics correspondant aux températures de fusion de chaque exon du gène RHD (Tableau 4).
Tableau 4 : Différents cas obtenus suite à la réalisation du test de GENOTROPY (Source : auteure d'après le manuel utilisateur de GENOTROPY):
C) Le positionnement réglementaire de GENOTROPY
1) Rôle exercé par l'entreprise dans le cadre de la directive et du Règlement
GENOTROPY possède le statut de « fabricant » au regard de la directive 98/79/CE et du règlement européen 2017/746 bien que l’entreprise sous-traite la fabrication de son kit car la définition n’exige pas qu’il fabrique son produit ; il peut aussi le faire fabriquer (définition à l’article 2.23 du RDM-DIV).
2) Certifications possédées par l'entreprise selon la directive
D’une part, GENOTROPY possède un Système de Management de la Qualité (SMQ) certifié EN ISO 13485 : 2016 par un ON depuis 2021 ; sa certification ayant été renouvelée en septembre 2023 pour un second cycle de 3 ans. Dans le cadre de la procédure d’évaluation de la conformité effectuée sous directive (Annexe IV, Assurance de la Qualité complète), ce SMQ est aussi couvert par un certificat UE relatif au système de gestion de la qualité.
D’autre part, le produit de GENOTROPY est considéré comme un DM-DIV : un « kit » composé d’un ensemble de « réactifs » (et non « instruments »), exclusivement destinés à un usage professionnel (et non « profane »). Ce produit a fait l’objet d’une documentation technique évaluée sous directive, à l’origine d’un autre certificat UE.
Par conséquent, aujourd’hui, GENOTROPY possède 3 certificats avec différentes durées de validité.
3) Statut du dispositif de GENOTROPY en période de transition
Selon le règlement européen 2017/746, un DM-DIV peut avoir 3 statuts différents. Alors que les deux premiers cas concernent des dispositifs qui existaient déjà sous directive, le troisième cas s’applique aux DM-DIV nouveaux :
- Cas n°1 : « Old device » (ou dispositif ancien) : il s’agit d’un dispositif qui a été mis sur le marché avant le 26 mai 2022 sous la directive et qui est resté présent sur le marché depuis cette date ;
- Cas n°2 : « Legacy device » : c’est un produit qui remplit deux conditions :
- Il est placé sur le marché après le 26 mai 2022 et jusqu’à la fin de sa période de transition conformément aux dispositions transitoires du Règlement ;
- Ce dispositif est :
- Soit couvert par un certificat UE valide délivré par un ON avant le 26 mai 2022 selon la directive ;
- Soit couvert par une déclaration de la conformité établie avant le 26 mai 2022 selon la directive et aura une procédure d’évaluation de la conformité au RDM-DIV qui nécessitera l’intervention d’un ON ;
- Cas n°3 : « IVDR device » : c’est un dispositif nouveau mis sur le marché après le 26 mai 2022 conformément au RDM-DIV.
Le produit de GENOTROPY fait donc partie de la deuxième catégorie : les « Legacy devices ».
4) Classe et procédure d'évaluation de la conformité du dispositif sous le Règlement
GENOTROPY a identifié la règle de classification 2 tiret 2 de l’Annexe VIII du règlement comme étant applicable à son produit : « Les dispositifs destinés à être utilisés pour déterminer les groupes sanguins (…) relèvent de la classe C, sauf s’ils sont destinés à la détermination d’un des marqueurs suivants : (…) – système Rhésus [RH1 (D) (…)] » [6].
Par conséquent, le produit commercialisé de GENOTROPY sera en classe D sous le RDM-DIV ; une classe de risque aux exigences renforcées par rapport à sa classe selon l’annexe II de la directive 98/79/CE : la liste A qui inclut à son premier tiret les « réactifs (…) pour la détermination des groupes sanguins suivants : (…) rhésus (C,c, D, E, e) » [7].
De plus, le produit de GENOTROPY, qui est en classe D sous règlement, est encadré par des spécifications générales communes (une possibilité existant en plus des normes harmonisées pour être conforme aux exigences réglementaires) sous ce même texte. Ces spécifications générales communes sont énoncées aux Annexes I et II du règlement d’exécution 2022/1107 de la Commission Européenne [9].
En conséquence, pour son DM-DIV, GENOTROPY devra suivre, la procédure d’évaluation de la conformité au RDM-DIV qui comprend les trois axes suivants (Figure 14):
Figure 14 : Illustration des trois composantes de la procédure d'évaluation de la conformité du dispositif de GENOTROPY au règlement 2017/746 (Source : auteure d'après l'article 48 du RDM-DIV):

III) Présentation et analyse des missions réalisées
A) Les dispositions transitoires prévues par le Règlement 2017/746
Ayant ainsi analysé comment le produit de GENOTROPY a été évalué sous la directive, quel est son statut en période de transition et comment il sera évalué selon le règlement 2017/746, ma première tâche en apprentissage a été de comprendre quelles sont les dates importantes liées à la transition au-delà du 26 mai 2022 ainsi que les conditions d’application de celles-ci.
1) Les articles 110, 112 et 113 du Règlement
On compte aujourd’hui plusieurs révisions du RDM-DIV depuis son entrée en vigueur en 2017 :
- Par des Règlements : par exemple, le règlement 2022/112 du 25 janvier 2022 ;
- Par des Règlement d’Exécution : par exemple, le règlement d’exécution 2023/607 du 15 mars 2023 ;
- Par des Règlements délégués : par exemple, le règlement délégué 2023/503 du 1er décembre 2022 ;
- Par des rectificatifs au règlement : par exemple, le rectificatif du 27 décembre 2019.
La version du RDM-DIV à utiliser est la version consolidée (version sans effet juridique) la plus récente qui rassemble toutes ses modifications, celle du 20 mars 2023 [25]. En particulier, l’article 110 fixe les dispositions transitoires applicables aux « Legacy devices ».
À partir d’une analyse de cette version consolidée, il est ainsi possible de répondre à plusieurs questions :
- Pour combien de temps sont encore valables les certificats délivrés selon la directive 98/79/CE (article 110.2 du RDM-DIV) ?
Le Règlement distingue 3 cas :
- Certificats délivrés avant le 25 mai 2017 : valides jusqu’à la fin de période indiquée dessus ;
- Certificats délivrés avant le 25 mai 2017 conformément à l’annexe VI : invalidés au plus tard le 27 mai 2025 ;
- Certificats délivrés après le 25 mai 2017 : invalidés au plus tard le 27 mai 2025.
- Jusqu’à quand le fabricant peut-il mettre son DM-DIV en service ou le mettre sur le marché en Europe ?
En règle générale, il est possible de continuer à mettre en service ou mettre sur le marché tant que le certificat produit (=certificat émis suite à l’évaluation de la Documentation Technique) délivré selon la directive 98/79/CE est valide. Le Règlement ne donne plus de date limite (article 110.4 du RDM-DIV).
Il est à noter que les termes « mis sur le marché » et « mis en service » sont respectivement définis aux points 21 et 22 de l’article 2 du RDM-DIV.
Toutefois, il y a une exception à cette règle générale : le cas des dispositifs pour lesquels (article 110.3 3ème paragraphe du RDM-DIV) :
- La procédure d’évaluation de la conformité prévue par la directive 98/79/CE ne nécessitait pas l’intervention d’un ON [25] ;
- La déclaration de conformité conformément à la directive 98/79/CE a été établie avant le 26 mai 2022 ;
- La procédure d’évaluation de la conformité au RDM-DIV nécessite l’intervention d’un ON.
Pour ces dispositifs, les dates limites de mise sur le marché ou mise sur le service sont les suivantes (Tableau 5) :
Tableau 5 : Périodes de transition de certains DM-DIV (Source : auteure d'après le RDM-DIV):
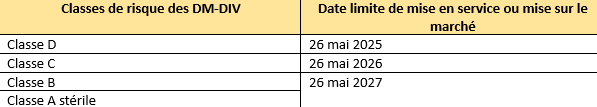
Ces différentes dates correspondent aux fins respectives des périodes de transition de ces DM-DIV vers le Règlement.
- À quelles conditions les DM-DIV peuvent-ils bénéficier de la prolongation des périodes de transition au-delà du 26 mai 2022 ?
Il y a 3 conditions (d’après le premier et quatrième paragraphe de l’article 110.3 du RDM-DIV) :
- Les DM-DIV continuent de respecter la directive 98/79/CE ;
- Il n’y a pas de changement significatif dans leur conception et leur finalité ;
- Les exigences du RDM-DIV relatives à la Surveillance Après Commercialisation, à la vigilance et à l’enregistrement des opérateurs économiques et des dispositifs sont appliquées en remplacement des exigences correspondantes de la directive 98/79/CE [25].
Il est à préciser que les notions de « surveillance après certification » et « surveillance du marché » sont des missions respectivement réalisées par les ON et Autorités Compétentes. Elles ne concernent pas les fabricants.
2) L'ordonnance n°2022-1186 (article 15)
Les fabricants français doivent effectuer une veille réglementaire et normative sur tous les textes pouvant impacter leur activité et leur produit : des textes d’origine internationale, européenne comme le RDM-DIV et française comme le Code de la Santé Publique.
À l’échelle française, il y a notamment l’ordonnance n°2022-1086 (cf. Annexe) qui a été adoptée afin de :
- Mettre en cohérence le droit français, plus précisément le Code de la Santé Publique, au Règlement européen 2017/746 [26] ;
- Abroger les articles de transposition de la directive ;
- Introduire des dispositions complémentaires au Règlement européen 2017/746.
Dans cette ordonnance entrée en vigueur le 31 juillet 2022, l’article 15 est à prendre en compte de façon particulière car il concerne les dispositions transitoires prévues à l’article 110 du RDM-DIV.
3) Résultats pour GENOTROPY et son Legacy Device
Les deux certificat UE possédés par GENOTROPY ont été émis avant le 25 mai 2017 conformément à l’Annexe IV de la directive 98/79/CE. Par conséquent, ils restent valables jusqu’à la fin de période indiquée dessus c’est-à-dire : le 26 mai 2025.
Le produit de GENOTROPY peut donc être mis en service ou mis sur le marché jusqu’au 26 mai 2025 avec ses certificats actuels. À cet effet, il doit obéir à l’ensemble des conditions réglementaires d’application obligatoire.
Dans ce cadre, l’objectif de ma mission globale durant l’année d’apprentissage a été le suivant :
| Mettre à jour le SMQ de la société conformément à la norme EN ISO 13485 : 2016 + A11 : 2021* ainsi qu’aux exigences réglementaires applicables Afin de pouvoir poursuivre la mise sur le marché du DM-DIV de l’entreprise jusqu’à la fin de sa période de transition |
Dans cette démarche, a été employé le guide MDCG 2022-8 dont l’annexe liste l’ensemble des exigences applicables aux DM-DIV « legacy devices »[27].
La mise à jour du SMQ selon l’EN ISO 13485 : 2016 n’est pas une obligation surtout chez GENOTROPY où le SMQ est déjà certifié selon la norme. Néanmoins, dans la start-up, dans un objectif d’amélioration continue, le choix a été fait de la faire en combinaison avec l’application des exigences règlementaires en matière de qualité (grâce à l’amendement A11) ainsi que des exigences purement réglementaires liées à la transition (Surveillance Après Commercialisation, vigilance…).
Par ailleurs, pour ce travail, a été employée la version européenne de la norme (qui est donc référencée ci-dessous) car l’ON de GENOTROPY évalue son SMQ selon cette version. Néanmoins, il est à noter que les exigences de cette version européenne de 2016 sont strictement les mêmes que celles de la version française de 2016.
4) Stratégie d'action globale pour l'implémentation des exigences
Une Planification Dynamique Stratégique (PDS) (Figure 15) a été réalisée afin de rechercher les enjeux liés à cette mission et les partager à l’ensemble de l’équipe de GENOTROPY :
Figure 15 : Planification Dynamique Stratégique associée à la mission (Source : auteure)
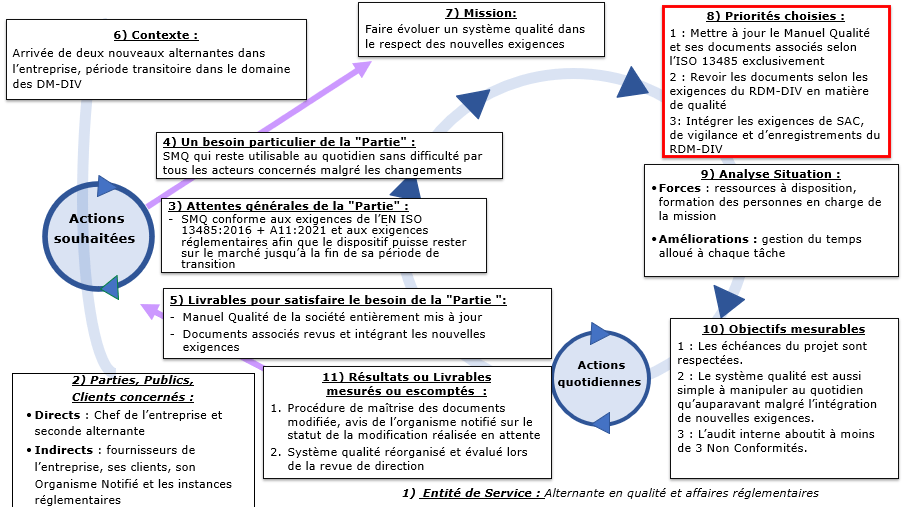
On notera l’ordre de priorité qui a été donné aux différentes tâches qui ont composé ma mission (Figure 16):
Figure 16 : Ordre de priorité dans la réalisation des tâches de la mission (Source : auteure):

Les tâches 1 et 2 sont présentées dans la partie B de ce rapport tandis que la tâche 3 se trouve dans la partie C.
B) Mettre à jour un Système de Management de la Qualité conformément à la norme EN ISO 13485 : 2016 et à son amendement A11
Les deux premières tâches (Tâche 1 et Tâche 2) de ma mission ayant fait appel à l’EN ISO 13485 : 2016 ainsi qu’à son amendement A11 de 2021, une présentation générale de cette norme s’impose.
1) Présentation générale de la norme
Fondée sur les exigences de la norme ISO 9001 : 2008 « Système de management de la qualité » qui est générique, la norme EN ISO 13485 : 2016 « Dispositifs Médicaux – Systèmes de management de la qualité- Exigences à des fins réglementaires » est une norme qui s’adresse à tout organisme (quel que soit sa taille ou sa nature) ayant une action sur une ou plusieurs étapes du cycle de vie d’un DM/DM-DIV, de la R&D à la réforme. Sont donc tous autant concernés le fabricant de DM/DM-DIV, le distributeur, le mandataire, l’importateur et tous les éventuels prestataires d’activités associées : par exemple, les prestataires de support technique.
L’objectif de la norme est de fournir à tous ces acteurs les exigences à respecter, pour leur SMQ, afin de démontrer qu’ils sont conformes aux exigences des clients et aux exigences règlementaires applicables.
La norme EN ISO 13485 : 2016 est une norme harmonisée au titre du RDM-DIV. Comme toute norme harmonisée, elle est identifiée par une décision d’exécution de la Commission Européenne : la décision (UE) 2022/729.
Être une norme harmonisée signifie qu’elle fournit une présomption de conformité à certaines des exigences du Règlement ; ici celles relatives à la qualité. Pour connaître ces exigences, le fabricant doit se référer à l’amendement A11 qui lui a été ajouté en septembre 2021. Cet amendement contient deux annexes Z (Z indique le fait que la norme est harmonisée au titre d’un règlement) : l’annexe ZA pour le règlement 2017/745 (RDM) et l’annexe ZB pour le règlement 2017/746 (RDM-DIV).
Dans chaque annexe Z, plusieurs tableaux relient la norme aux exigences des règlements : c’est-à-dire les exigences décrites à l’article 10, les annexes IX et XI. Chaque tableau décrit 3 cas possibles : « couvert », « partiellement couvert » et « non couvert » (Figure 17):
Figure 17 : Correspondance entre la norme EN ISO 13485 : 2016 et l'article 10 du Règlement 2017/746 (Source : Amendement A11 de la norme):
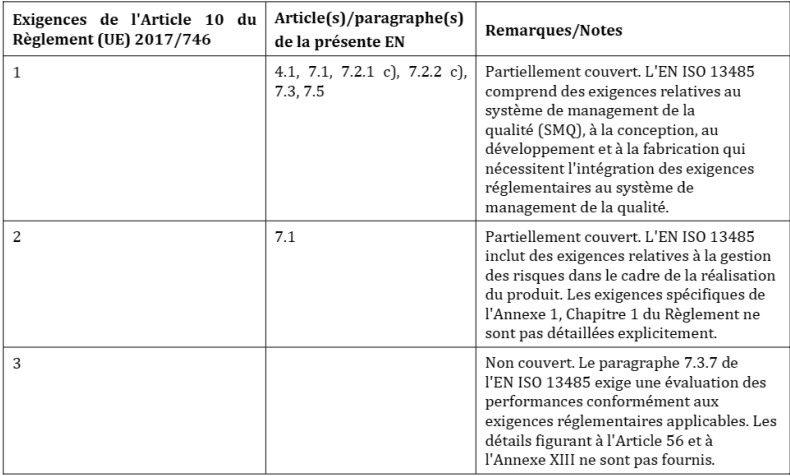
Pour les deux derniers cas, « partiellement couvert » et « non couvert », le fabricant doit donc mettre en place, en plus de la norme (quand il a fait le choix de l’appliquer), des « mesures supplémentaires » afin que son SMQ soit en conformité totale aux exigences du RDM-DIV.
Cette mise en place représente un travail conséquent de lecture et compréhension des textes qu’il faut donc implémenter par paliers pour ne pas se perdre (Figure 16).
2) Situation de départ chez GENOTROPY : un système qualité certifié mais améliorable
Pour avoir une idée d’ensemble de la compatibilité du SMQ de GENOTROPY avec l’EN ISO 13485 :2016, a été effectué, au début de la démarche, un autodiagnostic du SMQ en utilisant l’onglet « Évaluation rapide » (regroupement de 72 critères sur les 227 de la norme) d’un outil Excel réalisé par des étudiants de l’UTC en décembre 2017 [28].
À la suite de l’évaluation rapide, l’onglet résultat a figuré le graphe radar suivant (Figure 18):
Figure 18 : Résultat de l'autodiagnostic de départ du SMQ de GENOTROPY selon la norme EN ISO 13485 : 2016 (Source : auteure d'après l'outil Excel):
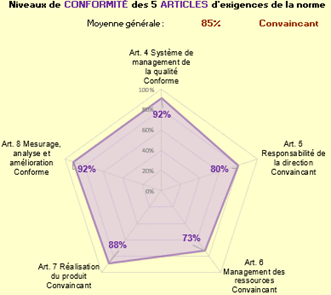
Le résultat global était « convaincant » ce qui est cohérent avec le fait que le SMQ est déjà certifié. Néanmoins, comme précisé par l’outil, « convaincant » signifie également que des améliorations peuvent encore être apportées.
À partir de ce résultat, la tâche 1 a correspondu à la mise à jour du système qualité de GENOTROPY conformément aux exigences de l’EN ISO 13485 : 2016.
Dans le cas de GENOTROPY, c’est le manuel qualité qui reprend ces exigences dans l’ordre, de la section 4 « Système de management de la qualité » à la section 8 « Mesurage, analyse et amélioration » de la norme. Celui-ci a par conséquent été mis à jour durant cette tâche.
3) Méthodologie choisie
Pour présenter la méthodologie employée à cet effet, un exemple concret est utilisé : celui du paragraphe 8.2.4 « Audit interne » de l’EN ISO 13485 : 2016.
i) Tâche n°1 : mise à jour du système qualité conformément à l'EN ISO 13485 : 2016
Pour la tâche n°1 (Figure 19), la méthodologie employée a compris 3 étapes.
Figure 19 : Illustration de la tâche n°1 de la mission (Source : auteure):

Étape 1 : répond-on aux bien exigences de l’EN ISO 13485 : 2016 dans le Manuel Qualité ?
Dans cette étape 1, il a été mené, pour chaque section du Manuel Qualité, une comparaison de l’existant par rapport aux exigences de la norme afin d’en déduire quels sont les écarts et de les corriger (Figure 20).
Figure 20 : Illustration de la méthodologie suivie pour mettre à jour le Manuel Qualité de GENOTROPY selon l'EN ISO 13485 : 2016 (Source : auteure):
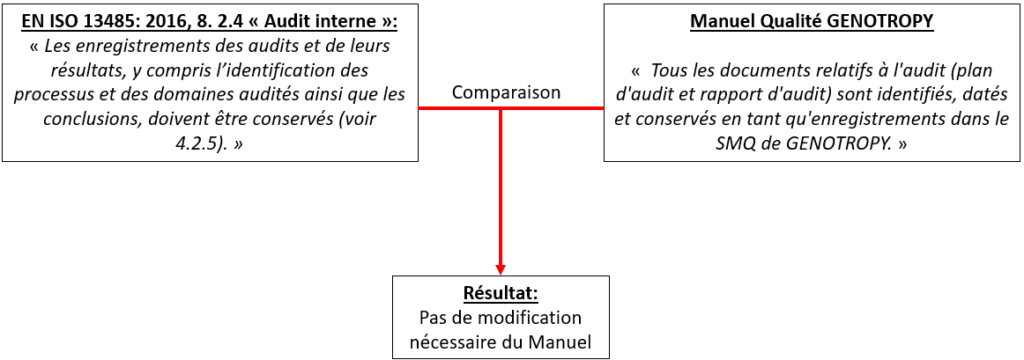
Étape 2 : possède t’on les documents demandés par la norme dans le système qualité (documents que l’on relie aux paragraphes correspondants du Manuel Qualité) ?
Dans cette étape 2, une attention particulière a été donnée, pour chaque section du Manuel Qualité, aux exigences documentaires de la norme grâce à l’analyse normative opérationnelle de celle-ci.
Une analyse normative opérationnelle est un processus qui peut être appliqué afin de simplifier la connaissance et la compréhension d’exigences normatives. Pour chaque sous-article du texte, un code couleur est utilisé pour identifier les éléments clefs de l’exigence :
- Les action à réaliser : ci-dessous en vert ;
- Sur quoi : ci-dessous en bleu ;
- Les documents demandés par la norme : ci-dessous en rouge ;
- Les éventuels éléments conditionnels à l’application des exigences : ci-dessous en violet.
Enfin, dans toute analyse normative opérationnelle, le sous paragraphe de la norme est aussi résumé en un objectif synthétique : en jaune ci-dessous.
La figure 21 montre le cas de l’analyse normative opérationnelle appliquée au sous paragraphe 8.2.4 « Audit interne » de l’EN ISO 13485 : 2016 (Figure 21) :
Figure 21 : Illustration du principe de l'analyse normative opérationnelle appliqué à un sous article de l'EN ISO 13485 : 2016 (Source : cours de Jean Matthieu Prot, UTC 2023):

Ainsi, à partir de cet exemple, s'est posée la question suivante. Le SMQ de GENOTROPY contenait-il :
- Une procédure pour les audits internes ;
- Un programme d’audit mis à jour annuellement ;
- Des enregistrements de tous les audits et de leurs résultats ?
Cela était bien le cas ici.
Étape 3 : le contenu des documents demandés est-il cohérent avec la section correspondante du Manuel Qualité ?
Dans l’étape 3, après avoir vérifié la présence des documents demandés par l’EN ISO 13485 : 2016 dans le système qualité, il a été vérifié que leur contenu était aligné avec la section correspondante du Manuel Qualité.
Pour le même exemple qu’au-dessus, le cas des audits internes, les enregistrements des audits et de leurs résultats ont ainsi été relus pour observer s’ils contenaient bien une « identification des processus et des domaines audités ainsi que les conclusions » des audits.
ii) Tâche n°2 : application des exigences réglementaires en matière de qualité grâce à l'A11 de l'EN ISO 13485:2016 :
La deuxième tâche a concerné la thématique suivante (Figure 22) :
Figure 22 : Illustration de la tâche n°2 de la mission (Source : auteure):

Pour cette deuxième tâche, il a été fait appel à l’amendement A11 de l’EN ISO 13485 : 2016. La dernière colonne « Remarques/Notes » de chaque tableau des annexes Z de cet amendement a été particulièrement utile car elle indique à quel degré chaque exigence réglementaire liée à la qualité est couverte par l’EN ISO 13485 et surtout ce qui est manquant et donc à implémenter en plus de la norme.
Dans l’exemple ci-dessous (Figure 23), on voit que le point 2.2 deuxième paragraphe (b) tiret 2 de l’annexe IX du Règlement est entièrement couvert par, entre autres, le paragraphe 8.2.4 « Audit interne » de l’EN ISO 13485 : 2016.
Figure 23 : Exemple d'exigence réglementaire couverte par le paragraphe 8.2.1 "Audit interne" de l'EN ISO 13485 : 2016 (Source : Amendement A11 de la norme):

Il n’y a donc pas de mesures supplémentaires à mettre en place par le fabricant pour ce point en particulier.
Pour cette tâche n°2, chez GENOTROPY, a été fait le choix de créer un enregistrement traçant la démarche de mise en conformité aux exigences réglementaires en matière de qualité ; cet enregistrement comprend le tableau suivant (Tableau 6) :
Tableau 6 : Extrait de la matrice de traçabilité crée chez GENOTROPY pour l'article 10 et l'annexe IX du RDM-DIV (Source : auteure):

De plus, pour respecter les délais liés aux audits, dans ce même document, les mesures supplémentaires à mettre en place ont été regroupées par document du système qualité impacté. En effet, la simple lecture de l’article 10 et l’annexe IX permet de voir qu’il s’agit de deux paragraphes du Règlement aux nombreux points communs.
Il est à noter que, pour GENOTROPY, cette tâche n°2 n’a pas concerné l’annexe XI du RDM-DIV puisque étant en classe D, la procédure d’évaluation de la conformité de son DM-DIV au nouveau Règlement se fera exclusivement selon l’annexe IX.
4) Résultats : un système qualité conforme
En fin de la démarche, un autodiagnostic du système qualité a été refait en utilisant le même outil qu’au départ (Figure 24).
Figure 24 : Résultat de l'autodiagnostic de départ du SMQ de GENOTROPY selon la norme EN ISO 13485 : 2016 (Source : auteure d'après l'outil Excel):
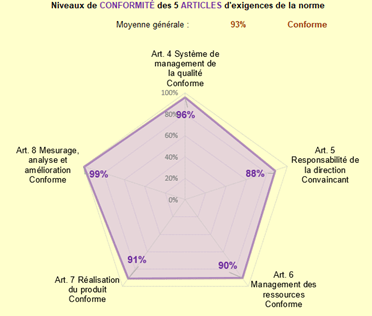
Par rapport à la situation de départ, cet autodiagnostic montre qu’il y a eu une progression dans la conformité du SMQ avec les exigences de l’EN ISO 13485 : 2016 de façon globale ainsi que pour chaque article de la norme. Ainsi, le SMQ de GENOTROPY est maintenant qualifié de « conforme » et non plus « convaincant ».
C) Les exigences réglementaires applicables pendant la période de transition
Les annexes IX et XI du RDM-DIV ainsi que l’article 10 ne sont toutefois pas les seuls paragraphes du Règlement à considérer en périodes de transition pour les DM-DIV « legacy devices ».
Ces paragraphes dont il est question ont fait l’objet de la tâche 3 de ma mission ; tâche qui est intitulée « application des exigences réglementaires propres à la transition » (Figure 25).
Figure 25 : Illustration de la tâche n°3 de la mission (Source : auteure):

Les exigences réglementaires liées à la transition des « legacy devices » sont retrouvées au paragraphe 3 de l’article 110 « Dispositions transitoires » du RDM-DIV, il s’agit des exigences relatives :
- À la SAC ;
- À la vigilance ;
- À l'enregistrement des opérateurs économiques et des dispositifs.
Ces exigences s'appliquent en remplacement des exigences correspondantes dans la directive 98/79/CE. Elles correspondent aux conditions qui sont aujourd’hui à respecter pour pouvoir bénéficier de la prolongation des périodes de transition au-delà du 26 mai 2022.
1) Le système de Surveillance Après Commercialisation sous le Règlement
i) Situation de départ : un système de SAC partiel face aux exigences réglementaires
La SAC n’est pas une nouveauté introduite par le Règlement ; il s’agit d’une exigence qui existait déjà sous la directive 98/79/CE ; en effet, d’après l’annexe III du texte : « Le fabricant met en place et tient à jour une procédure systématique d'examen des données acquises sur les dispositifs depuis leur production » [7].
De plus, cette exigence est aujourd’hui applicable à tous les DM-DIV sans exception.
Pour ces deux raisons, j’ai observé que GENOTROPY possédait déjà, à mon arrivée en septembre 2023, un système de SAC mis en place. Cependant, sa mise en place n’était que partielle (Figure 26):
Figure 26 : Diagramme d'Ishikawa réalisé afin de déterminer les causes de non achèvement du système de SAC de GENOTROPY (Source : auteure):
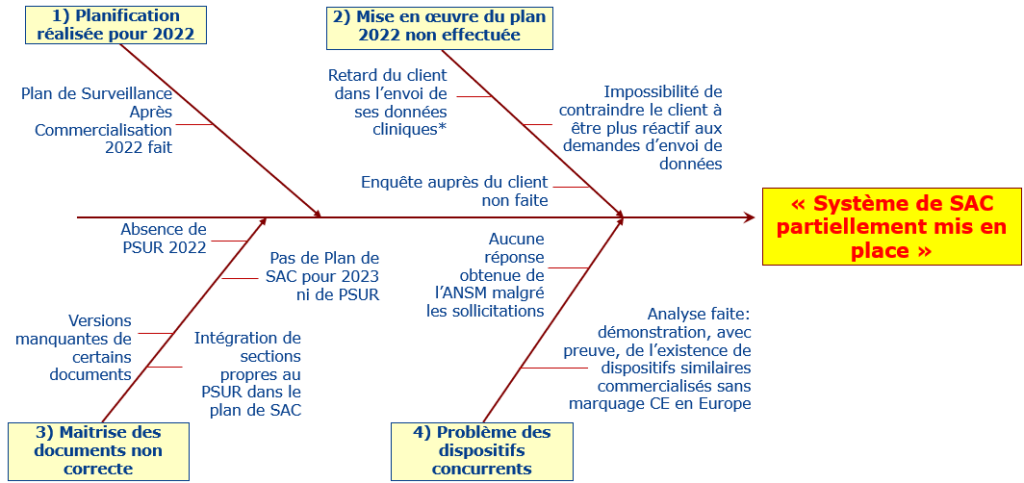
*Il est à noter que l’envoi de données cliniques par le client est crucial pour gérer la SAC et l’amélioration continue chez GENOTROPY car il s’agit d’une start-up avec un seul client qui n’a jamais eu ni de réclamations sur ses produits ni rencontré aucun incident de vigilance.
ii) Méthodologie choisie : se recentrer sur le Règlement 2017/746
Afin de parvenir à un système de SAC 100% compatible avec le RDM-DIV chez GENOTROPY, une méthodologie en 4 étapes a été suivie selon un cycle d’amélioration continue ou de DEMING. Comme la démarche envisagée est une activité stratégique proche de l’audit/inspection, a été fait le choix de débuter ce cycle par le « C » ou « Check » donnant ainsi un cycle CAPD (Figure 27) :
Figure 27 : Cycle de CAPD mise en œuvre chez GENOTROPY pour l'activité de SAC (Source : auteure):
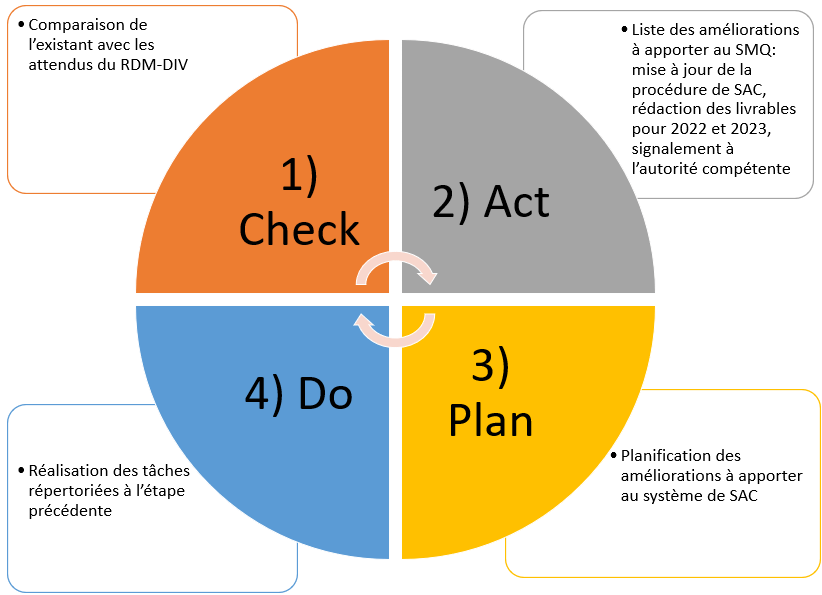
Focus sur le « Check » du cycle :
La première étape du cycle de CAPD a été cruciale puisqu’elle a consisté en une analyse du RDM-DIV afin de mieux comprendre le processus de SAC et les écarts de GENOTROPY par rapport à ces attendus.
La SAC (cf. article 78 du RDM-DIV) : définition et représentation du système, champs d’application :
Ainsi, le système de SAC, tel que défini dans le Règlement, est un processus dont l’objectif final est de surveiller et mesurer la qualité, la sécurité et des performances du DM-DIV à partir du moment où il est sur le marché de l’UE/dans la phase de post-production. Ce processus d’examen des données acquises sur le dispositif depuis sa production est :
- Systématique (à réaliser régulièrement) ;
- Proactif (et non réactif contrairement à la vigilance) ;
- Et documenté.
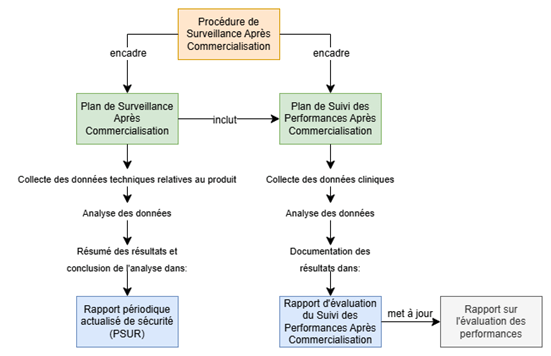
Une représentation du système de SAC à implémenter et mettre à jour sous le RDM-DIV peut être produite (Figure 28) :
Figure 28 : Le système de SAC à mettre en place selon le Règlement 2017/746 (Source : article 78 du RDM-DIV):
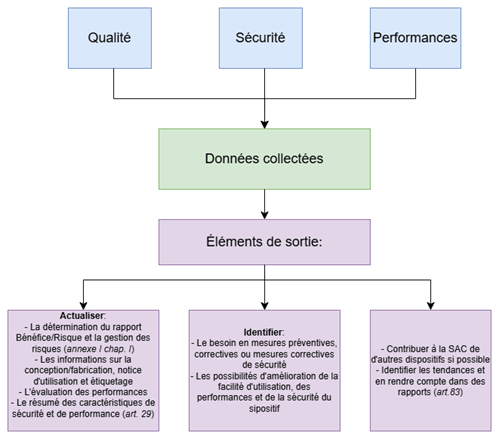
Il est à noter que le résumé des caractéristiques de sécurité et de performance (cf. article 29 du RDM-DIV) n’est pas applicable aux « Legacy devices » comme indiqué dans le guide MDCG 2022-8 [27].
Enfin, selon le RDM-DIV, un système de SAC doit être construit pour chaque DM-DIV ; proportionnellement à sa classe de risque (Tableau 7 ci-dessous) et de manière appropriée par rapport à son type (cf. article 78 du RDM-DIV).
iii) Résultats obtenus : un système de SAC complet dont le maintien de la conformité doit maintenant être assuré
À la fin de la mise en œuvre du cycle de CAPD, a été obtenu un système de SAC complet ; ce système ayant les caractéristiques suivantes :
1) Chez GENOTROPY, il s’agit maintenant d’un système lié directement au Manuel Qualité (auparavant mis à jour selon l’EN ISO 13485/l’annexe IX et l’article 10 du RDM-DIV) :
Tout d’abord, le système de SAC exigé par le RDM-DIV est lié directement au paragraphe 8.2.1 « Retours d’information » de la norme EN ISO 13485 : 2016 :
- En effet, en éléments d’entrée du processus de retours d’information, le fabricant doit recueillir et surveiller, en autres, les données provenant des activités de « postproduction » par conséquent des activités de SAC ;
- Au paragraphe 8.2.1, il s’agit de documenter les méthodes d’obtention et d’utilisation de ces informations : c’est ici qu’a été fait le lien avec le Plan de Surveillance Après Commercialisation car il liste précisément ces méthodes ;
- Au paragraphe 8.2.1, il s’agit enfin d’énoncer les éléments de sortie du processus de retours d’informations pour l’entreprise. Pour les données provenant des activités de SAC, il y a 2 types d’éléments de sortie :
- Le premier type englobe les exigences de l’EN ISO 13485 :2016 qui évoque, entre autres, la possibilité d’utiliser les retours d’information afin de mettre à jour le processus de gestion des risques liés au DM-DIV, processus repris de la norme NF EN ISO 14971 : 2019 « Dispositifs Médicaux – Application de la gestion des risques aux dispositifs médicaux » ;
- Le second type englobe les exigences réglementaires présentes à l’article 78 (Figure 28).
Ensuite, le système de SAC exigé par le RDM-DIV est aussi en lien avec le paragraphe 8.2.2 « Traitements des réclamations » de la norme EN ISO 13485 :2016 :
- En effet, parmi les retours d’information, certains peuvent être classés en tant que réclamations par le fabricant et les réclamations peuvent être séparées en deux groupes en fonction de leur origine ; il peut s'agir de :
- Réclamations liées à la vigilance ;
- Réclamations issues de la collecte et de l’analyse des données relatives aux activités de SAC. Le résultat et les conclusions de cette analyse sont présentes dans un autre document exigé par le RDM-DIV : le rapport de Surveillance Après Commercialisation ou le Rapport périodique actualisé de sécurité (en anglais : PSUR, "Periodic Safety Update Report").
2) Chez GENOTROPY, le système de SAC comprend désormais l’ensemble des livrables requis par le Règlement européen 2017/746 à leur emplacement requis :
Les livrables requis sont indiqués dans le RDM-DIV aux paragraphes suivants :
- Chapitre VII Section 1 « Surveillance après commercialisation » avec les articles 78 à 81 ;
- Annexe III « Documentation Technique relative à la surveillance après commercialisation » ;
- Annexe XIII partie B du RDM-DIV « Suivi des performances après commercialisation ».
Conformément à l’article 78 du RDM-DIV, ces livrables font partie intégrante du SMQ du fabricant : le système qualité et la documentation technique du produit pour le marquage CE sous le Règlement (Tableau 7).
Tableau 7 : Livrables requis en matière de SAC selon le RDM-DIV et leurs conditions d'applicabilité (Source : Chapitre VII du RDM-DIV):
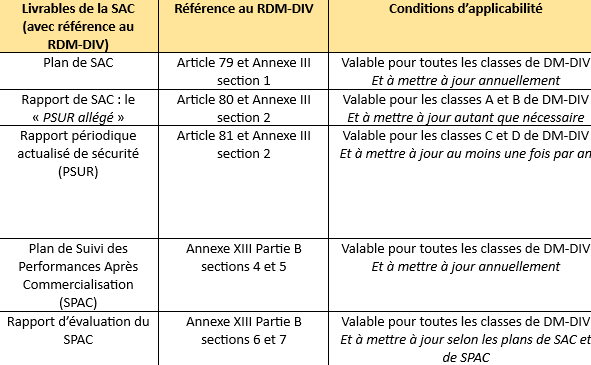
Pour le SPAC, il est à noter ce qui est écrit à l’Annexe XIII partie B section 8 du RDM-DIV : « Si le SPAC n'est pas jugé approprié pour un dispositif donné, une justification est fournie et documentée dans le rapport sur l'évaluation des performances » [6].
Chaque livrable du système de SAC peut être élaboré/mis à jour en utilisant les guides suivants (Tableau 8) :
Tableau 8 : Guides MDCG utilisables pour obtenir un système de SAC entièrement compatible avec le RDM-DIV (Source : auteure):

Enfin, il peut être utile de créer des liens entre les livrables du système de SAC (Figure 29) :
Figure 29 : Logigramme illustrant les interactions entre les livrables requis du système de SAC (Source : auteure):
3) Une tâche restante vis-à-vis du système de SAC :
Vis-à-vis du système de SAC, une tâche est restée en suspens chez GENOTROPY.
En effet, l’entreprise n’est pas parvenue à transmettre son PSUR à l’ON du fait de la non disponibilité du portail électronique de celui-ci qui est pourtant en principe dédié à sa soumission dans l’attente de la pleine fonctionnalité d’EUDAMED.
Cette situation peut sûrement s’expliquer par le fait que la rédaction d’un PSUR n’est que volontaire pour les fabricants de « Legacy devices » comme indiqué dans le guide MDCG 2022-8 : il précise en effet que seul le rapport de SAC est applicable aux "Legacy Devices" à condition que le fabricant, comme GENOTROPY, prépare volontairement un PSUR. Ainsi, la rédaction d’un PSUR ne devient obligatoire que lorsque le dispositif devient un « IVDR Device » c’est-à-dire conforme au Règlement européen 2017/746, avec passage en classe C ou D.
On peut donc supposer que la transmission du PSUR ne sera réalisable que lorsque la période de transition vers le Règlement européen 2017/746 sera terminée : elle devrait dès lors être possible soit par le portail électronique de l’ON soit via EUDAMED pour revue de l’ON qui y ajoutera son évaluation ; le PSUR et l’évaluation seront ensuite rendus disponibles aux autorités compétentes toujours via EUDAMED.
Dans la tâche n°3, la seconde exigence à mettre en œuvre fait partie d’une des méthodes réactives (et non proactives) listées dans le Plan de SAC afin de collecter les données sur la qualité, la sécurité et les performances du DM-DIV durant tout son cycle de vie : c’est la vigilance.
2) La vigilance sous le Règlement
i) Situation de départ : un système de vigilance présent dont la conformité avec le Règlement doit être vérifiée
Comme la SAC, la vigilance fait partie des responsabilités du fabricant de DM-DIV (cf. article 10.9 du RDM-DIV) indépendamment de sa localisation (le Règlement interdit à l’article 11.4 de déléguer, au représentant autorisé, les obligations en matière de vigilance).
Au sein de l’organisation du fabricant, c’est précisément la Personne Chargée de Veiller au Respect de la Règlementation (PCVRR) (cf. article 15 du RDM-DIV et guide MDCG 2019-7) qui assure la mise en place des exigences règlementaires de SAC et de vigilance. À noter que bien que les obligations de SAC et de vigilance soient applicables aux « Legacy devices » et plus généralement à tous les DM-DIV placés sur le marché, un PCVRR n’est pas exigé pour les « Legacy devices » comme indiqué dans le guide MDCG 2022-8.
Chez GENOTROPY, à mon arrivée, il y avait déjà un système de vigilance présent comprenant :
- Une procédure dédiée à la vigilance pour décrire les critères/délais réglementaires de notification des différents incidents avec leurs définitions légales ;
- Une PCVRR déclarée aux autorités par un formulaire de l’ANSM [29] ainsi que par EUDAMED (cf. point 3) suivant);
- Un dossier avec les différents formulaires des rapports de vigilance demandés par le RDM-DIV.
Avec la démarche engagée préalablement, mon objectif a été de m’assurer de la conformité totale de ce système avec le Règlement 2017/746. Il est à noter, qu’à l’heure actuelle, le guide MDCG 2023-3 « Questions and answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices » est inutilisable dans cette démarche, bien que central dans le cas des DM, car il ne comprend pas dans son champs d’application le Règlement européen 2017/746.
ii) Méthodologie choisie : appliquer l'outil de PDCA pour mieux comprendre les activités du processus de vigilance et leur enchaînement
La méthodologie choisie chez GENOTROPY afin d’aboutir à un système complet de vigilance reprend l’outil de PDCA à savoir PLAN-DO-CHECK-ACT.
Étape 1 : CHECK : Comparaison de l’existant avec les attendus du RDM-DIV [30] :
Cette étape a été réalisée en analysant le Règlement : plus précisément la section 2 du chapitre VII (articles 82 à 87). On retrouve aussi la notion de vigilance des DM-DIV à la section 2 « Organisation de la réactovigilance » (articles R5222-4 à R5222-11) du Code de la Santé Publique, les articles L.5222-2 à L.5222-4 inclus ainsi que l’article L. 5462 avec les sanctions (financières et pénales) prévues en cas de manquements aux obligations.
Après analyse du RDM-DIV, on comprend que la vigilance correspond aux signalements obligatoires de certains problèmes liés au DM-DIV et révélés au cours de leur phase d’après commercialisation. La vigilance entreprise par le fabricant est un processus en 4 activités (Figure 30):
Figure 30 : Les quatre activités du processus de vigilance selon le RDM-DIV ( Source : articles 82 à 87 du RDM-DIV):
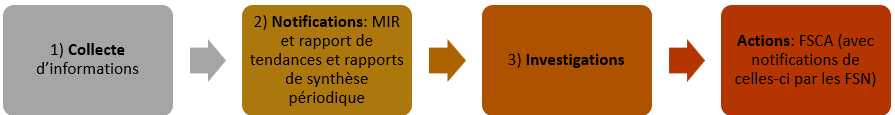
Activité 1 : Collecte d’informations :
La première activité est toujours celle de la collecte d’informations dans le cadre du Plan de Surveillance Après Commercialisation ; toutes les informations relatives à des incidents doivent être considérées. Le terme incident est défini à l’article 2.67 du RDM-DIV et ne doit pas être confondu avec celui d’évènement indésirable défini à l’article 2.60 (en anglais, « adverse event ») utilisé dans le contexte des études de performance menées avant la mise sur le marché (cf. article 76 du RDM-DIV). Malgré cette différence, l’utilisation de la terminologie pour les déclarations des effets adverses catégorisés de l’IMDRF est obligatoire par le fabricant dans les « Manufacturer Incident Report » (MIR).
Les informations peuvent être reçues des professionnels de santé ou des utilisateurs professionnels qui sont soumis, par l’article L.5222-3 du Code de la Santé Publique, à une obligation de déclaration à l’ANSM de tout incident grave dont ils connaissance. Les incidents liés au DM-DIV peuvent aussi être identifiées directement en interne au fabricant.
Activité 2 : Notifications des incidents graves :
La seconde activité est celle de la notification. Cette étape suit le principe suivant :
Si l’incident reçu est grave, alors il doit être obligatoirement notifié. En cas de doute sur sa nature, il faut également le notifier.
La notification est possible via plusieurs livrables dont les formulaires doivent impérativement être présents au sein du système de vigilance du fabricant afin d’être utilisables sans délai en cas de besoin :
- Cas n°1 : Le « Manufacturer Incident Report » (MIR) : cf. article 82.1 du RDM-DIV :
Les « incidents graves » qui impliquent des dispositifs mis sur le marché de l’UE sont à notifier dans le respect des délais fixés par le Règlement ; c’est-à-dire [30]:
- Immédiatement et au plus tard 2 jours après avoir eu connaissance : s’il s’agit d’une une menace grave pour la santé publique (définition à l’article 2.69) engendrée par un incident grave ;
- Immédiatement après l’établissement ou le soupçon d’un lien de causalité entre le dispositif et l’incident et au plus tard 10 jours après avoir eu connaissance de celui-ci : s’il s’agit d’un décès ou une détérioration grave de l’état de santé engendré par un incident grave ;
- Immédiatement après qu’un lien de causalité avéré ou raisonnablement envisageable soit établi entre l’incident et le dispositif et au plus tard 15 jours après avoir eu connaissance de l’incident : dans tous les autres cas d’incident grave.
La notification d’un incident grave doit être transmise à l’autorité compétente de l’État membre où a eu lieu cet incident par le biais du MIR. Un rapport initial incomplet peut être transmis dans un premier temps pour respecter les délais réglementaires, suivi ensuite d’un rapport complet.
L’ON du fabricant peut aussi être concerné par cette notification (cf. contrat liant la société à son ON).
- Cas n°2 : Le « rapport de tendances » : cf. article 83 du RDM-DIV :
Le rapport de tendances est un document à employer pour notifier les autorités compétentes des États Membres où l’incident grave a eu lieu en cas [25] :
- D’augmentation statistiquement significative :
- De la fréquence ou de la sévérité des incidents non graves :
- Qui pourraient avoir une incidence significative sur le rapport bénéfice/risque du dispositif ;
- Et qui ont entraîné ou pourraient entraîner des risques inacceptables pour la santé et la sécurité des patients, des utilisateurs ou d’autres personnes.
- Des résultats erronés attendus établie par comparaison avec les performances alléguées du dispositif comme indiqué dans la documentation technique et les informations relatives au produit.
- De la fréquence ou de la sévérité des incidents non graves :
L’objectif de ce rapport est d’empêcher des incidents non graves de causer des plus grands problèmes avec le temps.
- Cas n°3 : Le « rapport de synthèse périodique » : cf. article 82.9 du RDM-DIV :
Le rapport de synthèse périodique est un document qui ne peut être produit qu’en cas d’accord entre le fabricant et son autorité compétente coordinatrice (qui consulte les autres autorités compétentes référencées aux points a et b de l’article 87.8), selon un format, un contenu et une périodicité définie et dans les cas suivants [25] :
- Pour des incidents graves similaires ayant trait au même dispositif ou type de dispositif ;
- Dont la cause a été déterminée OU pour lesquels une même mesure de sécurité a été appliquée OU lorsque les Incidents sont communs et bien documentés.
L’objectif du rapport de synthèse périodique est de rassembler ce qui auraient été des rapports individuels d’incidents graves en un seul même document.
Il est à noter que tout autre incident ne rentrant pas dans l’une des catégories mentionnées au-dessus doit être impérativement documenté dans les rapports produits dans le cadre de la SAC.
Activité 3 : Investigations :
Après la notification d’un incident grave en application du RDM-DIV, intervient immédiatement la troisième activité dite d’analyse pour le fabricant ; il s’agit alors de :
- Conduire les investigations nécessaires ainsi qu’évaluer les risques consécutifs à l’incident ;
- Déterminer le besoin en mesures correctives de sécurité : les « Field Safety Corrective Actions » (FSCA) (terme défini à l’article 2.68 du RDM-DIV) [30].
Activité 4 : Actions : FSCA (avec notifications de celles-ci) :
Dans une quatrième activité, si elles ont auparavant été évaluées comme étant nécessaires, en application de l’article 82.1 du RDM-DIV, les FSCA doivent être notifiées sans retard injustifié et avant d’être prises sur les dispositifs mis sur le marché dans l’UE et ses pays tiers (seule le cas où la mise en œuvre immédiate des FSCA est nécessaire justifie une notification après son implémentation). Sont concernés par la notification :
- L’autorité compétente de l’État Membre où le fabricant a son siège ;
- Les autorités compétentes des États Membres où les FSCA ont été ou vont être mises en œuvre.
Les FSCA sont les mesures correctives prises par un fabricant, pour des raisons techniques ou médicales, afin de prévenir ou d’atténuer le risque d’incident grave [25] en rapport avec un dispositif mis à disposition sur le marché (cf. article 2.71). Selon le guide MEDDEV 2.12-1 rev. 8 [10], il peut s’agir par exemple de :
- Mesures de rappels du DM-DIV (cf. définition à l’article 2.65) ;
- Modifications, échanges, destructions du DM-DIV ;
- Conseils/recommandations données par le fabricant à l’utilisateur du dispositif ;
- Modifications/ ajouts à la notice d’utilisation du DM-DIV, de son étiquetage…
- Les « Field Safety Notice » (FSN) : cf. article 84.8 du RDM-DIV :
Par la suite, le fabricant informe les utilisateurs affectés des FSCA implémentées au moyen des FSN ou « Field Safety Notice ». Les FSN sont les « avis de sécurité » (cf. définition à l’article 2.72) dans le RDM-DIV et les « fiches d’avertissement » mentionnées dans la norme EN ISO 13485 : 2016. Comme indiqué dans le guide MDCG 2022-12 [31], les FSN ne sont pas transmises par le fabricant directement aux utilisateurs de ses dispositifs : il s’agit des autorités compétentes qui les ont reçues qui se chargeront de cette transmission.
Étape 2 : ACT : prendre les décisions qui s’imposent :
Après analyse du RDM-DIV, dans le cas de GENOTROPY, deux décisions se sont imposées.
D’une part, a été décidé de revoir entièrement la procédure de vigilance à cause de plusieurs écarts constatés par rapport aux attendus réglementaires ; notamment :
1- La structure choisie de la procédure qui rendait difficilement compréhensible le processus de vigilance : de plus, cette procédure ne présentait pas de liens avec le processus de SAC bien qu’elle lui est directement liée ;
2- L’absence d’une partie relative aux rapports de synthèse périodique alors qu’il s’agit pourtant d’une possibilité de signalement pour le fabricant instaurée par le Règlement ;
3- L’absence d’une partie relative aux dispositions d’EUDAMED en lien avec la vigilance.
D’autre part, a été décidé de s’assurer que le dossier du SMQ répertoriant les formulaires des rapports de vigilance était bien à jour : c’est-à-dire vérifier que tous les formulaires existants étaient bien disponibles dans le SMQ et dans leur version la plus récente.
Étape 3 : PLAN : planifier le travail à réaliser :
En raison des contraintes de temps liées à l’audit interne, cette étape n’a pu être appliquée ; il a fallu passer directement à la mise en pratique des décisions préalablement identifiées (étape suivante).
Étape 4 : DO : exécuter les tâches répertoriées à l’étape précédente :
Dans cette étape, la procédure et le dossier adjacent ont été entièrement revus menant aux résultats présentés ci-dessous.
iii) Résultats obtenus : un système de vigilance structuré comprenant toutes les modalités prévues par le RDM-DIV
Suite à la démarche mise en œuvre, les résultats suivants ont été obtenus :
1- Procédure avec une structure reprenant dans leur ordre les activités du processus de Vigilance. Cette procédure est désormais liée au Processus de SAC aux niveaux suivants (Figure 31):
Figure 31 : Interactions du processus de vigilance avec la SAC (en bleu sur la figure) (Source : auteure):
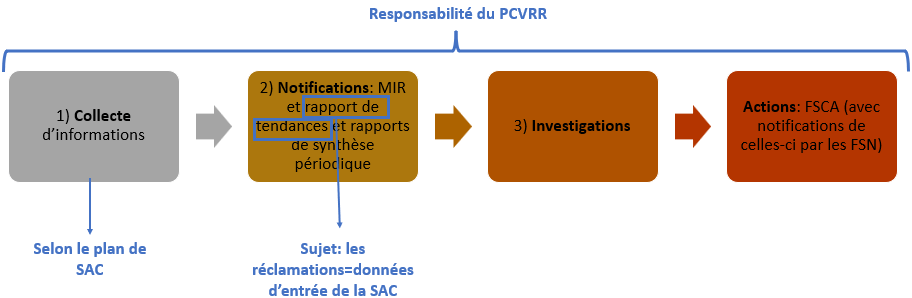
2- Une partie de la procédure est désormais dédiée aux rapports de synthèse périodique.
De plus, le dossier avec l’ensemble des formulaires utilisables pour reporter les cas de vigilance est complet avec les versions les plus récentes des documents disponibles ; ces formulaires sont tous présents sur le site web de l’ANSM.
Concernant ces formulaires, le fabricant doit cependant rester vigilant car certains n’ont pas encore été adaptés en fonction des exigences du RDM-DIV comme cela est précisé dans le guide MDCG 2022-12 [31] : cela est le cas du formulaire pour les FSCA, les rapports de synthèse périodique et les rapports de tendances. Par conséquent, pour ces cas, le guide MDCG 2022-12 [31] précise que le fabricant peut se servir des sections « Commentaires Généraux » des formulaires pour y indiquer ce qui est exigé par le RDM-DIV et manquant dans le reste du document.
Pour ce qui est des dispositions d’EUDAMED en lien avec la vigilance, celles-ci sont vues dans la partie suivante de ce rapport qui explique en détail le fonctionnement de la base de données européenne.
3) L'enregistrement des opérateurs économiques et des dispositifs
Selon l’article 110.3 du Règlement européen 2017/746, la dernière exigence réglementaire applicable aux « Legacy Devices » pendant la période de transition est celle relative à l’enregistrement des opérateurs économiques et des dispositifs.
Les exigences correspondantes sont présentes respectivement aux articles 26 « Enregistrement des dispositifs » et 28 « Système électronique d’enregistrement des opérateurs économiques » du RDM-DIV. Ces deux articles concernent évidemment EUDAMED.
Chez GENOTROPY, ma principale mission vis-à-vis de ces articles a été de vérifier si la société était bien dans le respect des règles qu’ils fixent.
i) Présentation générale d'EUDAMED
EUDAMED signifie “EUropean DATAbase on MEDical devices” ou banque de données européenne sur les Dispositifs Médicaux. Il s’agit d’un élément qui existait déjà sous les directives (mention par exemple dans la décision de la Commission du 19 avril 2010, [32]). Néanmoins, dans le Règlement 2017/746, sa prévalence a été renforcée puisqu’il y est mentionné dans près de 50 articles dont l’article 30 qui encadre sa gestion et le place sous la responsabilité de la Commission Européenne.
EUDAMED doit permettre la centralisation de l’ensemble des informations relatives au cycle de vie des dispositifs mis sur le marché de l’UE ; avec les opérateurs économiques qui leur sont associés. De cette manière, la plateforme doit permettre d’améliorer la transparence et la disponibilité des informations pour les patients et professionnels de santé ainsi que la coordination des différents États membres de l’UE.
Les spécifications fonctionnelles de la plateforme, définies par la Commission Européenne en collaboration avec le Groupe de Coordination des Dispositifs Médicaux (GDCM) (en anglais : MDCG, « Medical Device Coordination Group ») (cf. article 34 du RDM), ont été publiées pour la première fois en mars 2019. Leur dernière version date du 15 décembre 2022 (version 7.2) [33].
D’après ces spécifications, une fois achevé, EUDAMED sera composé de 6 modules interconnectés ; des modules qui sont communs à la fois aux DM et aux DM-DIV (Figure 32) :
- 1- « Enregistrement des opérateurs économiques » ;
- 2- « Enregistrement des dispositifs » ;
- 3- « Organismes Notifiés et certificats » ;
- 4- « Investigations cliniques et études de performances » ;
- 5- « Surveillance Après Commercialisation et Vigilance » ;
- 6- « Surveillance du marché ».
Figure 32 : Vue d'ensemble de l'architecture prévue de la base de données EUDAMED (Source : proposition de règlement modificatif au RDM-DIV, 23 janvier 2024):
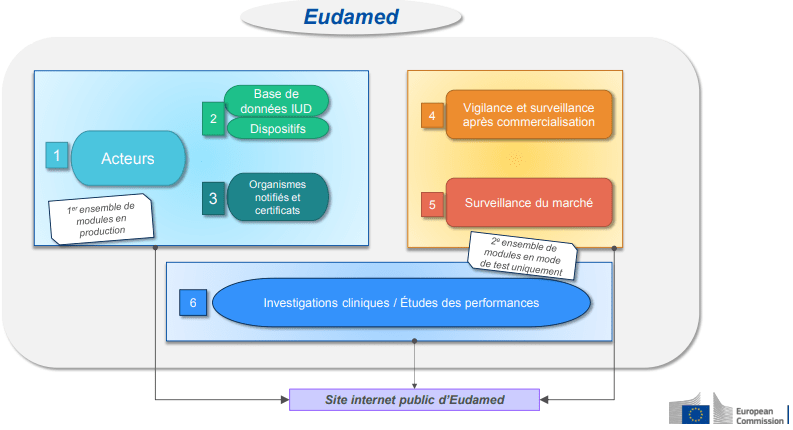
À terme, les six modules d’EUDAMED présenteront chacun des niveaux accessibles au public ; les établissements médicaux et la presse inclus [2].
Les principaux acteurs qui auront accès à EUDAMED resteront néanmoins [2] :
- Tous les opérateurs économiques : fabricants, mandataires, importateurs, PCVRR et assembleurs de systèmes et nécessaires (seuls les distributeurs ne sont pas concernés par l'obligation d'enregistrement sur EUDAMED) ;
- La Commission Européenne ;
- Les autorités compétentes nationales et de pays tiers à l’Union Européenne ;
- Les ON ;
- Le GDCM ;
- Les experts.
Chacun de ces acteurs est identifié dans EUDAMED grâce au « Single Registration Number » (SRN) ; numéro fourni par son autorité compétente nationale après son enregistrement initial dans EUDAMED. En fonction de l’activité exercée par une même entité, le SRN est différent car différents acteurs n’ont pas les même devoirs vis-à-vis d’EUDAMED [2]. Une même entité peut donc avoir plusieurs SRN si elle exerce plusieurs activités.
Un SRN est composé de trois parties (Figure 33) [34] :
- Partie 1 : Code en deux lettres du pays de l’acteur (d’après la norme ISO 3166-1) ;
- Partie 2 : Abréviation en deux lettres du statut de l’acteur ;
- Partie 3 : Code numérique à 9 chiffres.
Figure 33 : Exemple de code SRN pour un fabricant français (Source : Avanti Europe, octobre 2020):

ii) Etat des lieux de l'avancement d'EUDAMED
À l’heure actuelle, EUDAMED n’est pas complet [35].
En effet, seuls trois modules sont aujourd’hui disponibles (Figure 34) :
- 1- « Enregistrement des opérateurs économiques » (disponible depuis décembre 2020) : ce module doit permettre le regroupement des données d’identification des acteurs ayant accès à la base ;
- 2- « Enregistrement des dispositifs » (disponible depuis Octobre 2021) : ce module regroupe en principe toutes les données d’identification des dispositifs, notamment l’Identifiant Unique des Dispositifs (IUD) ;
- 3- « Organismes Notifiés et certificats » (disponible depuis Octobre 2021) : ce module regroupe en principe les données relatives aux ON, aux experts qui leurs sont affiliés ainsi qu’aux certificats de conformité CE qu’ils délivrent.
Figure 34 : Menu de recherche disponible sur EUDAMED en juillet 2024 (Source : auteure):

La moitié d’EUDAMED n’est donc pas encore prête. Par conséquent, l’utilisation d’EUDAMED n’est ni obligatoire ni exigée ; son utilisation n’est que volontaire aujourd’hui.
En accord avec les dispositions transitoires du RDM-DIV, le système, dans son entièreté, deviendra obligatoirement utilisable quand (cf. article 34 du RDM) :
- 1) Sur la base d’un rapport d’audit indépendant, EUDAMED aura été considéré comme pleinement opérationnel et conforme à ses spécifications fonctionnelles définies ;
- 2) La Commission Européenne aura ensuite consulté le GDCM ;
- 3) Une fois le 1) et le 2) remplis, la Commission aura publié un avis à cet effet dans le Journal Officiel de l’UE ;
- 4) 6 mois après cette publication : EUDAMED deviendra alors obligatoirement utilisable.
En attendant la pleine opérabilité de la base, les dispositions transitoires de l’article 112 et de l’article 113.3.f du Règlement sont applicables [6]. Le fabricant peut de plus s’appuyer sur le guide MDCG 2022-12 paru en juillet 2022 qui liste les dispositions alternatives pouvant être mises en œuvre avant la pleine fonctionnalité d’EUDAMED [31]. Enfin, le paragraphe III de l’article 15 de l’ordonnance 2022-1186 doit également être pris en compte (cf. Annexe).
iii) Les modules d'EUDAMED relatifs aux exigences réglementaires à implémenter durant la transition des Legacy Devices
La mise en œuvre d’EUDAMED reste lente : en effet, lors de son entrée en vigueur, l’article 34 du RDM indiquait que l’avis de la Commission annonçant sa pleine opérabilité serait publié au plus tard le 25 mars 2020 [6]. Plus de 4 ans après et la moitié de la base n’est pas encore déployée.
De plus, les trois modules déjà accessibles ne s’avèrent pas complètement opérationnels dans la pratique. On se concentre ci-dessous sur ceux relatifs à l’application des exigences réglementaires durant la période de transition des « Legacy devices » :
- Module 1 : « Enregistrement des opérateurs économiques » (cf. article 28 du RDM-DIV) :
Théorie : ce module sert à l’application de l’article 28 du RDM-DIV. Pour les fabricants, il leur permet de :
- Soumettre leurs informations (listées à l’annexe VI partie A section 1) avant la mise sur le marché de leur DM-DIV : une fois ces informations validées par l’autorité compétente nationale, EUDAMED crée un SRN obtenu par le fabricant. Le SRN sera, entre autres demandé pour la demande d’évaluation de la conformité de l’ON sous le Règlement (cf. article 28.1) ;
- Mettre à jour leurs informations sur EUDAMED dans un délai maximum d’une semaine après leur changement (cf. article 28.4) ;
- Confirmer l’exactitude de leurs informations un an après leur première transmission puis tous les deux ans (cf. article 28.5).
Pratique (sur la version 2.14.1 d’EUDAMED) :
Dans la pratique, ce module apparaît globalement complet par rapport aux attendus réglementaires. En effet, le fabricant peut aujourd’hui y enregistrer :
- Les informations nécessaires à son identification ;
- Son adresse ;
- Les informations relatives à la personne à contacter en interne avec son nom, son adresse et ses coordonnées ;
- Sa PCVRR et son autorité compétente nationale.
Toutefois, il est à préciser qu’au quotidien, l’accès à EUDAMED reste compliqué à cause de l’imbrication qui a été créé pour les droits et profils de ses utilisateurs.
En effet, prenons l’exemple d’un Opérateur Économique comme GENOTROPY qui est fabricant. GENOTROPY a son propre compte EUDAMED et cela est également le cas de chaque personne de l’entreprise. Chaque profil d’une personne de l’entreprise a un droit spécifique vis-à-vis de chaque module du compte de GENOTROPY. Il y a plusieurs droits possibles :
- « Local Actor Administrator » : il s’agit du profil le plus important vis-à-vis du compte de l’opérateur économique c’est pourquoi il possède les droits de tous les profils inférieurs ;
- « Local User Administrator » (optionnel) : il s’agit du profil en charge de la gestion des demandes d’accès de toute personne de l’entreprise au compte de GENOTROPY ;
- « Mandate manager » (optionnel) : il s’agit d’un profil spécifique aux fabricants situés en dehors de l’UE afin de soumettre leurs mandats aux représentants autorisés ;
- « Viewer » : il s’agit d’un profil par défaut, il permet simplement de voir les opérateurs économiques enregistrés dans EUDAMED et les informations d’identification de son propre opérateur économique.
Cette imbrication se complique.
En effet, il est obligatoire pour chaque opérateur économique d’avoir au moins deux LAA qui lui sont connectés dont 1 avec un profil actif (l’adresse email avec laquelle est faite l’enregistrement initial de l’opérateur économique devient automatiquement son premier LAA). Cette particularité n’est cependant pas adaptée au cas des entreprises avec 1 seul employé comme GENOTROPY l’était avant mon arrivée.
De plus, EUDAMED est organisé de telle sorte que chaque personne de l’entreprise qui souhaite mettre à jour ses droits vis-à-vis d’un module de l’opérateur économique doit être approuvée par un LAA ou LUA de ce même opérateur [36].
Alternative :
Aujourd’hui, s’il n’a pas fait d’enregistrement sur EUDAMED, le fabricant a toujours la possibilité d’enregistrer son activité via un formulaire national disponible sur le site de l’ANSM conformément à l’article 113 paragraphe 3.f du RDM-DIV. Cependant, il est à noter que ce formulaire n’a qu’une portée nationale contrairement à EUDAMED qui est valable à l’échelle européenne. Par conséquent, un réenregistrement sera à faire, dans EUDAMED, par tout fabricant qui aurait employé ce formulaire une fois que la base sera obligatoirement utilisable [29].
2. Module 2 : « Enregistrement des dispositifs » (article 26 du RDM-DIV) :
Pour ce module, un préalable doit être présenté : c’est le système IUD.
Le système IUD comprend toujours trois étapes.
Dans un premier temps, il y a pour les « IVDR devices », une obligation de production de l’IUD valable depuis l’entrée en application du RDM-DIV c’est-à-dire depuis le 26 mai 2022. Un IUD comprend toujours (Figure 35) [37] :
- L’IUD-ID de base qui est spécifique à un modèle de dispositif ; celui-ci est d’importance capitale pour l’accès à la base de données IUD au sein d’EUDAMED et il doit être référencé dans les documents réglementaires sous le RDM-DIV : déclaration UE de conformité, certificats, documentation technique et résumé des caractéristiques de sécurité et de performance ;
- L’IUD-ID qui est spécifique à un dispositif et à un fabricant : cette partie reste fixe ;
- L’IUD-IP qui indique la date d’expiration, le numéro de lot et/ou le numéro de série du DM-DIV : cette partie est dynamique.
Figure 35 : Exemple de code IUD délivré par l'entité GS1 sous format code 128 (Source : auteure d'après SNITEM, 2020):

D’après la décision d’exécution (UE) 2019/939 du 6 juin 2019, cette production de l’IUD passe obligatoirement par un des 4 organismes agréés par la Commission Européenne c’est-à-dire : le GS1, l’HIBCC, l’ICCBBA et l’IFA.
Dans un second temps, il y une obligation d’apposer l’IUD sur chaque niveau de conditionnement du DM-DIV (S’il y a plusieurs codes sur l’étiquetage, il faut également ajouter le logo UDI de la norme ISO 15223-1 à côté du code IUD) dans un format lisible par l’homme et un format lisible par la machine (=QR code, datamatrix ou code 128). D’après l’article 113.3.e du RDM-DIV, cette obligation posée par l’article 24.4 s’applique à des dates variables en fonction de la classe du DM-DIV :
- Classe D : 26 mai 2023 ;
- Classes B et C : 26 mai 2025 ;
- Classes A : 26 mai 2027.
Dans un troisième temps, dans le système IUD, la dernière étape est celle de la transmission des données rattachées à l’IUD-ID à EUDAMED, cette dernière étape restant volontaire aujourd’hui.
Aujourd’hui, les « Legacy devices » comme celui de GENOTROPY ne sont en revanche pas obligés de suivre ces trois étapes. En particulier, leur enregistrement dans EUDAMED n’est pour l’instant obligatoire que dans le cas où un incident grave se produit ou une FSCA est implémentée car l’enregistrement des dispositifs est une condition préalable à l’enregistrement éventuel de l’incident grave qui lui est lié dans EUDAMED.
Afin de permettre au fabricant l’enregistrement dans EUDAMED de son « Legacy Device », la base a créé deux outils en remplacement de l’IUD-ID de base et l’IUD ID : respectivement l’EUDAMED-DI et l’EUDAMED-ID.
Théorie : le module dédié à l’enregistrement des dispositifs dans EUDAMED doit permettre :
- Pour les « IVDR Devices » : de transmettre et visualiser l’IUD-ID de base du dispositif (définition à l’article 24.2) tel que défini en partie C de l’annexe VI du RDM-DIV, avant sa demande d’évaluation de la conformité au RDM-DIV auprès d’un ON, avec les données visées à l’annexe VI partie B du RDM-DIV (article 26.1) ;
- Pour les « IVDR Devices » et « Legacy devices » : de transmettre à EUDAMED les informations visées à l’annexe VI partie A section 2 du RDM-DIV et les mettre à jour suivant leurs changements (article 26.3).
Il est à noter que la plateforme doit également permettre des téléchargements automatiques des informations relatives à l’IUD d’après l’article 28.4 du RDM référencé à l’article 25 du RDM-DIV. En revanche, l’IUD-IP ne peut être transmis via EUDAMED.
Pratique (sur la version 2.14.1 d’EUDAMED) :
Le dispositif de GENOTROPY est un « Legacy Device ». Il n’est donc pas contraint de respecter toutes les obligations relatives à l’IUD. Pourtant, celles-ci ont tout de même été appliquées par l’entreprise : en particulier, l’ensemble des étapes décrites dans le guide intitulé « Management of Legacy devices » de la Commission Européenne [38] ont été suivies pour l’enregistrement du DM-DIV sur EUDAMED.
Alternative :
Aujourd’hui, s’il n’a pas encore fait d’enregistrement de ses dispositifs sur EUDAMED, le fabricant a la possibilité d’employer le même formulaire que celui nécessaire à une déclaration d’activité pour les déclarer à l’ANSM. Ce même formulaire sert aussi, par ailleurs, à la déclaration à l’agence de la personne en charge de la réactovigilance en interne au fabricant [29].
3. Module 5 : « Surveillance Après Commercialisation et Vigilance » (article 87 du RDM-DIV) :
Théorie :
Ce module d’EUDAMED doit permettre plusieurs éléments au fabricant :
- La soumission des :
- Rapports Périodique Actualisé de Sécurité (PSUR) pour les DM-DIV de classe D ;
- Rapports sur les incidents graves (incomplet puis complet) dans le respect des délais réglementaires ;
- Rapports sur les FSCA ;
- Rapports de synthèse périodique ;
- Rapports de tendances.
- La notification des :
- Incidents graves concernant des dispositifs mis sur le marché de l’UE ;
- FSCA prises à l’égard des dispositifs ;
- FSN relatives aux FSCA.
Pratique (sur la version 2.14.1 d’EUDAMED) :
Ce cinquième module fait partie des modules d’EUDAMED qui ne sont pas encore disponibles. Son achèvement est prévu au cours du deuxième trimestre de l’année 2024 d’après la Commission Européenne.
Alternative (d’après le guide MDCG 2022-12) :
Jusqu’à ce que EUDAMED soit pleinement opérationnel, pour assurer la transmission du PSUR à son ON (cas des classes D selon le RDM-DIV), le fabricant peut recourir à l’utilisation de mails sécurisés (d’après le guide MDCG 2022-12, [31]).
Selon le document MDCG 2022-12, la notification des incidents graves/FSCA/FSN et la soumission des différents rapports de vigilance doivent continuer à être faits par le biais des systèmes nationaux de vigilance par mail.
Par ailleurs, pour ce qui est du dernier module d’EUDAMED disponible à ce jour, le module 3- « Organismes Notifiés et certificats », celui-ci ne nécessite pas l’intervention des fabricants. Il est dédié à l’enregistrement des ON et des certificats délivrés par ceux-ci (cf. article 52 du RDM-DIV).
Aujourd’hui, cependant, ce module ne permet de voir aucune de ces informations (Figure 36) :
Figure 36 : Résultat obtenu sur le module 3 d'EUDAMED lorsque l'on veut visualiser les certificats émis par GMED (Source : auteure):
IV) Bilan professionnel à l'issue de l'apprentissage
1) Compétences acquises
Mon année d’apprentissage dans la qualité et les affaires réglementaires chez GENOTROPY m’a permis de :
- Acquérir des connaissances sur :
- Le monde professionnel d’entreprise : un milieu avec lequel je n’étais pas auparavant pas familière de par mes précédents stages ;
- Le domaine de la biologie médicale : notamment les techniques de PCR et d’ELISA, les équipements de surveillance et de mesure classiquement présents dans un environnement de laboratoire et l’incompatibilité rhésus D fœto-maternelle ;
- Le Règlement 2017/746 et les principales normes en qualité et dans le réglementaire : l’ISO 13485 pour le management de la qualité, l’ISO 14971 pour la gestion des risques, l’ISO 19011 pour l’audit de systèmes de management …
- La rédaction et la mise à jour de documents qualité ;
- Le processus de veille réglementaire et normative accompagné d’analyses d’écarts et d’analyses d’impacts ;
- Les méthodes d’assurance qualité opérationnelle : le suivi des indicateurs de performance liés au processus et aux objectifs qualité (les « Key Performance Indicators », KPIs), la gestion des changements « Change Control », le suivi des actions correctives et préventives « Corrective And Preventive Actions » (les CAPAs).
- Développer des compétences en matière de :
- Polyvalence : l’essentiel de mon quotidien a été la gestion de plusieurs projets en parallèle ce qui a été très stimulant pour moi ;
- Prise de responsabilités : notamment au travers de ma participation active aux différents audits qui ont rythmé les missions dans l’entreprise pendant l’année (audit fournisseur, audit interne et externe). Au départ intimidante du fait de la pression qu’elle générait chez moi, cette prise de responsabilités s’est progressivement transformée en une source de motivation car je l’ai assimilée au fait que j’étais pleinement intégrée dans le système qualité de l’entreprise ;
- Capacités d’adaptation : le quotidien en start-up étant évolutif et les aléas fréquents, j’ai dû faire preuve d’organisation, gérer les tâches à réaliser par ordre de priorité afin de respecter les délais ;
- Prise de recul : la plupart des situations que j’estimais au départ compliquées ont souvent été résolues en revenant au point de départ : c’est-à-dire la réglementation.
De par tous ses aspects, je pense que mon expérience professionnelle chez GENOTROPY a été enrichissante à tous les niveaux. J’ai désormais une meilleure idée du quotidien d’un chargé des affaires réglementaires et de la qualité. J’ai aussi gagné en confiance dans mes compétences professionnelles ainsi qu’en aisance dans mon anglais écrit et oral. Je suis donc particulièrement motivée à poursuivre dans cette voie après l’année d’apprentissage.
2) Compétences à acquérir
Il y a plusieurs compétences et connaissances me restant à acquérir à la suite de mon année d’apprentissage chez GENOTROPY.
Je pense notamment à la rédaction de la Documentation Technique d’un DM-DIV selon le Règlement, tâche que l’entreprise n’a pas encore pu développer puisque le système qualité est la priorité en période transition des « Legacy Devices ». Ainsi, en relation avec la Documentation Technique, je n’ai seulement pu participer qu’à son maintien de conformité selon la directive et dans la continuité des modifications apportées au système qualité.
J’ai aussi le souhait de développer ma connaissance des études de performances selon le Règlement. Enfin, je suis consciente de l’importance de consolider ma culture dans les domaines relatifs aux technologies des produits, pour la suite de ma carrière, car c’est d’eux dont dépendent ensuite les spécificités des activités réglementaires et qualité engagées.
3) Liens de l'apprentissage avec le Master Ingénierie de la Santé
Durant cette année d’apprentissage, le Master Ingénierie de la Santé que j’ai suivi à l’UTC m’a été utile sur plusieurs points :
- Tout d’abord, il m’a permis d’avoir une vision globale et solide sur les principaux Dispositifs médicaux rencontrés en milieu hospitalier ainsi que les métiers associés à leur cycle de vie, depuis leur conception jusqu’à leur mise sur le marché en passant par la période d’après commercialisation ;
- Ensuite, se sont avérées comme particulièrement nécessaires pour moi les différents éléments de la formation destinés à développer notre capacité à échanger avec des interlocuteurs variés ; notamment :
- Le choix de la formation de faire en partie appel à des interlocuteurs extérieurs pour ses enseignements,
- Le cours d’IDCB « Ingénierie de Projet » qui nous a permis de mener en groupe un projet abouti sur plusieurs mois ;
- Le cours d’IDCF « Organisation de la Santé » qui m’a fait comprendre l’importance de connaître l’environnement dans lequel on se situe en tant que chargé QARA ainsi que ses différentes parties prenantes notamment l’ANSM et les ON.
- Le cours d’IDCA « Management de la Qualité et des Organisations Biomédicales » m’a été primordial au quotidien dans mon entreprise d’accueil car il m’a fourni une boîte à outils auxquels faire appel pour la gestion de projets : en particulier, le Planning de GANTT ainsi que la Planification Dynamique Stratégique auquel j’ai eu plusieurs fois recours. Grâce à ces outils, je suis dorénavant capable de communiquer sur mon travail de façon plus efficace car je suis plus synthétique à l’écrit comme à l’oral et je me concentre davantage sur l’essentiel grâce à des supports visuels.
- Enfin, un cours auquel je suis également revenu de nombreuses fois durant mon apprentissage est celui d’IDCK « Audit et évaluation des organisations : normes et processus » pour comprendre son lien avec l’ISO 13485 ainsi que pour les nombreux audits auxquels j’ai pu participer lors de mon année : en particulier, l’audit fournisseur que j’ai pu mener en binôme avec ma collègue chez un fournisseur critique de GENOTROPY assurant la production de son kit.
Conclusion
Afin de bénéficier de la prolongation de la période de transition vers le Règlement, une démarche en trois points a été suivie chez GENOTROPY :
- Dans un premier temps, la démarche s’est concentrée sur la norme EN ISO 13485 : 2016 ;
- Puis, dans une seconde partie, une couche supplémentaire a été ajoutée avec la prise en compte de l’amendement A11 de la norme en lien avec l’article 10 et l’annexe d’évaluation de la conformité du dispositif dans le Règlement (Annexe IX ou XI) ;
- Pour finir, le troisième volet a été purement réglementaire : il a consisté en l’intégration des exigences réglementaires applicables en période de transition au système qualité.
Il est à préciser que cette démarche a été construite en fonction du cas de l’entreprise et du cadre réglementaire existant en matière de transition. Celui-ci sera bientôt modifié puisque, le 23 janvier 2024, la Commission a proposé de nouvelles mesures qui consistent notamment à accorder plus de temps aux entreprises pour faire leur transition, sous réserve de nouvelles conditions. Cette proposition qui a été adoptée par le Conseil de l’Union et le Parlement Européen fait l’objet d’une analyse dans un document séparé disponible sur la page WordPress de ce projet.
Il est également à remarquer que cette démarche n’a pas concerné la Documentation Technique du « Legacy Device » : seul le maintien de sa conformité à la directive a été assuré ainsi que sa mise à jour en continu en fonction des modifications apportées au système qualité.
Par ailleurs, la notion de changements n’a pas été abordée dans le rapport car jugée particulièrement dépendante de l’ON du fabricant. En effet, bien que la réglementation exige que seuls les changements considérés comme substantiels selon la directive soit reportés à l’ON avant leur implémentation et qu’aucun changement significatif en matière de conception et d’usage revendiqué (selon le Règlement 2017/746) soit mis en œuvre pendant la transition, l’ON peut, comme dans le cas de GENOTROPY, exiger que certains changements particuliers lui soient notifiés ; substantiels ou non, significatifs ou non.
Enfin, il est à souligner les coûts importants mobilisés par cette démarche : notamment en matière de ressources humaines, de finances et de temps. Cela est conséquent pour une TPE comme GENOTROPY et peut provoquer un ralentissement des activités de R&D de la société puisque les ressources que ces activités mobilisent sont désormais employées à la mise en œuvre des obligations liées au Règlement. L’innovation ralentit donc, ce qui peut être préjudiciable pour le bénéfice des patients.
Annexe
Analyse de l'article 15 de l'ordonnance n°2022-1186
L’ordonnance n°2022-1186 du 29 juillet 2022 [26] sert à l’adaptation du droit français au règlement européen 2017/746 afin d’assurer sa mise en œuvre. Dans cette ordonnance entrée en vigueur le 31 juillet 2022, un article est d’importance particulière : c’est l’article 15 car il porte sur les dispositions transitoires.
- Analyse des paragraphes I et II de l’article 15 (Tableau 9) :
Tableau 9 : Analyse des paragraphes I et II de l'article 25 de l'ordonnance n°2022-1086 (Source : auteure):
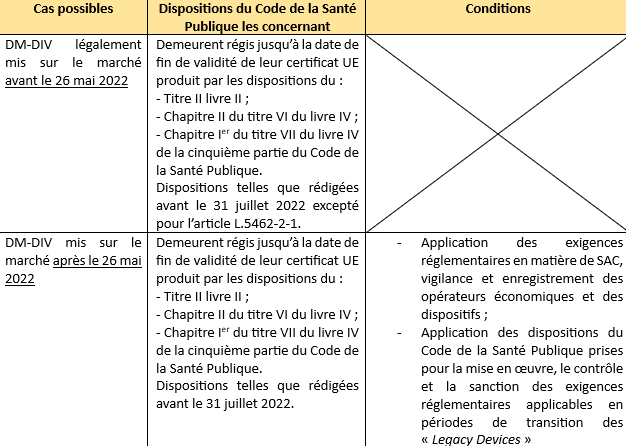
- Analyse du paragraphe III de l’article 15 :
Ce paragraphe concerne EUDAMED. Il fait la distinction entre :
- Les articles du Code de la Santé Publique qui restent applicables jusqu’à ce que EUDAMED soit obligatoirement utilisable par les opérateurs économiques ; c’est le cas des dispositions de :
- L’article L 1121-4 ;
- L’article L 1123-6 à L 1123-12 ;
- L’article L 5221-3 ;
- L’article L 5222-3.
- Les articles du Code de la Santé Publique qui deviennent applicables à partir de la date où EUDAMED devient obligatoirement utilisable par les opérateurs économiques ; c’est le cas des infractions mentionnées à :
- L’article L. 5462-2 ;
- L’article L. 5462-2-1 ;
- L’article L. 5462-4 ;
- L’article L. 5462-8 : aux points 1, 3, 7 et 8 ;
- L’article L. 1126-27.
- Analyse du paragraphe IV de l’article 15 :
Ce paragraphe concerne les études de performance des DM-DIV.
Celles-ci sont encadrées par les dispositions de la première partie du livre Ier du titre II du Code de la Santé Publique (dans leur rédaction antérieure au 31 juillet 2022) dans deux cas :
- Il s’agit de Recherches Impliquant la Personne Humaine (RIPH) mentionnées au point 1 de l’article L 1121-1 du Code de la Santé Publique :
- Qui ont obtenu, avant le 26 mai 2022, une autorisation de l’ANSM et l’avis positif d’un Comité de Protection des Personnes (CPP) pour être réalisées ;
- Dont la demande d’autorisation auprès de l’ANSM et la demande d’avis auprès du CPP étaient en cours d’instruction avant le 26 mai 2022.
- Il s’agit de RIPH mentionnées aux points 2 et 3 de l’article L 1121-1 du Code de la Santé Publique :
- Qui ont obtenu, avant le 26 mai 2022, l’avis positif du CPP pour être réalisées ;
- Dont la demande d’avis auprès du CPP était en cours d’instruction avant le 26 mai 2022.
Pour ces deux cas, les dispositions de l’article L. 1123-10 ne sont pas applicables. De plus, tout évènement indésirable grave et défectuosités de DM-DIV survenant dans le cadre de ces recherches doit être notifié conformément à l’article 76 du RDM-DIV.
- Analyse du paragraphe V de l’article 15 :
Ce paragraphe concerne les DM-DIV « in house » c’est-à-dire les DM-DIV fabriqués et utilisés au sein même des établissement de santé. Pour ces dispositifs sont aujourd’hui applicables :
- Le point III de l’article L. 5221-3 du Code de la Santé Publique ;
- Le paragraphe 5 de l’article 5 du RDM-DIV (points b et c ainsi que d à i) ;
Le point d du paragraphe 5 de l’article 5 du RDM-DIV ne deviendra applicable qu’à partir du 26 mai 2028. Le seul point restant du paragraphe 5 de l’article 5 du RDM-DIV, le point a, n’est pas mentionné.
- Analyse du paragraphe VI de l’article 15 :
Ce paragraphe fait référence à l’ordonnance n°2022-582 portant adaptation du droit français au Règlement (UE) 2017/745 [39].