
IDS249 - Outils d’accompagnement à la mise en conformité des dispositifs médicaux de diagnostic in vitro legacy devices au Règlement (UE) 2017/746
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...




Contacts
- AGBODJALOU Générose : agbodjalougenerose@gmail.com
- EL ATTAOUI Fatima-Zahra : fatimazahra.elattaoui98@gmail.com
- KUETE LONTSI Hermann:hermannkuete91@gmail.com
- KWEKEU KWEDJIN Audrey : majouaudrey@gmail.com
Citation
A rappeler pour tout usage : G. AGBODJALOU, F.-Z. EL ATTAOUI, H. KUETE LONTSI et A. KWEKEU KWEDJIN, "Outils d’accompagnement pour la mise en conformité des dispositifs médicaux de diagnostic in vitro existants (legacy devices) au Règlement européen (UE) 2017/746", Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositif Médical et Affaires Règlementaires, Mémoire de Projet, Janvier 2025, https://travaux.master.utc.fr/, réf n°249, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids249, https://doi.org/10.34746/ids249.
Résumé
Depuis son entrée en vigueur le 5 mai 2017, le Règlement (UE) 2017/746 sur les dispositifs médicaux de diagnostic in vitro impose des défis considérables aux fabricants et aux organismes notifiés, impactant indirectement la disponibilité des dispositifs sur le marché.
D’une part, les fabricants doivent désormais répondre à des exigences accrues, nécessitant une interprétation précise et une intégration rigoureuse dans leurs pratiques. D’autre part, le nombre d’organismes notifiés, déjà limité, a encore diminué en raison des nouvelles conditions d’accréditation prévues par le règlement (UE) 2017/746. Ces organismes, confrontés à un afflux massif de dossiers à traiter, peinent à respecter les délais fixés par le Conseil et le Parlement européens pour les évaluations de conformité. Pour faciliter la transition des fabricants vers cette nouvelle règlementation, des guides élaborés par le Medical Device Coordination Group (MDCG) apportent des recommandations précieuses. Dans ce contexte, notre projet vise à soutenir les fabricants en leur fournissant deux outils : d'une part, une cartographie qui permet d’expliquer rapidement au fabricant les conditions pour bénéficier des périodes transitoires et en quoi consiste la mise en conformité au règlement (UE) 2017/746 ; d'autre part, un outil
d’autodiagnostic permettant de faire ressortir les écarts entre les pratiques des fabricants de dispositifs médicaux de diagnostic in vitro (DMDIV) legacy devices sous la directive et les exigences du règlement. Ces outils reposent sur une analyse approfondie des exigences règlementaires et la prise en compte des besoins exprimés par les professionnels du secteur et collectés lors d’une enquête terrain.
Mots clés : Dispositifs médicaux de diagnostic in vitro, Règlement 2017/746, Directive 98/79/CE, Dispositions transitoires, Outils d'autodiagnostic
Abstract
Since its entry into force on May 5, 2017, Regulation (EU) 2017/746 on in vitro diagnostic medical devices has imposed considerable challenges on manufacturers and notified bodies, indirectly impacting the availability of devices on the market.
On the one hand, manufacturers are now required to meet increased demands, necessitating precise interpretation and rigorous integration into their practices. On the other hand, the number of notified bodies, already limited, has further decreased due to the new accreditation conditions stipulated by the regulation (UE) 2017/746. These bodies, overwhelmed by a massive influx of files to process, struggle to meet the deadlines set by the European Council and Parliament for conformity assessments. To facilitate the transition of manufacturers to this new regulation, guides developed by the Medical Device Coordination Group (MDCG) provide valuable recommendations. In this context, our project aims to support manufacturers by providing two tools : a chronological template-style mapping that quickly explains the conditions for benefiting from transitional periods and what compliance with Regulation (EU) 2017/746 entails. These tools will enable manufacturers to optimally plan their compliance before the end of the transitional period.these tools are based on an in-depth analysis of regulatory requirements and take into account the needs expressed by industry professionals, gathered through a field survey.
Key words : Regulation 2017/746, Manufacturer, regulatory requirements, Transition to Regulation (EU) 2017/746, Self-diagnostic tools
Téléchargements
Error ! This download item is not available ! It may be unpublished, private, or you may not have permission to view it.
Error ! This download item is not available ! It may be unpublished, private, or you may not have permission to view it.
Error ! This download item is not available ! It may be unpublished, private, or you may not have permission to view it.
Error ! This download item is not available ! It may be unpublished, private, or you may not have permission to view it.
Outils d’accompagnement à la mise en conformité des dispositifs médicaux de diagnostic in vitro legacy devices au Règlement (UE) 2017/746
Introduction générale
Le marché mondial des dispositifs médicaux de diagnostic in vitro “DMDIV” est estimé à 82,52 milliards d’euros de chiffre d'affaires en 2024 [1]. Ce marché pourrait représenter un taux de croissance annuel de 3,13 % et, par estimation, 96,28 milliards d’euros d’ici 2029 [1]. En Europe, ce marché est estimé cette année à 20,46 milliards d’euros [2] et 2,43 milliards d’euros en France [3]. Cette croissance rapide s'accompagne d'une demande accrue de validation rigoureuse et d'homologation des dispositifs pour garantir leur sécurité et leur efficacité. C’est ainsi qu’en 1990, la Directive 98/79/CE relative au DMDIV est entrée en vigueur.
L'affaire PIP (Poly Implant Prothèse), une société française créée en 1991, a déclenché en 2010 un grand intérêt médiatique [4]. Le fabricant produisait des prothèses mammaires avec un gel non conforme aux normes. En effet, le gel indiqué dans le dossier technique était différent du gel commercialisé, ce qui a engendré un scandale sanitaire. Un rapport de 2012 dressait un tableau accablant d'un manque de réactivité de la part de l'AFSSAPS (aujourd'hui ANSM) [4]. En 2016, le fondateur a été condamné à quatre ans de prison, assortis d'une amende de 75 000 euros [5]. L'organisme notifié a été condamné en 2013 puis en 2021 pour manquement à ses devoirs de contrôle. Environ un million de prothèses défectueuses ont été vendues, et 400 000 victimes dans le monde ont été dénombrées [5]. Bien que ce scandale n’est pas directement impliqué les DMDIV, il a servi de signal d’alarme pour repenser l’ensemble du cadre règlementaire qui régit les DMDIV. Aussi, le magazine Le Monde dénonce même l’absence de contrôle « Ces dispositifs médicaux bénéficient facilement du certificat « Conformité Européenne » permettant de les vendre dans toute l’Europe… Et ce, quasiment sans aucun contrôle » [6]. La capacité de la directive à assurer la sécurité a été ainsi remise en cause ces dernières années. Afin de combler ces lacunes, le Règlement (UE) 2017/746 a été adopté. Il renforce les exigences de la Directive (sur la classification, les exigences essentielles, les exigences en matière de sécurité et de performance…) et s'applique de manière uniforme dans l’Union Européenne.
Les DMDIV désignés « legacy devices » sont mis sur le marché après le 26 mai 2022, à l’issue du délai d’application du Règlement (UE) 2017/746 [7]. Cette date, initialement prévue pour l’entrée en application du règlement, a été repoussée à plusieurs reprises par voie d'amendement [9]. Ces dispositifs sont toujours couverts par des certificats délivrés sous la Directive 98/79/CE [8]. Une période de transition a été accordée à ces dispositifs afin de maintenir leur mise sur le marché, permettant aux fabricants d’effectuer les ajustements requis par la mise en conformité au règlement [9]. En effet, la Directive 98/79/CE était relativement succincte, ce qui offrait aux fabricants une certaine flexibilité pour définir eux-mêmes le niveau de preuves qu’ils estimaient suffisant pour démontrer la conformité de leurs dispositifs [15]. En revanche, sous le Règlement (UE) 2017/746, les fabricants doivent produire davantage de preuves cliniques, mettre à jour la documentation technique et mettre en place une gestion des risques plus rigoureuse [7]. L'un des effets les plus notables de l’entrée en application du Règlement (UE) 2017/746 est que 80 % des DMDIV doivent désormais faire l’objet d’un contrôle par des organismes notifiés [14]. Aussi pour répondre à ces exigences, des ressources humaines importantes et du temps doivent être investis, associé à des délais à respecter.
Dans un tel contexte, la problématique suivante est soulevée :
Comment faciliter la transition des dispositifs médicaux "legacy devices" de la Directive 98/79/CE au Règlement (UE) 2017/746, tout en optimisant l'efficience des processus de mise en conformité ?
Pour y répondre, des outils seront réalisés à destination des fabricants de DMDIV afin de les accompagner dans leur processus de mise en conformité au Règlement (UE) 2017/746.
1. Les legacy devices face au Règlement européen relatif aux Dispositifs Médicaux de Diagnostic In Vitro
1.1 Panorama des dispositifs concernés
En France, selon les données de l'année 2023, parmi les 1393 entreprises fabriquant des dispositifs médicaux (DM), 814 produisent des DM à usage individuel et des consommables, 449 des DM dits d'équipement (respirateur, pousse-seringue, réchauffeur de patient), 150 des DM de diagnostic in vitro (DIV), et 168 des DM numériques [10]. Le secteur des DM compte donc le plus grand nombre d'entreprises, tandis que le secteur des DMDIV en regroupe la plus petite proportion. En dépit de cette répartition, quel type de dispositif médical détient actuellement la plus grande part de marché ?
Nos recherches bibliographiques révèlent plusieurs tendances significatives concernant le marché des dispositifs médicaux de diagnostic in vitro (DMDIV), tant au niveau européen que mondial. Ce secteur a connu une forte croissance entre 2020 et 2021, avec des hausses respectives de 25 % et 44 % par rapport à l'année précédente [11]. La figure 1 ci-dessous présente l’évolution sur le plan mondial du chiffre d'affaires des dispositifs de diagnostic in vitro dans le monde entre 2007 et 2024.
Figure 1 : Evolution du chiffre d’affaires mondial de l’industrie du diagnostic in vitro de 2007 à 2021, avec des prévisions pour 2024 [11]
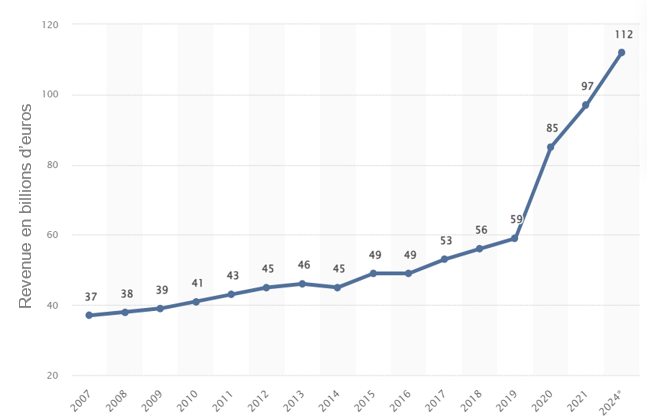
Selon le Panorama du SNITEM 2021, le chiffre d'affaires de l'industrie des DM en France a légèrement progressé en 2021, passant de 30,2 milliards à 30,7 milliards d'euros. Cette augmentation est majoritairement attribuée au secteur du diagnostic in vitro qui affichait une croissance annuelle de 9,7 % tandis que l'industrie des DM en France a diminué de 0,5 % par an entre 2019 et 2021 [12].
En 2023, le Panorama du SNITEM confirme encore que près de la moitié de la croissance du chiffre d'affaires de l'industrie des DM est portée par le diagnostic in vitro. La décroissance observée chez les DM dans le secteur entre 2019 et 2021, principalement due à la crise sanitaire, s’est inversée, avec une reprise de 0,5 % en 2023 alors que les entreprises du DMDIV continuent de présenter une dynamique de croissance positive (entre 5 et 10 % par an depuis 2019) [10]. Ce secteur, bien que comptant un nombre limité d'entreprises fabricantes comparé aux DM traditionnels, représente désormais une part significative du marché global.
Cependant, la mise en conformité au Règlement (UE) 2017/746 représente un enjeu crucial pour les fabricants de DMDIV et les organismes notifiés. Ce processus est marqué par des exigences réglementaires plus strictes, complexes et souvent sujettes à diverses interprétations. Par exemple, la classification des tests de dépistage peut varier : certains peuvent être classés en classe C ou D en fonction de l'évaluation du risque associé, entraînant des obligations de conformité distinctes pour chaque catégorie [7,8].
En effet, la Directive 98/79/CE était relativement succincte, ce qui offrait aux fabricants une certaine flexibilité pour définir eux-mêmes le niveau de preuves qu’ils estimaient suffisant pour démontrer la conformité de leurs dispositifs [15]. En revanche, sous le Règlement (UE) 2017/746, les fabricants doivent produire davantage de preuves cliniques, mettre à jour la documentation technique et une gestion des risques plus rigoureuse [7]. L'un des effets les plus notables de l’entrée en application du Règlement (UE) 2017/746 est que 80 % des DMDIV doivent désormais faire l’objet d’un contrôle par des organismes notifiés [14]. Aussi pour répondre à ces exigences des ressources humaines importantes et en temps doivent être mobilisés, associé à des délais à respecter.
1.2 Echéances des dispositions transitoires
Dans ce contexte, la Commission européenne a repoussé par voie d'amendement les délais de mise en conformité des dispositifs anciens ou legacy device prévue à l’article 120 du Règlement (UE) 2017/746 [9]. La période de transition des DMDIV s’étend jusqu’au 26 mai 2025 pour les diagnostics in vitro à haut risque, au 26 mai 2027 pour les diagnostics in vitro à faible risque et au 26 mai 2028 pour certaines dispositions concernant les dispositifs fabriqués et utilisés dans les établissements de santé [9]. La figure 2 ci-dessous présente ces dates butoirs.
Figure 2 : Chronologie schématique de la transition vers le règlement (UE) 2017/746 des dispositifs médicaux de diagnostic in vitro legacy devices (Source : Auteurs)
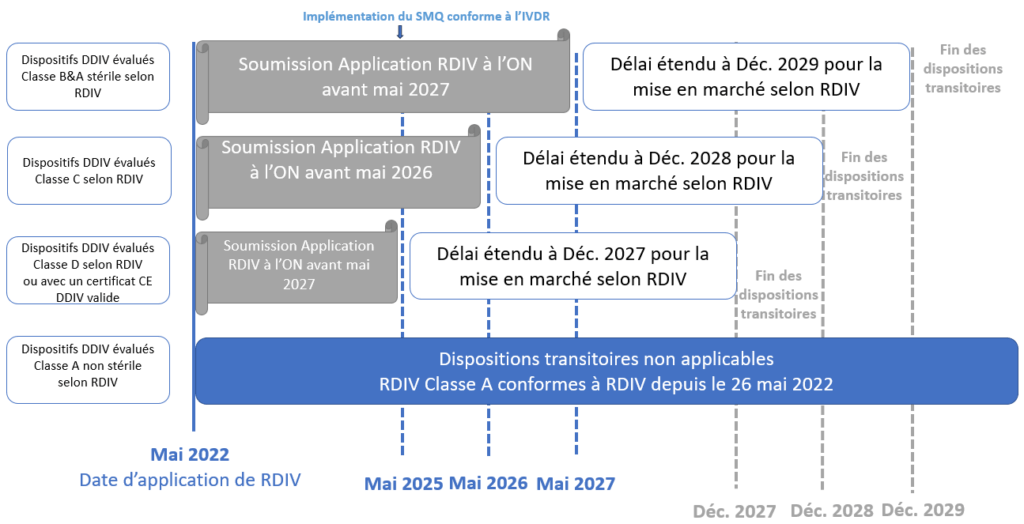
Les fabricants de legacy devices concernés par les dispositions transitoires peuvent bénéficier des nouveaux délais de transition du règlement moyennant certaines conditions cumulatives, à savoir :
- “Leurs dispositifs doivent rester conformes aux exigences de la Directive 98/79/CE” ;
- “Les dispositifs ne subissent pas de modification significative de la conception et de la destination. Cependant, les exigences du Règlement (UE) 2017/746 relatives à la surveillance après commercialisation, à la surveillance du marché, à la vigilance, à l'enregistrement des opérateurs économiques et des dispositifs s'appliquent et remplacent les exigences correspondantes de ladite directive » ;
- « Au plus tard le 26 mai 2024, le fabricant a mis en place un système de gestion de la qualité (SGQ) conformément au Règlement (UE) 2017/746. »
- « Au plus tard le 26 mai 2024, le fabricant, ou son mandataire, doit introduire une demande formelle d’évaluation de la conformité » [9].
1.3 Changements majeurs introduits par le Règlement (UE) 2017/746 et leurs implications pratiques
Un règlement européen, contrairement à une directive, est directement applicable et uniforme dans tous les États membres de l’Union européenne. Une directive, en revanche, doit être transposée en droit national, laissant ainsi une certaine marge d’interprétation à chaque pays. Ce caractère directement applicable explique le niveau de détail du règlement, qui définit précisément les processus et éléments nécessaires pour prouver la conformité des dispositifs.
En plus d’être plus précis, le Règlement apporte de nombreuses nouveautés par rapport à la Directive, avec pour objectif principal de renforcer la protection des patients et des utilisateurs. Ces évolutions reflètent une volonté claire d’améliorer la sécurité, la performance et la transparence dans le domaine des DMDIV. L’analyse qui suit mettra en lumière les différences majeures entre la Directive 98/79/CEE et le Règlement (UE) 2017/746, en se concentrant sur les obligations spécifiques des fabricants.
Le Règlement (UE) 2017/746 a renforcé les exigences et a posé un cadre plus rigoureux et détaillé sur les points présents sur la figure 3 ci-après :
Figure 3 : Représentation des exigences visant les fabricants renforcées par le Règlement (UE) 2017/746 (Source : Auteurs)
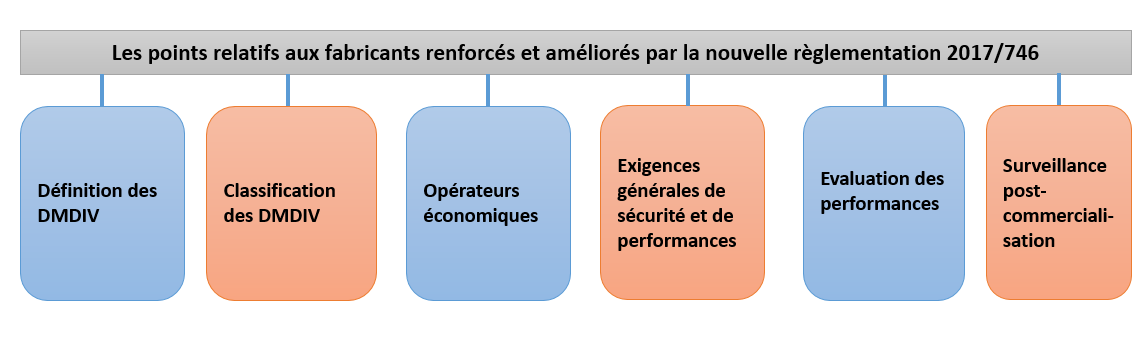
❖ Evolution de la définition du DMDIV de la Directive 98/79/CE au Règlement (UE) 2017/746
Le paragraphe 2 de l'article 2 du Règlement (UE) 2017/746 apporte de nouvelles spécifications en ce qui concerne la définition d’un DMDIV. En effet, le Règlement étend la définition du DMDIV au logiciel. De plus, est considéré également comme DMDIV au tout dispositif concernant le point c)” qui est la prédisposition à une affection ou à une maladie et au point e) permettant de prévoir la réponse ou les réactions à un traitement” [7,8].
- Classification des dispositifs
Le Règlement (UE) 2017/746 introduit un nouveau système de classification des DMDIV, basé sur sept règles définies dans l’annexe VIII, avec une évaluation proportionnelle aux risques. Il tient compte de critères généraux : la destination prévue du dispositif, les risques pour la santé publique, les patients, les utilisateurs et les tiers. En fonction de ces critères, ils sont classés en quatre catégories de risque : A, B, C et D, allant du risque le plus faible (A) au risque le plus élevé (D). Les modalités d’évaluation de la conformité dépendent de cette classification : les dispositifs de classe A sont généralement auto-certifiables par le fabricant, sauf s’ils sont stériles, tandis que ceux des classes B, C et D nécessitent une évaluation obligatoire par un organisme notifié. Ce cadre remplace l’approche de la Directive 98/79/CE, qui se basait uniquement sur deux listes (A et B) de l’annexe II, et vise à renforcer la sécurité et la transparence en tenant compte des niveaux de risque associés à chaque dispositif [7,8,15].
- Opérateurs économiques et leurs obligations
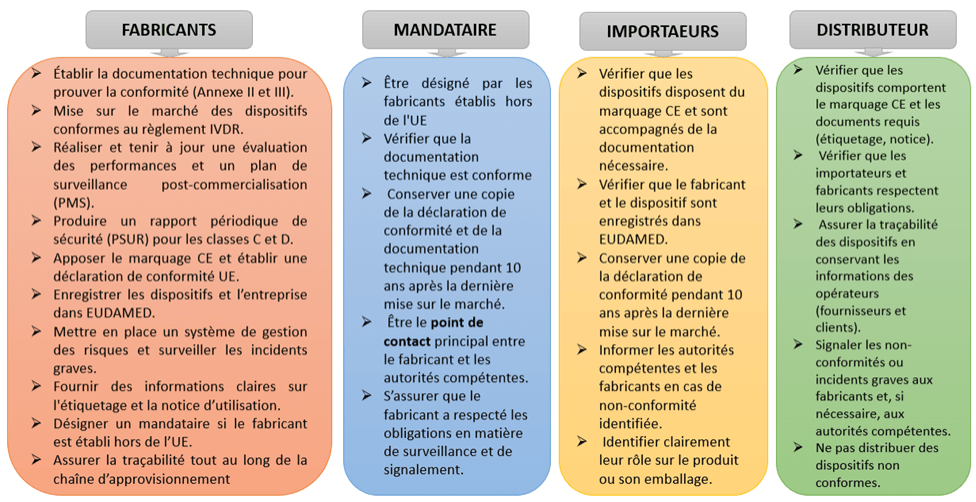
Le Règlement (UE) 2017/746 établit clairement dans les articles 10 à 15 les rôles et renforcent les exigences qui incombent aux différents opérateurs économiques impliqués dans le cycle de vie d’un DMDIV [7]. Il s’agit du :
Fabricant : "Toute personne physique ou morale qui fabrique ou remet à neuf un dispositif, ou qui fait concevoir, fabriquer ou remettre à neuf un dispositif, et le commercialise sous son nom ou sa marque" [7].
Mandataire : "Toute personne physique ou morale établie au sein de l'Union qui a reçu et accepté un mandat écrit d'un fabricant, situé en dehors de l'Union, pour agir en son nom en ce qui concerne des tâches déterminées liées aux obligations incombant à ce fabricant en vertu du présent Règlement" [7].
Importateur : "Toute personne physique ou morale établie dans l'Union qui met sur le marché de l'Union un dispositif provenant d'un pays tiers" [7].
Distributeur : "Toute personne physique ou morale de la chaîne d'approvisionnement, autre que le fabricant ou l'importateur, qui met un dispositif à disposition sur le marché, jusqu'au stade de sa mise en service" [7].
Tous ces opérateurs collaborent pour signaler tout problème de sécurité ou de conformité pour garantir la traçabilité et la sécurité des DMDIV. Cette collaboration implique que chaque acteur, en fonction de son rôle, transmette les informations appropriées (par exemple, réclamations d'utilisateurs, défauts identifiés ou incidents graves) aux autres parties concernées, y compris les autorités compétentes si nécessaire [7]. La figure 4 ci-après présente les exigences portées sur les opérateurs économiques selon le Règlement (UE) 2017/746 :
Figure 4 : Synthèse des exigences des opérateurs économiques selon le Règlement (UE) 2017/746 (Source : Auteurs)
- Exigences générales en matière de sécurité et de performance
Les exigences de l’annexe I du Règlement (UE) 2027/746 existaient déjà dans la Directive 98/79/CE mais leurs contenus sont plus détaillés et poussés dans le règlement pour renforcer la sécurité des DMDIV. Les changements les plus notables se trouvent dans :
- Gestion des risques
Le premier chapitre de cette annexe est consacré à la gestion des risques. Il impose aux fabricants d’implanter un système couvrant toutes les étapes du cycle de vie du dispositif, incluant l'identification, l'évaluation et la maîtrise des risques, ainsi que le suivi post-commercialisation. Ce système doit être documenté, mis à jour régulièrement en se basant sur les données collectées après la mise sur le marché, ce qui permet de garantir une gestion continue et traçable des risques, assurant un rapport bénéfices/risques favorable [7].
- Performance, conception et fabrication
Les exigences en matière de performance, de conception et de production des dispositifs sont renforcées. Elles sont distinguées en deux dimensions : analytique (capacité du dispositif à mesurer avec précision les paramètres visés) et clinique (aptitude à apporter des résultats cliniquement pertinents). Le fabricant doit prouver que son dispositif répond à ces deux critères, en tenant compte des normes et pratiques technologiques généralement acceptées [7]. Par ailleurs, le Règlement (UE) 2017/746 impose des limites strictes sur l’utilisation de substances dangereuses, nécessitant une justification détaillée et l'exploration d’alternatives plus sûres. Il introduit également de nouvelles exigences pour les dispositifs utilisant des matériaux biologiques, incluant des procédures de maintenance renforcées pour garantir une sécurité accrue [7].
- Evolution du contenu de la documentation technique :
L’annexe II du Règlement (UE) 2017/746 énumère avec détails la constitution de la documentation technique. De nouveaux éléments y sont ajoutés et le contenu de certaines exigences ont été renforcés [6,14]. Le tableau 1 ci-dessous les présente, en rappelant ce qui était demandé par la directive et ce qui est maintenant exigé par le règlement.
Tableau 1 : Renforcement du contenu de la documentation technique [7,8]
| Eléments de la documentation technique | Directive 98/79/CE | Règlement 2017/746 |
| 1. Description générale du dispositif | La description du dispositif, son fonctionnement et son utilisation prévue. | La description est plus détaillée, incluant les classes de risque, le groupe cible, et l'utilisation prévue avec une justification clinique. |
| 2. Exigences essentielles | La démonstration de conformité avec les exigences générales de l'annexe I. | La démonstration de la conformité avec les exigences générales de l’annexe I et une partie dédiée à la gestion des risques et des preuves approfondies. |
| 3. Analyse et gestion des risques | La réduction des risques est mentionnée de manière moins approfondie. | Un processus complet de gestion des risques, avec une documentation continue tout au long du cycle de vie du DMDIV. |
| 4. Données de conception et de fabrication | Les spécifications du dispositif et procédés de fabrication généraux. | La documentation inclut les détails de conception, les matériaux utilisés, le processus de fabrication et la traçabilité des lots. |
| 5. Données de performances analytiques | Les résultats d'essais étaient limités aux performances analytiques. | Les résultats d'essais sont détaillés, incluant des tests approfondis de performances analytiques (sensibilité, spécificité, etc.). |
| 6. Données de performances cliniques | Rarement requis, sauf pour certains dispositifs critiques tels que : les tests de dépistage du VIH (VIH-1 et VIH-2) ainsi que les tests de dépistage des hépatites B, C et D. | L’évaluation des performances cliniques est requise, incluant des études cliniques et preuves fondées sur les données collectées. |
| 7. Surveillance post-commercialisation (PMS) | Aucune exigence explicite sur la PMS. | Un plan de surveillance comprenant le suivi du rapport bénéfices/risques et les incidents après la mise sur le marché doit être ajouté. |
| 8. Étiquetage et instructions d'utilisation | Conformité aux exigences générales (annexe I, section 8). | Conformité détaillée avec des informations étendues, adaptées aux professionnels et aux profanes, selon le dispositif. |
| 9. Justification du rapport bénéfices/risques | Faiblement détaillé, basé sur une analyse simple. | Exigence approfondie d'évaluation et de documentation, avec mise à jour régulière en fonction des données collectées au fur et à mesure avec la surveillance post-commercialisation. |
- Documentation relative à la surveillance post-commercialisation
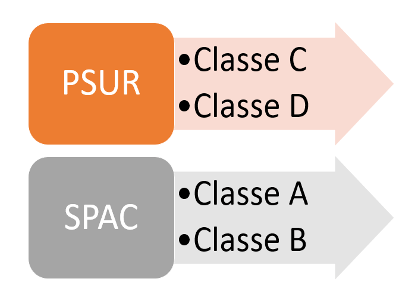
La surveillance post-commercialisation, renforcée par le Règlement et détaillée dans l'annexe III, impose aux fabricants d’élaborer un plan structuré pour collecter et analyser les données de sécurité et de performance des dispositifs. Ces données alimentent deux types de rapports : le rapport périodique actualisé de sécurité (PSUR) et le rapport de surveillance après commercialisation (SPAC), permettant une évaluation continue des performances, la détection rapide des risques et leur correction [7]. La figure 5 présente précise les types de rapports exigés pour chaque classe :
Figure 5 : Documentation relative à la surveillance post-commercialisation exigée par le Règlement (UE) 2017/746 pour chaque classe de DMDIV (Source : Auteurs)
❖ Évaluation des performances, études des performances et suivi des performances après commercialisation
- Évaluation des performances :
Le Règlement (UE) 2017/746 impose une évaluation systématique des performances analytiques (précision et fiabilité des mesures) et cliniques (pertinence et utilité pour les patients), avec une documentation rigoureuse et continue. Ces performances doivent être démontrées tout au long du cycle de vie du dispositif, grâce à des données issues d'études ou de la littérature scientifique [7].
- Études des performances
Le Règlement (UE) 2017/746 exige également des études des performances, qui consistent en des investigations systématiques réalisées pour confirmer les performances revendiquées d’un dispositif dans des conditions d’utilisation réelles. Elles visent à valider les performances analytiques et cliniques, notamment pour les dispositifs de classes C et D qui présentent un risque modéré à élevé [7].
- Suivi des performances
Le Règlement (UE) 2017/746 introduit le suivi des performances après commercialisation, qui est une exigence clé pour garantir la sécurité et la performance continue des dispositifs après leur mise sur le marché. Ce processus consiste à collecter, analyser et évaluer des données sur les performances analytiques et cliniques des dispositifs, afin d’identifier les risques émergents, de vérifier le maintien des performances revendiquées et à mettre à jour, si nécessaire, la documentation technique [7].
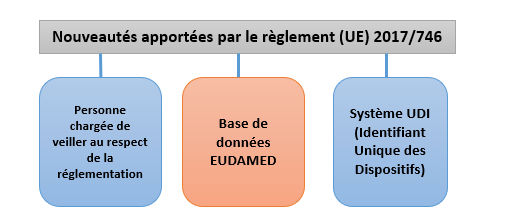
❖ Nouveautés apportées par le Règlement (UE) 2017/746
Le Règlement (UE) 2017/746 a apporté des changements significatifs pour garantir une meilleure sécurité, traçabilité et transparence dans le domaine des DMDIV. Ces nouveautés visent à renforcer la conformité réglementaire des acteurs économiques tout en améliorant le suivi des dispositifs sur le marché. Comme illustré dans la figure 6, trois éléments clés ressortent de ces nouvelles exigences [7] :
Figure 6 : Représentation des nouveautés apportées par le Règlement (UE) 2017/746 (Source : Auteurs)
- Identification des dispositifs dans la base de données EUDAMED
L’article 30 du Règlement porte sur la base de données européenne sur les dispositifs médicaux EUDAMED qui intègre plusieurs systèmes électroniques pour rassembler et traiter les informations relatives aux dispositifs présents sur le marché et aux opérateurs économiques concernés. La base de données permet d'accroître la transparence générale, notamment grâce à un meilleur accès à l'information pour le public et les professionnels de santé, d'éviter les obligations de notification multiples et de renforcer la coordination entre les États membres. La base de données EUDAMED comprend six modules interconnectés et un site web public : 1) l’enregistrement des acteurs, 2) l’enregistrement des IUD/dispositifs, 3) les organismes notifiés et certificats, 4) les vigilance et surveillance des dispositifs après commercialisation, 5) les investigations cliniques et études de performances, 6) la surveillance du marché et la section EUDAMED "public" qui est la partie accessible au grand public, permettant à tous, y compris les patients et les professionnels de santé, de consulter certaines informations essentielles sur les DM [7,16].
Figure 7 : Illustration des éléments à mettre en place pour EUDAMED (Source : Auteurs)

- Système d’Identification Unique du Dispositif
Une des nouveautés introduites par le règlement (UE) 2017/746est la mise en place de la base de données pour l’Identifiant Unique du Dispositif (IUD).Ce système repose sur deux composantes principales : le IUD-ID (Dispositif) et le IUD-IP (Production) pour améliorer la traçabilité de tous les DMDIV dans l’UE, comme illustré dans la figure 8 .
Son contenu est décrit dans l’annexe VI, partie C du règlement (UE) 2017/746. « L’IUD-ID est un code numérique ou alphanumérique unique propre à un modèle de dispositif qui sert également de « clé d’accès » aux informations stockées dans une base de données IUD » et « L'IUD-IP est un code numérique ou alphanumérique unique identifiant l'unité de production d'un dispositif ». Les différents types d'IUD-IP sont le numéro de série, le numéro de lot, l'identifiant de logiciel et la date de fabrication ou d'expiration ou ces deux types de date [7].
Avant sa mise sur le marché, et conformément aux règles de l'entité d'attribution visée à l'article 24, paragraphe 2 du règlement, les fabricants doivent attribuer au dispositif un IUD-ID de base tel qu'il est défini à l'annexe VI, partie C, et le transmettre à la base de données IUD avec les autres principaux éléments de données visés à l'annexe VI, partie B, en rapport avec le dispositif en question.
L’annexe VI, partie C, section 3.9 relève qu’un « Un nouvel IUD-ID est requis chaque fois qu'une modification est susceptible de susciter une erreur d'identification du dispositif et/ou une ambiguïté dans sa traçabilité » [7].
Figure 8 : Illustration de la mise en place de l’IUD-ID et l’IUD-IP [13]
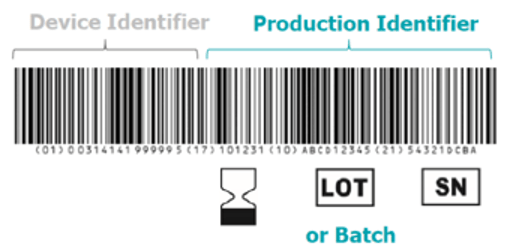
L'UDI doit figurer au moins sur l'étiquette du dispositif, sur tous les niveaux de conditionnement supérieurs, sur les rapports de vigilance, sur la déclaration de conformité UE. Il est utilisé pour signaler les incidents graves et les actions correctives de sécurité [7,13].
- Personne chargée de veiller au respect de la réglementation
Le règlement (UE) 2017/746impose que chaque fabricant désigne une personne chargée de veiller au respect de la réglementation (PRRC). La fonction de PRRC peut être externalisée dans le cas des petites entreprises. La PRRC a différentes fonctions notamment : la vérification de la conformité des dispositifs, l’établissement et le maintien de la documentation technique et de la déclaration de conformité UE, la vérification des obligations en matière de surveillance après commercialisation [7].
2.Démarche mise en oeuvre pour l’élaboration des solutions facilitant la transition des legacy devices au règlement (UE) 2017/746
La démarche a d'abord consisté en la conduite d’entretiens avec des professionnels afin de recueillir les besoins des professionnels, suivis de l'analyse des outils existants cette étape a permis de réaliser l’état de l’art des outils qui existent déjà pour s’en inspirer, pour aboutir finalement à l'élaboration du cahier des charges comme le résume la figure 9 ci-dessous :
Figure 9 : Présentation des étapes suivi pour élaborer les solutions proposées (Source : Auteurs)

2.1Résultats des enquêtes auprès des professionnels de dispositifs médicaux de diagnostic in vitro
Les entrevues que nous avons menées tout au long du projet ont joué un rôle crucial dans la définition graduelle du champ d'action et dans la direction précise de la création d'outils spécifiques destinés à faciliter le respect du règlement (UE) 2017/746 par les fabricants de DMDIV. Ces entrevues dont le récapitulatif est visible dans le tableau 2 ci-dessous, réalisées aussi bien en personne que par visioconférence, nous ont permis de collecter divers points de vue d'experts du domaine. Nous avons mené des entretiens avec divers acteurs réglementaires des dispositifs médicaux notamment : des responsables affaires réglementaires, des consultants.
Tableau 2 : Résumé des interviews (Source : Auteurs)
| Type d’entreprise | Poste | Taille de l’entreprise | Chiffre d’affaires |
| Petite et Moyenne Entreprise | Responsable affaires réglementaires | 20 à 49 salariés | 2 863 898 € |
| Petite et Moyenne Entreprise | Chargé d'affaire réglementaire | 6 à 9 | 65 000 € à 98 000 € |
| Petite et Moyenne Entreprise | Chargé d'affaire réglementaire | 20 à 49 | 10 702 455 € |
| Petite et Moyenne Entreprise | Consultante | 1 à 2 | |
| Petite et Moyenne Entreprise | Alternante affaires réglementaires | 20 à 49 | 2 563 964 € |
| Petite et Moyenne Entreprise | Alternant | 100 à 199 | 22 484 952 € |
Plusieurs défis ont été soulignés lors des entretiens concernant la nouvelle règlementation concernant les dispositifs médicaux de diagnostic in vitro. Selon une chargée d’affaires réglementaires, les exigences en matière de documentation technique sont complexes et peuvent varier en fonction des entreprises et des dispositifs. Elle a suggéré des approches afin d'optimiser le temps
requis pour leur mise en conformité. Un chargé d’affaires réglementaires et responsable PCVRR (Personne Chargée de Veiller au Respect de la Règlementation), qui travaille dans une entreprise cumulant les rôles de distributeur, fabricant et importateur a suggéré d’intégrer dans le projet un rappel des dispositions transitoires à respecter pour les "legacy devices" afin de bénéficier de la
période de transition. Une autre interlocutrice a proposé d'inclure des outils concrets tels que des formations, des bases de données pour des articles scientifiques et des guides pour la création des dossiers d'évaluation clinique. Finalement, une professionnelle du domaine des dispositifs médicaux de diagnostic in vitro a souligné la difficulté des fabricants de DMDIV de passer d'une autocertification à une évaluation par un organisme notifié. Elle a suggéré d'inclure une section sur la classification des dispositifs, la gestion des risques et la gestion des modifications des dispositifs.
2.2Cahier des charges des solutions proposées
Les différents retours obtenus lors des entrevues avec les professionnels ainsi que nos recherches bibliographiques sur l’état de l’état nous ont permis de définir le cahier des charges des outils d’aide à la mise en conformité des legacy devices au règlement (UE) 2017/746.
Il s’agit de guider les utilisateurs à travers les étapes critiques pour rester conformes durant la période transitoire et se mettre en conformité au règlement (UE) 2017/746. Ils doivent être conçus pour fournir des informations claires et précises, accessibles aussi bien aux professionnels qu’aux non-experts du secteur.
Les recherches bibliographiques ont permis d’identifier des travaux d’étudiants des promotions précédentes du master ingénierie de la santé. Des éléments que nous avons trouvés intéressants dans ses travaux sont utilisés pour les outils
Le 1er outil est une cartographie interactive, inspirée d’un projet des travaux de master [18] qui présente les conditions requises pour bénéficier des périodes transitoires et les éléments à mettre en place pour se mettre en conformité au règlement (UE) 2017/746. La cartographie intègre des liens et des référencements aux articles et annexes du règlement (UE) 2017/746. Des liens pour avoir accès aux différents guides disponibles qui donne des éclaircissements sur comment faire la classification des DMDIV, la gestion des modifications significatives du DMDIV, la gestion des risques et l’évaluation des performances cliniques, des liens fiables pour collectées des données de la littérature scientifique, les liens pour identifier des sociétés de recherche sous contrat (ou CRO pour Contract Research Organization)[19] pour réaliser les essais cliniques requis par le règlement, les liens vers les sites des autorités compétentes qui interviennent dans le processus d’évaluation des dispositifs. Ainsi les utilisateurs de la cartographie perdront moins de temps sur les moteurs de recherche pour retrouver ces informations car elles sont d’ores et déjà capitalisées dans la cartographie.
D’autres part le projet intitulé “ Passage de la Directive 93/42 au Règlement Européen 2017/745 relatif aux dispositifs médicaux ” [17] qui propose un outil d’autodiagnostic conçu sur Excel® pour permettre aux fabricants de DMDIV de savoir les éléments à mettre en place en plus pour rentrer en conformité aux nouvelles exigences et définir des plans d’actions de mise en conformité prioritaires qu’ils auraient identifiés grâce à l’outil. Il a servi de base et a été exploité pour l’élaboration du 2ème outil d’autodiagnostic de conformité des DMDIV legacy devices au règlement (UE) 2017/746. En utilisant notre outil d’autodiagnostic, les professionnels n’auront plus besoin de procéder à une analyse des écarts entre la directive 98/79/CE et le règlement (UE) 2017/746. Cette ambition requiert que le contenu de l’outil d’autodiagnostic soit rédigé rigoureusement et conformément aux textes réglementaires.
2.3 Méthodologie adoptée pour réaliser les outils
Pour atteindre ces objectifs, plusieurs ressources ont été exploitées pour garantir une approche rigoureuse et adéquate. D'abord, la bibliothèque de l'UTC a été exploitée pour mener une recherche bibliographique exhaustive. Ceci a rendu facile l'exploration des bases de données scientifiques, des textes spécialisés et des publications sur le sujet.
- La cartographie interactive
- Recenser les conditions à respecter pour bénéficier des dispositions transitoires afin de se préparer à la mise en conformité au règlement (UE) 2017/ 746.
- Utiliser les informations recueillies lors des entrevues et nos recherches bibliographiques pour recenser les guides, liens, textes réglementaires, adresse de CRO qui pourraient être utiles pour répondre aux exigences du Règlement.
- Présenter les premiers éléments mise en place dans la cartographie aux professionnels lors des interviews pour avoir leurs avis pour correction et amélioration
- L’outil d’autodiagnostic
- Exploiter les commentaires et critiques des professionnels pour améliorer l’outil et pour le rendre réellement utilisable sur le terrain ;
- Recenser les articles et annexes propres aux fabricants dans la directive 98/79/CEE abrogée et la dernière version du 9 juillet 2024 du règlement (UE) 2017/746 : une étude détaillée des textes est menée, en particulier la directive 98/79/CE et le règlement (UE) 2017/746. Cette étude facilite la détection et le regroupement des documents spécifiques pour les fabricants ;
- Confronter la directive 98/79/CE et le règlement (UE) 2017/746 dans le but de souligner les aspects consolidés par le nouveau règlement et les nouveautés introduites.
- Transformer les exigences extraites de la confrontation en affirmation dans l’outil d’autodiagnostic ;
- Insérer des graphiques qui feront ressortir un visuel des écarts entre les deux textes ;
- Présenter l’outil d’autodiagnostic à des professionnels pour recueillir leurs retours sur l’efficience et l’efficacité de l’outil ;
- Exploiter les commentaires et critiques des professionnels pour améliorer l’outil et pour le rendre réellement utilisable sur le terrain ;
3. Réalisation des outils
3.1. Mode d’emploi de la cartographie interactive
La cartographie interactive a été réalisée à l'aide de l'application de conception Canva®, sous la forme d'une présentation interactive. Elle est structurée en deux grandes parties :
- La deuxième partie : Cette section présente nos recommandations pour réussir la transition vers le règlement (UE) 2017/746.
- La première partie : Cette section rappelle le calendrier de transition entre la directive 98/79/CE et le règlement (UE) 2017/746. Elle inclut également les conditions nécessaires pour bénéficier de la période transitoire.
La page de garde de la cartographie interactive sert de présentation de l’outil (contexte d’utilisation et objectifs). Un bouton interactif intitulé "Start" est placé au bas de la page pour permettre à l'utilisateur de commencer la navigation dans la cartographie et d'explorer les deux grandes sections :
- Partie 1 : Calendrier de transition de la directive 98/79/CE au Règlement (UE) 2017/746 :
Le calendrier de transition présenté dans cette cartographie interactive illustre les principales étapes et dates clés pour le passage de la directive 98/79/CE au règlement (UE) 2017/746, chaque segment de la chronologie est interactif : en cliquant sur une étape, l’utilisateur accède aux détails spécifiques des conditions à remplir pour cette étape particulière. Cela inclut les critères à respecter pour maintenir la mise sur le marché des legacy devices pendant la période transitoire, comme illustré dans la figure 10 :
Figure 10 : Calendrier de transition de la directive 98/79/CE au Règlement (UE) 2017/746 (Source : Auteurs)
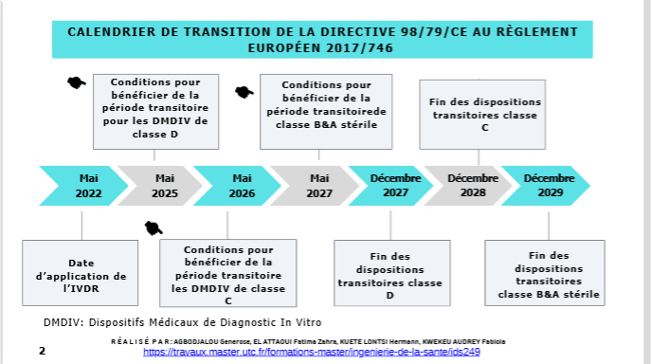
En cliquant sur une phase du calendrier, l’utilisateur est redirigé vers une page dédiée qui présente les conditions nécessaires pour bénéficier de la période transitoire sous le règlement (UE) européen 2017/746. Sur cette page, les conditions sont clairement présentées sous forme de blocs :
-Mise à jour d’un Système de Management de la Qualité (SMQ).
-Soumission d’une demande de certification auprès d’un Organisme Notifié (ON).
-Maintien de la conformité aux exigences de la directive 98/79/CE.
-Surveillance post-commercialisation et vigilance.
Et en cliquant sur chaque condition, l’utilisateur accède à des détails approfondis et spécifiques concernant cette exigence.
Une fois consultées, l’utilisateur peut facilement revenir au calendrier grâce à un bouton interactif placé en bas de la page nommé Calendrier, comme illustré dans la figure 11 :
Figure 11 : Conditions pour bénéficier de la période transitoire sous le règlement (UE) 2017/746 (Source : Auteurs)

Chaque bloc de condition est associé à une page détaillée, offrant des informations approfondies et des ressources pratiques pour guider les utilisateurs dans leur démarche de mise en conformité au règlement (UE) 2017/746. Ces pages incluent des liens interactifs utiles spécifiques à chaque condition.
Par exemple, le bloc intitulé "Soumission d'une demande de certification auprès d'un Organisme Notifié (ON)" présente en détails les conditions requises pour la soumission. Cette page comprend également un renvoi externe direct vers la liste des organismes notifiés européens accrédités, habilités à traiter les demandes relatives aux DMDIV.
Figure 12 : Conditions pour la soumission d'une demande de certification auprès d'un Organisme Notifié (ON) telles que présentées dans la cartographie proposée (Source : Auteurs)

- Partie 2 : Recommandations à suivre pour réussir la transition vers le règlement européen 2017/746
Cette page présente les recommandations à suivre pour réussir la transition vers le règlement européen (UE) 2017/746 . nous avons choisi de nous concentrer sur trois étapes essentielles, qui constituent les piliers de la conformité au règlement :
- La Classification
Déterminer correctement la classe du dispositif conformément aux règles définies dans le règlement (UE) 2017/746. Cette étape est essentielle, car elle conditionne les exigences applicables et le processus d’évaluation de la conformité.
- La Documentation Technique
Rassembler toutes les informations techniques nécessaires sur le dispositif, notamment les preuves de sécurité, d'efficacité, et de performance clinique.
Figure 13 : Recommandations à suivre pour réussir la transition vers le règlement (UE) 2017/746 telles que présentées dans la cartographie proposée (Source : Auteurs)

3.2. Mode d’emploi de l’outil d’autodiagnostic
Cet outil est conçu sur Excel et comporte 5 onglets :
-Le premier onglet étant le mode d’emploi : Cette feuille explique le mode de fonctionnement de l’outil et l’échelle d’évaluation. Il est possible d'évaluer chacune des exigences en utilisant un taux de véracité de « 100% », « 35% », et « 0% », qui représentent respectivement « Fait », « En Cours », et « Non Fait ». Dans le cas où une exigence n'est pas applicable à un dispositif évalué, le fabricant a la possibilité de sélectionner la véracité « Non Applicable », ce qui sera pris en considération dans les résultats globaux de l'évaluation, comme illustré dans la figure 14 :
Figure 14 : Échelles d’évaluation des exigences (Source : Auteurs)

Cet onglet présente également un entête où le fabricant pourra remplir les données en rapport avec son entreprise tel que le nom, le type d’entreprise et le nom du dispositif tel que présenté dans la figure 15 :
Figure 15 : Onglet du mode d’emploi (Source : Auteurs)
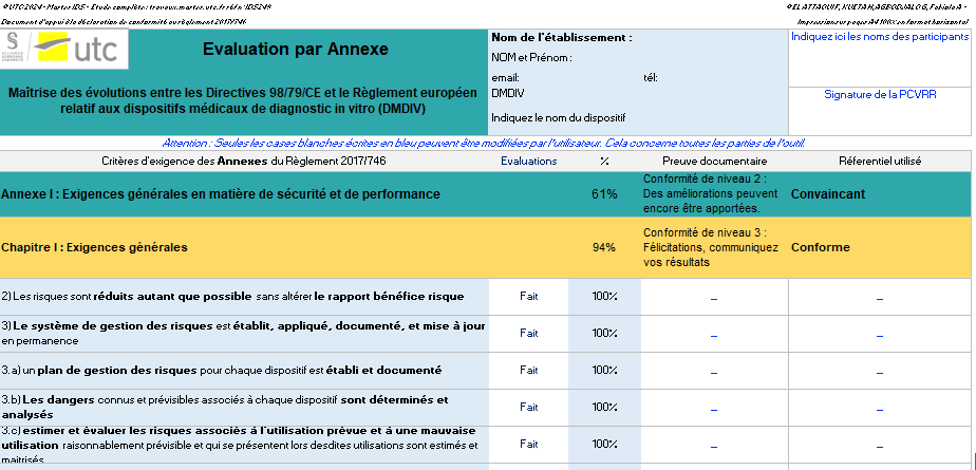
- L’évaluation des exigences par article et l’évaluation des exigences par annexe. Les articles des chapitres et les exigences générales des annexes pertinentes pour le fabricant de DMDIV legacy devices sont contenus dans ces onglets comme indiqué sur la figure 16. Après avoir rempli une exigence, le fabricant va mentionner le document de preuve qui démontre la conformité à l'exigence, ainsi que le référentiel utilisé (s'il existe) sur lequel le fabricant s'appuie pour prouver la conformité, comme illustré dans la figure 16 :
Figure 16 : Onglet de l'évaluation par annexe (Source : Auteurs)
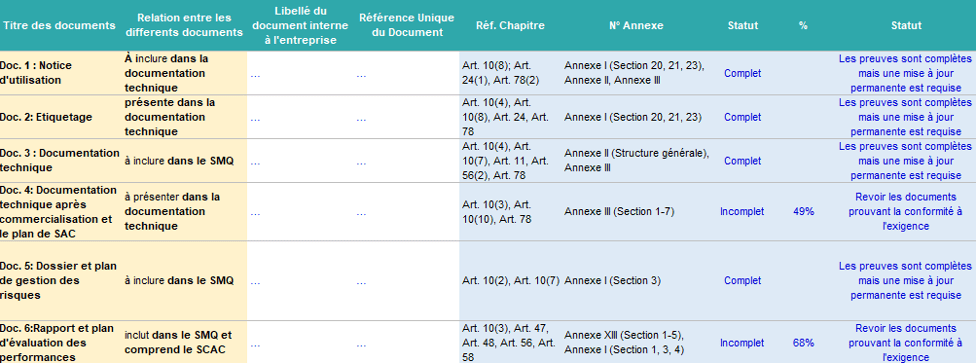
Le 4ème onglet porte sur la maîtrise documentaire : après l'évaluation, cette feuille expose un bilan des exigences et une synthèse des preuves documentaires requises par la réglementation. De façon notable, l’onglet permet ainsi à l’utilisateur de repérer tous les documents requis pour prouver sa conformité au règlement (UE) 2017/746. Cet onglet permet à l’utilisateur de vérifier le statut de chaque document s’il est complet ou pas, comme illustré dans la figure 17
Figure 17 : Onglet de la maîtrise documentaire (Source : Auteurs)
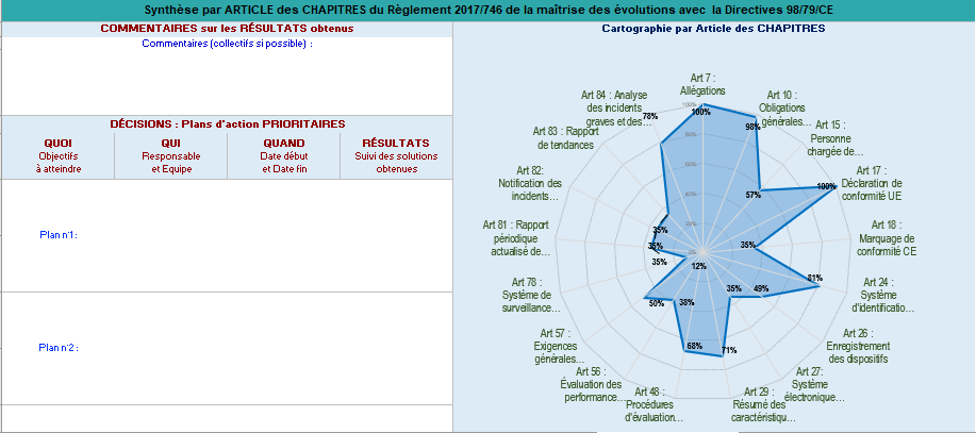
Le dernier onglet présente les résultats globaux sous forme de graphes qui mettent en évidence le taux de conformité aux exigences des différents articles et annexes. Par exemple, dans le cas représenté par la figure 18 ci-dessous le fabricant doit prioriser la mise en conformité aux exigences de l’article 78 au taux d’avancement le plus faible.
Figure 18 : Synthèse par article des chapitres du Règlement 2017/746 (Source : Auteurs)
3.3. Perspectives et limites des outils proposés
Les outils proposés ont été soumis à des professionnels du secteur des DMDIV pour une première évaluation.
L’outil d’autodiagnostic a été apprécié comme complet, la transcription des exigences du règlement (UE) 2017/746 fidèle évitant ainsi de dénaturer les textes du règlement. Un auditeur d’un ON a ainsi relevé que “le côté redondant des éléments figurant dans l'outil est à l’image de la documentation technique ”.
L’ergonomie de l’outil d’autodiagnostic peut être améliorée par un travail de synthèse additionnel visant à regrouper les exigences en plusieurs catégories à l’image de la documentation technique que les organismes notifiés examinent pour donner l’attestation de conformité.. Les différentes exigences peuvent être classées en 4 parties : la partie administrative, la partie préclinique, la partie clinique ainsi que la partie relative à la surveillance post-commercialisation. Chaque partie figurera dans différents onglets. Ceci permettra de réduire les nombreuses lignes figurant dans l’outil d'autodiagnostic dans son état actuel.
Quant à la cartographie interactive, elle peut être mise à jour au vu des amendements et des nouveaux guides qui pourraient être publiés par la suite.
3.3.1 Limites des outils proposés
Sachant que le règlement (UE) 2017/746 est entré en vigueur depuis l’année 2022, les retours des différents entretiens ont permis de remarquer que les outils proposés seront particulièrement utiles pour les Petites et Moyennes Entreprises (PME) commercialisant des DMDIV de classe A et B dont les périodes de transition s’étendent jusqu’en décembre 2029. Ils guideront également les jeunes diplômés en affaires réglementaires appelés à intervenir sur le sujet. Par exemple, l’outil d’autodiagnostic proposé leur permettra en situation professionnelle de se situer sur ce qui leur reste à mettre en œuvre et de mieux se planifier.
Conclusion
Le marché des DMDIV est en pleine croissance confrontée à l’imminence de l’échéance prochaine de la mise en conformité de tous les DMDIV legacy devices au règlement (UE) 2017/746 qui renforce les exigences liées à la performance et la sécurité des dispositifs, augmentant les coûts de mise en conformité règlementaire sur le marché européen pour les fabricants.
Pour accompagner les professionnels du secteur dans leurs démarches et optimiser leur coûts, deux outils sont proposés : une cartographie interactive regroupant les informations essentielles, ressources et guides utiles pour optimiser la mise en conformité et un outil d’autodiagnostic permettant d’identifier les écarts entre la directive 98/79/CE et le règlement (UE) 2017/746.
Leur utilisation permet une compréhension plus fine des exigences réglementaires et de gagner du temps dans la préparation et la planification des étapes de mise en conformité. L’expérience des premiers professionnels utilisateurs est positive et ouvre la voie à l’intégration de fonctionnalités complémentaires à des fins de personnalisation.
La transition vers le règlement (UE) 2017/746 est un processus plus exigeant mais indispensable pour garantir la sécurité et l’efficacité des dispositifs. Les outils développés représentent une première étape vers une transition plus précise et adaptée, nécessitant des améliorations constantes pour mieux répondre aux exigences pratiques du terrain et maintenir la confiance des utilisateurs.