
IDS298 – Analyse du Règlement (UE) 2021/2282 – élaboration d’un guide d’appropriation pour les fabricants de dispositifs médicaux
DOI mémoire
https://doi.org/10.34746/ids298Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- CHALVIDAL Lisa : Chalvidal.lisa@gmail.com
- KADIRI Chourouk : Kadirichorouk@gmail.com
- NASSOH Kenza : Kenzanassoh20@gmail.com
- NZENGUI Steeven : Braggynzengui@gmail.com
- OUHAB Sabrina : Sabrinaouhab02@gmail.com
Citation
A rappeler pour tout usage : L.CHALVIDAL, C.KADIRI, K.NASSOH, S.OUHAB, S.NZENGUI, "Analyse du Règlement (UE) 2021/2282 – élaboration d’un guide d’appropriation pour les fabricants de dispositifs médicaux". Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositif médical et affaires réglementaires, Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS298, janvier 2026, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids298/, lien DOI : https://doi.org/10.34746/ids298
Remerciements
De prime abord, notre groupe souhaite remercier l’Université de Technologie de Compiègne et son corps professoral pour l’accompagnement, le soutien et leur expertise tout au long de ce semestre. Leur enseignement de qualité et les différents conseils prodigués ont largement contribué à notre développement personnel et professionnel.
Nos remerciements sont adressés les plus sincères aux responsables du Master Ingénierie de la Santé de l’Université de Technologie de Compiègne, Madame Isabelle CLAUDE, Madame Julie FOLLET et Monsieur Jean-Matthieu PROT, pour leur engagement et leur disponibilité en dépit de leurs multiples occupations. Leur soutien constant a été une source de motivation et a joué un rôle clé dans l’avancement de ce travail.
Nous tenons également à exprimer une reconnaissance particulière envers notre suiveur de projet, Madame Julie FOLLET, pour son sens de la critique constructive, ses conseils éclairés et son suivi attentif qui nous ont guidés à chaque étape et ont fortement contribué à structurer et à enrichir notre travail.
Une sincère reconnaissance envers tous les intervenants qui, à travers des entretiens sur site et par appels téléphoniques, ont contribué à l’élaboration de ce travail.
Enfin, nous souhaitons exprimer notre profonde gratitude à toutes les personnes qui ont, de près ou de loin, contribué à la réalisation de ce projet.
Résumé
Ce travail analyse le nouveau cadre européen d’évaluation des technologies de santé, défini par le Règlement (UE) 2021/2282, et son articulation avec le Règlement (UE) 2017/745 relatif aux dispositifs médicaux. Alors que le Règlement (UE) 2017/745 encadre la mise sur le marché et l’évaluation clinique, le Règlement (UE) 2021/2282 introduit une évaluation clinique commune (ECC) harmonisée au niveau européen afin de centraliser et structurer les données cliniques fournies par les fabricants.
L’étude met en évidence les critères de sélection des dispositifs éligibles, les acteurs impliqués, ainsi que les deux productions scientifiques clés du règlement : les Consultations Scientifiques Communes (CSC) et les Évaluations Cliniques Communes (ECC). Une analyse des processus, complétée par des retours de terrain, montre que les fabricants rencontrent des difficultés d’interprétation du texte et un manque de clarté sur les preuves supplémentaires à fournir.
Pour répondre à ces besoins, un guide interactif a été conçu. Ce guide propose une visualisation simplifiée des étapes réglementaires, des logigrammes explicatifs et une comparaison des exigences entre les deux règlements. Il vise à aider les fabricants de dispositifs médicaux à mieux comprendre, anticiper et appliquer le Règlement (UE) 2021/2282. L’outil constitue une base pratique pour faciliter l’appropriation du règlement et accompagner les futurs développements réglementaires dans le domaine des dispositifs médicaux.
Abstract
This work examines the new European framework for health technology assessment defined by Regulation (EU) 2021/2282, and its interaction with Regulation (EU) 2017/745 on medical devices. While Regulation (EU) 2017/745 governs market access and clinical evaluation, Regulation (EU) 2021/2282 introduces a Joint Clinical Assessment (JCA) designed to harmonize, centralize, and structure the clinical evidence provided by manufacturers at the European level.
The study highlights the eligibility criteria for selected devices, the key stakeholders involved, and the two main scientific outputs of the regulation : Joint Scientific Consultations (JSC) and Joint Clinical Assessments (JCA). Process analysis, complemented by field feedback, shows that manufacturers face difficulties in interpreting the regulation and understanding the additional clinical evidence required.
To address these challenges, an interactive guide was developed. This guide offers simplified visual representations of regulatory steps, explanatory flowcharts, and a comparative analysis of requirements between the two regulations. Its purpose is to support medical device manufacturers in better understanding, anticipating, and implementing Regulation (EU) 2021/2282. This tool serves as a practical resource to facilitate adoption of the regulation and to support future regulatory developments in the medical device sector.
Téléchargements



Analyse du Règlement (UE) 2021/2282 – élaboration d’un guide d’appropriation pour les fabricants de dispositifs médicaux
Introduction - Aperçu général du cadre réglementaire appliqué aux dispositifs médicaux
Les dispositifs médicaux (DM) occupent aujourd’hui une place cruciale dans le parcours de soins. Ils participent au diagnostic, au traitement et à l’accompagnement quotidien des patients. Leur cycle de vie est soumis à un cadre réglementaire strict défini par le Règlement (UE) 2017/745. Entré en application en 2021, ce règlement est la référence en Europe pour garantir la sécurité, les performances et l’évaluation clinique des DM [1].
Le règlement (UE) 2017/745 a profondément renforcé l’évaluation clinique des dispositifs médicaux en Europe. Il exige la production de preuves cliniques solides, proportionnelles au risque que le DM fait peser sur la santé des patients et des utilisateurs, compte tenu de son usage prévu, de sa classe, de sa nouveauté et de son impact clinique, et produites par une méthodologie scientifique rigoureuse. Il prévoit également un suivi clinique après commercialisation (SCAC) structuré et continu, afin de confirmer, dans des conditions d'utilisation réelles, la sécurité, les performances et les bénéfices cliniques du dispositif tout au long de son cycle de vie. La preuve clinique ne peut pas se reposer uniquement sur des données théoriques ou des comparaisons à l’état de l’art clinique d’un DM équivalent. Pour les dispositifs présentant un risque élevé, notamment les dispositifs implantables et ceux de classe III, le règlement exige la réalisation d'investigations cliniques (IC). Ces études visent à produire des données issues de l'utilisation du dispositif chez des patients afin de démontrer sa conformité et d'obtenir le marquage CE médical [1, 2].
Une fois le marquage apposé, le dispositif médical peut être commercialisé sur le marché européen. Cette étape n’implique pas un remboursement systématique par un État membre. Pour obtenir le remboursement du DM, des démarches complémentaires doivent être effectuées auprès de chaque État Membre, et des données relatives au bénéfice clinique et au positionnement du DM dans la stratégie thérapeutique sont attendues. Pour ce faire, il est essentiel de prendre en compte, dès le début du développement clinique du DM, l'ensemble des attentes relatives aux évaluations cliniques, qu'il s'agisse de celles demandées par le Règlement (UE) 2017/745 ou de celles nécessaires à une demande de remboursement [2]. Dans cette optique, l'Union Européenne a mis en place un nouveau règlement, le Règlement (UE) 2021/2282 relatif à l'évaluation des technologies de la santé, afin de réduire la duplication des efforts des fabricants et des autorités nationales d'évaluation des technologies de la santé (EST) en harmonisant et centralisant le dépôt des données cliniques [3, 4]. Toutefois, certaines difficultés surviennent quant à l’application pratique de ce nouveau règlement, entre autres, la compréhension et l'interprétation des exigences qui l’accompagnent, ce qui peut constituer un problème pour les fabricants des dispositifs médicaux.
Dès lors, par l’analyse de ces deux règlements et de leurs interactions, un outil pratique a été développé afin de faciliter la mise en œuvre du Règlement (UE) 2021/2282. Cet outil permettra l’accompagnement des fabricants de DM dans leurs démarches réglementaires pour la compréhension de ce règlement et mettra en lumière les preuves supplémentaires (comparées au Règlement 2017/745) à fournir.
Chapitre 1 : Cadre réglementaire des dispositifs médicaux
1. Le Règlement (UE) 2017/745 : Mise sur le marché des dispositifs médicaux
1.1 Cadre général
Le Règlement (UE) 2017/745 relatif aux DM s'applique directement et uniformément dans toute l'Union Européenne (UE) [1] . Il vise non seulement à assurer la mise sur le marché de DM sûrs et performants et la libre circulation dans l’UE, mais aussi à établir un environnement réglementaire renforcé [1].
Afin de prouver la sécurité et la performance des DM, le règlement impose entre autres d’effectuer une évaluation clinique des DM qui conduit à la rédaction d’un rapport d’évaluation clinique regroupant les données obtenues, et à insérer dans un dossier technique imposé au fabricant conformément à l’Annexe II dudit règlement.
Au-delà des exigences réglementaires relatives à l’évaluation clinique, le règlement (UE) 2017/745 établit des dispositions s’étendant sur tout le cycle de vie des DM, allant de la définition des responsabilités des acteurs économiques à la surveillance après commercialisation et la vigilance tout en passant par l’encadrement des procédures d’évaluation de la conformité. Globalement, son objectif est de garantir et maintenir un niveau élevé de fiabilité du marché européen des dispositifs médicaux.
1.2 Processus de mise sur le marché des dispositifs médicaux
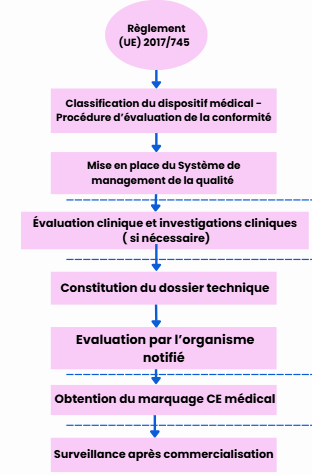
a) Classification d’un dispositif médical et mise en place du système de management de la qualité
Le processus de mise sur le marché d’un DM est initié par la détermination de sa classe de risque. Elle se fait conformément aux règles de l’annexe VIII du Règlement (UE) 2017/745 [1] et pose les bases des exigences réglementaires applicables et le chemin à suivre jusqu’à l’obtention du marquage CE. Une fois la classe définie, le fabricant des DM doit mettre en place un système de management de la qualité (SMQ), aligné sur la norme ISO 13485 : 2016, un processus de gestion des risques en conformité avec l’ISO 14971 : 2019, un contrôle des phases de conception et production, ainsi qu’une gestion documentaire [1].
b) Evaluation clinique
Après la mise en place d’un SMQ, le fabricant doit monter un dossier technique afin de compiler les preuves prouvant la conformité du dispositif aux exigences de sécurité et de performance. Parmi les preuves à fournir, il y a l’évaluation clinique du dispositif, et cette dernière permet de prouver la sûreté du dispositif. Elle débute par l’élaboration d’un plan d’évaluation clinique (CEP) (article 61 et annexe XIV du Règlement (UE) 2017/745) [1], dont l’objectif est de définir la méthodologie, les objectifs et les critères d’évaluation [1] (voir figure 1).
Le fabricant procède alors au recensement et à l’analyse critique de la totalité (ou presque) des données cliniques disponibles, en particulier les données issues de la littérature scientifique, de données précliniques, d’investigations cliniques antérieures ou provenant d’un retour d’expérience clinique après commercialisation existante, dans le cas échéant [1].
Dans le cas d’un DM mis pour la première fois sur le marché, une investigation clinique est obligatoire, exception faite, si les conditions spécifiques d’exemption prévues sont dûment justifiées notamment la démonstration de l’équivalence et l’accès à des données suffisantes d’un dispositif équivalent déjà présent sur le marché (article 61(6) du Règlement (UE) 2017/745 [1].
Par ailleurs, si le dispositif est déjà sur le marché et n’a subi aucune modification susceptible d’impacter sa sécurité ou son niveau de performance, le fabricant peut avoir recours aux données provenant de l’évaluation clinique précédente, complétées par les données de
surveillance après commercialisation et, le cas échéant, le plan de suivi clinique après commercialisation (annexe XIV Règlement (UE) 2017/745) [1].
Une fois l’ensemble des données cliniques nécessaires recensées, le fabricant de DM rédige le rapport d’évaluation clinique (REC). Ce rapport synthétise l’ensemble des preuves, les confronte au regard des exigences du Règlement (UE) 2017/745 et conclut sur le bénéfice clinique attendu. L’évaluation clinique terminée, le dossier technique est ainsi constitué, conformément aux annexes II et III du Règlement (UE) 2017/745. Il comprend notamment la description détaillée du dispositif, sa destination, ses critères de performance, les preuves précliniques, ainsi que le CEP et CER [1].
c) Évaluation par l’organisme notifié et mise sur le marché
Une fois la constitution du dossier technique terminée, l’évaluation du DM conformément aux exigences du Règlement (UE) 2017/745 est réalisée par l’organisme notifié (ON). Si la conformité est démontrée, le dispositif obtient alors le marquage CE médical, autorisant sa mise sur le marché européen [1] (Cf. articles 19 et 20 du Règlement (UE) 2017/745) (voir figure 1).
d) Surveillance après commercialisation
Après la mise sur le marché, tous les dispositifs doivent faire l’objet d’une surveillance post- commercialisation, incluant la mise à jour continue des données cliniques, la vigilance et les actions de suivi clinique post-commercialisation (PMCF), afin de garantir la sécurité et la performance tout au long du cycle de vie du dispositif [1] (voir figure 1).
Figure 1 : Processus général de mise sur le marché des DM selon le Règlement (UE) 2017/745 - source : auteurs
Au-delà de la conformité des DM aux exigences du Règlement (UE) 2017/745, les autorités de santé prêtent également attention à l'évaluation de la valeur clinique et médico-économique des innovations dans le secteur de la santé.
Après l’obtention du marquage CE médical, l’étape suivante pour un dispositif médical est celle du remboursement, qui vise à évaluer la stratégie thérapeutique et la valeur ajoutée du dispositif par rapport aux technologies existantes sur le marché. Contrairement à l’évaluation clinique réalisée pour le marquage CE, l’évaluation dans le cadre du remboursement ne se concentre pas uniquement sur la preuve clinique, mais sur l’intérêt thérapeutique global, le bénéfice clinique relatif et la place du dispositif dans la prise en charge des patients. Dans ce processus, le fabricant doit généralement soumettre des demandes auprès des autorités compétentes de chaque État membre dans lesquelles ils souhaitent avoir le remboursement, ce qui entraîne une duplication des preuves cliniques et des efforts administratifs, tant pour le fabricant que pour les autorités. Cette distinction montre que le remboursement est un processus indépendant du marquage CE, propre à chaque pays. C’est dans ce contexte qu'a vu le jour le Règlement (UE) 2021/2282, relatif à l’évaluation des technologies de santé. Ce dernier s’inscrit dans un souci d’harmonisation des méthodes d’évaluation clinique dans l’UE. La section qui suit est dédiée à l'étude de ce nouveau règlement et met en évidence ses objectifs et son articulation avec le Règlement (UE) 2017/745.
2. Le Règlement (UE) 2021/2282
2.1 Cadre général
Le Règlement (UE) 2021/2282 vise à améliorer le niveau de preuves cliniques à fournir par les fabricants en vue de constituer le dossier de remboursement de leur produit au sein des Etats membres (EM) de l’Union. Il permet ainsi d’aider les EM à prendre des décisions sur le remboursement ou non du dispositif, fondé sur des données cliniques probantes et conformes à leurs attentes. Ce règlement touche les technologies de santé, ce qui, au sens du Règlement (UE) 2021/2282, concerne les nouveaux médicaments et dispositifs médicaux [3].
Le calendrier d’application du Règlement (UE) 2021/2282 est progressif. Après son entrée en vigueur en 2022, il connaît une phase de mise en œuvre qui débute en 2025 et s’appliquera à une liste de dispositifs définis par le groupe de coordination de l’évaluation des technologies de santé (ou HTACG pour Health Technology Assessment Coordination Group). Chaque année n, le groupe de coordination élabore le programme de travail pour l’année suivante n+1, et les décisions prises par la Commission via les actes d’exécution suivent ce même calendrier et de nouveaux dispositifs sont sélectionnés afin d'être évalués conformément à ce règlement.
Il se concentre exclusivement sur les preuves cliniques à fournir par le fabricant pour le dossier de remboursement, tandis que les dimensions économiques telles que la tarification et le remboursement relèvent toujours de la compétence propre à chaque EM. Par exemple en France, la réévaluation clinique est conduite par la Haute Autorité de Santé (HAS) [5] tandis que la décision de remboursement relève du ministère de la santé après négociation du prix entre le CEPS et le fabricant. De fait, même si le Règlement (UE) 2021/2282 harmonise à l’échelle de l’Union la manière de produire et de structurer les données cliniques nécessaires à l’évaluation des DM, la décision finale concernant le remboursement reste nationale. Ce fonctionnement permet à chaque pays de conserver ses spécificités organisationnelles tout en facilitant l’accès aux innovations grâce à une base clinique commune et partagée [3].
Le Règlement (UE) 2021/2282 désigne de nouveaux rôles. Les fabricants des DM concernés par le Règlement (UE) 2017/745 sont appelés développeurs de technologie de santé dans le Règlement (UE) 2021/2282. Ce terme comprend les développeurs de médicaments et de DM. Dans un souci d’harmonisation du travail, notre travail utilisera la terminologie de fabricants de DM.
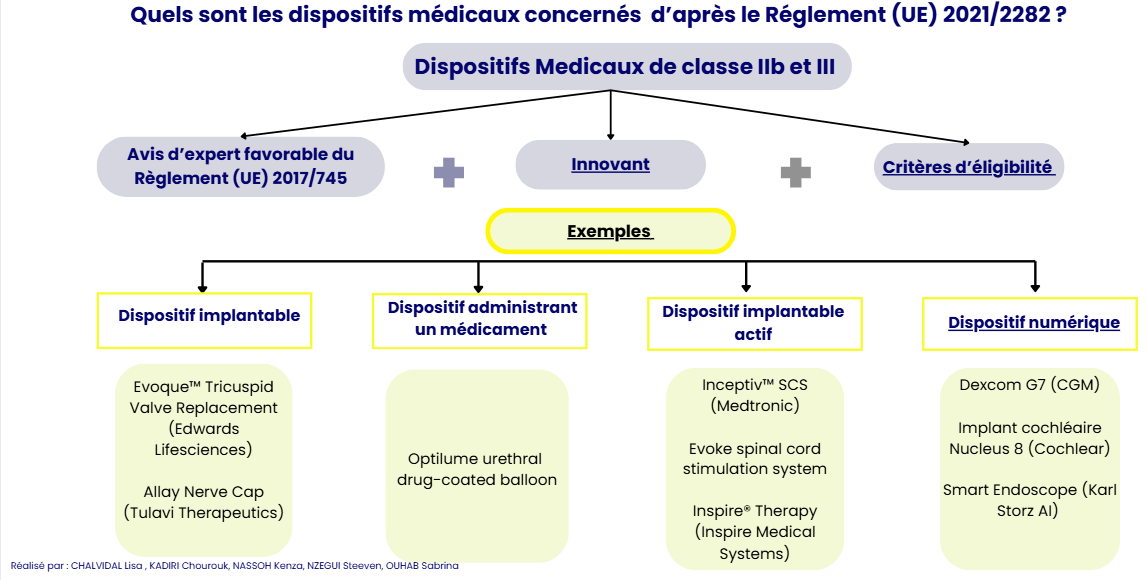
2.2 Périmètre d’application du Règlement (UE) 2021/2282 : Les dispositifs médicaux concernés
Le Règlement (UE) 2021/2282 s’applique, pourvu qu’ils remplissent plusieurs conditions cumulatives, à certains DM de classes IIb et III tels que définis par le Règlement (UE) 2017/745 (toutes les conditions doivent être remplis simultanément pour qu’un dispositif de classe IIb ou III soit éligible). Ils reçoivent un avis favorable des panels d’experts européens tels que mentionnés dans l’article 106 du Règlement (UE) 2017/745, avis émis dans le cadre de la procédure de consultation de l’évaluation clinique (CECP). De plus, le dispositif doit présenter un caractère innovant et satisfaire certains critères d’éligibilité, définis par le Règlement (UE) 2021/2282 [6], qui seront présentés dans cette section. Il est donc important de noter que la sélection de ces DM relève du Groupe de coordination, qui à travers son programme de travail annuel, sélectionne les technologies à évaluer tel que prévu à l’article 6 dudit règlement. En outre, les critères qui seront décrits dans cette section sont des critères attestant de l’éligibilité des DM à cette sélection (voir figure 2).
Figure 2 : Caractéristiques des dispositifs médicaux concernés par le Règlement (UE) 2021/2282 - source : auteurs
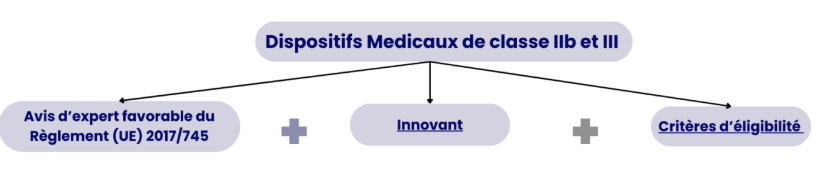
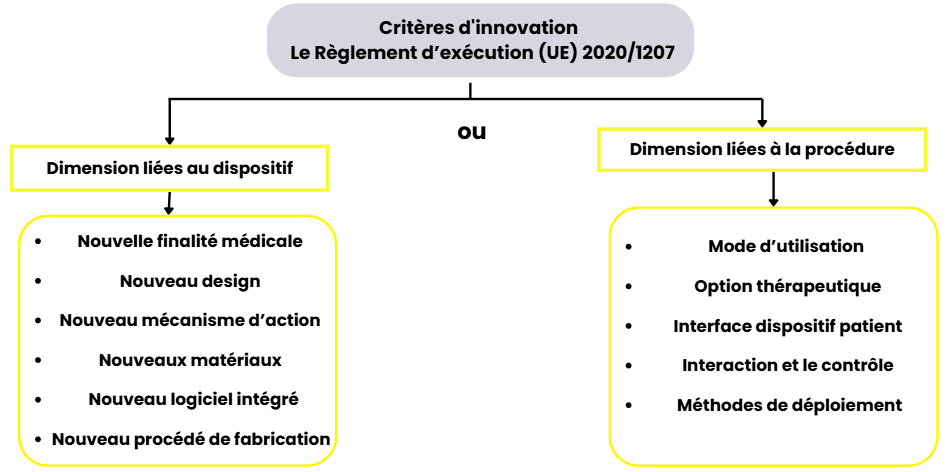
Un caractère innovant spécifié selon le Règlement d'exécution (UE) 2020/1207 et comprenant deux volets : un lié aux dispositifs incluant notamment une nouvelle finalité médicale, un nouveau design, de nouveaux matériaux, un nouveau logiciel intégré, et un deuxième volet lié à la procédure d’utilisation (voir figure 3) [6].
Figure 3 : Les critères d'innovation des dispositifs médicaux concernés par le Règlement (UE) 2021/2282 tels que mentionnés dans le Règlement d’exécution (UE) 2020/1207 - source : auteurs
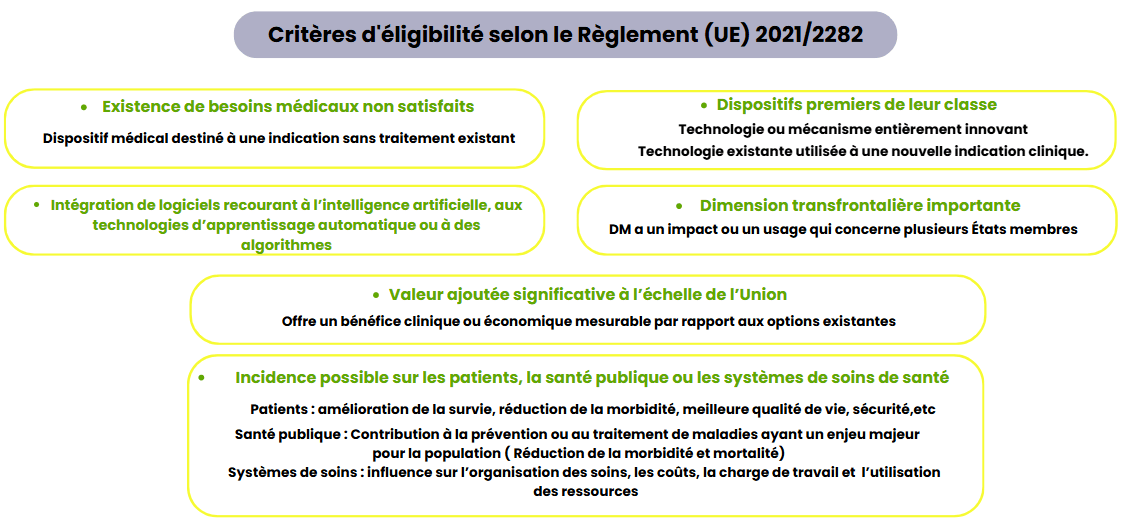
Les critères d’éligibilité des DM au Règlement (UE) 2021/2282 sont listés dans la figure suivante (voir figure 4) [7] :
Figure 4 : Les critères d’éligibilité des dispositifs médicaux au Règlement (UE) 2021/2282 - source : auteurs
Après avoir mentionné les critères cumulatifs attestant de l’éligibilité d’un DM au champ d’application du Règlement (UE) 2021/2282, il convient de mettre en lumière les acteurs clés impliqués dans sa mise en œuvre. En comprenant à quel niveau précis intervient tel acteur et quelles responsabilités lui sont assignées, on peut saisir toute la logique du règlement et comprendre comment les évaluations cliniques conjointes sont organisées, validées et mises à disposition des États membres. La section suivante sera, par conséquent, consacrée à l’étude de tous ces acteurs essentiels.
2.3 Les acteurs clés de l’évaluation des technologies de la santé selon conformément au Règlement (UE) 2021/2282
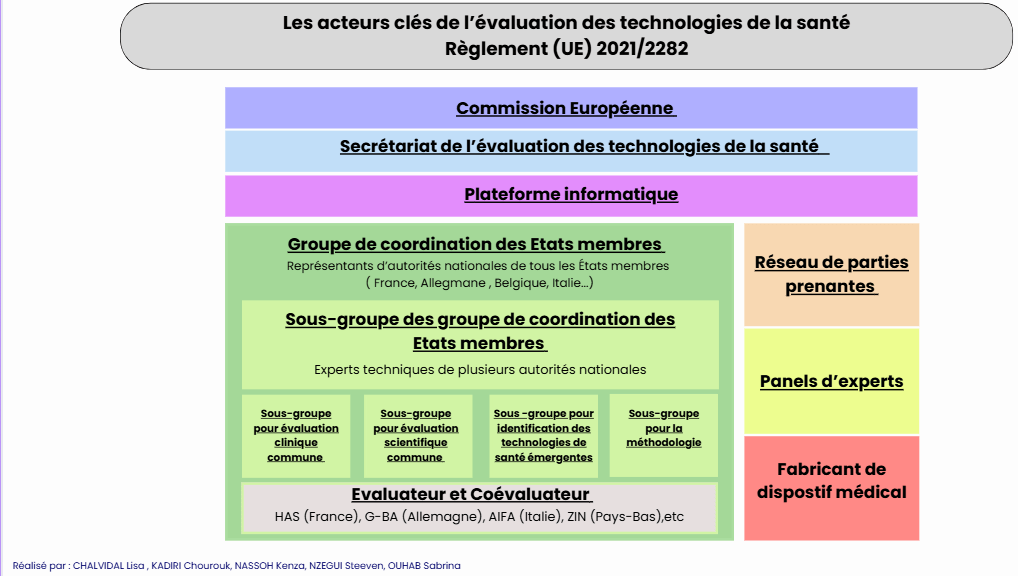
Dans le cadre du Règlement (UE) 2021/2282, l’évaluation des technologies de santé repose sur une organisation européenne structurée autour de plusieurs acteurs clés (voir figure 5) [3, 8].
Figure 5 : Les acteurs clés de l’évaluation des technologies de la santé sur le Règlement (UE) 2021/2282 - source : auteurs

La Commission européenne exerce un rôle central de pilotage, d’harmonisation et de supervision du système. Elle met en place le cadre réglementaire, assure la gestion des bases de données, la transparence des processus et encadre la sélection et la coordination des experts et panels cliniques. Elle est appuyée par le Secrétariat de l’évaluation des technologies de la santé (ETS), chargé de coordonner les échanges d’informations nécessaires à la préparation, à la mise à jour et au suivi des évaluations cliniques communes, en lien avec les organismes notifiés, les fabricants de DM, les patients et l’ensemble des parties prenantes (voir figure 5) [3, 8].
Le Groupe de coordination des EM sur l’évaluation des technologies de la santé se compose de représentants désignés par les EM et occupe une place stratégique dans la gouvernance du Règlement (UE) 2021/2282. En effet, il oriente les travaux, valide les nominations des entités impliquées dans les évaluations et supervise la réalisation des consultations scientifiques communes (CSC) ainsi que les évaluations cliniques communes (ECC). Il s’appuie pour cela sur un sous-groupe de coordination, qu’il nomme en son sein, un évaluateur et un co-évaluateur, issus des EM également. L’évaluateur pilote l’ensemble du travail et rédige les rapports, tandis que le co-évaluateur apporte un second avis, vérifie les analyses et contribue à garantir la robustesse scientifique des conclusions. Leur sélection repose sur des critères d’expertise clinique, d’expérience en évaluation des technologies de la santé, d’indépendance, de disponibilité et d’équilibre géographique au sein de l’UE [3, 8]. Par ailleurs, il convient de noter qu’à ce jour la désignation formelle de l’ensemble des membres du groupe de coordination n’est pas encore totalement aboutie, et il n’existe publiquement aucun calendrier fixé de nomination de ces derniers. La Commission européenne a indiqué à cet effet que les désignations devront intervenir progressivement pour faciliter la pleine application du Règlement (voir figure 5) [9].
Plus tard, un panel d’experts intervient afin d’apporter une expertise clinique spécialisée dans le domaine visé par l’évaluation. Le domaine ici fait allusion au champ spécifique thérapeutique, pathologique ou technologique auquel se rapporte la technologie de la santé ou le DM. On peut citer en guise d’exemple, une spécialité médicale telle que la cardiologie ou la neurologie, un type de dispositif comme des prothèses orthopédiques ou des stimulateurs cérébraux. Le panel est composé d’experts consultés dans le cadre du Règlement (UE) 2017/745 sélectionnés par le fabricant, ainsi que d’experts individuels, sélectionnés par le groupe de coordination et le sous-groupe de coordination. Ces experts contribuent à l’analyse scientifique et peuvent être sollicités lors des consultations scientifiques communes (voir figure 5) [3, 8]. Point important à noter, tout comme le groupe de coordination, il n’existe pas à ce jour de liste exhaustive des membres du panel d’experts rendue publique.
Pour finir, le réseau de parties prenantes, créé et encadré par la Commission européenne, regroupe des associations de patients, de consommateurs, des organisations non gouvernementales (ONG) actives dans le domaine de la santé telles que des associations luttant contre des maladies spécifiques ou promouvant l'accès équitable aux soins, des professionnels de santé et des fabricants de DM. Ce réseau soutient, à la demande, les travaux du groupe de coordination et de ses sous-groupes, garantissant une participation large, représentative et transparente dans l’ensemble du processus d’évaluation (voir figure 5) [3, 8].
Pour faciliter la réalisation des travaux communs et l’échange d’informations entre les auteurs, la commission met en place et tient à jour une plateforme informatique de l'ETS : Elle est un outil facilitant la coopération entre les différents acteurs impliqués dans l’évaluation des technologies de santé. Développé par la Commission européenne, cet outil représente l’infrastructure centrale du Règlement (UE) 2021/2282, est garant de la fluidité des échanges, facilite la circulation harmonisée des données ainsi que de la transparence des procédures entre les acteurs.
On retrouve en son sein : une page publique accessible, destinée à diffuser les informations essentielles, un intranet sécurisé réservé au groupe de coordination et à ses sous-groupes, un système sécurisé permettant les interactions entre les évaluateurs, les fabricants de DM, les experts et les groupes d’experts du Règlement (UE) 2017/745, ainsi qu’un espace d’échange dédié au réseau des parties prenantes [3, 8].
Il est important de noter qu’actuellement, il n’existe aucun lien formellement explicité dans les textes entre la plateforme informatique et la base de données Eudamed.
Après avoir présenté les critères d’éligibilité des DM concernés par le Règlement (UE) 2021/2282 ainsi que les principaux acteurs impliqués dans sa mise en œuvre, il convient désormais de s’intéresser particulièrement aux productions apportées par ledit règlement. Ces productions constituent le cœur opérationnel même du Règlement (UE) 2021/2282 : elles matérialisent l’expertise propre des panels et des évaluateurs, fournissent les preuves ainsi que les recommandations destinées à soutenir les décisions des autorités nationales. La section suivante détaillera leurs objectifs, caractérisera leurs attributs et expliquera leur rôle dans le cycle d’évaluation des DM entrant dans le champ d’application dudit règlement.
2.4 Les productions scientifiques du Règlement (UE) 2021/2282
Les productions apportées par le nouveau règlement sont planifiées et réalisées par le groupe de coordination dans le cadre de son programme de travail annuel. Il s’agit des Consultations scientifiques communes (CSC) et des Évaluations cliniques communes (ECC). Le groupe de coordination fixe chaque année le nombre et le type d’ECC de même que le nombre de CSC à mener pour la prochaine année (Cf. article 6 du Règlement (UE) 2021/2282). Le processus commence par les CSC qui serviront de base pour les ECC à mener par la suite.
a) La Consultation scientifique commune (CSC)
Les CSC interviennent en amont des ECC et permettent au fabricant de DM qui en formule la demande, et sous condition que le DM réponde aux critères d’éligibilités (Cf.chapitre 1, section 2.2 [3]) d’être susceptible de faire l’objet d’une ECC (et les essais cliniques doivent être au stade de la planification [10]), et de bénéficier de l’avis du sous-groupe CSC sur son plan d’évaluation clinique ou l’investigation clinique. Ceci afin de faciliter la production de preuves cliniques ciblées et pertinentes et de clarifier les attentes d’une ECC [3].
Les CSC ne sont toutefois pas contraignantes : elles aident à comprendre les attendus des ECC sans engendrer de difficulté réglementaire directe pour le fabricant de DM qui en fait la demande et pour le groupe de coordination [3]. Ces consultations peuvent être réalisées en parallèle de la consultation des panels d'experts européens mobilisés dans le cadre du Règlement (UE) 2017/745 et du Règlement (UE) 2021/2282 lors du processus de la constitution du dossier technique tel qu’exigé par le Règlement (UE) 2017/745 (voir figure 1). La procédure entière durera approximativement 4-5 mois à partir de la réception de la demande de CSC déposée par le fabricant, sachant que le nombre de CSC sera défini annuellement par le groupe de coordination [11]. Si le nombre de dossiers éligibles est supérieur au nombre de CSC prévu, le groupe de coordination sélectionnera les dossiers prioritaires sur la base de critères d’éligibilité (définis dans les articles 5 et 7 du Règlement (UE) 2021/2282, Cf. chapitre 1 section 2.2 les DM concernés [3]).
Le processus de consultation scientifique commune (CSC), tel que prévu par le Règlement (UE) 2021/2282, s’intègre dans le parcours de développement et de mise sur le marché des DM concernés (Cf chapitre 1, section 2.2 Les DM concernés) [3].
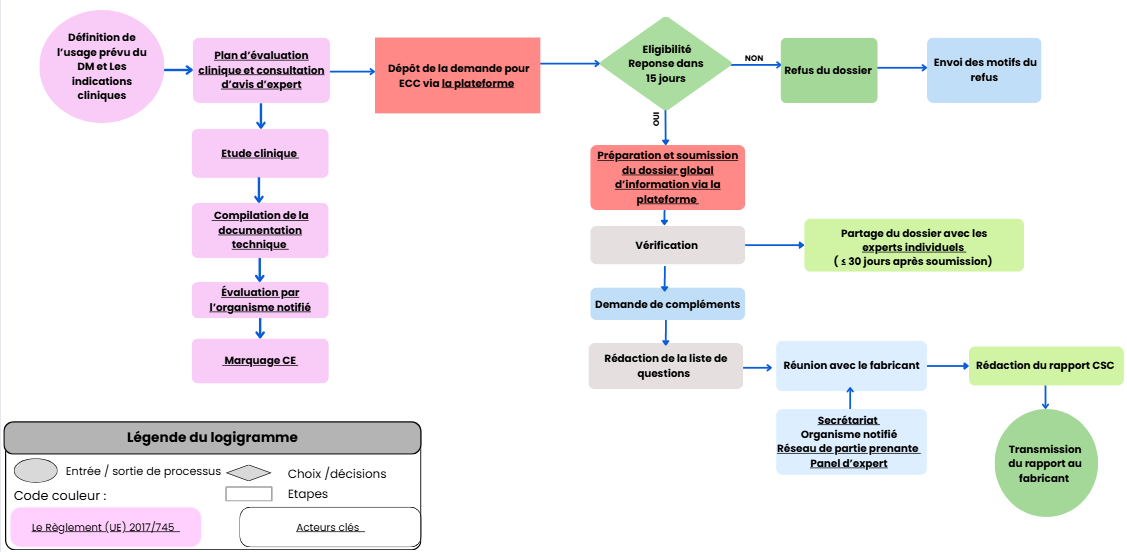
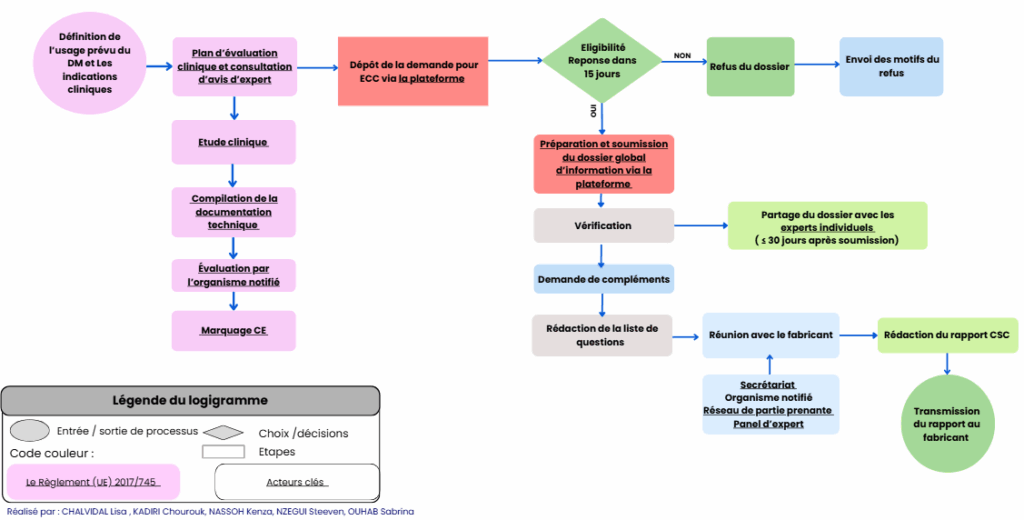
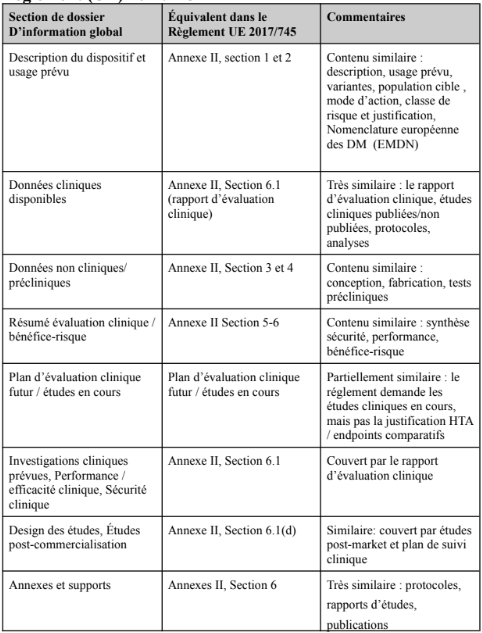
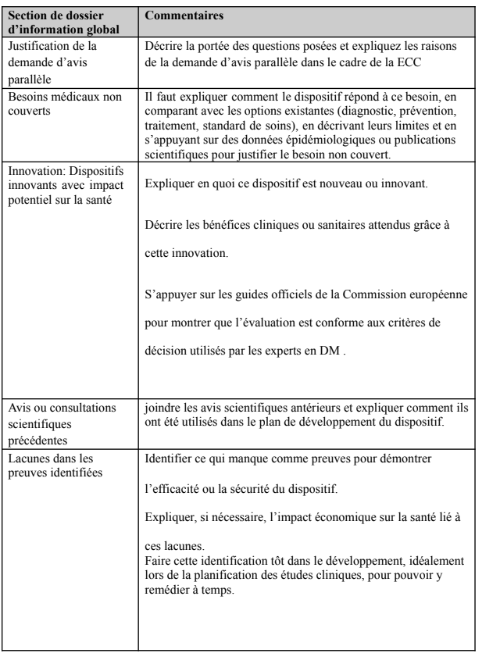
Lorsque le fabricant de DM souhaite bénéficier d’une CSC, il dépose sa demande sur la plateforme informatique de l'ETS durant les périodes prévues à cet effet [8, 10]. Il faut noter que le groupe de coordination définit, au plus tard le 30 novembre de chaque année, un programme de travail annuel et c’est au cours de cet évènement que les périodes de dépôt de demande des CSC sont établies. Les périodes varient et le groupe de coordination exige au moins 3 périodes durant l’année (Cf. article 6 du Règlement (UE) 2021/2282 [3]). La demande est alors traitée, et une réponse sur l’éligibilité est fournie dans un délai de 15 jours. En cas de refus, les motifs sont transmis au fabricant [11]. Si la demande est acceptée, le développeur doit préparer et soumettre un dossier global d’information via la plateforme [3]. Un tableau comparatif des sections du dossier global d’information entre le Règlement (UE) 2017/745 et le Règlement (UE) 2021/2282 est présenté en Annexe I. Les informations supplémentaires à fournir sont détaillées dans l’Annexe II [12]. Ce dossier fait l’objet d’une vérification pouvant conduire à des demandes de compléments par le sous-groupe de la consultation scientifique commune. Une fois la conformité du dossier confirmée, une liste de questions est rédigée par les évaluateurs [10].
Le dossier est ensuite partagé avec les experts individuels, dans un délai maximal de 30 jours après sa soumission. Une réunion est organisée entre le fabricant, les experts, le secrétariat de l’ETS, les parties prenantes et, si nécessaire, l’ON pour clarifier les éléments scientifiques. À l’issue de cette étape, le rapport de consultation scientifique commune (CSC) est rédigé. Le document final est ensuite transmis par la Commission européenne au fabricant, marquant l’aboutissement du processus [13].
Tout ce processus est illustré dans la figure 6 qui représente le processus de la consultation scientifique commune et son articulation avec le Règlement (UE) 2017/745.
Figure 6 : Schéma du processus de Consultation Scientifique Commune (CSC) - source : auteurs
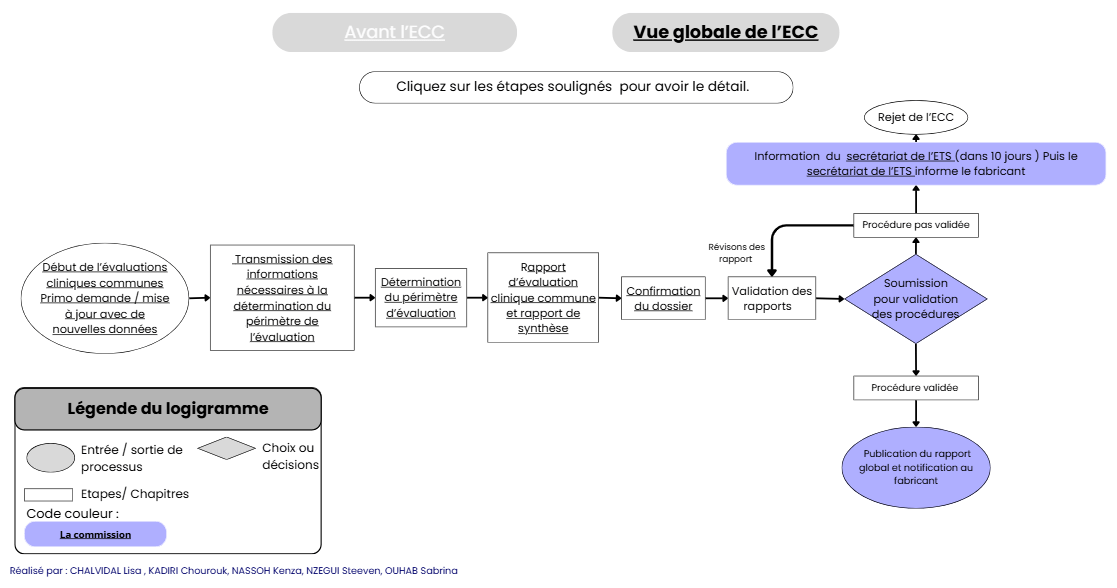
b) L’Évaluation Clinique Commune (ECC)
L’évaluation clinique commune est réalisée après l’obtention du marquage CE médical. Elle est réalisée afin de fournir une évaluation scientifique harmonisée et rigoureuse de la sécurité, des performances et des bénéfices cliniques d’un DM. Elle aboutit à la constitution de deux rapports, un rapport d’évaluation clinique et un rapport de synthèse, qui délivrent respectivement une analyse scientifique claire des données cliniques disponibles pour le dispositif évalué et du degré de certitude de ces données, et un résumé des points clés de l’ECC de façon claire et compréhensible (Cf. article 9 du Règlement (UE) 2021/2282) [3].
Les rapports ne contiennent aucune évaluation médico-économique, ni appréciation de la valeur thérapeutique de la technologie évaluée, et se limitent à une description de l’analyse scientifique (Cf. article 9 du Règlement (UE) 2021/2282 [3]. Même si une technologie de santé fait l’objet d’une évaluation clinique au niveau européen, les décisions relatives à la prise en charge, à la tarification et au remboursement restent de la compétence des autorités nationales de chaque État membre [2].
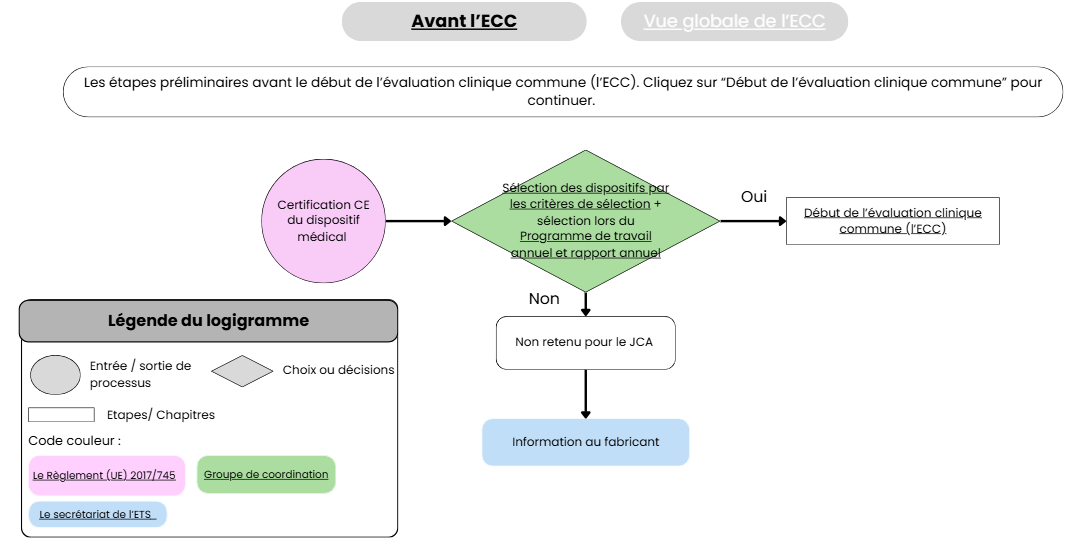
Le processus commence après la certification CE du DM, et s’il est sélectionné dans le programme de travail annuel de la Commission européenne et du groupe de coordination comme expliqué au point 2.1. Dans ce cas, le secrétariat informera le fabricant de la sélection de son produit pour l’ECC.
En revanche, si un dispositif n’est pas retenu pour une ECC, la décision est formalisée dans un avis de non-sélection, et est transmise par le secrétariat de l’ETS au fabricant de DM. Afin que celui-ci connaisse les raisons de la non-sélection et puisse, le cas échéant, améliorer ou compléter les données cliniques pour une éventuelle demande future [13].
Ce mécanisme garantit que seuls les dispositifs présentant un potentiel clinique significatif ou une innovation majeure font l’objet d’une évaluation coordonnée au niveau européen, tout en assurant une communication claire et structurée avec les fabricants des DM [7]. Ce processus est présenté par la figure 7, ci-dessous.
Figure 7 : Initiation de la procédure d’évaluation clinique- source : auteurs
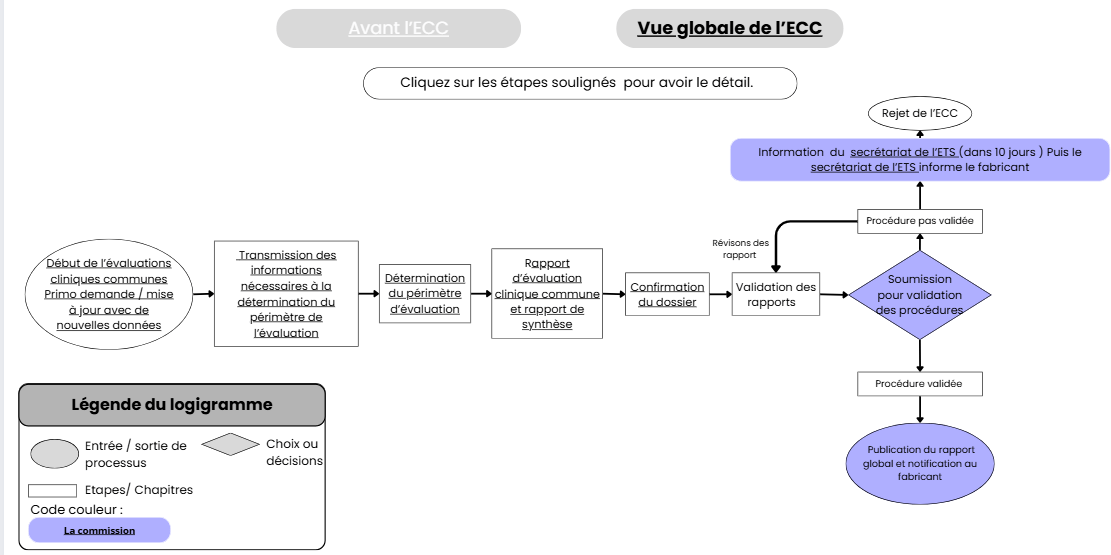
Le processus d’une ECC débute par la transmission, par le fabricant de DM, des informations nécessaires (Cf. article 3 du Règlement 2025/2086, règlement d’exécution relatif aux procédures détaillées applicables aux évaluations communes des DM et DM de diagnostic in vitro) à la définition du périmètre de l’évaluation [17]. Sur la base de ces éléments, les évaluateurs et les Co-évaluateurs vont mettre en place un sondage sous la forme PICO qui permet de définir précisément le périmètre de l’évaluation [14, 15]. Concrètement, PICO correspond à la Population concernée (P), l’Intervention ou l’usage du dispositif (I), le Comparateur retenu (C) et les principaux Résultats de santé attendus (O) [14, 16]. Par exemple, pour un dispositif de télésurveillance de l’insuffisance cardiaque, la question PICO pourrait être formulée ainsi : Chez des patients adultes atteints d’insuffisance cardiaque chronique (P), la télésurveillance par dispositif implantable X (I), comparée au suivi habituel en consultation (C), permet‑elle de réduire les hospitalisations pour décompensation et d’améliorer la qualité de vie (O) ? [15,16]...
Une fois que le ou les PICO ont été recueillis par l'enquête, l'évaluateur et le co évaluateur les convertissent en un ensemble de PICO qui définissent les données attendues par le fabricant. Par la suite, le secrétariat de l’ETS demande aux fabricants de fournir un certain nombre de justificatifs en fonction des PICO définis. Ces pièces sont détaillées dans l'Annexe I du Règlement (UE) 2025/2086 [17]. Toutefois, la majorité des informations demandées sont déjà présentées par le Règlement (UE) 2017/745 dans la description et spécification du DM, le plan et rapport d’évaluation clinique. Les informations non couvertes par le Règlement (UE) 2017/745 sont détaillées dans l’Annexe III [3, 17].
Après analyse des informations soumises par le fabricant, l’évaluateur et le co évaluateur rédigent le rapport d’évaluation clinique commune et le rapport de synthèse. Ces rapports sont ensuite soumis à la Commission Européenne, qui peut rejeter l’évaluation ou la soumettre à révision en cas de non conformité des procédures. En cas d’approbation, les rapports finalisés sont validés, puis publiés sur la plateforme informatique de l’ETS [17]. Il n’y a pas d’indications plus précises sur la partie de la commission chargée de cela.
L’ensemble de ce processus est illustré dans la figure 8, qui montre les différentes étapes à effectuer lors de l’évaluation clinique commune.
Figure 8 : Schéma du processus d'Évaluation clinique commune (ECC) - source : auteurs
2.5 Études pilotes : DM évalués dans le cadre du Règlement (UE) 2021/2282
L’EUnetHTA est un projet mis en place par l’UE afin de tester une méthode d’évaluation clinique des technologies de la santé commune afin de centraliser, harmoniser et mutualiser les évaluations entre les États membres. Sont présentés à titre illustratif deux DM soumis à une évaluation pilote réalisée dans le cadre du projet EUnetHTA 21 par un groupement d’agences d’évaluation des technologies de la santé, conformément au Règlement (UE) 2021/2282 [18].
a) Optilume® Urethral Drug-Coated Balloon (DCB)
L’Optilume® est un cathéter-ballon endoscopique recouvert de paclitaxel, molécule utilisée en chimiothérapie pour bloquer la prolifération des cellules cancéreuses. C’est un dispositif conçu pour dilater l’urètre et libérer localement le médicament par contact direct avec la paroi urétrale afin de réduire la récidive des sténoses [19].
Il est de classe III, donc dispositif à risque élevé pour le patient et est marqué CE. Il vise la population d’hommes adultes. L’Optilume® a été sélectionné dans le cadre du projet pilote EUnetHTA 21 en raison de son caractère innovant, de son usage implantatoire temporaire et de son potentiel impact clinique dans une pathologie fréquente et invalidante [19, 20].
Deux principales études ont été menées, à savoir l'étude Robust I sur 35 patients suivis pendant trois (3) ans, et l'étude Robust III sur 127 patients suivis pendant deux (2) ans. Les études ont témoigné d’une amélioration significative de la perméabilité urétrale, la réduction des symptômes, et la réduction de récidives, avec une sécurité acceptable par rapport à la dilatation standard de l’urètre. Toutefois, la durée du suivi restait limitée (2 à 3 ans) et nécessitait des données complémentaires. Globalement, l’ECC de l’optilume® reconnaît un bénéfice clinique modéré, c’est-à-dire une amélioration observée de l’état des patients, sans toutefois obtenir une amélioration radicale et exponentielle du pronostic de la pathologie. Bien que modéré, le bénéfice clinique obtenu reste pertinent par rapport aux alternatives existantes (stents urétraux, traitements constructifs comme l'urétroplastie, ou encore les traitements endoscopiques standards tels que la dilatation mécanique de l’urètre) justifiant son intégration dans la procédure d’évaluation clinique du Règlement (UE) 2021/2282 [20].
b) Système de stimulation de la moelle épinière Evoke® (SCS)
C’est un dispositif de classe III implantable actif. Ce neurostimulateur de la moelle épinière est capable d’ajuster automatiquement la stimulation électrique grâce à la mesure continue des potentiels évoqués (ECAP), permettant une thérapie en boucle fermée de la douleur chronique. Il vise les adultes et la sélection des patients éligibles nécessite une évaluation par un spécialiste de la douleur encore appelé algologue, un neurochirurgien ou un anesthésiste-réanimateur spécialisé en neuromodulation, et tient compte de certains critères, notamment la présence d’une douleur neuropathique chronique rebelle datant d’au moins 6 mois ou encore d’un échec des traitements conventionnels (médicaments, chirurgie par exemple). Il est commercialisé dans de nombreux pays Européen (relatif au marquage CE médical) et a été approuvé par la Food and Drug Administration (FDA) en février 2022 [21].
Le dispositif Evoke (Spinal Cord Stimulation-SCS) a également été sélectionné dans le projet pilote EUnetHTA 21 en raison de son caractère hautement innovant (premier système de stimulation de la moelle épinière à boucle fermée), de son impact potentiel important sur la prise en charge de la douleur chronique (la douleur neuropathique chronique touche environ 20 % des adultes européens, et entre 20 à 40 % des patients restent insuffisamment soulagés malgré les traitements standards cités précédemment), et de son statut de dispositif implantable de classe III. Les études ont été réalisées sur des patients adultes présentant une douleur neuropathique chronique réfractaire, c’est-à-dire ne répondant pas aux traitements standards, majoritairement lombosciatalgies post-chirurgicales et ayant une durée moyenne de la douleur comprise entre 3 et 8 ans. La douleur était mesurée principalement par échelle d’évaluation numérique de la douleur, échelle de 0 signifiant une absence de douleur à 10 correspondants à la douleur maximale [21].
Les évaluations ont montré que, comparé aux systèmes SCS en boucle ouverte précédents, le système Evoke produisait de meilleurs résultats. En effet, à 12 mois on pouvait observer que 82,3 % des patients Evoke® présentaient une réduction supérieure ou égale à 50 % du score d’évaluation numérique de la douleur contre 60,3 % pour le groupe SCS en boucle ouverte. Le bénéfice clinique persistant à 24 mois avec respectivement 74 % contre 51 % [20].
Le système Evoke® présentait aussi une meilleure stabilité de la stimulation. La stimulation se définit ici par la capacité qu’a le DM à maintenir un niveau de stimulation électrique constant, en dépit des mouvements qui altèrent la distance électrode–moelle. Les études ont montré que les ECAP restent dans la plage thérapeutique 89 à 99 % du temps avec Evoke® contre 35 à 40 % avec les systèmes en boucle ouverte conventionnels. Néanmoins, les organismes d'évaluation des technologies de santé dont la HAS recommandent de poursuivre la collecte de données en vie réelle compte tenu du caractère innovant de la technologie (boucle fermée basée sur les ECAP) et du niveau de risque associé à un dispositif implantable actif de classe III. Les conclusions du l’ECC soutiennent donc un intérêt clinique élevé, cohérent avec les exigences du Règlement (UE) 2021/2282 pour les dispositifs innovants à fort impact [21].
3. Enjeux du Règlement (UE) 2021/2282 sur l’évaluation des technologies de santé
L’application du Règlement (UE) 2021/2282 soulève des enjeux spécifiques pour chacun des acteurs concernés.
Pour les autorités nationales de santé, l’enjeu majeur est que chaque pays, sur la base de l'évaluation des technologies de santé, puisse se baser pour réaliser le remboursement des technologies de santé tout en ayant l’interdiction de demander des pièces supplémentaires déjà̀ présentes dans l’ECC. Pour les fabricants des DM, le Règlement (UE) 2021/2282 vise à réduire la duplication des preuves cliniques à fournir pour le remboursement du DM, ce qui pourrait se traduire par une potentielle réduction des coûts liés à la préparation de dossiers nationaux multiples et par un raccourcissement des délais d’accès au marché [3]. Pour les patients, l’harmonisation promise devrait se traduire par un accès plus rapide et plus équitable aux technologies innovantes, grâce à la diminution des délais nécessaires à l’évaluation clinique. Elle offre également une meilleure transparence sur les preuves scientifiques démontrant la sécurité et l’efficacité des dispositifs grâce à la centralisation des données cliniques [5]. À ce stade, ces gains restent des bénéfices attendus et n’ont pas été quantifiés de façon publique. Aucune estimation fiable et publiée n’était disponible au moment de la rédaction.
Pour l’UE, l’enjeu est double, d’un côté, il s’agit de renforcer la souveraineté sanitaire et de consolider un marché unique plus attractif pour les innovations médicales. De l’autre, il faut parvenir à un consensus politique entre 27 Etat membres aux contextes économiques et sanitaires variés, ce qui représente un défi important. Cela est réalisé par le groupe de coordination dont l'objectif est de réduire les divergences de jugement et de limiter les évaluations multiples, ce qui permettrait de gagner du temps et d’économiser des ressources humaines et financières. Cette harmonisation implique toutefois de surmonter des différences importantes dans les standards de soins, les pratiques d’évaluation et les systèmes de remboursement propres à chaque pays [13].
Le Règlement (UE) 2021/2282 vise à renforcer la coopération entre les EM en instituant une évaluation clinique commune des DM lors du processus de remboursement, appuyé par productions scientifiques : ECC, CSC. Cette organisation permet de mutualiser l’expertise et d’améliorer la qualité scientifique des rapports d’évaluation cliniques. Cependant, cette coopération peut s’avérer complexe à mettre en œuvre, en raison des différences entre systèmes nationaux, des priorités parfois divergentes et de la coordination d’acteurs multiples dans des contextes variés. Ainsi ce nouveau règlement soulève donc des questions tant sur son application que sur son efficacité [3].
La question centrale de ce travail est de déterminer comment améliorer la compréhension et l’appropriation du Règlement (UE) 2021/2282 sur l’évaluation des technologies de la santé par les fabricants de dispositifs médicaux. Il s’agit également d’examiner si ce règlement introduit des exigences supplémentaires en matière de preuves cliniques par rapport au cadre établi par le Règlement (UE) 2017/745.
Chapitre 2 : Méthodologie de travail amenant à la réalisation du guide pour les fabricants de DM
2.1 Analyse de l’appropriation du Règlement (UE) 2021/2282 par les acteurs concernés
Une enquête de terrain a été menée auprès de personnes concernées ou intéressées par la mise en œuvre du Règlement (UE) 2021/2282. L’objectif de cette enquête était de recueillir les perceptions et les besoins concrets des professionnels vis-à-vis du Règlement (UE) 2021/2282, ainsi que leurs avis sur les opportunités, les risques associés à sa mise en œuvre et les stratégies ou recommandations permettant d’anticiper son intégration dans les pratiques de développement des DM. Les personnes sollicitées ont été identifiées selon leur expérience en affaires réglementaires. Au total, 25 personnes ont été contactées, issus principalement de services affaires réglementaires, qualité ou évaluation clinique de différents types d’organisations (fabricants de DM, filiales pharmaceutiques, cabinets de conseils et autorités d’évaluation (comme la HAS). Parmi eux, 4 ont répondu à notre sollicitation. Les quatre répondants exercent tous dans les affaires réglementaires et évoluent au sein de structures intervenant soit dans la fabrication de DM, soit dans l’accompagnement réglementaire des entreprises du secteur.
2.2 Outils et modalités de collecte des informations
Les entretiens ont été réalisés par appels téléphoniques et réunions à distance, après une présentation du projet et de notre démarche. Chaque entretien débutait par une brève présentation du contexte et durait en moyenne une quinzaine de minutes.
Exemples de questions posées :
Avez-vous des exemples de dispositifs médicaux qui, selon vous, entreront directement dans le périmètre d’évaluation prévu par le Règlement (UE) 2021/2282 ?
Quelle est votre perception générale de ce nouveau Règlement (UE) 2021/2282 ?
Pensez-vous que les entreprises du DM sont déjà bien préparées à cette nouvelle réglementation ?
Quelles seront, selon vous, les principales conséquences de ce règlement sur les entreprises du secteur de la santé ?
Quels aspects du Règlement (UE) 2021/2282 vous semblent encore difficiles à comprendre ou à interpréter ?
Selon vous, le règlement pourrait-il devenir un levier pour attirer des entreprises de santé étrangères vers le marché européen ?
Avez-vous des exemples de dispositifs médicaux qui, selon vous, entreront directement dans le périmètre d’évaluation prévu par le Règlement (UE) 2021/2282 ?
2.3 Constats initiaux sur la connaissance du règlement
Les résultats de notre enquête montrent que le Règlement (UE) 2021/2282 est récent et que les informations disponibles sont limitées, avec peu d’experts connaissant sa mise en œuvre concrète pour les DM. Certaines personnes interrogées, bien qu’ayant connaissance du règlement, n’ont pas pu fournir d’informations supplémentaires par rapport au texte officiel. De plus, la complexité réglementaire, notamment la nécessité de synchroniser le Règlement (UE) 2021/2282 avec le Règlement (UE) 2017/745, complique la compréhension et l’appropriation du texte. Ces éléments montrent qu’il est nécessaire de concevoir un guide interactif et pédagogique afin de faciliter la compréhension et l’appropriation du règlement.
2.4 Développement d’un guide pour soutenir l’appropriation du règlement (UE) 2021/2282
Sur la base des documents réglementaires disponibles sur le Règlement (UE) 2021/2282, et en s’inspirant des travaux de collègues de l’UTC qui avaient conçu un guide pédagogique interactif sous forme de diapositives groupe IDS251 [22], nous avons conçu un outil de forme similaire intégrant des logigrammes permettant :
- De visualiser les liens entre les deux règlements Règlement (UE) 2021/2282 et le Règlement (UE) 2017/745,
- De clarifier les acteurs impliqués (Commission européenne, groupes de coordination, EM, développeurs, etc.),
- D’expliquer les étapes de la CSC et de l’ECC, et les documents supplémentaires (comparés au Règlement 2017/745) à fournir par le développeur de DM lors de ces étapes.
Cette démarche a permis de construire un outil répondant aux besoins réels des utilisateurs, rendant la réglementation accessible même à ceux qui ne sont pas familiers avec le Règlement (UE) 2021/2282.
2.5 Cahier des charges du guide
Le guide doit répondre aux besoins identifiés lors de l’enquête en proposant :
- Une présentation simplifiée du Règlement (UE) 2021/2282, centrée sur les éléments jugés pas clairs par les professionnels interrogés.
- Des explications structurées et progressives, permettant à un utilisateur novice de comprendre les notions clés (CSC, ECC, rôles des acteurs, liens avec le Règlement (UE) 2017/745).
- Des parcours de lecture clairs, incluant des schémas ou étapes logiques pour faciliter la compréhension.
- Une organisation intuitive permettant de trouver rapidement les informations essentielles.
- Un format numérique interactif, permettant une navigation simple et une consultation aisée.
Ce travail pourra s’inscrire dans une ambition plus large : rendre le règlement plus accessible et compréhensible, faciliter l’appropriation par les fabricants de DM, et servir de base pour de futures formations ou outils interactifs.
Chapitre 3 : Présentation du guide d’appropriation du Règlement (UE) 2021/2282 par les fabricants de dispositifs médicaux
3.1 Vue d’ensemble du guide
Un guide d’application du Règlement (UE) 2021/2282 destiné aux fabricants a été créé, en mettant l’accent sur des recommandations applicables et adaptées aux réalités du terrain, ce guide vise à soutenir les fabricants de DM dans leurs démarches réglementaires, en les aidant à mieux anticiper, organiser et documenter leurs évaluations cliniques dans le cadre du Règlement (UE) 2021/2282. La figure 9 montre la page de garde du guide.
Figure 9 : Page de garde du guide pratique pour l’évaluation clinique des dispositifs médicaux (Règlement UE 2021/2282) – source : auteurs

La seconde page est dédiée à une présentation des fonctionnalités et des modalités de navigation (voir figure 10).
Figure 10 : Aperçu page d’accueil du guide d’appropriation du Règlement (UE) 2021/2282 - source : auteurs
3.2 Structure générale et navigation du guide
Le guide se compose de 5 volets qui sont présentés dans une barre de menu en haut de chaque diapositive. Dans cette barre menue, il y a aussi le bouton accueil qui est représenté par la maison et le bouton ampoule qui permet d’aller sur la page d’aide du guide. Ce guide est interactif, ainsi que de nombreuses cases sont cliquables afin d’en savoir davantage (voir figure 11).
Figure 11 : Interface interactive et navigation des volets du guide d’appropriation du Règlement (UE) 2021/2282 – source : auteurs

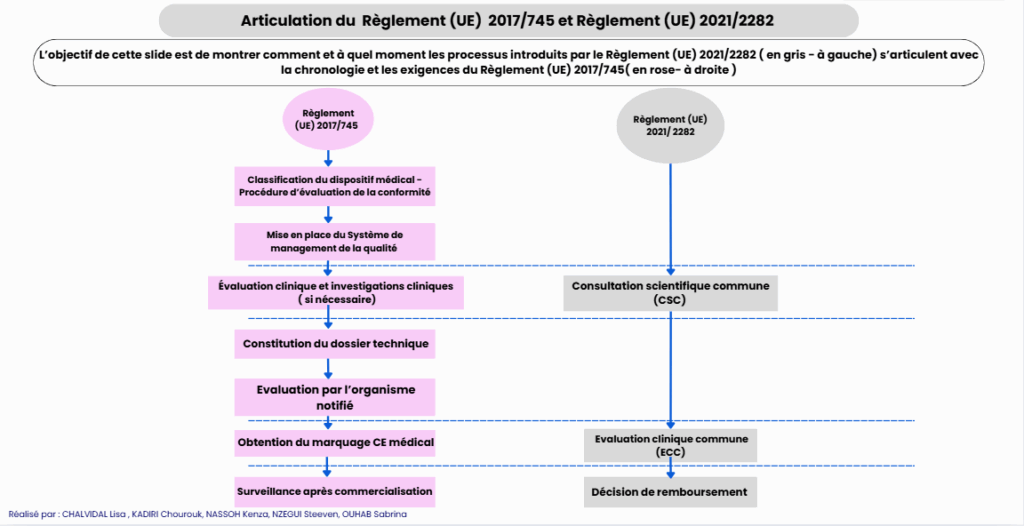
Le premier volet représente les étapes permettant l’obtention du marquage CE médical en synchronisant les processus de mise en conformité au Règlement (UE) 2017/745 ainsi qu’au Règlement (UE) 2021/2282 (voir figure 12 ci-dessous).
Figure 12 : Articulation des étapes d’obtention du marquage CE médical selon les Règlement (UE) 2017/745 et Règlement (UE) 2021/2282 - source : auteurs
Dans le second volet, (les acteurs) les principaux acteurs du Règlement (UE) 2021/2282 sont présentés (voir figure 13 ci-dessous).
Figure 13 : Volet du guide d’appropriation du Règlement (UE) 2021/2282 dédié à la présentation des principaux acteurs impliqués dans sa mise en œuvre - source : auteurs
Dans le troisième volet (Les dispositifs médicaux), le périmètre d’application et les critères de sélection des DM concernés sont expliqués et illustrés avec des exemples d'entreprises et de dispositifs qui peuvent rentrer dans ces critères (voir figure 14 ci-dessous).
Figure 14 : Volet du guide d’appropriation du Règlement (UE) 2021/2282 dédié à la présentation des critères de sélection des dispositifs médicaux - source : auteurs
Le quatrième volet présente les étapes du processus de consultation scientifique commune sous la forme d’un logigramme. Les documents à fournir par le fabricant au titre du Règlement (UE) 2021/2282 mais non exigés par le Règlement (UE) 2017/745 sont identifiés (voir figure 15 ci-dessous).
Figure 15 : Volet du guide d’appropriation du Règlement (UE) 2021/2282 dédié à la présentation du processus de Consultation Scientifique Commune - source : auteurs
Enfin dans le volet (Évaluation clinique commune), les étapes de l'évaluation clinique commune sont représentées sous la forme d’un logigramme et les documents à fournir par le fabricant de DM qui ne sont pas demandés par le Règlement (UE) 2017/745 sont identifiés (voir figure 16 ci-dessous).
Figure 16 : Volet du guide d’appropriation du Règlement (UE) 2021/2282 dédié à la présentation du processus d’Évaluation Clinique Commune - source : auteurs
3.3 Retours des premiers utilisateurs du guide
Afin d’évaluer la pertinence, ainsi que la facilité de navigation et d’utilisation du guide, celui-ci a été transmis, accompagné d’un questionnaire, à un panel de personnes travaillant dans le secteur des affaires règlementaires dispositifs médicaux.
Les avis des camarades de promotion ont été recueillis, en particulier ceux de la filière « Technologies biomédicales et Territoires de santé » qui ne possèdent aucune connaissance préalable de ce Règlement. Leurs retours permettent d’évaluer l’utilisabilité du guide par des personnes profanes, et ont surtout porté sur l’amélioration de la navigation au sein du guide et la précision de certaines informations, suggérant des ajustements mineurs pour rendre le document plus clair et accessible.
Le guide a été diffusé sur le réseau LinkedIn afin de recueillir les avis d’un panel composé de cadres travaillant dans les affaires réglementaires, de fabricants et de personnes profanes, où il a reçu un accueil encourageant sous forme de likes et de soutiens, ce qui témoigne de l’intérêt porté au projet. Bien que nous n’ayons pas obtenu de retours détaillés sur le contenu lui-même, cette diffusion a permis de donner de la visibilité au guide et de valoriser le travail réalisé. Il a également été transmis aux participants des entretiens réalisés, dans l’optique d’obtenir des retours et d’identifier d’éventuels axes d’amélioration.
Toutefois, le nombre de retours obtenus a été limité, avec quatre réponses reçues.
Conclusion
Cette étude met en lumière les exigences clés du cadre réglementaire européen pour la mise sur le marché des dispositifs médicaux (DM), en particulier la phase d’évaluation clinique selon le Règlement (UE) 2017/745, augmentée des spécificités requises par le Règlement (UE) 2021/2282, relatif à l’évaluation des technologies de santé, entré en vigueur le 11 janvier 2022 et appliqué à partir de 2026 pour les dispositifs médicaux. Elle souligne l’importance pour les fabricants concernés par le Règlement (UE) 2021/2282 d’anticiper dès les premières étapes de développement de leurs produits, les démarches liées aux Consultations Scientifiques Communes et aux Évaluations Cliniques Communes, afin de garantir la conformité réglementaire tout en optimisant la collecte de données probantes et la valeur clinique de leurs dispositifs.
Les entretiens menés auprès de professionnels du secteur ont mis en évidence un besoin clair d’outils pédagogiques accessibles pour comprendre et appliquer un cadre réglementaire encore récent et peu connu en Europe. Pour répondre à cette demande, l’élaboration de logigrammes explicatifs a été entreprise, sur la base d’une analyse détaillée des textes législatifs européens, afin de clarifier des étapes réglementaires souvent complexes et peu documentées dans la littérature.
L’analyse comparative des exigences des Règlement (UE) 2017/745 et Règlement (UE) 2021/2282 en matière d’évaluation clinique des DM, l’élaboration de logigrammes synthétiques sur les processus impliqués, ainsi que d’un guide pédagogique interactif dédié au Règlement (UE) 2021/2282, ont permis de répondre aux les points critiques rencontrés par les fabricants nécessitant un accompagnement ciblé.
Dans la mesure où l’application du Règlement (UE) 2021/2282 s’établit de manière progressive (jusqu’en 2030) et que la première liste de DM concernés sera publiée en 2026, cet outil pour servir de base aux promotions futures afin d’avoir les grandes lignes de ce nouveau règlement dans le cadre de leurs travaux, de l’adapter à des dispositifs concrets et, le cas échéant, de partager leurs résultats ou analyses avec les entreprises concernées. D’ici là, le guide reste un outil de référence pouvant être enrichi et ajusté, et il est prévu que, lorsque la liste complète sera disponible, les étudiants pourront bénéficier de retours plus nombreux et variés de la part des professionnels et des fabricants.
Références bibliographiques
[1] Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n°178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra
[2 ] Haute Autorité de Santé, « Parcours du dispositif médical en France », Haute Autorité de Santé. Consulté le : 13 décembre 2025. [En ligne]. Disponible sur : https://www.has-sante.fr/jcms/p_3213810/fr/parcours-du-dispositif-medical-en-france
[3] Règlement (UE) 2021/2282 du Parlement européen et du Conseil du 15 décembre 2021 concernant l’évaluation des technologies de la santé et modifiant la directive 2011/24/UE (Texte présentant de l’intérêt pour l’EEE) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, Bruxelles, déc. 2021. [En ligne]. Disponible
[4] Haute Autorité de Santé, « Règlement Européen sur l’évaluation des technologies de santé », HAS, Haute Autorité de Santé. Consulté le : 17 septembre 2025. [En ligne]. Disponible sur : https://www.has-sante.fr/jcms/p_3545447/en/reglement-europeen-sur-l-evaluation-des-technologies-de-sante
[5] Member State Coordination Group on Health Technology Assessment, « EU HTA Regulation for Patients and Clinical Experts », mai 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/document/download/40feaa77-58a6-4a4b-9a77-2fc032e284b3_en?filename=hta_20250516_co01_en.pdf
[6] Official journal of EU, « Commission guidance for the medical devices expert panels on the consistent interpretation of the decision criteria in the clinical evaluation consultation procedure (Text with EEA relevance) 2020/C 259/02 - Publications Office of the EU ». Consulté le : 17 novembre 2025. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52020XC0807(01)
[7] Directorate-General for Health and Food Safety, « Guidance for the selection of Medical Devices (MD) and In Vitro Diagnostic Medical Devices (IVD) for Joint Scientific Consultations (JSC) - Public Health ». Consulté le : 15 novembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/publications/guidance-selection-medical-devices-md-and-vitro-diagnostic-medical-devices-ivd-joint-scientific_en
[8] European Commission, « Implementation of the Regulation on health technology assessment - Public Health ». Consulté le : 17 novembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/health-technology-assessment/implementation-regulation-health-technology-assessment_en
[9] European Commission, « Mise en œuvre du règlement concernant l’évaluation des technologies de la santé - Public Health ». Consulté le : 13 décembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/health-technology-assessment/implementation-regulation-health-technology-assessment_fr
[10] Commission Implementing Regulation (EU) 2025/117 of 24 January 2025 laying down rules for the application of Regulation (EU) 2021/2282 with regard to the procedures for joint scientific consultations on medical devices and in vitro diagnostic medical devices », janv. 2025. Consulté le : 17 novembre 2025. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg_impl/2025/117/oj
[11] Haute Autorité de santé, Webinaire | Ajustements 2024 du référentiel et témoignages d’établissements de santé - 12/09/23, (15 septembre 2023). Consulté le : 5 avril 2024. [En ligne Vidéo]. Disponible sur : https://www.youtube.com/watch?v=L-TvrD5FDjo
[12] Directorate-General for Health and Food Safety, « Briefing document template for parallel HTACG/Expert Panels JSC for medical devices - Public Health ». Consulté le : 15 novembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/publications/briefing-document-template-parallel-htacgexpert-panels-jsc-medical-devices_en
[13] European Commission, « Évaluation des technologies de la santé. Synthèse du document : Règlement (UE) 2021/2282 concernant l’évaluation des technologies de la santé », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, juill. 2024. Consulté le : 23 septembre 2025. [En ligne]. Disponible sur : https://eur-lex.europa.eu/FR/legal-content/summary/health-technology-assessment.html
[14] J. Stornello, « Guides thématiques : Pratique factuelle : Poser une question clinique ». Consulté le : 30 novembre 2025. [En ligne]. Disponible sur : https://libguides.biblio.usherbrooke.ca/pratiquefactuelle/question_clinique
[15]G. Promé, « Évaluation clinique des Dispositifs Médicaux », Qualitiso. Consulté le : 30 novembre 2025. [En ligne]. Disponible sur : https://www.qualitiso.com/evaluation-clinique-dispositif-medical/
[16] Médecine Générale Travaux de Fin d’Études MGTFE, « La question de recherche : la méthode PICO ». Consulté le : 30 novembre 2025. [En ligne]. Disponible sur : https://mgtfe.be/guide-de-redaction/5-recherche-bibliographique/5-3-la-question-de-recherche-methode-pico/
[17] European Commission, « Commission Implementing Regulation (EU) 2025/2086 of 17 October 2025 laying down, pursuant to Regulation (EU) 2021/2282 on health technology assessment, procedural rules for the interaction during, exchange of information on, and participation in, the preparation and update of joint clinical assessments of medical devices and in vitro diagnostic medical devices at Union level. » 20 octobre 2025. Consulté le : 8 novembre 2025. [En ligne]. Disponible sur : https://eur-lex.europa.eu/eli/reg_impl/2025/2086/oj/eng
[18] I. Urbina, R. Adams, J. Fernandez, A. Willemsen, N. Hedberg, et A. Rüther, « Advancing cooperation in Health Technology Assessment in Europe : insights from the EUnetHTA 21 project amidst the evolving legal landscape of European HTA », International Journal of Technology Assessment in Health Care, vol. 40, no 1, p. e75, janv. 2024, https://doi.org/10.1017/S0266462324004689
[19] Laborie Medical Technologies, « Optilume® Urethral Drug Coated Balloon for the treatment of Anterior Urethral Stricture », Laborie – Urology Products. Consulté le : 17 novembre 2025. [En ligne]. Disponible sur : https://www.laborie.com/product/optilume-drug-coated-balloon-for-urethral-stricture-treatment
[20] European Network for Health Technology Assessment (EUnetHTA 21), « JCAMD001 Assessment Report – Optilume Urethral Drug-Coated Balloon », EUnetHTA 21, juin 2023. Consulté le : 13 novembre 2025. [En ligne]. Disponible sur : https://fr.scribd.com/document/725606960/EUnetHTA-21-JCAMD001-optilume-assessment-report-v1-1-2#content=query:JCA,pageNum:2,indexOnPage:1,bestMatch:false
[21] U.S. Food and Drug Administration (FDA), « Evoke Spinal Cord Stimulation (SCS) System – P190002 », FDA. Consulté le : 17 novembre 2025. [En ligne]. Disponible sur : https://www.fda.gov/medical-devices/recently-approved-devices/evoke-spinal-cord-stimulation-scs-system-p190002
[22] M. Bouramdane, K. C. Dzegang, H. Kadiri, C. C. Ngomeni Yanze, et H. Slika, « Mise sur le marché des dispositifs médicaux en Europe et aux Etats-Unis : Guide d’accompagnement pour les dispositifs médicaux logiciels de Classe II », Université de Technologie de Compiègne, janv. 2025. doi : 10.34746/IDS251.
Annexes
Annexe I : Tableau comparatif : Similarités avec la documentation du Règlement (UE) 2017/745
Annexe II : Points à développer pour le dossier d’information global
Annexe III : Données demandées lors de la constitution du dossier d’évaluation clinique et non demandées par le Règlement (UE) 2017/745
Extrait issu de l’Annexe I Règlement (UE) 2025/2086.
- 1.3. Synthèse [paragraphe 1, points j) à m) de l’annexe II du règlement (UE) 2021/2282]: Cette section fournit une brève synthèse du dossier en mettant l’accent sur le périmètre de l’évaluation tel que défini à l’article 8, paragraphe 6, du règlement (UE) 2021/2282 et partagé avec le DTS dans le cadre de la première demande de la Commission visée à l’article 10, paragraphe 1, du règlement (UE) 2021/2282 (ci-après le « périmètre de l’évaluation »). La synthèse comprend : a) la mention de tous les PICO pour lesquels aucune donnée n’a été fournie ; b) un résumé des données analysées (par exemple, mesures des effets avec précision statistique pour chaque résultat) en ce qui concerne le périmètre de l’évaluation, indiquant si les résultats étaient fondés sur des données probantes directes ou indirectes. Les données sont fournies séparément pour chaque PICO ; c) le degré de certitude concernant le ou les PICO.
- 2.2.3.Statut réglementaire du dispositif médical :Cette section : a) précise le statut réglementaire du dispositif médical dans l’indication prise en considération pour cette ECC en Australie, au Canada, en Chine, au Japon, au Royaume-Uni, aux États-Unis d’Amérique et dans d’autres pays, le cas échéant ; b) indique la date à laquelle le dispositif médical a été mis sur le marché de l’UE, le cas échéant ; c) détaille les programmes d’accès précoce/d’usage compassionnel en cours ou prévus dans les États de l’EEE ; d) précise si le dispositif médical a été certifié au titre du règlement (UE) 2017/745 pour d’autres indications que l’indication prise en considération pour la présente ECC. Les références des déclarations doivent être fournies. Le texte intégral des références doit figurer à l’appendice D.1.
- 2.3. CSC liée à l’ECC [paragraphe 1, point g) de l’annexe II du règlement (UE) 2021/2282]: Lorsque le dispositif médical a fait l’objet d’une CSC dans le cadre du règlement (UE) 2021/2282, la présente section explique tout écart par rapport à la proposition de production de données probantes recommandée. Les recommandations doivent être consignées à l’appendice D.8.
- 3. Périmètre de l’évaluation :[paragraphe 1, point j) de l’annexe II du règlement (UE) 2021/2282] Cette section : — reproduit le périmètre de l’évaluation dans le format partagé avec le DTS dans le cadre de la première demande de la Commission visée à l’article 10, paragraphe 1, du règlement (UE) 2021/2282 ; — identifie clairement tout PICO pour lequel des données n’ont pas été transmises et explique les raisons de cette omission
- 4.1. Critères de sélection des études envisagées pour l’ECC : Cette section précise les critères d’inclusion et d’exclusion pour les études à prendre en considération dans le cadre de la présente ECC compte tenu du périmètre de l’évaluation. Le DTS consulte, si elles sont disponibles, les orientations méthodologiques adoptées par le groupe de coordination conformément à l’article 3, paragraphe 7, point d), du règlement (UE) 2021/2282. Les spécifications relatives aux critères d’inclusion et d’exclusion sont fournies pour chaque PICO, le cas échéant.
- 4.2.2. Sélection des études pertinentes :Dans cette section est consignée la méthode de sélection des études pertinentes sur la base des résultats de la recherche d’informations effectuée en fonction des critères d’inclusion et d’exclusion définis à la section 4.1. Cette spécification est fournie pour chaque PICO, le cas échéant. Le DTS consulte le processus de sélection suggéré dans les orientations méthodologiques adoptées par le groupe de coordination conformément à l’article 3, paragraphe 7, point d), du règlement (UE) 2021/2282.
- 4.3.2. Comparaisons directes par méta-analyse par paire :Le protocole relatif aux synthèses de données probantes, y compris le plan d’analyse statistique pertinent, est fourni à l’appendice D.5.
- 4.3.3. Comparaisons indirectes : Le protocole relatif aux synthèses de données probantes, y compris le plan d’analyse statistique pertinent, est fourni à l’appendice D.5.
- 4.3.4. Analyses de sensibilité : Cette section décrit et justifie les méthodes utilisées pour toutes les analyses de sensibilité effectuées. Elle décrit la finalité vers laquelle tend l’analyse de sensibilité, ou le paramètre méthodologique auquel elle répond, ainsi que les hypothèses sous-jacentes.
- 4.3.5. Analyses des sous-groupes et autres modificateurs d’effets
- 5.1.7. Liste des études incluses globalement et par PICO : Cette section définit la liste des études incluses dans le dossier, qui servent de base à chaque PICO. Si aucune donnée probante n’est disponible pour une question PICO relevant du périmètre de l’évaluation, il convient de l’indiquer clairement dans le dossier (« Aucune probante fournie par le DTS ») et de fournir la justification appropriée
- 5.2. Caractéristiques des études incluses : Conformément au paragraphe 1, point m) de l’annexe II du règlement (UE) 2021/2282, la présente section fournit une vue d’ensemble, sous forme de tableau, de la conception de l’étude et de la population de l’étude pour toutes les études incluses dans le dossier afin d’aborder tout PICO. Des informations précises sont fournies sur : a) le type d’étude et sa conception ; b) la date et la durée de l’étude ; c) la population participant à l’étude, y compris les principaux critères d’éligibilité et lieux ; d) les caractéristiques de l’intervention et du ou des comparateurs ; e) les résultats de l’étude ; f) le cas échéant, la date d’arrêté des données ; g) la taille de l’échantillon ; h) les méthodes d’analyse. Les interventions mentionnées dans l’étude doivent être caractérisées et des informations sur le déroulement de l’étude (c’est-à-dire les délais de suivi prévus et réels par résultat) doivent être fournies. Les études incluses dans le dossier sont brièvement décrites. Une description détaillée de la méthodologie de l’étude doit être fournie à l’appendice A.
- 5.3. Analyse des données : Conformément à l’article 9, paragraphes 2 et 3, du règlement (UE) 2021/2282, la présente section présente les données analysées pour répondre à chaque question de recherche relevant du périmètre de l’évaluation. Les données sont ventilées par PICO. Elles sont présentées de manière à respecter les normes internationales en matière de médecine factuelle. Le DTS consulte, si elles sont disponibles, les orientations méthodologiques adoptées par le groupe de coordination conformément à l’article 3, paragraphe 7, point d), du règlement (UE) 2021/2282, et décrit et justifie tout écart par rapport à ces orientations. Cette section fournit également toutes les informations nécessaires pour évaluer le degré de certitude concernant le ou les PICO, en tenant compte des points forts et des limites des données probantes disponibles, y compris, sans toutefois s’y limiter, le risque de biais. Les informations détaillées sont fournies dans les appendices correspondants.