
IDS041 - Fabricants : Passage de la Directive 93/42 au Règlement Européen 2017/745 relatif aux dispositifs médicaux
DOI mémoire
https://doi.org/10.34746/eqf1-xp88Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs

KABBABI Kaouter 
NYAGAM KEMAJOU Olivier Donald 
MHAMDI Salma
Contacts
Citation
A rappeler pour tout usage : KABBABI Kaouter, NYAGAM KEMAJOU Olivier Donald, MHAMDI Salma « Fabricants : Passage de la directive93/42 CEE au Règlement Européen 2017/745 relatif aux dispositifs médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs Médicaux et Affaires Réglementaires (DMAR), Mémoire de projet, janvier 2020, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids041 ; https://doi.org/10.34746/eqf1-xp88
Article publié
Suite à ces travaux, un article a été publié : ID interne : 2021_07_idsap
Résumé
Le secteur des dispositifs dans l’union européenne passe par une période très importante. En effet la directive 93/42/CEE va être remplacé par le règlement 2017/745 relatifs aux dispositifs médicaux.
Ce rapport porte sur les évolutions réglementaires à prendre en compte pour la transition de la directive 93/42/CEE au règlement européen 2017/745 qui sera d'application obligatoire à partir du 26 mai 2020. Ce règlement va améliorer la traçabilité et la transparence pour l’ensemble des membres de l’Union Européenne.
Ce travail consiste dans un premier temps à identifier les nouveautés apportées par le règlement européen 2017/745 relatif aux dispositifs médicaux (DM), puis de proposer une démarche de transition qui va aider les fabricants à implémenter ce règlement, et enfin présenter l'outil d’autodiagnostic pour aider les fabricants à s’auto évaluer par rapport aux évolutions règlementaires du règlement.
Abstract
The device sector in the European Union is going through a very important period. Directive 93/42/EEC will be replaced by Regulation 2017/745 on medical devices.
This report deals with the regulatory developments to be taken into account for the transition from Directive 93/42/EEC to European Regulation 2017/745, which will be mandatory from 26 May 2020. This regulation will improve traceability and transparency for all members of the European Union.
This work consists first of all in identifying the novelties brought by the European regulation 2017/745 relating to medical devices (MD), then in proposing a transition approach that will help manufacturers to implement this regulation, and finally in presenting the self-diagnostic tool to help manufacturers to self-assess themselves in relation to the regulatory evolutions of the regulation.
Téléchargements

Fabricants : Passage de la directive 93/42 CEE au Règlement Européen 2017/745 relatif aux dispositifs médicaux
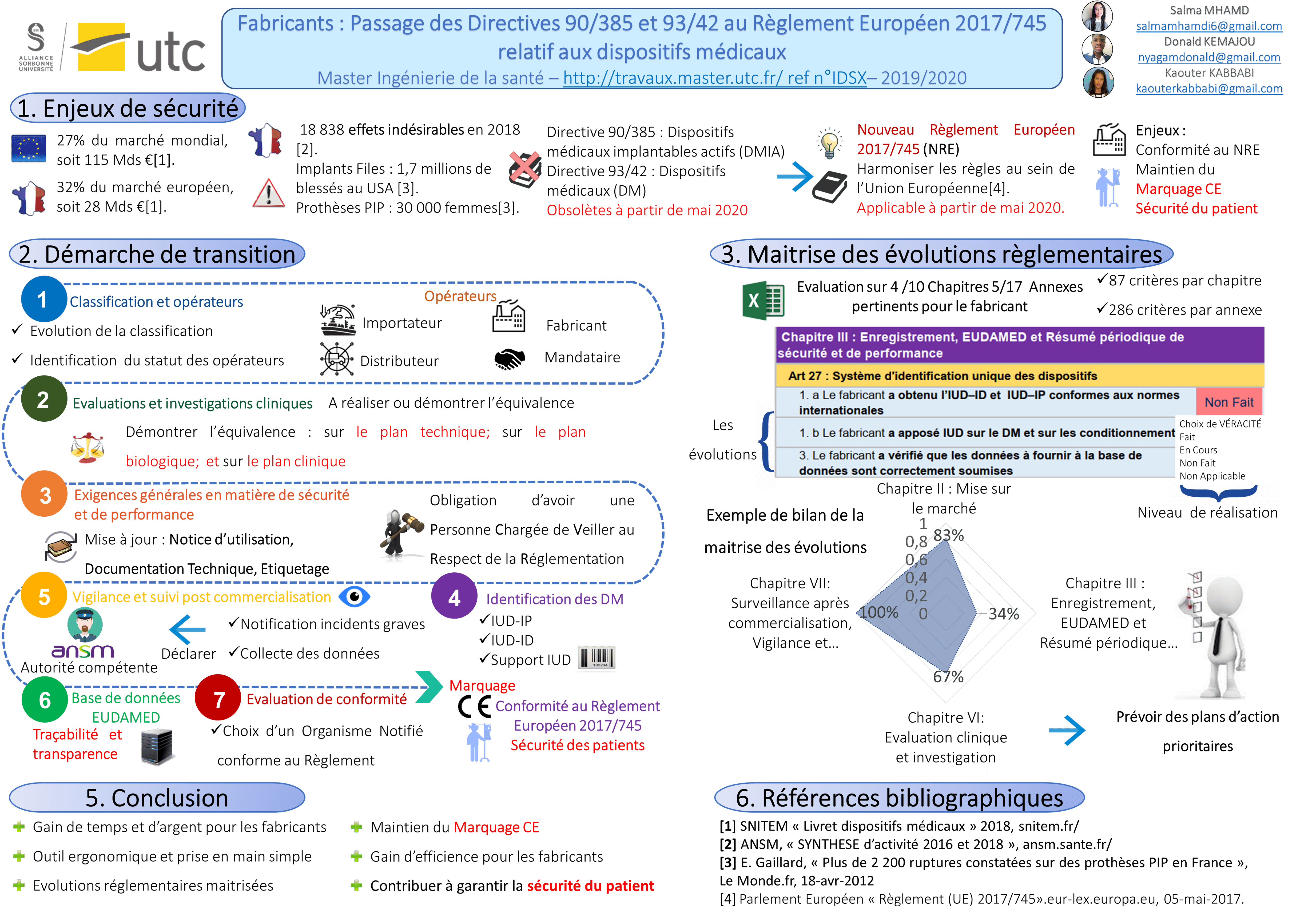
Fabricants : Passage de la directive 93/42 CEE au Règlement Européen 2017/745 relatif aux dispositifs médicaux

Maîtrise des évolutions entre les Directives 90/385 et 93/42 et le Règlement Européen 2017/745 relatif aux dispositifs médicaux (DM) et aux dispositifs médicaux implantables actifs (DMIA)
Mémoire complet :
Fabricants : Passage de la Directive 93/42 au Règlement Européen 2017/745 relatif aux dispositifs médicaux
Remerciements
L'élaboration de ce rapport a été le fruit de la contribution de plusieurs personnes que nous tenons à remercier.
Nous exprimons notre reconnaissance particulièrement à M Dan ISTRATE, Enseignant-Chercheur à l'Université de Technologie de Compiègne (UTC) dans le laboratoire Biomécanique et Bio ingénierie pour sa disponibilité, ses recommandations qui nous ont permis de mieux comprendre notre sujet.
Merci à M Gilbert FARGES, Enseignant-chercheur en génie biomédical et management de la qualité à l’UTC, pour sa disponibilité, ses explications et ses recommandations. Grâce à lui nous avons obtenu des informations nécessaires et des réponses pertinentes à nos questions.
Merci à nos Responsables de formation pour leur disponibilité et leurs recommandations tout au long du déroulement de ce projet. Grâce à eux nous avons eu l’opportunité de prendre contact avec les intervenants extérieurs pour des éclaircissements.
Merci à Kaouthar OUISSA, Rihab OMRANI, Silyana SALMI, Ghita MOUTIE, et Cédric Chrispy PETNTANG DJONGANG pour leur dynamisme, leur sérieux, et leur motivation tout au long de ce projet.
Enfin nous tenons à remercier tous les étudiants qui ont participé de près ou de loin à la réalisation de ce rapport notamment avec leurs remarques pertinentes qui nous ont permis de mieux étayer notre sujet.
Contexte et Enjeux réglementaires
Le marché européen des technologies médicales représente en 2017 environ 115 M€, soit 27 % du marché mondial. La France est le 4ème acteur mondial et le 2ème au niveau européen, derrière l'Allemagne et devant le Royaume-Uni. Le marché français des dispositifs médicaux s’élève à 28M€ du chiffre d’affaires, dont 8 milliards à l’export indique le Syndicat National de l'Industrie des Technologies Médicales (SNITEM) [1].
Cette activité très dynamique et innovante prend de l’ampleur avec près de 27 000 entreprises impliquées, soit 650 000 emplois au niveau de l’Union Européenne [2]. Durant ces dix dernières années, des scandales liés à certains dispositifs médicaux ont été relevés mettant ainsi en danger la sécurité des patients à l'échelle nationale et internationale. Il s’agit notamment des « Implant Files » qui ont causé aux Etats-Unis 82 000 morts, 1.7 millions de blessés, 3.6 millions de défaillances de 2009 à 2017 [3], et des prothèses « PIP » qui ont causé des dommages à 30 000 femmes en France [4], remettant ainsi en cause le système de traçabilité des dispositifs médicaux.
D’après l’agence nationale de sécurité du médicament et des produits de santé (ANSM), il y a une croissance des effets indésirables liés aux dispositifs médicaux, soit 15 961 de déclarations en 2016 [5], et 18 838 déclarations en 2018[6]. Les fabricants ont été imprudents face aux exigences règlementaires de la directive 93/42, c’est ainsi qu’elle sera abrogée au bénéfice du règlement 2017/745 relatif aux dispositifs médicaux.
Toutefois, il faut noter que la directive se distingue du règlement par sa transposition en droit national, contrairement au règlement qui entre en vigueur dès sa publication au Journal Officiel de l’Union Européenne (JOUE). Ceci étant l’objectif de la Commission Européenne à travers la mise en place de ce règlement qui va permet « de renforcer la sécurité sanitaire toute en favorisant l’innovation, et d’harmoniser l’application des règles sur le territoire de l’Union Européenne » [7].
Face à ce grand changement, les entreprises biomédicales conformes à la directive 93/42 doivent démontrer l’efficacité de leurs produits afin de satisfaire les exigences générales du règlement et d’assurer la sécurité des patients.
Chapitre 1 : Comparaison entre la directive 93/42 et le règlement 2017/745
I. Définitions et champ d’application
Le règlement européen 2017/745 relatif aux dispositifs médicaux « établit des règles concernant la mise sur le marché, la mise à disposition sur le marché ou la mise en service de dispositifs médicaux à usage humain et de leurs accessoires dans l'Union ».
Il s’applique à compter du 26 mai 2020 et abroge la directive 93/42. La définition du dispositif médical est étendue dans le règlement avec les termes manquants dans la directive. Ainsi, selon le règlement, un dispositif médical est « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article, destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes :
— diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
— diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
— investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
— communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus,
Et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens [7].»
Le règlement donne plus de précisions sur certaines définitions comme : accessoire d’un dispositif médical, d’un dispositif sur mesure et dispositif implantable. Il faut également noter qu’il y a 58 nouvelles définitions, dont dispositif falsifié, étiquette, notice d’utilisation, identifiant unique, nanomatériau, surveillance après commercialisation…
La Commission, après avoir consulté le GCDM détermine si un produit est considéré dispositif médical ou accessoire selon les définitions données par le présent règlement. En ce qui concerne, les produits incorporant des médicaments, des tissus et cellules humains, des biocides ou des produits alimentaires, la Commission consulte selon le cas l'Agence européenne des médicaments, l'Agence européenne des produits chimiques et l'Autorité européenne de sécurité des aliments.
Le règlement européen 2017/745 s’applique à tout dispositif proposé à une personne physique ou morale au moyen de services de la société de l'information, il s’agit d’une « société qui fait un usage intensif des réseaux d’information et de la technologie de l’information et possède une industrie de contenus diversifiée ». Il s’applique également aux dispositifs qui sont exploités pour un but commercial sans être mis sur le marché. §Article 6
Il est interdit de tromper l’utilisateur ou le patient avec des illustrations et signes, du texte, des noms, des marques, des images et des signes sur l’étiquette ou la documentation fournie, par exemple en donnant au produit, des fonctionnalités qu’il n’a pas, ou en ne mentionnant pas qu’il existe un risque lors de son utilisation.
II. Les opérateurs économiques
Désormais, pour un dispositif implantable, une nouvelle obligation s’impose, c’est la carte d'implant. Le fabricant doit obligatoirement fournir au patient les informations nécessaires à la bonne utilisation du DM, particulièrement son identification (nom, IUD, adresse, site web…) et sa durée de vie, les précautions et mesures à prendre et toute information qui assure la bonne utilisation du DM en toute sûreté.
Si un fabricant de dispositif n'est pas présent dans un État membre, il doit désigner un mandataire unique pour mettre son DM sur le marché européen. Le mandataire et le fabricant sont responsables des dispositifs défectueux. La désignation constitue le mandat du mandataire, elle n'est approuvable que si elle est acceptée par écrit par le mandataire.
Une copie du mandat doit être donnée à l’autorité compétente. Si le mandataire met fin à son mandat, il doit prévenir l’autorité compétente et l’organisme notifié en citant les causes de sa décision. Le changement de mandataire se fait par contrat entre le fabricant, si possible le mandataire sortant et le nouveau. Ainsi les obligations des opérateurs économiques mentionnées dans le règlement, sont illustrées dans le tableau ci-dessous.
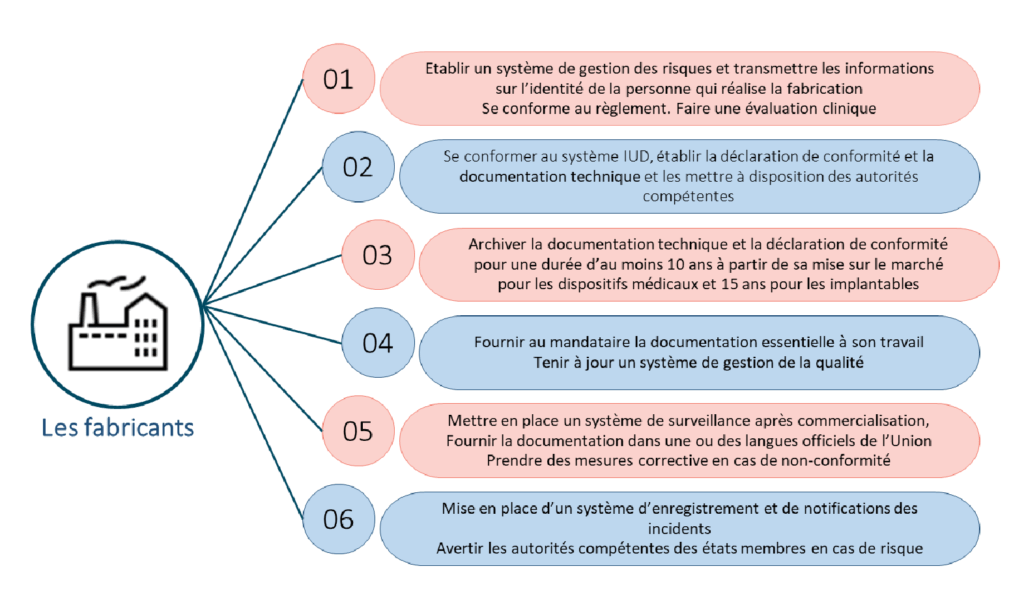
Figure 1 : Obligations du fabricant (source : auteurs et d'après [7])
Tableau 1 : Obligations de Mandataires, Importateurs, et Distributeurs (source : auteurs et d'après [7])
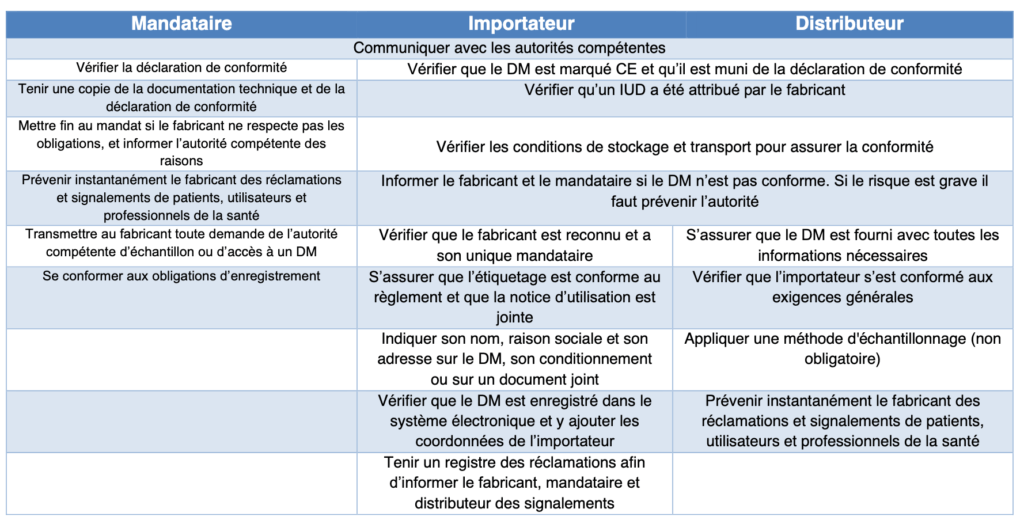
Les obligations des fabricants s'appliquent aux distributeurs et aux importateurs s’ils mettent à disposition sur le marché un dispositif médical sous leurs propres noms, leurs raisons sociales ou sous leurs marques ou modifient la destination du DM.
III. Personne chargée de veiller au respect de la réglementation
Le nouveau règlement apporte une nouvelle obligation pour les fabricants et mandataires non mentionnée dans la directive concernant une personne chargée de veiller au respect de la réglementation. En effet, il faut qu’il y ait au moins un employé chargé de la réglementation en vigueur, il doit soit :

Figure 2 : Formation du PCVRR (source : auteurs et d'après [7])
Les micros et petites entreprises peuvent ne pas recruter une personne qualifiée au sein de leur organisation, toutefois elles doivent dans ce cas externaliser cette obligation et faire appel à des boites de conseil.
Les missions principales de la personne chargée des affaires réglementaires sont les suivantes :

Figure 3 : Les missions du PCVRR (source : auteurs et d'après [7])
La personne chargée des affaires réglementaires ne subit aucune pression et obstacle pour le bon accomplissement de son travail. Dans le cas où l’entreprise dispose de plusieurs personnes chargées de la réglementation, le domaine et la responsabilité de chacun doivent être énoncés par écrit § Article 15 du REDM.
- Systèmes et nécessaires & Parties et composants
Afin de mettre sur le marché des dispositifs médicaux ayant la même destination et marqués CE sous forme d’un système ou d’un nécessaire, il faut établir une déclaration qui les assemblent. La vérification de la compatibilité et le conditionnement du système ou nécessaire doivent être assurés tout en fournissant les informations nécessaires aux utilisateurs.
« Toute personne physique ou morale qui met à disposition sur le marché un article spécifiquement destiné à remplacer une partie intégrante ou un composant identique ou similaire d'un dispositif défaillant ou usé afin de maintenir ou de rétablir la fonction du dispositif sans en altérer les performances, les caractéristiques de sécurité ou la destination, veille à ce que l'article ne compromette pas la sécurité et les performances du dispositif. Des pièces justificatives sont tenues à la disposition des autorités compétentes des États membres. » §Article 22
- Normes harmonisées
Le contenu de l’article 5 de la directive « renvoi aux normes » est différent de l’article 8 du règlement qui spécifie que s’il n'existe pas de normes harmonisées ou si celles applicables ne suffisent pas, après avoir consulté le GCDM la Commission peut déterminer des spécifications communes par rapport à l’évaluation clinique, la documentation technique, et les exigences relatives aux investigations cliniques.
- Dispositifs à usage unique et leur retraitement
Plus détaillés dans le règlement, il est précisé qu’il faut une autorisation par législation nationale afin de retraiter ou réutiliser les DM à usage unique. Conformément au règlement, toute personne physique ou morale qui retraite un dispositif à usage unique afin de le réutiliser dans l'Union est nommée « fabricant du dispositif retraité » et est considérée producteur.
Le retraitement se fait en établissant et en assurant les exigences relatives à la gestion des risques, la validation des procédures, la traçabilité, le système de gestion de la qualité et la déclaration des incidents.
IV. Le système IUD
Le Système IUD permet d’attribuer un identifiant unique à chaque dispositif. Il est composé de 2 parties :

Figure 4 : Système IUD (source : auteurs et d'après [7])
Le DM est inscrit selon le système IUD qui repose sur 4 concepts :
- La production d'un IUD comprenant le numéro Identifiant Dispositif et le numéro Identifiant Production ;
- L’apposition de cet identifiant sur l’étiquette du DM ou le conditionnement ;
- L’enregistrement de l'IUD par les opérateurs économiques, les établissements de santé et les professionnels de la santé ;
- L’intégration des IUD dans la base de données.
a. Le support IUD
Il s’agit d’un IUD en marquage clair (interprétation lisible des caractères d'information encodés dans le support IUD) et au format lisible par la machine qui utilise la technologie identification et saisie automatiques des données AIDC (cartes à puce, codes à barres). Ce support IUD doit apparaître sur l'étiquette, emballage ou sur le dispositif lui-même et sur chaque niveau de conditionnement supérieur. §Annexe VI partie C
- Les règles d’apposition des IUD à certains dispositifs
L’apposition des IUD sur certains dispositifs médicaux prévue dans le règlement figure dans le tableau ci-dessous :
Tableau 2 : Les règles d’apposition des IUD à certains dispositifs (source : auteurs et d'après [7])
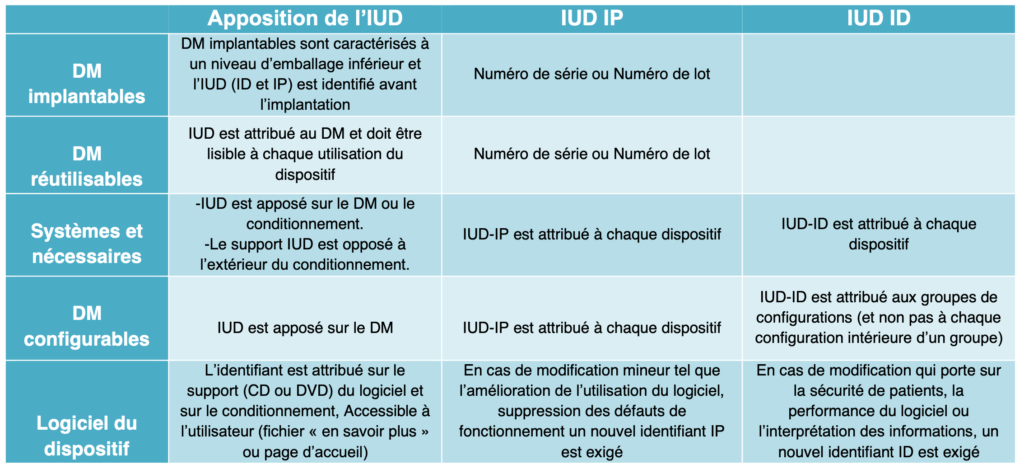
b. La base de données
Pour avoir une traçabilité du cycle de vie des dispositifs médicaux, le règlement 2017/745 a mis en place une base de données où l’enregistrement des DM est obligatoire : il s’agit de la base de données « EUDAMED ». Cette base de données est actuellement en cours de construction et sera accessible d’ici mai 2022[9]. Elle est présentée dans toutes les langues des états membre et les données sont enregistrées par le fabricant.
Cette nouvelle base de données EUDAMED permet aux autorités compétentes d’avoir des informations précises quant à la fabrication et la surveillance du dispositif en toute transparence, puisque cette base comportera tous les systèmes d’informations.
Cette base de données ne sera pas utilisée seulement par les autorités nationales compétentes et la Commission européenne, mais y accèderont aussi le groupe de coordination des dispositifs médicaux (GCDM), les organismes notifiés (ON), les opérateurs économiques (OE), des experts, des autorités compétentes non européennes et le public, incluant les établissements médicaux et la presse.
Les utilisateurs du système EUDAMED peuvent charger, extraire et modifier des données, ainsi que créer des rapports et des requêtes sur la base de données EUDAMED. Cette dernière est gérée par la commission (valide, rassemble, traite et mettre à disposition du public les données).
c. Informations à saisir pour enregistrer les opérateurs économiques et les dispositifs médicaux
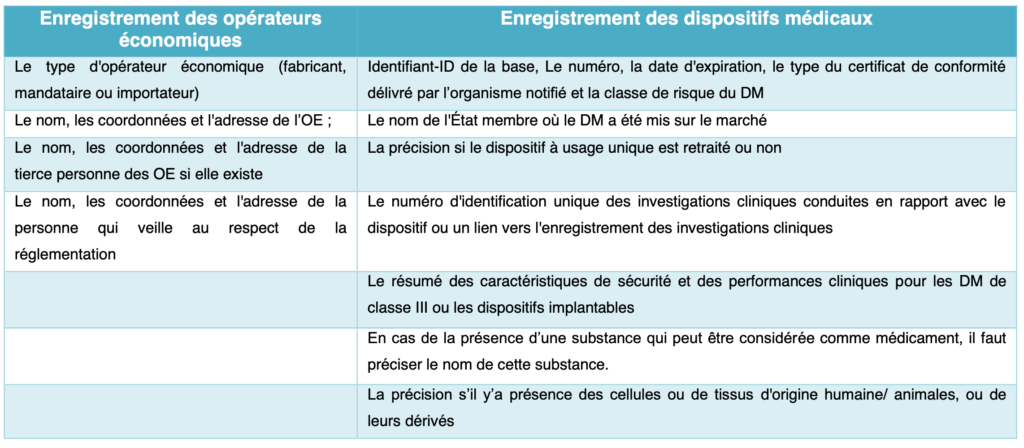
Le présent règlement désigne une nomenclature des DM internationalement reconnue et accessible gratuitement à d'autres parties prenantes.Le fabricant, le mandataire et l’importateur doivent s’enregistrer dans la base de données en fournissant des informations sur leurs coordonnés, mais aussi sur leurs dispositifs. Les différentes informations à enregistrer sont mentionnés ci-dessous :
Tableau 3 : Enregistrement des opérateurs économiques et des dispositifs médicaux (source : auteurs et d'après [7])
V. Organismes Notifiés
L’accès au marquage CE nécessite d’évaluer la conformité du dispositif médical aux exigences de mise sur le marché prévues par la réglementation. Cette évaluation sera réalisée par un organisme notifié qui est un organisme de contrôle indépendant, désigné par les Etats membres de l’UE pour réaliser les missions d’évaluation de conformité, de contrôle et d’essais. Ces organismes doivent respecter certains critères d’indépendance, d’intégrité, d’impartialité, de formation et de compétence.
Les organismes notifiés sont spécialisés dans des domaines dans lesquels ils fournissent leur savoir-faire et expertise afin d’évaluer la conformité d’un produit. D’autre part ils sont aussi en mesure d’évaluer des dossiers fournis par le fabricant et effectuer des audits du système qualité mis en place. Le fabricant est amené à vérifier si son ON couvre l’ensemble des DM et s’il est conforme au règlement 2017/745. La liste des ON est publié par la commission dans la base de données NANDO (New Approach Notified and Designated Organisations).
VI. Classification des dispositifs médicaux
§ Annexe VIII et article 51 du REDM vs Annexe IX et article 9 de la directive
Le règlement ajoute un groupe d’experts qui peut être consulté en cas de litige entre le fabricant et l’organisme notifié. La durée d’utilisation des dispositifs médicaux n’a pas changé dans les deux textes, il s’agit de :
- L’utilisation temporaire pour une durée de moins de 60 minutes en continu ;
- L’utilisation court terme pour une durée de 30 jours ;
- L’utilisation long terme pour une durée de plus de 30 jours.
Cependant, au niveau des règles de classification des dispositifs, le règlement propose 22 règles et 80 critères au détriment de la directive qui ne mentionne que 18 règles et 56 critères. Il existe donc 5 nouvelles règles de classification ajoutées par le règlement à savoir : § Annexe IX règle 11, et 19 à 22.

Figure 5 : Nouvelles règles de classification (source : auteurs et d'après [7])
La définition de peau ou muqueuse lésée apparait dans le règlement comme étant « une peau ou une muqueuse présentant une altération pathologique ou consécutive à une maladie ou à une blessure. » § Annexe VIII point 2.8, et l’affirmation qu’un dispositif est réputé permettre un diagnostic direct, lorsqu’il fournit lui-même le diagnostic de la maladie, des informations décisives pour l’établissement du diagnostic : chose qui ne figure pas dans la directive.
Une spécificité du règlement est la classification des accessoires d’un dispositif médical et d’un produit (Annexe XVI et Annexe VIII points 3.2). Cette classification est faite indépendamment des dispositifs avec lesquels les accessoires sont utilisés. Il apparait également la classification des logiciels (règle 11) :
Les critères ajoutés dans le règlement concernent :
- Les dispositifs non invasifs consistant en une substance ou mélange de substance et aux organes humains § règle 3 ;
- Les dispositifs implantables actifs, leurs accessoires, les implants mammaires, filets chirurgicaux, les prothèses articulaires totales ou partielles, les prothèses discales, et les dispositifs implantables entrant en contact avec la colonne vertébrale § règle 8 ;
- Les dispositifs actifs destinés à émettre des rayonnements ionisants à usage thérapeutiques, y compris les dispositifs qui les commandent, les contrôlent et qui agissent sur ceux -ci § règle 9 ;
- Les dispositifs actifs destinés à commander, à contrôler ou à agir directement sur les performances des dispositifs implantables actifs § règle 9.
VII. Evaluations cliniques et investigations cliniques
a. Evaluations cliniques
§ Article 61 + Annexe XIV section 1 du REDM vs Annexe X de la Directive
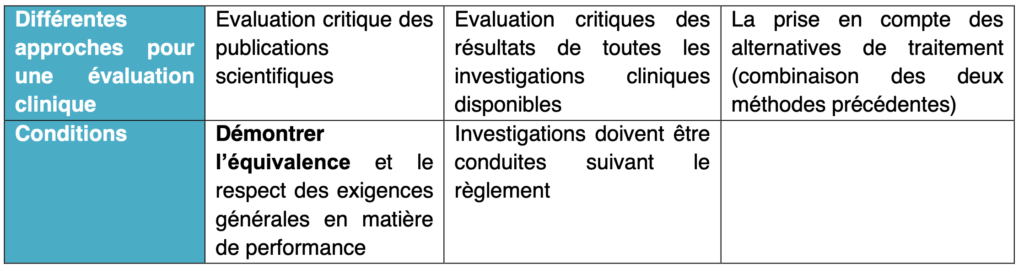
Les évaluations cliniques ont été renforcées dans le règlement, avec beaucoup de précision sur les performances à atteindre pendant l’évaluation clinique. Comme l’affirme les deux textes réglementaires, les évaluations cliniques sont fondées sur les données cliniques, sur l’évaluation des effets secondaires indésirables et sur le caractère acceptable du rapport bénéfice / risque. Néanmoins, dans le règlement il apparait le groupe de coordination des dispositifs médicaux qui joue le rôle de consultant en matière de stratégie de développement clinique pour les dispositifs de classe IIb et III. Concernant la démarche à suivre pour mener une évaluation clinique, le fabricant a trois choix possibles sous conditions. Ces conditions ont été renforcées et bien exprimées dans le règlement contrairement à la directive. Il s’agit de :
Tableau 4 : Approches pour établir une évaluation clinque (source : auteurs et d'après [7])
L’évaluation clinique peut se baser sur la notion
d’équivalence au dispositif en question à condition que les caractéristiques
techniques, biologiques et cliniques suivantes soient similaires (pas
différence significative en ce qui est la sécurité et les performances
cliniques) :

Figure 6 : Principe d'équivalence (source : auteurs et d'après [7])
Selon le règlement, les dispositifs n’ayant pas de destination finale ont l’obligation de démontrer l’existence d’un bénéfice clinique et la performance du dispositif. Pour les évaluations cliniques de ces dispositifs, elles doivent être fondées sur données pertinentes concernant la sécurité, et des données provenant de la surveillance clinique après commercialisation ou d’investigation clinique spécifique.
Des investigations cliniques ne sont pas conduites si les données cliniques existantes provenant d’un dispositif similaire sont justifiées. Les détails sur le contenu du plan d’évaluation clinique sont mentionnés dans le règlement. Les notions de rapport de SCAC, de plan de SCAC et de résumé des caractéristiques de sécurité et des performances cliniques pour les dispositifs implantables et de classe III sont également détaillées. Ces documents doivent être mis à jour de façon permanente durant tout le cycle de vie du dispositif.
b. Suivi clinique après commercialisation
Effectué sur la base d’un plan de suivi après commercialisation établit par le fabricant, Il s'entend comme un processus continu de mise à jour de l'évaluation clinique. Le fabricant collecte et évalue de manière proactive les données cliniques résultant de l'utilisation chez ou sur les humains d'un dispositif qui porte le marquage CE et est mis sur le marché ou mis en service conformément à sa destination.
Ceci étant dans le but de confirmer la sécurité et les performances pendant toute la durée de vie prévue du dispositif, d'assurer le caractère constamment acceptable des risques identifiés et de détecter les risques émergents sur la base d'éléments de preuve concrets.
Le fabricant analyse les résultats du SCAC et les documente dans un rapport d'évaluation du SCAC, qui fait partie du rapport sur l'évaluation clinique et de la documentation technique. Les conclusions du rapport d'évaluation du SCAC sont prises en compte pour l'évaluation clinique ainsi que pour la gestion des risques. Si le SCAC met en évidence la nécessité de mesures préventives et/ou correctives, le fabricant met en place de telles mesures
c. Investigations cliniques
§ Article 62 à 82 + Annexe XV du RDM vs Article 8 et 15 de la Directive
Comme évoqué dans les deux textes, toute investigation clinique doit se faire dans les conditions normales d’utilisation du dispositif. Elle doit garantir la protection des droits, de sécurité, de dignité, et du bien-être des participants. Ses objectifs sont les suivants :

Figure 7 : Objectifs d'une investigation clinique (source : auteurs et d'après [7])
L’investigation clinique se fait sur la base d’un protocole d’investigation, appelé plan d’essai dans la directive, où sont définies les affirmations du fabricant concernant la sécurité, la performance et les aspects relatifs au rapport bénéfice/risque.
Les dispositifs de classe III et implantables ont l’obligation de faire des investigations clinques. Sauf si :
Tableau 5 : Cas exceptionnels relatifs aux investigations cliniques (source : auteurs et d'après [7])

La notion de promoteur est évoquée dans le règlement. Ce promoteur a une grande responsabilité pour la bonne conduite des investigations cliniques. Quant à la notion d’investigateur dans le règlement, c’est le corolaire de médecin dans la directive. Le règlement met un accent particulier sur la fiabilité et la robustesse des données cliniques provenant des investigations cliniques.
En ce qui concerne les conditions à remplir pour conduire une investigation, elles sont similaires aux deux textes réglementaires. Mais le règlement spécifie et détaille toutes les informations à fournir aux participants de l’investigation clinique, ainsi que les informations sur les personnes vulnérables visées dans l’investigation clinique (les participants incapables, les mineurs, les femmes enceintes ou allaitantes etc…).
Contrairement à la directive, le règlement évoque les démarches à suivre en cas d’investigation clinique en situation d’urgence, et les informations sur la compensation de dommages (mise en place par l’Etat membre concerné par l’investigation). Les procédures relatives à la demande d’investigation clinique auprès des états membres et la méthode d’évaluation de ceux-ci sont très bien détaillées dans le règlement.
Cependant, Le promoteur doit transmettre aux états membres via le système électronique relatif aux investigations cliniques qui obtient un numéro d’identification unique à l’investigation toutes les informations nécessaires. Cette demande sera accompagnée de la documentation relative aux investigations cliniques. L’Etat membre concerné et le comité d’éthique valident la demande avant le début de l’investigation.
Pour les dispositifs médicaux implantables et de classe III, l’état membre notifie l’autorisation au promoteur de débuter dans un délai de 45 jours (60 jours dans la directive), voire 20 jours supplémentaires après consultation du groupe de coordination des dispositifs médicaux (GCDM).
Une nouvelle procédure énoncée dans le règlement est l’investigation clinique après commercialisation d’un dispositif déjà marqué CE. Les dispositions en termes de modifications affectant la sécurité des participants, ou la fiabilité des données cliniques sont également mentionnées, ainsi que les mesures à prendre en cas d’interruption temporaire ou anticipé d’une investigation clinique. Un délai d’au plus 12 jours est précisé dans le règlement concernant la mise à disposition les informations complémentaires demandées par un Etat membre concerné, au le promoteur.
La documentation relative aux investigations cliniques établie par le fabricant ou promoteur qui comporte le protocole de l’investigation clinique, la brochure de l’investigateur, et le formulaire de la demande, est minutieusement détaillée contrairement à la directive.
d. Système électronique relatif aux investigations cliniques
Le règlement prévoit pour les investigations un système électronique relatif aux investigations cliniques qui est en charge de :

Figure 8 : Système électronique relatif aux investigations clinique (source : auteurs et d'après [7])
Tous ces informations sont accessibles au public exceptés l’échange d’informations entre les états membres et la Commission. Le résumé et le rapport relatifs aux investigations cliniques sont mis à dispositif du public sauf en cas de confidentialité via le système électronique avant la mise sur le marché du dispositif et au plus tard lors de l’enregistrement du dispositif (dans le cas contraire alors l’accessibilité au public après un an). En cas d’arrêt anticipé ou interruption, ils sont accessibles immédiatement après leur transmission.
VIII. Exigences générales en matière de sécurité et de performances
§ Annexe I RDM vs Annexe I Directive
Dans l’ensemble, les exigences générales en matière de sécurité et de performances n’ont pas subi de grandes modifications, sauf pour les nouveaux dispositifs médicaux qui apparaissent dans la classification selon le règlement où il existe de nouvelles exigences à respecter. Comme le mentionne les deux textes, le dispositif médical conçu et fabriqué doit atteindre les performances prévues par le fabricant dans les conditions normales d’utilisation, adapté à sa destination, et doit présenter des bénéfices au patient au regard des risques.
Le règlement vient rajouter qu’il faut mettre en place un plan et un système de gestion des risques qui sont documentés et mises à jour en permanence. Il rajoute également l’évaluation de l’incidence (dangers, fréquence de répétition, estimation des risques, rapport B/R) des informations issues du système de surveillance après commercialisation. Pour donner suite aux résultats de l’analyse des risques, le fabricant doit si nécessaire former les utilisateurs sur la mise en œuvre des mesures de protection.
En ce qui concerne les dispositifs n’ayant pas de finalité médicale mentionnés dans le règlement, ils doivent présenter un risque nul ou un risque inférieur au risque maximum acceptable lié à l’utilisation du produit. Lors de la conception, selon les deux textes, le fabricant doit faire attention aux choix des matériaux, aux substances utilisées, et à leur compatibilité compte tenu de la destination du dispositif, et si nécessaire des recherches biophysiques validées.
- Les substances
Les deux textes obligent le fabricant à réduire les risques liés aux substances ou aux particules y compris les débris dus à l'usure, les produits de dégradation et les résidus de transformation, susceptibles d'être libérés d'un dispositif. Le règlement vient ajouter que les dispositifs ou les parties de dispositifs ou matériaux utilisés en contact direct avec le corps humain, ou destinés à introduire ou prélever un médicament, substance ou gaz dans le corps, ou destinés à transporter, stocker des médicaments ou gaz dans le corps ne contiennent les substances CMR pour la reproduction de catégorie 1A/1B, ou des substances PE, dans une concentration supérieure à 0,1% en fraction massique (m/m) que lorsque cela est justifiée.
Cependant, pour les dispositifs contenant des substances CMR et PE, une justification portant sur la présence de ces substances doit être documentée indiquent les deux textes. Le règlement fournit en plus les détails sur cette justification. Il s’agit d’une analyse et une estimation de l’exposition potentielle du patient, analyse des substances matériaux ou conceptions de substitution possibles, l’avis scientifiques des comités et les arguments expliquant l’inclusion des personnes vulnérables dans le traitement s’il y a lieu. Cette justification doit être disponible dans la notice d’utilisation comme dans la documentation technique.
De plus il faut réduire au maximum les risques associés à la taille et aux propriétés des particules qui sont libérées dans le corps du patient en ce qui concerne les nanomatériaux
- Les dispositifs contenant une substance considérée comme médicament et dispositifs composés de substances ou de combinaisons de substances qui sont absorbées par le corps humain.
Vérifier la qualité, la sécurité et l'utilité de la substance qui, utilisée séparément, est considérée médicament comme prévu dans la procédure d'évaluation de la conformité applicable du règlement. Pour les aspects ne relevant pas du règlement, ces dispositifs se conforment aux exigences applicables de l'annexe I de la directive 2001/83/CE, en ce qui est de l'absorption, la distribution, le métabolisme, l'excrétion, la tolérance locale, la toxicité, des interactions avec d'autres dispositifs, médicaments ou substances et les risques d'effets indésirables, comme le prévoit la procédure d'évaluation de la conformité du règlement.
- Les dispositifs contenant des matières d’origine biologique
Pour les dispositifs fabriqués à partir de dérivés de tissus ou de cellules d'origine humaine, non viables ou rendus non viables : le don, l'obtention et le contrôle des tissus, ainsi que des cellules sont conformes à la directive 2004/23/CE. Le système de traçabilité de ces dispositifs doit être complet et respecte les exigences en matière de traçabilité et de protection des données des directives 2004/23/CE et 2002/98/CE.
Pour les dispositifs fabriqués à partir de tissus ou de cellules d'origine animale, ou de leurs dérivés, non viables ou rendus non viables, en fonction de l'espèce animale, si possible, les tissus et cellules d'origine animale, ou leurs dérivés, proviennent d'animaux qui ont été soumis à des contrôles vétérinaires adaptés à leur utilisation prévue. Le fabricant doit conserver les documents sur l'origine géographique des animaux. Les dispositifs fabriqués à partir de tissus ou de cellules d'origine animale, ou de leurs dérivés, se conforme à l’exigence du règlement (UE) no 722/2012.
Pour les dispositifs fabriqués à partir de dérivés de tissus ou de cellules d'origine humaine, ou de cellules d'origine animale, non viables ou rendus non viables, les dispositifs fabriqués à partir de substances biologiques non viables autres, le fabricant garantit la sécurité (élimination des virus l’application de méthodes validées d’élimination) optimale du patient lors du traitement, la conservation, le contrôle et la manipulation de ces substances.
- Dispositifs ayant une fonction de diagnostic ou de mesurage
Les exigences appliquées aux dispositifs ayant une fonction de mesurage seront également appliquées aux dispositifs ayant une fonction de diagnostic. Il s’agit notamment de garantir l’exactitude et la précision, et la stabilité de ces dispositifs en tenant compte des méthodes scientifiques et techniques appropriées. Le fabricant doit également indiquer les limites de précision du dispositif.
- Protection contre les rayonnements
Les dispositifs émettant les rayonnements dangereux doivent désormais avoir dans leur notice d’utilisation des informations concernant les essais d’acceptation et de performances, les critères d’acceptation et procédure de maintenance. Ces dispositifs doivent également respecter les exigences de la directive 2013/59 Euratom fixant les normes de base relatives à la protection aux rayonnements ionisants, indique le règlement. Dans la mesure du possible, le fabricant doit choisir des méthodes qui réduisent l’exposition du patient et l’utilisateur aux rayonnements.
- Les systèmes électroniques programmables
Les fabricants de logiciels doivent respecter les exigences en matière de sécurité et des performances. Il s’agit de tenir en compte lors de leur fabrication et de leur conception des logiciels destinés à être utilisés en combinaison avec les plateformes mobiles (taille et contraste de l’écran), les caractéristiques spécifiques de celle-ci et des facteurs externes liés à leur utilisation (luminosité). Le fabricant doit également énoncer les exigences minimales concernant le matériel informatique, les caractéristiques des réseaux informatiques et des mesures de sécurité informatique (les habilitations).
- Les dispositifs actifs et dispositifs raccordés à des dispositifs actifs
Pour ces dispositifs, les deux textes exigent qu’ils doivent se munir d’un moyen de vérification de l’état de la source d’énergie interne dont dépend la sécurité du patient. Ces dispositifs doivent également garantir le niveau d’immunité intrinsèque contre les inférences électromagnétiques, de plus ils doivent disposer des limitations d’accès pouvant empêcher le dispositif de fonctionner, et les moyens pour éliminer ou réduire les risques résultant en condition de premier défaut.
- Protections contre les risques mécaniques et thermiques
Les erreurs susceptibles d’être commises pendant le montage ou remontage de certaines pièces, pouvant engendrer des risques, doivent être rendues impossibles par la conception, ajoute le règlement. Des indications apparaissent sur les éléments mobiles du dispositif ou son enveloppe lorsqu’il est nécessaire de connaitre le sens d’un mouvement pour éviter un risque.
- Protection contre les risques émanant des dispositifs destinés par le fabricant à des profanes
Une nouveauté du règlement, ce sont les dispositifs destinés à des profanes qui doivent fonctionner en tenant compte de l’aptitude et des moyens dont disposent les personnes visées. Ils doivent donner des informations facilitant la compréhension des profanes, garantir une utilisation correcte après une formation appropriée toute en réduisant les erreurs de manipulation et d’interprétation des résultats. Le fabricant doit également prévoir une procédure permettant au profane de vérifier les performances du dispositif et des avertissements en cas de résultat non valide.
- Informations figurant sur l'étiquette
L’étiquetage comporte en plus des informations reconnues par la machine (RFID ou code-barres), un support UID ou tout code permettant de tracer le dispositif. Pour les dispositifs composant des substances et combinaison de substances qui sont destinées à être introduites dans le corps humain par un orifice du corps ou par application sur la peau et qui sont absorbées par le corps humain ou dispersées localement dans celui-ci, la composition qualitative globale du dispositif et des informations quantitatives sur les composants principaux permettant d’obtenir l’action principale voulue.
Les informations signalant la présence de ces substances sont apposées sur le dispositif lui-même et/ou sur le conditionnement de chaque unité ou, s'il y a lieu, sur le conditionnement de vente pour les dispositifs contenant des substances CMR et PE.
- Les informations suivantes figurent sur le conditionnement stérile
Les informations figurant sur le conditionnement stérile ne sont pas mentionnées dans la directive. Il s’agit notamment :
- Des indications permettant de reconnaître le conditionnement stérile, et montrant que le dispositif est en état stérile, ainsi que la méthode de stérilisation ;
- Le nom, l’adresse du fabricant et la description du dispositif en question ;
- Dans le cas des dispositifs faisant l’objet d’une investigation clinique, la mention « exclusivement pour des investigations cliniques », de même pour les dispositifs sur mesure ;
- Le mois et l'année de fabrication, ainsi que la date d’expiration ;
- Des instructions indiquant qu'il convient de se reporter à la notice d'utilisation afin de savoir comment procéder lorsque le conditionnement stérile est endommagé ou involontairement ouvert avant utilisation.
- Informations figurant dans la notice d'utilisation
Les notices doivent désormais avoir un lien vers le résumé des caractéristiques de sécurité et des performances, une description des bénéfices cliniques escomptés, des informations permettant au professionnel de santé de vérifier l’adéquation du dispositif, le choix du logiciel et des accessoires adaptés. Le règlement ajoute également qu’il faut prévoir une « mention à l'intention de l'utilisateur et/ou du patient indiquant que tout incident grave survenu en lien avec le dispositif devrait faire l'objet d'une notification au fabricant et à l'autorité compétente de l'État membre dans lequel l'utilisateur ou le patient est établi ».
Pour les logiciels, les exigences minimales concernant le matériel informatique, les caractéristiques des réseaux informatiques et les mesures de sécurité informatique, ainsi que la protection contre l'accès non autorisé doivent être indiquées.
Les fabricants doivent prévoir des mises en garde et précautions des dispositifs qui sont composés de substances ou de combinaisons de substances, si nécessaire le profil général d'interaction entre le dispositif et les produits de son métabolisme et d'autres dispositifs, médicaments et substances. Le règlement mentionne également qu’il faut renseigner sur les contre-indications, les effets secondaires indésirables et les risques liés au surdosage. La composition qualitative globale du dispositif et les informations quantitatives sur les composants principaux permettant d’obtenir l’action principale voulue seront également mentionnées dans la notice.
Les informations étiquetées signalant la présence de ces substances sont apposées sur le dispositif lui-même et/ou sur le conditionnement de chaque unité ou, s'il y a lieu, sur le conditionnement de vente pour les dispositifs contenant des substances cancérogènes, mutagènes ou toxiques ou substances possédant des propriétés perturbant le système endocrinien. Pour les dispositifs destinés aux profanes, le fabricant indique dans la notice d’utilisation les circonstances dans lesquelles le profane doit consulter un professionnel de la santé.
XI. Documentation technique
§ Annexe II RDM vs Annexe VII point 3 Directive
- Description et spécification du dispositif, y compris les variantes et les accessoires
Par rapport à la directive, le règlement vient ajouter dans la documentation technique l’UID-ID, ou un code permettant la traçabilité d’un dispositif. Le fabricant doit également inclure dans la documentation la description, la liste complète des différentes configurations mises à disposition sur le marché produites par le fabricant et une présentation des dispositifs similaires sur le marché.
- Exigences générales en matière de sécurité et de performances
Le fabricant mentionne dans la documentation technique ou dans le résumé de la documentation, une référence précisant les documents fournissant les documents de preuves du respect de chaque norme harmonisée ou spécification commune.
- Vérification et validation du produit
Un plan de suivi clinique après commercialisation et un rapport d’évaluation de la SCAC sont intégrés dans la documentation. Pour les logiciels, le fabricant établit un résumé de vérification et de validation qui comprend la description du logiciel, sa conception, et son processus de développement, tel qu’il est utilisé dans le dispositif. Ce résume contient également toutes les configurations du matériel informatiques, et des différents systèmes d’exploitation.
- Les autres informations
Une mention dans le cas où un dispositif incorpore comme partie intégrante une substance. Cependant, la documentation indique la source de la substance et fournit des données et des essais effectués pour évaluer la sécurité, la qualité et l'utilité de la substance, compte tenu de sa destination du dispositif.
Une indication pour le dispositif incorporant comme partie intégrante des tissus ou cellules d'origine humaine ou animale, ou leurs dérivés, dont l'action est accessoire à celle du dispositif. Ainsi la documentation renseigne sur tous les matériaux d'origine humaine ou animale utilisés et fournit des informations détaillées relatives au respect des exigences applicables.
En ce qui concerne les dispositifs qui sont composés de substances ou de combinaisons de substances, des informations détaillées doivent être introduites notamment celles sur la conception, sur les protocoles d'essai (métabolisme, absorption, tolérance locale, toxicité génotoxicité etc…) ou sur les études complètes des méthodes d'analyses de données. En absence de ces études le fabricant doit se justifier.
Dans le cas des dispositifs mis sur le marché à l'état stérile, une description des conditions environnementales pour les étapes de fabrication et les rapports de validation (test de bio charge, des essais de recherche de résidus de stérilisation).
X. Dispositifs sur mesure et Dispositifs n'ayant pas de finalité médicale
§ Respectivement l’annexe XIII RDM vs Directive 8 et l’annexe XVI et article 61 RDM
a. Dispositifs sur mesure
Pour la déclaration à faire par la fabrication de dispositifs sur mesure, il doit ajouter des indications selon laquelle le dispositif contient ou incorpore une substance médicamenteuse, y compris un dérivé du sang ou du plasma humain ou des tissus ou des cellules d'origine humaine ou animale. La déclaration doit comporter également le nom et l’adresse de tous les lieux de fabrication, l’acronyme ou code numérique du dispositif. Comme affirmés par les deux textes, le fabricant documente sur les données acquises dans le cadre de la SCAC et met en œuvre des mesures correctives.
b. Dispositifs n’ayant pas de finalité médicale
Ces dispositifs se conforment aux spécifications communes prévues, et doivent conduire des investigations cliniques sauf si les données cliniques existantes sont suffisantes pour le dispositif similaire. Pour démontrer leur performance, ils doivent présenter un bénéfice clinique, et ont l’obligation de faire un suivi clinique après commercialisation. Lors de l’enregistrement des dispositifs, le fabricant doit préciser si la destination du dispositif est autre qu’une destination médicale, et leur notice d’utilisation doit avoir des informations concernant l’absence des risques liés à leur utilisation.
Il s’agit donc de : lentille de contact, les lasers et équipements à lumière intense (pour supprimer un tatouage, ou pour l’épilation), équipements destinés à la stimulation cérébrale (pour modifier l’activité neuronale du cerveau), équipements destinés à réduire les tissus adipeux ou modifier l’anatomie. Les produits pour les tatouages et piercings ne figurent pas parmi ces dispositifs. §Annexe XVI
XI. Surveillance après commercialisation et vigilance
a. Surveillance après commercialisation
Le règlement européen exige la surveillance après commercialisation (SAC) des DM. En se basant sur le type et la classe de risque d’un dispositif médical, les fabricants doivent concevoir, établir, documenter, appliquer, maintenir et mettre à jour un système de surveillance de leurs produits. Ce système doit être mis en place et actualisé afin de faire la collecte, l’enregistrement et l’analyse des données liées à la sécurité, la qualité et la performance des DM. Il faut ainsi établir un plan déterminé de surveillance afin de faire l’actualisation des points suivants : le rapport bénéfice/risque, l’évaluation clinique, l’étiquetage, les caractéristiques de sécurité, les besoins en termes de mesures de sécurité, et les mesures correctives et préventives.
b. La documentation après commercialisation
C’est un document qui doit être présenter de manière de manière claire et consultable. Il doit comporter un plan de surveillance après commercialisation et un PSUR.

Figure 9 : Le plan de surveillance après commercialisation , Source : Auteurs et d'après [7]
c. Vigilance
- Notification des incidents graves et des mesures correctives de sécurité
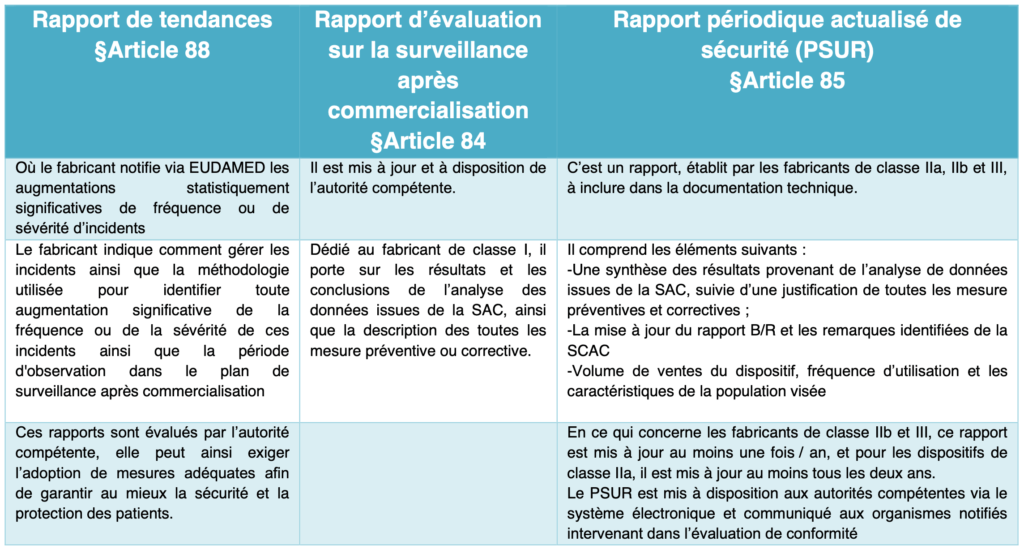
Le règlement oblige les fabricants des DM de notifier auprès des autorités compétentes les incidents graves et les mesures correctives, visant la sécurité avant leurs applications, liés aux produits mis à disposition dans le territoire de l’Union Européenne. La notification doit respecter les délais suivants :
- 15 jours au maximum après connaissance de l’incident pour le cas des incidents graves.
- 10 jours au maximum après connaissance de l’incident dans le cas de détérioration grave non attendue de l’état de santé du patient ou dans le cas d’un décès.
- 2 jours au maximum après connaissance de l’incident pour le cas d’une menace grave pour la santé publique.
Pour des cas d’incidents graves pareils ou déjà documentés, le fabricant peut se mettre d’accord avec les autorités compétentes en question pour transmettre à la place des rapports d’incidents des rapports de synthèse. Dans les autres cas, il faut présenter le rapport d’incident avec la procédure du suivi envisagée.
Ce règlement laisse au choix des états membres l’encouragement des professionnels, l’enregistrement national des déclarations reçues en termes de vigilance, l’information des fabricants par les déclarations reçues sur un incident grave.
Tableau 6 : Documentation relative à la surveillance après commercialisation (source : auteurs et d'après [7])
d. Analyse des incidents graves et des mesures correctives de sécurité
Après avoir notifié un incident grave, le fabricant doit mener le plutôt possible les investigations essentielles liées à cet acte et au dispositif en question. En coopération avec l’AC (autorité compétente) ou l’ON (organisme notifié) aucune investigation pouvant changer le DM n’est entrepris au point d’appliquer l’ensemble des évaluations sur les causes de cet incident.
Les états membres évaluent avec les fabricants ou les ON tous types d’information liés aux incidents graves ou aux mesures correctives survenant dans leur territoire. Lors de cette évaluation l’AC évalue les risques conséquents des incidents et de leurs mesures correctives en prenant en considération un certain nombre de critères. Pour toutes les mesures correctives appliquées par le fabricant, ce dernier est censé fournir les documents essentiels aux évaluations des risques sur demande de l’AC.
L’autorité nationale compétente fait le suivi des investigations faites par le fabricant. Elle peut même intervenir dans ces investigations.
Les conclusions des investigations sont à présenter sous forme de rapport aux AC via un système de la BDD EUDAMED. L’AC qui effectue l’évaluation communique aux autres AC les mesures correctives que les fabricants envisagent ou obligent à suivre.
Les utilisateurs doivent être informés des mesures correctives le plutôt possible. L’AC ou l’autorité compétente coordinatrice formule ses observations par rapport à cet avis qui doit être normalement uniforme pour tous les états de l’Union. Il s’agit d’un avis qui permet de bien identifier le dispositif en question à travers l’IUD et le numéro d’enregistrement délivré par le fabricant.
Il déclare aussi la raison des mesures correctives, le niveau de risque ainsi que les dispositions que doit prendre un utilisateur. L’avis en question doit être enregistré dans le système de la BDD EUDAMED et accessible par le public. Les fabricants sont informés des risques modifiant le rapport bénéfice/risque, pour qu’ils puissent prendre les mesures correctives nécessaires.
e. Système électronique relatif à la vigilance et à la surveillance après commercialisation
Les rapports traitant les incidents graves sont transmis via le système de la BDD (base de données) EUDAMED à l’AC de l’état membre en question, il s’agit des rapports de tendances et des mesures correctives visant la sécurité.
f. Confidentialité, protection des données et sanctions
§ Chapitre 9 RDM
Cette partie traite l’obligation de confidentialité de tous types d’informations comme la propriété intellectuelle, les données à caractère commercial et les résultats obtenus lors des audits, des inspections et des investigations par les autorités compétentes, les commissions et les organismes notifiés. La diffusion ou l’échange de ce type d’information n’est autorisé que sous accord entre les états membres ou les organismes concernés.
Le nouveau règlement décide de déléguer toujours la partie protection des données à la directive 95/46/CE et le règlement 45/2001.
Chaque état membre définit ses règles de sanction contre toute violation de ce règlement. Ces règles ou ce régime de sanction est à déposer par les états membres auprès de la commission avant le 25 Février 2020.
XII. Evaluation de conformité
§Annexe IX, X, XI et chapitre 5 section B du RDM vs §Annexe III, IV, V, VI et VII de la directive
Le fabricant doit, lors de la mise sur le marché d’un DM ou avant la mise en service d’un DM qui n’est pas mis sur le marché, faire une évaluation de conformité. Cette évaluation se fait toujours sur la base du système de gestion de la qualité accompagnée d’une documentation technique, ou sur la base d’un examen de type ou en encore sur la base de la vérification de la conformité du produit dans les deux textes réglementaires. Ces procédures sont mentionnées dans les annexes respectives IX, X et XI du règlement.
L’établissement de la documentation technique après commercialisation est l’un des documents à fournir lors de la déclaration de conformité des dispositifs de classe IIa. Il existe désormais dans le règlement au détriment de la directive une procédure d’évaluation de conformité pour les DM de classe I mis sur le marché à l’état stérile (Is), et également une procédure concernant les DM ayant une fonction de mesurage (Im) et les instruments chirurgicaux réutilisables (Ir) (§ article 52 point 7). La figure ci-dessus illustre cette procédure de conformité.
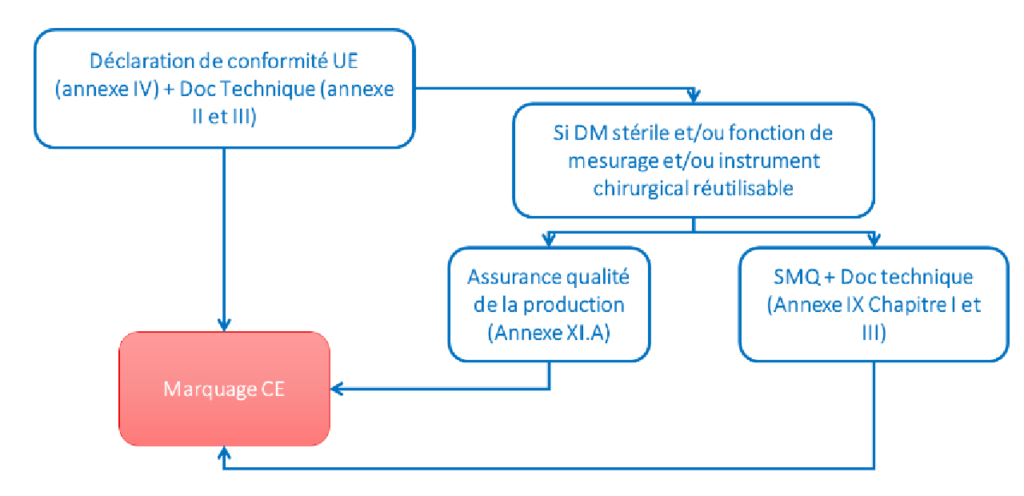
Figure 10 : Procédures d'évaluation de conformité pour les classe I spécifiques (Ir, Is, Im) , (source : auteurs et d'après [7])
De plus, l’intervention de l’organisme notifié est obligatoire lors de ces procédures d’évaluation de conformité concernant ces dispositifs. Des procédures à l’égard des dispositifs ayant un médicament comme partie intégrante, des dispositifs intégrant des tissus ou de cellules d'origine humaine ou animale, non viables ou leurs dérivés et des dispositifs composés de substances ou de combinaisons de substances destinées à être introduites / absorbées / dispersées par un orifice ou par application cutané, sont également prévues dans le règlement (article 52 point 9, 10, et 11).
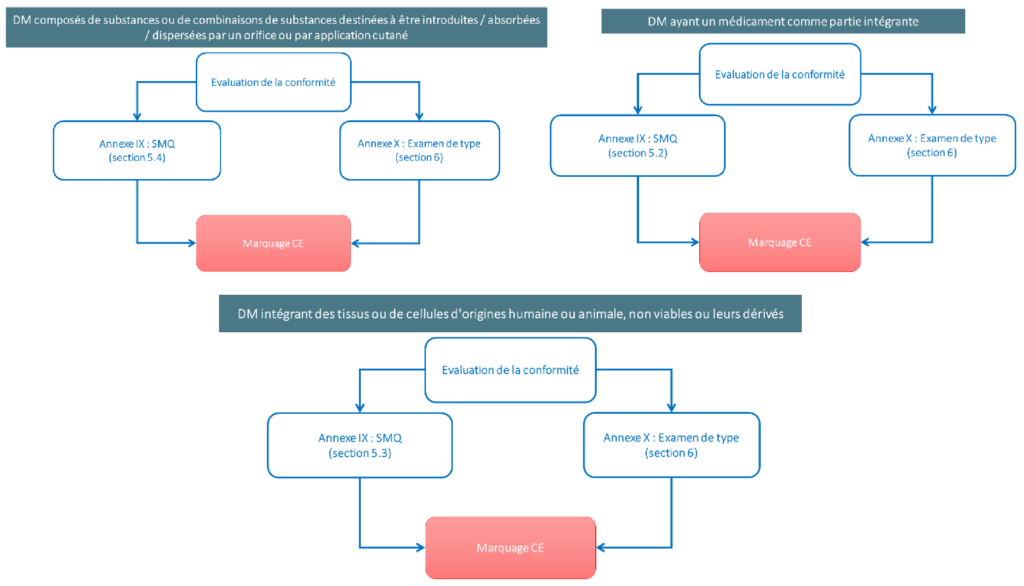
Figure 11 : Proécures d'évaluation de conformité des nouveaux dispositifs médicaux de classe III (source : auteurs et d'après [7])
Le règlement précise que lorsque la procédure d’évaluation nécessite l’intervention d’un organisme notifié, le fabricant introduit une demande unique auprès d’un organisme notifié habilité. En dehors des procédures d’évaluation pour les DM implantables de classe III et les DM actifs de classe IIb destinés à administrer dans l’organisme et/ou à retirer de l’organisme un médicament, une procédure d’évaluation dans le cadre de l’évaluation clinique doit être suivie par l’organisme notifié, soit conformément à l’annexe IX, section 5.1, soit par l’annexe X, section 6 qui sont des procédures dites spéciales. Ces procédures ne s’appliquent pas au DM en cas de renouvellement du certificat délivré au titre du règlement, également lorsque le dispositif a été conçu en modifiant un dispositif déjà commercialisé (aucune incidence sur le rapport B/R) par le même fabricant pour la même destination, et enfin si les principes de l'évaluation clinique pour le type ou la catégorie du dispositif font l'objet de spécifications communes pertinente, avec approbation de l’ON.
L’organisme notifié peut exiger aux fabricants d’entreprendre des études spécifiques de SCAC. Désormais, à l’opposé de la directive, les informations relatives aux certificats délivrés, suspendus, rétablis, annulés ou refusés, ainsi que les certificats assortis des restrictions seront rendus accessible au public. Un système électronique relatif aux organismes notifiés et aux certificats de conformité sera disponible. De plus, une procédure est prévue dans le règlement pour informer le fabricant qui désire changer d’organisme notifié. Ainsi il doit alors :

Figure 12 : Procédure pour changer un ON (source : auteurs et d'après [7])
En ce qui concerne le certificat de libre vente, il doit comporter l’IUD-ID de base du dispositif tel qu’il a été transmis à la base de données IUD, et également le numéro unique permettant d'identifier le certificat délivré par l'organisme notifié.
Les modifications aux niveaux des procédures d’évaluation de conformité sont les suivantes :
- Procédure d’évaluation de conformité sur la base d’un SMQ et d’une documentation technique (§ Annexe IX RDM vs Annexe II directive)
Dans la demande émise par le fabricant et soumise à un organisme notifié pour l’évaluation du système de gestion de la qualité, elle doit comporter de plus :
- Si le fabricant n’a pas son siège social dans l’Union, alors il renseigne les coordonnées de son mandataire (nom et adresse de son siège) ;
- Un projet de déclaration de conformité UE (visée à article 19 et annexe IV du RDM) du DM concerné ;
- Une description des procédures en place pour maintenir l’efficacité du SMQ ;
- Une description et documentation de SAC et si nécessaire de SCAC ;
- Des procédures garantissant le respect des obligations en matière de vigilance ;
- Une documentation relative au plan d’évaluation clinique et d’une description qui garantit la mise à jour en permanence du plan d’évaluation clinique, en prenant en compte l’état de l’art.
Un document d’orientation a été émis le 14 novembre 2018, illustrant la relation entre l’ISO 13485 et deux règlements 2017/745 et 2017/746[10].
Dans la documentation présentée pour l’évaluation de conformité du SMQ, le fabricant doit :
- Décrire le procédures et techniques pour la surveillance, la vérification, la validation et le contrôle, qui portent sur la stratégie mise en place pour respecter la règlementation, l’identification des exigences pertinentes, la qualification et le traitement d’équivalence et la classification, le choix et le respect des procédures d’évaluation de la conformité, une description de la gestion des risques et du suivi clinique après commercialisation, des solutions retenues pour respecter les exigences relatives à l’établissement de la notice utilisation et de l’étiquetage, et enfin ces procédures portent sur la gestions es modification de la conception ou du SMQ.
- Renseigner sur la traçabilité de l’étalonnage des équipements d’essai, et mettre à disposition aux organismes notifiés la documentation technique et celle relative au SAC
En ce qui concerne la surveillance des DM de classe IIa, IIb, et III, le fabricant fournit les informations pertinentes sur la documentation des conclusions et remarques ressortissant du plan de SAC et du plan de SCAC, pour un échantillon représentatif du DM accompagné de toutes dispositions sur la vigilance.
Cette évaluation de la surveillance des DM de classe III comprend un essai des pièces et des matériaux indispensables à l’intégrité du DM, au cas contraire la vérification que la quantité de matériaux et pièces achetées correspondent aux quantités de DM fini (cela en est de même pour la procédure d’évaluation de la conformité sur la base de la vérification de conformité du produit de l’annexe XI du REDM).
Des procédures spéciales sont prévues dans le règlement pour les DM de classe III et IIb ( §Annexe IX point 5), et également des procédures pour les nouveaux dispositifs incorporant une substance médicamenteuse, et incorporant des tissus ou des cellules d’origine humaine ou animale, ou leurs dérivés non viable.
De plus pour ce qui est des dispositions administratives, il n’y a pas eu de modifications majeures, mais toute fois les documents mentionnés dans ces dispositions doivent être accessibles aux autorités compétentes lorsqu’une entreprise fait faillite ou cesse son activité.
- Procédure d’évaluation de conformité sur la base d’un examen de type (§Annexe X REDM vs Annexe III de la directive)
Comme précédemment, la demande comporte en plus de la documentation technique, une documentation relative au SAC. Concernant le certificat, l’ON délivre un certificat d’examen UE de type comportant l’identification du fabricant, les conditions de validité et des conclusions de l’évaluation. Toutefois les documents mentionnés dans les dispositions administratives sont accessibles aux autorités compétentes pendant une période d’au moins 10 ans (5 ans dans la directive) après la commercialisation du DM, et avant la cessation d‘activités du fabricant.
XIII. Démarche à suivre pour implémenter le règlement 2017/745

Figure 13 : Démarche à suivre pour implémenter le règlement 2017/745 (source : auteurs)
Chapitre 2 : Outil d’autodiagnostic sur les écarts entre le Règlement Européen 2017/745 et la Directive 93/42
Pour aider les entreprises dans leur transition des directives 90/385 et 93/42 au règlement européen 2017/745, un outil d’autodiagnostic a été développé. Cet outil a été conçu sur l’Excel et permet aux fabricants de s’évaluer sur les écarts entre les deux textes réglementaires. Cependant, l’outil comporte 6 feuilles parmi lesquelles nous avons :
- {Mode d’emploi}
Cette feuille explique le fonctionnement de l’outil, décrit également le contenu de chaque feuille, et échelles d’évaluation choisies pour l’élaboration de l’outil. Chacune des exigences peut être évaluer suivant un taux de véracité de « 100% », « 35% », et « 0% », correspondant respectivement à « Fait », « En Cours », et « Non Fait ». Si une exigence n’est pas applicable à un dispositif faisant l’objet de l’évaluation, alors le fabricant peut choisir la véracité « Non Applicable », et ce sera pris en compte dans les résultats globaux de l’évaluation. Pour encourager l’utilisateur, le niveau de conformité aux articles ici est apprécié comme suit :

Figure 14 : Echelles d'évaluation (source : auteurs)
Etant un texte réglementaire, l’outil prévoit un niveau de preuve documentaire rigoureux avec deux niveaux de véracité « Complet » lorsque les toutes les preuves sont fournies (100%) et « Incomplet » en cas d’insuffisances de preuves documentaire (inférieure à 100%). Le fabricant pourra choisir « Dispositif implantable actif » ou « Dispositif médical » dans la cellule type de dispositif selon son besoin, et indiquer le nom proprement dit du dispositif.

Figure 15 : Mode d'emploi (source : auteurs)
- {Evaluation par chapitre} et {Evaluation par annexe}
Ces feuilles renferment respectivement les articles des chapitres et les exigences générales des annexes pertinentes pour le fabricant de dispositifs médicaux. Une fois qu'une exigence est satisfaite, il est nécessaire de mentionner le document de preuve qui prouve la conformité à l'exigence, ainsi que le référentiel utilisé (s'il existe) sur lequel le fabricant s'appuie pour démontrer la conformité.
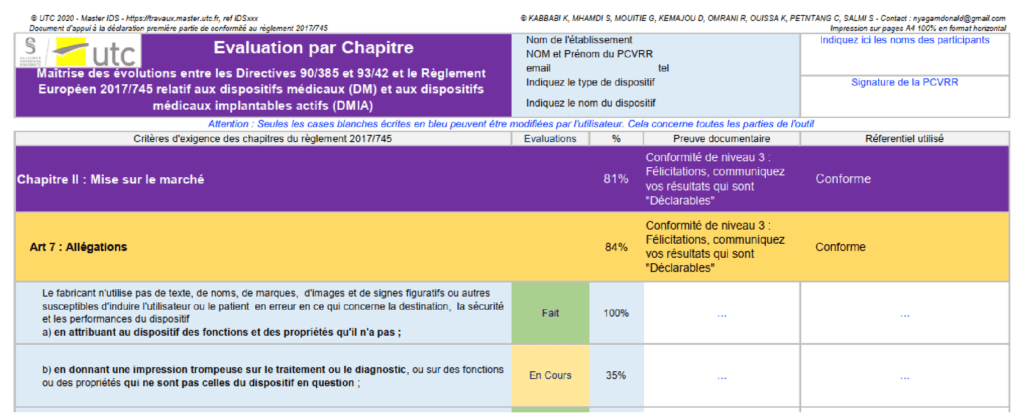
Figure 16 : Evaluation (source : auteurs)
- {Résultats globaux}
Grâce à un graphe radar, le fabricant visualise les résultats de son évaluation sur la base des chapitres et des annexes sur lesquels le dispositif a été évalué. Un tableau de synthèse et des zones d'élaboration des plans d'amélioration sont prévus. Dans ce cas, la conformité aux évolutions du « Chapitre III : Enregistrement, EUDAMED, et résumé périodique de sécurité et de performance » sont insuffisantes, il est donc nécessaire de prévoir des plans d’actions prioritaires pour maitriser cette évolution.
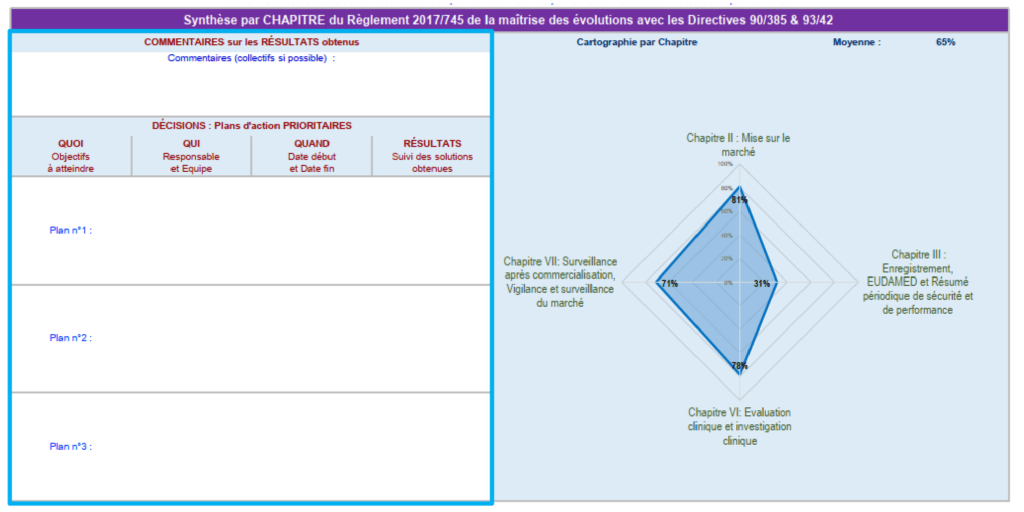
Figure 17 : Résultats globaux (source : auteurs)
- {Maitrise documentaire}
Une fois l’évaluation terminée, un bilan et une cartographie des preuves documentaires exigées par la réglementation sont présentés sur cette feuille. C’est une partie très importante car elle va permettre aux fabricants de suivre toute la documentation nécessaire pour l’implémentation du Règlement Européen. Une fois l’ensemble des exigences relatives à une documentation est respecté, une moyenne est calculée pour obtenir le pourcentage relatif à ce document.

Figure 18 : Maitrise documentaire (source : auteurs)
- {Déclaration ISO 17050}
L’auto déclaration de conformité qui est établit, datée et signée par un responsable interne à l’entreprise. Cependant, c’est uniquement lorsqu’une exigence a un taux de conformité supérieur ou égale à la valeur choisie par l’évaluateur qu’elle est jugée déclarable. Le niveau minimal « Déclarables » peut être personnalisé 50% à 90%. La déclaration reprendra toutes têtes de chapitre et des annexes, avec les pourcentages de conformité associés.

Figure 19 : Déclaration de conformité selon la norme ISO 17 050 (source : auteurs)
Conclusion
Le Règlement Européen 2017/745 impose de nouvelles obligations aux différents opérateurs économiques et aux organismes notifiés et apporte plus de précision que la Directive 93/42 afin d’assurer la sécurité du patient et la performance des dispositifs médicaux pour une prise en charge sûre et de qualité. Ainsi, tout fabricant doit démontrer l’efficacité de ses produits en mettant en œuvre les meilleures pratiques pour respecter les exigences réglementaires relatives aux dispositifs médicaux.
De plus, le règlement apporte une meilleure traçabilité et transparence pour garantir le suivi du dispositif médical durant tout son cycle de vie de la conception, passant par la livraison jusqu’à la réforme, ceci grâce à la base de données EUDAMED et l’enregistrement qui donne accès à différentes données.
L’outil d’autodiagnostic développé à cet effet permettra au fabricant de s’évaluer sur les évolutions entre la Directive et le Règlement afin de se conformer au règlement. Il permettra également aux personnes chargées de veiller au respect de la réglementation de gagner du temps, grâce à la prise en main simple et l’ergonomie de l’outil. A la suite de l’évaluation, cette personne qualifiée devra mettre en place des plans d’amélioration si nécessaires, et suivre les résultats obtenus afin de maintenir le marquage CE et garantir un niveau plus élevé de la sécurité du patient.
Références bibliographiques
[1] E. Leroy, « Le secteur des dispositifs médicaux », snitem.fr, 28-mai-2018. [En ligne]. Disponible sur : https://www.snitem.fr/le-snitem-en-action/les-publications/tout-savoir-sur-le-dispositif-medical. [Consulté le : 22-janv-2020].
[2] J. Doe, « Medical Devices », Internal Market, Industry, Entrepreneurship and SMEs - European Commission, 21-nov-2018. [En ligne]. Disponible sur : https://ec.europa.eu/growth/sectors/medical-devices_en. [Consulté le : 17-déc-2019].
[3] A. Lagadec, « Implants Files » : les 10 implants qui ont causé le plus d’incidents aux Etats-Unis », Le Monde.fr, ICIJ, 25-nov-2018. [En ligne]. Disponible sur : https://www.lemonde.fr/implant-files/article/2018/11/25/implants-files-les-10-implants-qui-ont-cause-le-plus-d-incidents-aux-etats-unis_5388419_5385406.html
[4] E. Gaillard, « Plus de 2 200 ruptures constatées sur des prothèses PIP en France », Le Monde.fr, 18-avr-2012. [En ligne] Disponible sur : https://www.lemonde.fr/implant-files/article/2018/11/25/le-manque-de-controle-des-dispositifs-medicaux-met-en-peril-la-securite-de-millions-de-patients_5388424_5385406.html
[5] ANSM, « SYNTHESE d’activité 2016 ». [En ligne]. Disponible sur : https://ansm.sante.fr/var/ansm_site/storage/original/application/efc41a539ed75517a2f3e1886ff64bc5.pdf. [Consulté le : 17-déc-2019].
[6] ANSM, « SYNTHÈSE D’ACTIVITÉ 2018 ». [En ligne]. Disponible sur : https://ansm.sante.fr/var/ansm_site/storage/original/application/1e7d5025d0ceefcf17a2be10892402a9.pdf. [Consulté le : 17-déc-2019].
[7] Parlement Européen, « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) ». Ed. JO L 117, http://data.europa.eu/eli/reg/2017/745/oj, 05-mai-2017. [Consulté le : 17-déc-2019]
[8] Parlement Européen, « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux ». Ed JO L 169, http://data.europa.eu/eli/dir/1993/42/oj, 12-juill-1993. [Consulté le : 17-déc-2019]
[9] « European database on medical devices (EUDAMED) | Internal Market, Industry, Entrepreneurship and SMEs ». [En ligne]. Disponible sur : https://ec.europa.eu/growth/sectors/medical-devices/new-regulations/eudamed_en. [Consulté le : 17-déc-2019].
[10] Editions Afnor, « FD CEN/TR 17223 : Document d’orientation sur la relation entre l’EN ISO 13485 :2016 (Dispositifs médicaux — Systèmes de management de la qualité — Exigences à des fins réglementaires) et le Règlement européen relatif aux dispositifs médicaux ainsi que le Règlement relatif aux dispositifs médicaux de diagnostic in vitro », 14-nov-2018. [En ligne]. Disponible sur : www.afnor.org. [Consulté le : 17-déc-2019].
[11] G. Promé, « Règlement Dispositifs Médicaux (UE) 2017/745 : guide de survie à destination des fabricants », Qualitiso, 14-mai-2017. [En ligne]. Disponible sur : https://www.qualitiso.com/reglement-ue-2017-745-guide/?fbclid=IwAR3LXDq4Vrsmav1Yi52VH0go6NGYwAm0SjjElQe6LP6GjQCsbvrdSpr5tFg
[12] ANSM, « Fiche du dégré de nouveauté d’un dispositif médical ». [En ligne]. Disponible sur : https://ansm.sante.fr/var/ansm_site/storage/original/application/569e94a19945e1f0eb6e6d0d0fff8c21.pdf. [Consulté le : 17-déc-2019].
[13] Commission européenne, « Le Guide bleu relatif à la mise en œuvre de la réglementation de l’Union européenne sur les produits », 2016. [En ligne]. Disponible sur : https://eur-lex.europa.eu/legal-content/FR/TXT/?uri=OJ:C:2016:272:TOC. [Consulté le : 17-déc-2019].