IDS297 – Stratégies et leviers pour accélérer l’intégration des outils numériques dans les Affaires Réglementaires des dispositifs médicaux
DOI mémoire
https://doi.org/10.34746/ids297Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur.e.s





Contacts :
- KENGNIE Diane Price Aout : dianekengnie05@gmail.com
- GUIHIA Hamza : hamzaguihia6@gmail.com
- BENCHAOUCH Hafsa : hafsafleur442@gmail.com
- NOHRA Sarita : saritanohra1@gmail.com
- OUHASSOU Oumaima : Oumaimaouhassou2003@gmail.com
A rappeler pour tout usage : D. KENGNIE, H. GUIHIA, H. BENCHAOUCH, S. NOHRA, O. OUHASSOU « STRATEGIES ET LEVIERS POUR ACCELER L'INTEGRATION DES OUTILS NUMERISATION DANS LES AFFAIRES REGLEMENTAIRES », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, janvier 2026, https://travaux.master.utc.fr/, réf n° IDS297, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids297/, https://doi.org/10.34746/ids297
Résumé
La numérisation des affaires réglementaires est devenue un enjeu central pour les fabricants de dispositifs médicaux face aux exigences accrues du Règlement (UE) 2017/745 (MDR) et du Règlement (UE) 2017/746 (IVDR). Ce mémoire analyse les principaux outils numériques utilisés dans ce domaine, notamment les ERP, PLM, eQMS, EUDAMED et l’intelligence artificielle, et leur rôle tout au long du cycle de vie du dispositif médical.
Une étude terrain, fondée sur des entretiens et un questionnaire auprès de professionnels des affaires réglementaires, met en évidence une adoption encore partielle de ces outils, en particulier au sein des PME. Les principaux freins identifiés concernent le coût de mise en place, le manque d’interopérabilité et les doutes liés à la conformité réglementaire.
Ce travail propose enfin des leviers concrets pour accélérer la digitalisation, reposant sur la formation, la sensibilisation et un accompagnement progressif. La numérisation apparaît ainsi comme un levier essentiel pour renforcer la conformité, la traçabilité et l’efficacité des processus réglementaires.
Abstract
The digitalization of regulatory affairs has become a key challenge for medical device manufacturers in response to the increasing requirements of the EU Medical Device Regulations (MDR/IVDR). This thesis analyzes the main digital tools used in regulatory affairs, including ERP, PLM, eQMS, EUDAMED, and artificial intelligence, and their role throughout the medical device lifecycle.
A field study based on expert interviews and a questionnaire highlights a still limited and uneven adoption of these tools, particularly among small and medium-sized enterprises. The main barriers identified are implementation costs, lack of system interoperability, and concerns related to regulatory compliance.
This work also proposes practical levers to accelerate digital integration through training, awareness, and progressive implementation. Overall, digitalization emerges as a key factor for improving compliance, traceability, and regulatory efficiency.
Téléchargements

Stratégies et leviers pour accélérer l'intégration des outils numériques dans les affaires réglementaires des dispositifs médicaux.

Mémoire Complet
Stratégies et leviers pour accélérer l’intégration des outils numériques dans les Affaires Réglementaires des dispositifs médicaux
Introduction
L’évolution des réglementations dans le domaine des dispositifs médicaux s’est opérée depuis plusieurs décennies. À l’origine, le département des affaires réglementaires gérait les dossiers réglementaires au format papier et traitait les demandes de manière artisanale. La demande des marchés s’ouvrant à l’international et en raison de la complexification des exigences qualité (ISO 13485:2016 et FDA 21 CFR Part 11), les fabricants ont dû acquérir de nouveaux outils. Dans le but de s’adapter aux exigences du règlement européen 2017/745, les entreprises du secteur des DM(Dispositifs médicaux) font en effet appel à différentes solutions digitales, dont l’ERP (Enterprise Resource Planning), le PLM (Product Lifecycle Management) et l’eQMS (Electronic Quality Management System) [1,2]. En centralisant la gestion de la qualité ou encore la gestion de la documentation et des processus, celles-ci permettent effectivement de diminuer les coûts liés aux non-conformités. Une étude Mckinsey de 2021 révèle que la transition vers un « système qualité digitalisé » fait gagner un temps significatif aux équipes qualité en automatisant les tâches documentaires et les vérifications manuelles [3]. En parallèle, la montée en puissance de l’intelligence artificielle (IA) bouleverse l’approche réglementaire, en ouvrant de nouvelles possibilités : automatisation de la production de documents d’expertise, anticipation des risques par des analyses prédictives, repérage même minimal des signaux de vigilance après la mise sur le marché, fluidification des échanges documentaires.
Sur la base des données publiées dans le rapport du marché Global Healthcare Compliance Software, édité par Spherical Insights en l’année 2024, le marché global des logiciels de compliance et réglementaires dans la santé a été évalué à 3,1 milliards USD dans le monde en 2023, avec une prévision de 9,25 milliards USD à l’horizon 2033, soit un taux de croissance annuel composé d’environ 11,55% entre 2023 et 2033 (10 ans) [4]. Dans ce contexte, la digitalisation des affaires réglementaires constitue un levier stratégique pour anticiper les évolutions réglementaires, intégrer les innovations numériques et garantir la conformité dans un contexte globalisé.
Ce projet a pour objectif d’établir un état des lieux des solutions numériques existantes et actuellement utilisées par l’industrie du dispositif médical pour suivre le cycle de vie d’un produit (de la conception jusqu’aux activités de suivi post-commercialisation). Il s’agira également de recenser les solutions numériques qui favorisent la traçabilité et la gestion multiservices des documents, afin de les rendre accessibles et intelligibles lors de la constitution de la documentation technique et du système qualité. Enfin, le projet vise à mettre en évidence les perspectives du secteur ainsi que l’impact de ces outils sur les activités des services qualité et affaires réglementaires, en analysant les pratiques actuelles et les pistes d’innovation attendues par l’industrie du dispositif médical.
Pour mener à bien ce projet, le mémoire contient trois chapitres : la première partie est centrée sur l'état de l’art des outils numériques existants, puis la deuxième partie sur l’état des lieux de la numérisation, et pour finir, la troisième partie sur les stratégies et leviers pour faciliter l’utilisation et l’implémentation des outils numériques en affaires réglementaires au sein des entreprises (les PME en particulier).
Chapitre 1 - La numérisation des affaires réglementaires (fondements, enjeux et acteurs clés)
1. Pourquoi la numérisation est-elle devenue incontournable ?
Conformément au Règlement européen (UE) 2017/745 (RDM), qui impose une documentation traçable et stricte depuis la conception du produit jusqu’à la surveillance post-commercialisation, le secteur des dispositifs médicaux connaît une transformation majeure. En effet, la traçabilité d’un produit nécessite la capacité de suivre précisément son historique, sa localisation ainsi que l’ensemble des modifications qu’il a subies.
Au-delà du respect réglementaire, cette exigence de traçabilité joue un rôle essentiel : elle permet à l’entreprise de réagir efficacement en cas d’incident, de réclamation ou d’audit, en identifiant rapidement l’origine du problème et les actions correctives à mettre en place. Pour les petites et moyennes entreprises (PME), qui constituent la majorité du tissu industriel, cet impératif représente un enjeu d’autant plus critique.
L’exigence accrue de traçabilité imposée par le RDM crée un défi majeur pour les fabricants, et l’ampleur de ce défi apparaît clairement à travers les chiffres disponibles. Selon une enquête menée en 2023 par 35 organismes notifiés européens (Notified Bodies), 14 539 demandes de certificats RDM ont été déposées, mais seuls 4 873 certificats ont été délivrés, laissant ainsi plus de 10 000 produits en attente [5].
Ce retard significatif illustre la difficulté des entreprises à répondre aux exigences documentaires, à assurer la cohérence des données cliniques et à démontrer la conformité de leurs produits.
Dans ce contexte, « Regulatory Science of Medical Devices » rapporte que 40 % des fabricants de dispositifs à risque moyen et 45 % de ceux à haut risque rencontrent des incohérences dans leurs données cliniques [6]. Les lacunes en gestion documentaire et en qualité des preuves cliniques deviennent alors particulièrement visibles, soulignant la nécessité d’outils permettant d’améliorer la fiabilité et la conformité du dossier technique.
Une étude RAPS/KPMG confirme cette tendance : 78 % des entreprises ne maîtrisent pas entièrement les exigences du RDM [7]. Ces résultats montrent que le risque de non-conformité demeure élevé. Pour y faire face, l’accompagnement des PME se révèle indispensable, notamment via des solutions numériques et des formations adaptées.
2. Les cinq outils de la transformation réglementaire
Pour répondre aux difficultés rencontrées par les PME dans la gestion documentaire, la conformité réglementaire et la mise en œuvre des exigences du MDR, plusieurs solutions numériques peuvent les accompagner efficacement. Il s’agit principalement de logiciels intégrés, tels que les ERP, les PLM et les eQMS, dont l’utilisation est fortement recommandée dans l’industrie des dispositifs médicaux pour structurer les données, centraliser les documents et automatiser les processus qualité. À cela s’ajoutent les technologies d’intelligence artificielle, également recommandées, qui viennent compléter ces outils.
Ces outils, bien documentés dans la littérature scientifique, contribuent à améliorer la fiabilité des informations, à renforcer la conformité réglementaire et à optimiser les processus internes, un enjeu majeur pour des structures souvent limitées en ressources humaines et techniques[8].
À l’inverse, EUDAMED n’est pas un outil interne mais une base de données européenne obligatoire pour le dépôt des informations relatives aux dispositifs médicaux. Bien qu’elle joue un rôle central dans la transparence et la surveillance du marché, elle ne doit pas être considérée comme une solution de gestion interne comparable à l’ERP, au PLM ou à l’eQMS.
Afin de mieux comprendre le rôle et la complémentarité de ces systèmes, il est nécessaire de préciser leurs spécificités et les besoins auxquels chacun répond.
a) Enterprise Resource Planning (ERP)
Un ERP est un système logiciel intégré qui centralise et automatise les principaux processus opérationnels d’une entreprise (production, stocks, approvisionnements, finance, ressources humaines, etc.). Il offre un référentiel de données unique, ce qui améliore la visibilité, réduit les redondances et facilite la planification. L’ERP est aujourd’hui considéré comme une solution de référence pour l’intégration et l’optimisation des processus internes [6].
b) PLM (Product Life cycle Management):
Le Product Life cycle Management désigne à la fois un ensemble de méthodes et un système logiciel visant à gérer toutes les données et processus associés au cycle de vie d’un produit, depuis sa conception, ses phases d’ingénierie, la fabrication, la validation, les modifications, jusqu’à sa fin de vie [9].
c) electronic Quality Management System (eQMS)
Un eQMS est la version numérique d’un système de management de la qualité (SMQ), qui automatise et centralise les processus qualité : contrôle des documents, gestion des écarts, actions correctives et préventives (CAPA), audits, formation, etc. Il permet une meilleure traçabilité, un contrôle des versions, des signatures électroniques et assure que les processus qualité sont exécutés, contrôlés et améliorés [10].
d) European Database on Medical Devices (EUDAMED)
EUDAMED est la base de données européenne mise en place par la Commission européenne pour appliquer le Règlement (UE) 2017/745 relatif aux dispositifs médicaux (MDR) et le Règlement (UE) 2017/746 relatif aux dispositifs médicaux de diagnostic in vitro (IVDR). Cette base de données vise à centraliser l’ensemble des informations relatives au cycle de vie des dispositifs médicaux sur le marché de l’UE : enregistrement des acteurs économiques, des dispositifs, des organismes notifiés, des certificats, des études cliniques, de la vigilance post-commercialisation et de la surveillance du marché [11]. EUDAMED est structurée autour de six modules interconnectés, dont certains sont déjà opérationnels sur une base volontaire et d’autres en cours de déploiement [11].
e) L’intelligence artificielle (IA)
L’intelligence artificielle désigne l’ensemble des techniques informatiques permettant à des systèmes de réaliser des tâches qui exigent normalement l’intelligence humaine comme apprendre, raisonner, résoudre des problèmes, interpréter des données ou prendre des décisions [12]. Dans le contexte des affaires réglementaires, l'IA est utilisée pour automatiser et optimiser divers processus, notamment la surveillance des évolutions réglementaires, la préparation des soumissions réglementaires, la gestion de la conformité et l'analyse des risques [13].
Par exemple la FDA (Food and Drug Administration) qui est l’agence américaine qui contrôle la sécurité des médicaments, des dispositifs médicaux et de nombreux produits de santé au Etat Unis, utilise depuis 2025 une intelligence artificielle appelée Elsa pour aider ses employés dans les tâches réglementaires. Elle sert concrètement à résumer les gros dossiers envoyés par les entreprises, vérifier plus rapidement les protocoles d’essais cliniques, repérer des problèmes de sécurité dans les rapports d’événements indésirables, comparer les notices et étiquetages proposés avec ceux déjà approuvés, et aider à décider quels sites doivent être inspectés en priorité. L’objectif est simplement de faire gagner du temps aux équipes, mais toutes les décisions restent prises par des experts humains. “FDA launches agency-wide AI tools to optimize performance for the American people” (2025) [14].
3. Adoption et impact des outils numériques et de l’IA
L'adoption de ces outils est devenue une pratique courante, qui se justifie par le grand impact stratégique de ces outils. Le marché mondial des logiciels ERP pour la fabrication de dispositifs médicaux est estimé à 2,59 milliards USD en 2024, et pourrait atteindre 5,1 milliards USD d’ici 2032, avec un taux de croissance annuel de près de 9% [15]. Cette augmentation montre que les sociétés et les entreprises reconnaissent l’intérêt économique et opérationnel qu’apportent ces systèmes numériques, intérêt qui se traduit par la réduction des erreurs, ou encore la création de processus plus sécurisés et d'une réglementation conforme. L'utilisation d’un PLM basé sur le cloud par les entreprises européennes a atteint 37%, et la part des PME dans ces déploiements a connu une augmentation de 22% en 2021 à 30% en 2023 . Ces chiffres montrent que l’intégration numérique est en train de devenir un levier concret d’efficacité et de compétitivité, particulièrement pour les structures moins dotées en ressources. De surcroît, l’intelligence artificielle s’ajoute à ces outils. De nouveaux défis se manifestent suite à son utilisation, couvrant l’interopérabilité entre les systèmes, l’acceptation organisationnelle (au sein de l’équipe), ou encore la sécurité et la confidentialité des données relatives aux normes de cybersécurité. Pour surmonter ces limites humaines et organisationnelles, l’IA, ainsi que d’autres outils numériques, semblent être les moyens les plus appropriés. Toutefois, ils nécessitent une intégration réfléchie pour générer réellement des bénéfices en termes de traçabilité, de conformité et d’efficacité. Au final, les statistiques précitées ne servent pas seulement à quantifier le problème : elles montrent les risques économiques et réglementaires, justifient le recours aux outils numériques et à l’IA, et permettent la mesure de l’impact potentiel de leur adoption sur l’efficacité, la conformité et la compétitivité des PME dans le secteur des dispositifs médicaux.
4. Les acteurs clés de la numérisation des affaires réglementaires
La numérisation des affaires réglementaires dans le secteur des dispositifs médicaux implique une collaboration coordonnée entre plusieurs acteurs internes et externes (Tableau 1). Chacun joue un rôle essentiel dans la conformité au Règlement européen 2017/745 (MDR) et dans l’intégration cohérente des outils numériques tels que l’ERP, le PLM, l’eQMS, EUDAMED ou encore l’intelligence artificielle. Cette transformation repose donc sur une architecture humaine, organisationnelle et technique complexe, où chaque acteur contribue à la qualité, à la traçabilité et à la sécurité des données réglementaires .
Tableau 1 : Les acteurs cibles d’utilisation des outils numériques [source : auteur.e.s]
| Acteurs internes | Acteurs externes |
| Direction des Affaires Réglementaires (RA): - Assure la conformité du dispositif médical. - Veille réglementaire MDR. - Prépare et met à jour la documentation technique. - Suivi du cycle de vie du produit. - Dépend de la qualité des données dans eQMS, ERP, PLM [7]. | Autorités compétentes : - Définissent le cadre réglementaire. - Supervisent la conformité. - Gèrent des plateformes comme EUDAMED pour centraliser les données [11]. |
| Département R&D : - Fournit les données techniques et cliniques nécessaires à la conformité : conception, tests, analyse des risques, modifications produit. - Utilise PLM pour structurer et tracer les données [9]. | Organismes Notifiés : - Évaluent la conformité MDR et délivrent les certificats CE. - Utilisent les portails numériques et dossiers électroniques pour accélérer les revues [5]. |
| Département Qualité (QA): - Gère la documentation, audits, CAPA, non-conformités et traçabilité [10]. | Consultants et Experts QARA : - Aide à mettre en place les outils numériques tels que ERP, PLM, eQMS. - Préparent les dossiers MDR et forment les équipes. -Facilitent la transition numérique [8]. |
| Département Informatique (IT) : - Intègre et maintient les outils numériques - Assure l’interopérabilité et la cybersécurité des données. - Effectue la validation logicielle (Computer Software Assurance) . | Fabricants de solutions numériques (SAP, Siemens, Oracle, Greenlight Guru): - Fournissent les outils pour automatiser la documentation et améliorer la qualité des données [3]. |
| Plateformes et bases réglementaires internationales : - Fournissent l’accès à EUDAMED, FDA CDRH, ClinicalTrials.gov, bases de vigilance. - Facilitent la transparence, la traçabilité et l’harmonisation des pratiques [11]. |
5. Enjeux de la Numérisation des Affaires Réglementaires
Du fait du renforcement de la réglementation européenne et des attentes accrues en matière de transparence, de sécurité et de traçabilité, la numérisation des affaires réglementaires dans le secteur des dispositifs médicaux connaît une évolution croissante et incontournable . L’intégration d’outils numériques dans ce domaine a le potentiel de transformer les processus liés à la conformité réglementaire. Dans un environnement externe impliquant les autorités compétentes, la numérisation contribue à optimiser les processus et à renforcer l’efficacité des échanges. Elle favorise également une harmonisation des pratiques réglementaires, un système plus prévisible et efficace, une standardisation des critères d’évaluation appliqués par les organismes notifiés, ainsi qu’une traçabilité simplifiée [16].Pour les organismes notifiés, les enjeux concernent notamment : la capacité à évaluer et traiter des dossiers numériques, la cohérence d’interprétation des données réglementaires et une amélioration de la structure documentaire, permettant un gain de temps dans la vérification des preuves d’exigence[17].
Du côté des fabricants, les technologies de numérisation documentaire présentent plusieurs avantages : elles permettent notamment de rationaliser les processus de mise en conformité et d’améliorer le time-to-market des dispositifs médicaux. Plusieurs études indiquent que l’automatisation de tâches répétitives (gestion documentaire, versioning, traçabilité) peut réduire significativement les délais de validation interne, selon le degré d’intégration du système utilisé. Ces gains restent variables d’une entreprise à l’autre, mais ils contribuent globalement à une meilleure structuration de l’approche réglementaire et à un suivi post-commercialisation plus efficace. Pour le public, cette transformation s’inscrit dans un objectif de transparence, notamment via l’accès facilité aux données réglementaires et l’alimentation de bases de données comme EUDAMED .
Ainsi, ces outils numériques deviennent des instruments essentiels pour sécuriser la conformité réglementaire et optimiser les processus, en particulier pour les PME, qui peuvent en tirer un avantage stratégique considérable.
6. Problématique du sujet
Aujourd’hui, de nombreuses entreprises, et surtout les PME, continuent de recourir à des systèmes papier pour assurer la traçabilité et la documentation nécessaires à leur conformité. Comme l’a souligné un expert du secteur lors de nos échanges : « Beaucoup d'entreprises aujourd’hui fonctionnent encore sur du papier, alors que ces outils permettent un gain de temps considérable et une mise en conformité rapide, facilitant ainsi l’arrivée sur le marché des produits. » C’est dans ce contexte que nous avons décidé d’explorer la problématique suivante :
« Comment accélérer l’intégration des outils numériques dans le secteur des affaires réglementaires des dispositifs médicaux au sein des PME, tout en garantissant la conformité et la traçabilité exigées par le règlement européen, et en permettant une meilleure compréhension des grands types d’outils mobilisables à chaque phase du cycle de vie du produit ? »
Chapitre 2 - Étude terrain sur la numérisation des affaires réglementaires
Afin de compléter l’état de l’art présenté dans le chapitre précédent, ce chapitre propose une analyse terrain visant à comprendre la maturité numérique actuelle des entreprises du secteur des dispositifs médicaux, en particulier les PME, ainsi que les pratiques, freins et attentes des acteurs en charge de la conformité réglementaire. Cette étude repose sur :
- Des entretiens réalisés avec des professionnels du domaine
- L’analyse des réponses d’un questionnaire adressé aux chargés d’affaires réglementaires
- Et un travail de réflexion mené en groupe pour identifier les causes profondes des difficultés rencontrées dans la digitalisation
Cette démarche permet de dresser un état des lieux réaliste et d’identifier les besoins majeurs pour la suite du projet.
1. Méthodologie de l’étude terrain
a) Entretiens avec des experts réglementaires
Un entretien a été organisé avec une Senior QARA Consultant, experte MDR / ISO 13485 / Marquage CE, ainsi qu’avec une chargée des affaires réglementaires. L’objectif principal de cette rencontre était de recueillir des informations permettant de cadrer et structurer le questionnaire destiné aux entreprises, afin d’identifier avec précision les besoins, difficultés et pratiques actuelles liées à la numérisation des affaires réglementaires. Cet échange a permis :
D’identifier les problématiques quotidiennes rencontrées par les services RA, telles que :
- La dispersion et la multiplicité des documents (PDF, Word, Excel…)
- L’absence d’interopérabilité entre les différents systèmes utilisés.
- La dépendance à certaines personnes clés connaissant les anciens processus.
- Le manque de formation aux outils numériques et aux exigences réglementaires.
De comprendre les écarts entre exigences réglementaires et capacités numériques actuelles :
- Les obligations de traçabilité et de conformité réglementaire sont souvent difficiles à satisfaire avec des outils partiellement digitalisés ou des processus papier.
- La maturité numérique des entreprises varie fortement selon leur taille et leurs ressources, ce qui influence la pertinence et le format des questions à poser.
D’obtenir une vision opérationnelle du terrain, essentielle pour la construction du questionnaire :
- Les informations recueillies permettent de formuler des questions ciblées, compréhensibles par les professionnels, et de hiérarchiser les thèmes à aborder : outils utilisés, freins à l’adoption, compétences internes, besoins en interopérabilité, etc.
- L’entretien a également permis d’identifier des bonnes pratiques et des points sensibles à explorer pour évaluer l’impact réel de la numérisation sur le travail quotidien.
En synthèse, ces entretiens fournissent un cadre structurant pour la rédaction du questionnaire, en garantissant que les questions soient pertinentes, adaptées aux profils des répondants et alignées avec les objectifs de l’étude : mesurer la maturité numérique, identifier les obstacles et recenser les besoins spécifiques des services RA.
b) Questionnaire adressé aux experts réglementaires
Afin de compléter les entretiens réalisés avec des experts du domaine, un questionnaire anonyme a été conçu (voir annexe 3) et distribué auprès des professionnels du secteur des dispositifs médicaux. Sa rédaction s’est appuyée directement sur les enseignements tirés des entretiens, qui ont permis d’identifier les problématiques quotidiennes, les écarts entre exigences réglementaires et capacités numériques, ainsi que les attentes en matière de digitalisation.
Le questionnaire avait pour objectif de recueillir des données quantitatives permettant d’évaluer :
- Le niveau de maturité numérique des entreprises,
- Les outils actuellement utilisés (tableurs, eQMS, logiciels de gestion documentaire, plateformes de soumission, outils d’IA, etc.)
- Les difficultés rencontrées dans le cadre de la digitalisation
- Les attentes et besoins futurs concernant la digitalisation
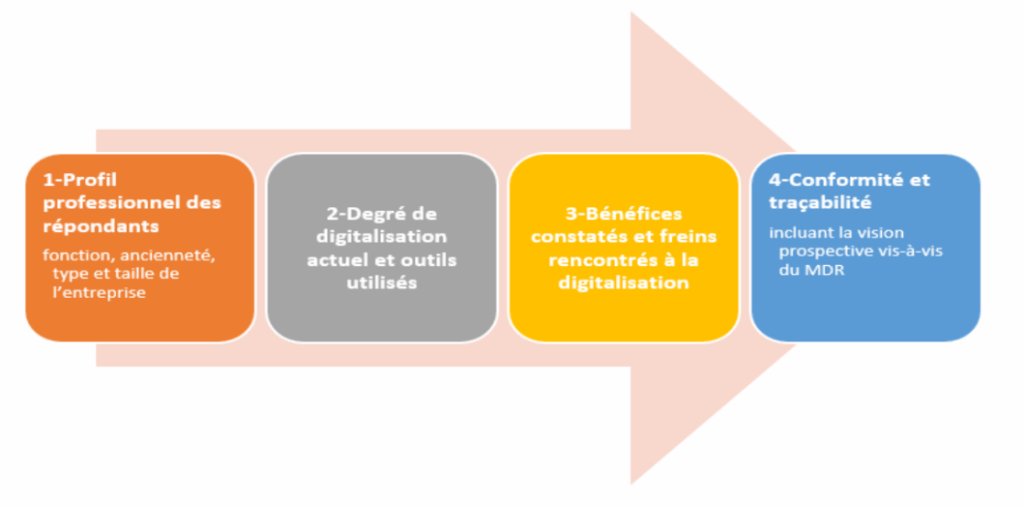
Il a été structuré en quatre parties(Figure 1) :
Figure 1 : Découpage thématique du questionnaire en quatre parties [source : auteur.e.s]
Le questionnaire a été envoyé à différents professionnels des services qualité et affaires réglementaires, et 24 réponses ont été recueillies, offrant une première base d’information significative. Les répondants présentaient des profils variés en termes d’ancienneté (moins d’un an à plus de dix ans) et provenaient d’entreprises de différentes tailles, avec une présence systématique de TPE et PME. Les données ainsi collectées ont alimenté les analyses et ont permis de dégager des tendances récurrentes concernant l’état de la digitalisation au sein des entreprises étudiées, constituant un complément essentiel aux observations issues des entretiens.
2. Niveau actuel de digitalisation dans les services réglementaires
Les entretiens et les résultats du questionnaire montrent que la situation est très hétérogène selon les entreprises. Néanmoins, plusieurs constats reviennent de manière récurrente :
a) Une majorité de PME encore peu digitalisées(Figure 2)
Les chargés RA signalent que :
- Une large partie des activités est réalisée via des outils dispersés,
- Les documents sont stockés dans des dossiers ou plateformes non centralisés,
- Les processus restent en grande partie manuels (gestion documentaire, signatures, versions…).
Figure 2 : Résultats du questionnaire sur le niveau de digitalisation des services réglementaires [source : auteur.e.s]
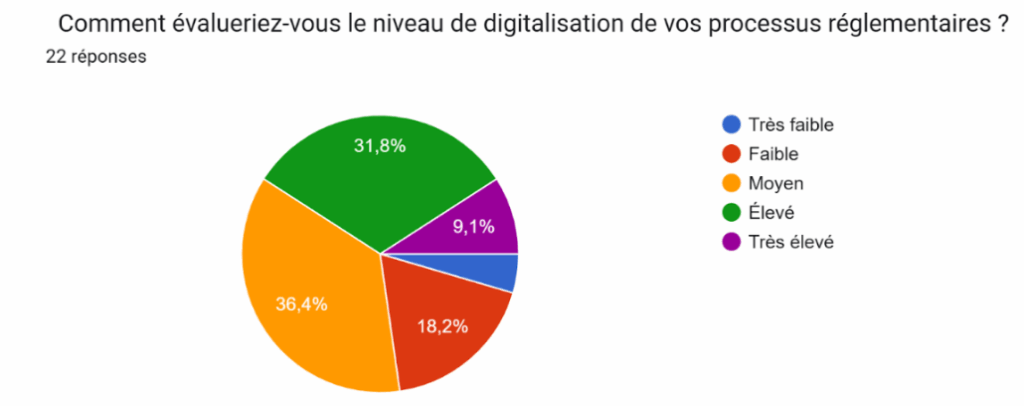
Ces observations sont confirmées par les données du questionnaire : 4,5 % des répondants considèrent le niveau de digitalisation de leur entreprise comme très faible, 18,2 % comme faible, et 36 % comme moyen. Ces résultats rejoignent également les données du Chapitre I, qui indiquent que 78 % des entreprises ne maîtrisant pas encore le RDM fonctionnent avec des processus dispersés et non centralisés.
b) Une adoption partielle des outils numériques
L’analyse des réponses au questionnaire montre que l’adoption des outils numériques par les entreprises reste partielle et hétérogène(Figure3).
Figure 3 : Résultats du questionnaire sur les outils numériques adoptés [source : auteur.e.s]
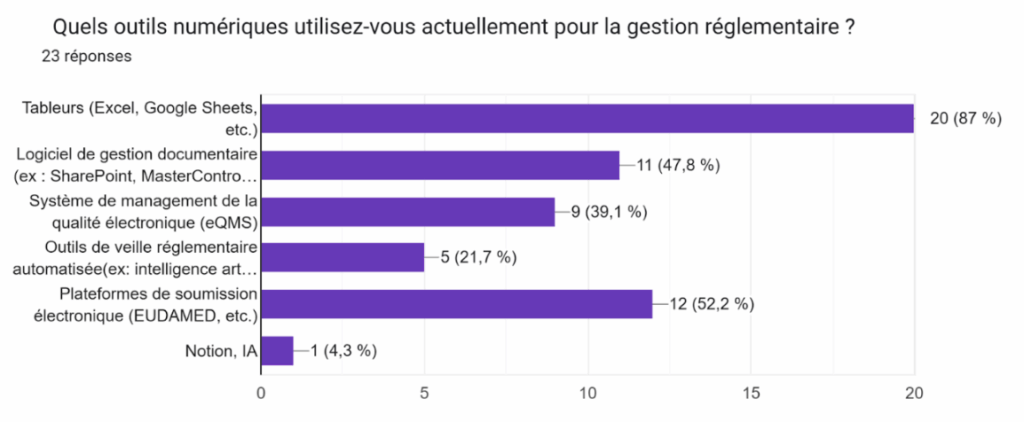
- 87 % des répondants utilisent encore des tableurs (Excel, Google Sheets, etc.) pour gérer leurs activités et documents.
- 52 % ont recours à des plateformes de soumission électronique (comme EUDAMED) cela implique que certaines PME n’ont pas encore finalisé leur transition vers le MDR qui l’oblige.
- 47 % utilisent un logiciel de gestion documentaire (ex. SharePoint, MasterControl…).
- 39 % disposent d’un système de management de la qualité électronique (eQMS).
Ces chiffres montrent que si certains outils numériques sont déjà intégrés, la majorité des entreprises continue de s’appuyer sur des solutions simples ou non centralisées, ce qui reflète une adoption progressive et partielle. Cette situation met en évidence le besoin d’une stratégie d’intégration plus cohérente pour assurer l’efficacité, la traçabilité et la conformité réglementaire dans l’ensemble des processus.
3. Principaux freins identifiés sur le terrain
L’analyse croisée des données issues des entretiens, du questionnaire et des discussions en groupe a permis de dégager les principaux obstacles rencontrés par les entreprises dans la digitalisation des affaires réglementaires. Parmi l’ensemble des freins identifiés, les trois plus fréquemment mentionnés ont été sélectionnés pour une analyse approfondie. Ces freins ont été synthétisés à l’aide des diagrammes d’Ishikawa, permettant de mettre en évidence les causes profondes et les liens entre différents facteurs.
a) Frein n°1 : Le coût de mise en place des outils numériques
Le coût global de la transformation numérique constitue le frein majeur avec un taux de 60,9%, particulièrement pour les PME. Il comprend l’acquisition de licences logicielles, le matériel informatique, la formation du personnel, la maintenance, la mise à niveau des réseaux, la cybersécurité ainsi que l’accompagnement au changement et l’adaptation des processus internes.
Les retours des entretiens et du questionnaire montrent que les PME sont fréquentes à reporter ou limiter la digitalisation en raison de ces coûts. Les investissements financiers constituent ainsi un obstacle majeur à l’adoption des outils numériques, impactant directement le niveau de maturité digitale et la capacité à atteindre une conformité optimale avec le MDR/IVDR. L’effet principal se traduit par un ralentissement de l’implémentation des outils numériques stratégiques et une limitation de l’efficacité des processus internes.
Synthèse des impacts identifiées grâce aux diagrammes d’Ishikawa (voir annexe 1), :
- Multiplicité des formats documentaires (PDF, Excel, Word)
- Procédures non harmonisées entre départements
- Dépendance à des consultants externes coûteux
- Besoin de nouveaux matériels et licences logicielles
- Réglementations strictes (RGPD, MDR) et faible concurrence sur certaines solutions
Cette synthèse met en évidence la nécessité de solutions modulaires, évolutives et financièrement accessibles, permettant aux entreprises de progresser graduellement vers une digitalisation complète tout en maîtrisant les dépenses.
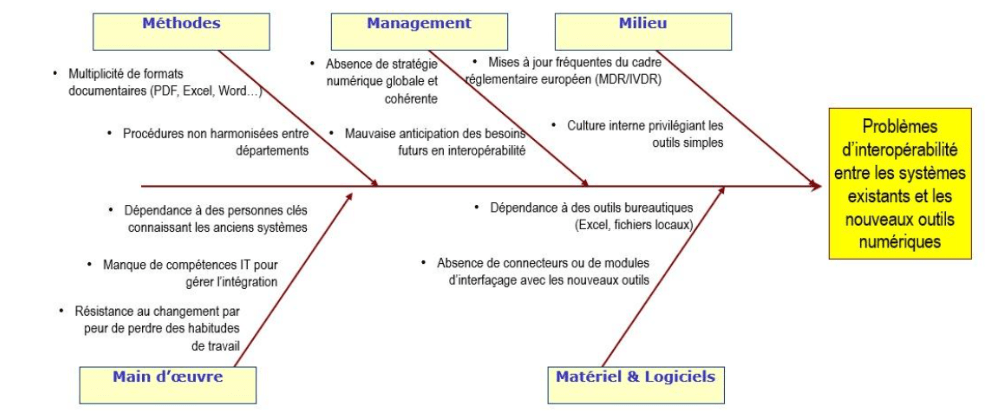
b) Frein n°2 : Le manque d’interopérabilité entre les systèmes
Le second frein concerne l’interopérabilité, qui affecte la cohérence et la fluidité des échanges de données entre systèmes existants et nouveaux outils numériques. Les causes identifiées (voir annexe 2) incluent la multiplicité de formats documentaires, des procédures non harmonisées, l’absence de stratégie numérique globale et une mauvaise anticipation des besoins futurs. S’y ajoutent les mises à jour fréquentes du cadre réglementaire MDR/IVDR, une culture interne privilégiant les outils simples, la dépendance à des personnes clés maîtrisant les anciens systèmes, le manque de compétences IT, la résistance au changement et l’absence de connecteurs ou modules d’interfaçage. Ce frein engendre des difficultés dans la centralisation des données, la traçabilité et le suivi réglementaire.
c) Frein n°3 : Doute sur la conformité réglementaire (sécurité des données et traçabilité)
Le troisième frein, bien que moins prioritaire, est le doute sur la capacité à respecter les exigences réglementaires, en particulier en matière de sécurité des données et de traçabilité. Il découle principalement du manque de formation et de compétences numériques du personnel. Ce déficit freine l’adoption des outils et expose les entreprises à un risque de non-conformité autre élément renforçant ce frein concerne la validation des solutions digitales, exigée par l’ISO 13485:2016, qui stipule que tout logiciel utilisé dans le système de management de la qualité doit être validé avant utilisation et après toute modification (§4.1.6) . Les principes de l’IEC 62304:2006, applicable au cycle de vie des logiciels, imposent également des activités formelles de vérification et validation proportionnées aux risques. Pour de nombreuses PME, cette étape représente un investissement important en temps, en compétences et en ressources, constituant ainsi un frein supplémentaire à l’adoption d’outils numériques.
4. Analyse transversale (tendances et constats clés)
L’analyse des entretiens et du questionnaire met en lumière plusieurs limites concrètes (Tableau2) des outils largement employés (tableurs, SharePoint, etc.), qui freinent une digitalisation pleinement efficace :
Tableau 2 : limites concrètes des outils employés [source : auteur.e.s]
| Limites des tableurs (Excel, Google Sheets, …) | Limites de SharePoint |
| - Ces outils ne garantissent pas une traçabilité robuste ni une piste d’audit fiable. Les processus manuels inhérents aux tableurs les rendent vulnérables aux erreurs humaines et à la perte de version, ce qui pose un problème quand il s’agit de conformité réglementaire[18]. -Les tableurs ne sont pas conçus pour l’automatisation des workflows qualité : pas de déclenchement automatique d’actions (CAPA, validation, escalades), ce qui les rend peu adaptés aux processus réglementaires structurés [18]. - Sur le plan de la sécurité des données, Excel offre des contrôles limités : un fichier peut être facilement copié, partagé et modifié, sans journalisation fiable des modifications, ce qui peut compromettre l’intégrité et la confidentialité des données sensibles[19]. | - Il ne gère pas bien les PDF scannés ou les images sans OCR intégré) et la gestion fine des droits d’accès, ce qui peut poser des problèmes de sécurité et de structuration documentaire [20]. - L’administration de SharePoint pour des usages réglementaires ou SMQ nécessite des paramétrages complexes (métadonnées, taxonomie, configuration des droits), ce qui peut rendre son adoption lourde et coûteuse . |
L’analyse approfondie des freins montre que certaines causes se répètent fréquemment et constituent des motifs récurrents affectant plusieurs obstacles : la multiplicité des formats documentaires et les procédures non harmonisées apparaissent à la fois dans le frein lié aux coûts et dans celui de l’interopérabilité, tandis que le manque de compétences et de formation revient dans le doute sur la conformité et dans l’adoption des outils. Ces motifs communs révèlent des enjeux structurants, tant techniques que organisationnels, qui nécessitent une attention particulière.
Cette étude terrain met en évidence que la digitalisation des affaires réglementaires est non seulement nécessaire, mais également attendue par les professionnels. Toutefois, plusieurs freins majeurs persistent, en particulier le coût des outils, l’absence d’interopérabilité et le manque de formation.
Les analyses internes du groupe et les retours du questionnaire convergent vers les mêmes constats, confirment la pertinence des causes identifiées dans les diagrammes d’Ishikawa présentés en annexe. Ces résultats constituent une base essentielle pour le Chapitre III, dans lequel nous analyserons les stratégies et leviers permettant d’accélérer l’intégration des outils numériques, tout en garantissant la conformité et la traçabilité réglementaire.
Chapitre 3 - Stratégies et leviers pour accélérer l’intégration des outils numériques
Après avoir synthétisé les freins à l’adoption numérique, l’analyse révèle que le manque de formation et l’inconscience de l’importance des outils numériques constituent le frein principal. Ce chapitre se concentre donc sur l’illustration concrète de la valeur ajoutée de ces outils à chaque étape du cycle de vie du dispositif médical, depuis la conception jusqu’au suivi post-commercialisation. Il met en lumière les leviers permettant une adoption efficace, tels que la formation, la sensibilisation et l’interopérabilité entre systèmes. Enfin, ce chapitre montre comment ces outils numériques surpassent les méthodes traditionnelles basées sur des documents dispersés, qui présentent de multiples limites, notamment en termes de traçabilité, de conformité et d’efficacité opérationnelle.
1. Descriptions des outils numériques mobilisables à chaque phase du cycle de vie du dispositif médical(Tableau3)
a) Introduction au cycle de vie d’un dispositif médical
Le cycle de vie d’un dispositif médical regroupe l’ensemble des étapes allant de la conception à la mise sur le marché, puis à la surveillance post-commercialisation (Figure4). Selon le Règlement (UE) 2017/745 (MDR) et la norme ISO 13485, chaque étape doit être encadrée par une gestion continue des risques et par un Système de Management de la Qualité (SMQ) garantissant la sécurité, la performance et la conformité du produit.
Figure 4 :Schéma illustrant le cycle de vie d’un dispositif médical [source : auteur.e.s]
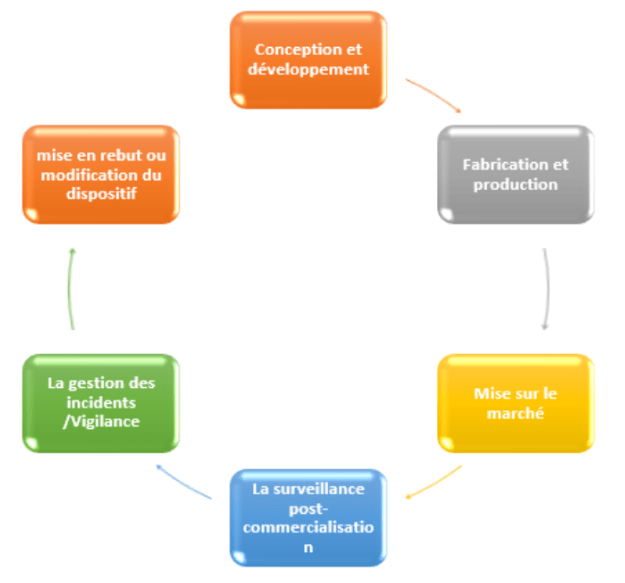
La numérisation joue un rôle déterminant dans ce cadre : elle améliore la traçabilité, facilite la gestion documentaire, renforce la cohérence réglementaire et réduit les erreurs humaines. Les outils numériques apportent ainsi un soutien essentiel pour structurer les informations, accélérer la conformité au MDR et préparer efficacement les audits.
b) Phase de conception et développement
Dès la phase de conception, l’intégration d’outils numériques dédiés aux affaires réglementaires permet de mieux organiser les informations, de garantir une traçabilité claire et d’anticiper plus facilement les exigences du MDR/IVDR. Des solutions comme les systèmes PLM, les eQMS, les ERP, les bases réglementaires telles que EUDAMED, ou encore les outils d’intelligence artificielle, offrent un véritable soutien pour structurer et harmoniser l’ensemble du développement.
Les plateformes PLM, par exemple, rassemblent toutes les données liées aux exigences de conception, dossiers de conception, dossiers maîtres, nomenclatures ou encore modifications et les connectent directement aux activités réglementaires comme l’analyse de risques ou les tests de vérification et validation. Cela facilite l’application de la maîtrise de la fabrication exigée par l’ISO 13485 et le MDR. De leur côté, les eQMS permettent de gérer efficacement les procédures qualité, la documentation, les enregistrements de développement, les CAPA et les revues de conception, ce qui rend le processus prêt pour un audit dès les premières étapes du projet.
Les ERP, bien que largement utilisés pour la gestion de la production, interviennent également dans la planification, les achats, la gestion des stocks et parfois la qualité., nécessitent que les données produit telles que les nomenclatures, les versions et la codification soient définies dès la conception pour assurer une parfaite correspondance entre le produit imaginé et celui effectivement fabriqué, point essentiel pour la conformité et la traçabilité. Quant à EUDAMED, même si la saisie des données intervient plus tard, les informations nécessaires (UDI, caractéristiques, classification, indications, etc.) doivent être préparées dès la conception pour éviter des retours en arrière sur le design ou la documentation technique.
Enfin, les outils d’intelligence artificielle apportent une aide précieuse pour automatiser l’identification des dangers selon l’ISO 14971, relier les exigences du MDR/IVDR aux éléments du développement ou encore générer plus rapidement une partie de la documentation technique comme les analyses de risques ou les Instructions d’Utilisation. Ces technologies permettent de réduire les erreurs humaines, d’accélérer la rédaction du dossier technique et d’assurer une meilleure cohérence entre toutes les étapes.
L’intégration de ces outils numériques dès la conception transforme profondément la gestion des affaires réglementaires : la conformité devient un processus continu, fluide et parfaitement intégré au développement du dispositif, plutôt qu’une étape réalisée en fin de projet [8].
c) Phase de fabrication et industrialisation
- Contribution de l’ERP à la production
L’utilisation de l’ERP dans les environnements réglementés permet d’optimiser la coordination des activités de fabrication et d’améliorer la cohérence des informations entre les différents services de l’entreprise. Son intégration dans la phase industrielle renforce le contrôle des opérations et la traçabilité des produits.
Gestion des ordres de fabrication : L’ERP permet une planification plus précise des ordres de fabrication en offrant une visibilité complète sur les ressources, les matières premières et les capacités de production. Les opérations sont mieux synchronisées, ce qui réduit les retards et améliore la fiabilité du planning de production [21].
Traçabilité des lots : L’ERP assure une traçabilité complète et automatisée des matières premières, des composants, des opérations de fabrication et des produits finis. Cette traçabilité ascendante et descendante est essentielle pour répondre aux exigences du règlement européen MDR 2017/745, notamment pour les audits, les rappels, et la gestion des actions correctives (CAPA) .
Intégrité des données (Data Integrity) : L’intégrité des données est garantie grâce à l’enregistrement automatique des actions, à la sécurisation des accès et à la réduction des manipulations manuelles. L’ERP soutient les principes ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate), indispensables pour la conformité aux normes ISO 13485 et FDA 21 CFR 820[ 29].
- Automatisation du contrôle qualité
La digitalisation du contrôle qualité constitue un levier majeur d’amélioration dans la fabrication des dispositifs médicaux. Les systèmes numériques facilitent la surveillance continue des processus, la gestion des documents et la détection précoce des écarts.
Systèmes numériques de contrôle qualité (eQMS) :Les eQMS centralisent la documentation, les enregistrements de contrôle, les audits internes, les CAPA et les validations. Ils permettent de réduire le recours au papier et d’accélérer les cycles de vérification, améliorant ainsi la réactivité du service qualité et la préparation aux audits réglementaires [29, 30].
Enregistrement des non-conformités : L’enregistrement numérique des non-conformités facilite l’identification rapide des écarts, la gestion des risques et la mise en place d’actions correctives. L’intégration ERP–eQMS assure une liaison directe entre la production et la qualité, garantissant un traitement plus efficace des incidents et une meilleure maîtrise des performances du processus [25]. Il est toutefois important de noter que cette interopérabilité n’est pas automatique et dépend de la capacité des systèmes à être intégrés et validés. Conformément à l’ISO 13485:2016, toute interface logicielle utilisée dans le système qualité doit être validée [26]. Les recommandations de la FDA sur la Computer Software Assurance (2022) rappellent également que les connexions entre logiciels doivent être évaluées en fonction des risques [14].
d) Phase de gestion qualité et conformité réglementaire
La gestion des risques, encadrée par la norme ISO 14971:2019 [27], s’inscrit pleinement dans la phase de gestion qualité et de conformité réglementaire, en lien direct avec les activités des équipes Qualité et Affaires Réglementaires (QARA). La numérisation de cette activité via des plateformes eQMS permet de structurer l’analyse des dangers tout au long du cycle de vie du dispositif, depuis la conception jusqu’au suivi post-commercialisation.
Concrètement, l’eQMS centralise les analyses de risques, assure la mise à jour continue des matrices et établit des liens traçables entre chaque danger identifié, les mesures de maîtrise associées, les exigences réglementaires et les essais de vérification correspondants. Cette structuration facilite le travail des équipes QARA en réduisant les manipulations manuelles et en fiabilisant la cohérence des données réglementaires [28].
L’automatisation du calcul des indices de criticité, combinée à l’interconnexion avec le PLM et le dossier technique, permet d’assurer une synchronisation en temps réel des informations. Les équipes QARA disposent ainsi d’une vision consolidée et actualisée du profil de risque, facilitant la préparation des audits, la justification des choix réglementaires et la continuité documentaire exigée par le RDM [29].
L’utilisation de l’intelligence artificielle marque une évolution importante dans cette phase. Les solutions basées sur l’IA analysent automatiquement les exigences réglementaires, identifient les éléments manquants dans les documents et assistent la préparation du dossier technique, du rapport d’évaluation clinique ainsi que des documents PMS et PMCF, conformément aux lignes directrices MDCG. L’IA facilite aussi la veille réglementaire en détectant les mises à jour du MDR, des MDCG ou de la FDA et en évaluant leur impact sur le dispositif [30].
e) Phase de mise sur le marché
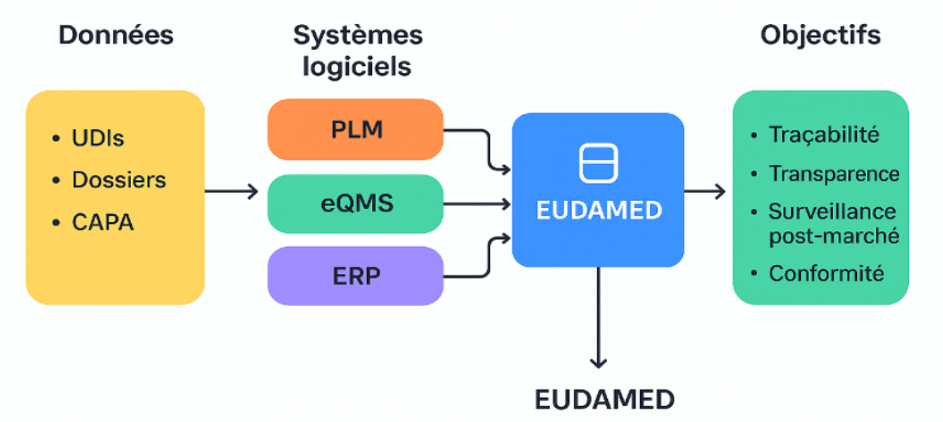
La phase de mise sur le marché d’un dispositif médical repose sur l’enregistrement précis des données réglementaires et l’assurance de leur conformité[31]. Dans l’Union européenne, cette étape est centrée sur EUDAMED. Les fabricants y enregistrent les dispositifs, les UDI (Unique Device Identifier), ainsi que les informations relatives aux acteurs économiques (fabricants, mandataires, importateurs) afin d’assurer la traçabilité et la transparence des produits sur le marché [2].
Chaque dispositif se voit attribuer un Basic UDI-DI et un UDI-DI, transmis à EUDAMED conformément aux spécifications de l’Annexe VI du MDR[2]. Ces identifiants garantissent le suivi des dispositifs tout au long de la chaîne logistique, depuis le fabricant jusqu’à l’utilisateur final.
Pour simplifier et sécuriser la transmission des données, les systèmes internes tels que PLM, eQMS et ERP sont interconnectés avec EUDAMED [8]:
- PLM centralise les informations techniques et les versions des documents, et collecte les métadonnées UDI
- eQMS valide et contrôle les données via des workflows automatisés, garantit l’audit trail et la conformité aux exigences de l’Annexe 11 et du 21 CFR Part 11 [2].
- ERP fournit les informations logistiques et de production, telles que les numéros de lots et les numéros de série, assurant la cohérence avec les informations réglementaires.
Ces systèmes sont reliés via des API ou middleware qui formatent les données pour EUDAMED (XML/JSON), permettant l’export automatique des informations, la réduction des erreurs manuelles, la synchronisation continue et une meilleure réactivité face aux mises à jour réglementaires [2].
L’enregistrement des acteurs dans EUDAMED est également obligatoire. Chaque acteur obtient un identifiant unique (SRN ou Actor ID) qui relie les dispositifs, les certificats, les événements de vigilance et les rapports post-market à leur propriétaire légal [32]. Cette interconnexion assure la cohérence des données et facilite la surveillance réglementaire.
Pour les dispositifs déjà commercialisés avant l’application du MDR (legacy devices), l’enregistrement dans EUDAMED reste nécessaire, avec des identifiants spécifiques, afin de garantir la traçabilité et la conformité [32].
Enfin, la veille réglementaire numérique, souvent assistée par l’intelligence artificielle, permet de détecter automatiquement les mises à jour des lignes directrices MDCG, les nouvelles exigences du MDR ou de la FDA, et d’évaluer leur impact sur les dispositifs enregistrés [13]. Cette automatisation offre une approche proactive pour maintenir la conformité tout au long de la vie commerciale des dispositifs.
Figure 5 :Processus d’interconnexion des systèmes internes avec EUDAMED [source : auteur.e.s]
f) Phase de surveillance post commercialisation (PMS/PMCF/Vigilance)
- eQMS pour le suivi post-commercialisation
Dans la phase post-commercialisation d’un dispositif médical, un eQMS est déterminant pour assurer la conformité, la traçabilité et la réactivité. Certains systèmes d’eQMS permettent de gérer de manière automatisée des aspects tels que les dossiers, les CAPA, les plaintes et les déclarations de vigilance tout en assurant la conformité à l’ISO 13485 et au MDR 2017/745.
Au moyen de l’eQMS, les plaintes des utilisateurs ou des professionnels de santé sont enregistrées et structurées via des workflows de manière à permettre leur analyse pour une meilleure identification éventuelle de signaux de sécurité ou de performance, déclencher des investigations et mettre en œuvre des actions correctives (CAPA). Lors des incidents graves, les déclarations et les actions de sécurité (FSCA) peuvent être documentées en un lieu unique, permettant de générer les rapports, d’assurer la traçabilité de tous les événements et la production des Periodic Safety Update Reports (PSUR) exigés par le MDR [33]. Quand un incident est constaté, l’eQMS gère les CAPA, planifie les actions, assure leur mise en œuvre, contrôle leur efficacité et archive les preuves. Grâce à cette méthode, la justification, la communication et le suivi des mesures sont systématiquement conduits.
L’intégration de la gestion des plaintes, de la vigilance et des CAPA au sein de l’eQMS structure toutes les informations relatives aux incidents et les actions correctives mises en place, tout en garantissant la conformité sur l’ensemble du cycle de vie du produit [34].
- IA appliquée à la surveillance post-market
La surveillance post-commercialisation des dispositifs médicaux peut bénéficier de manière significative de l’intelligence artificielle, qui va automatiser et optimiser des processus critiques : pour commencer, elle permet d’analyser automatiquement les événements indésirables, car les algorithmes du machine learning (et du deep learning) vont pouvoir traiter de façon automatique les rapports d’événements pour en extraire les éléments les plus pertinents en termes d’information, puis en classe et priorise, une enquête plus précise sera diligentée sur ceux qui le nécessitent tout particulièrement grâce au Natural Language Processing (NLP), qui traite les textes des formulaires ou des courriers relatifs aux événements indésirables [35]. Ensuite, l’IA va faciliter la détection des tendances : les algorithmes, par leur capacité à traiter et analyser de très grands volumes de données de vigilance, vont permettre de mettre plus rapidement en évidence des tendances récurrentes ou émergentes (clusters) que ne le permet l’analyse humaine et manuelle traditionnelle[35]. Enfin, grâce à la prédiction des risques, ces modèles peuvent permettre de modéliser des scénarios sur la base des données historiques afin d’anticiper des défaillances : ainsi, une étude concernant les pompes à perfusion a montré que des approches de machine learning (Random Forest, Decision Tree, etc.) peuvent prédire avec une grande puissance le risque de défaillance avant qu’il ne se soit manifesté [36]. Ces technologies qui intègrent un PMS, permettent une approche plus proactive si ce n’est prédictive de la surveillance, qui passe d’une approche résolument réactive du type « réparer après l’incident » à une approche préventive intelligente, qui accroît la sécurité des dispositifs ainsi que la confiance des parties prenantes[37].
- Outils de reporting et dashboards
Pour utiliser judicieusement les données issues de l’eQMS et des systèmes IA, il est nécessaire de se doter d’outils permettant le reporting et la visualisation (dashboards) des données. Des plateformes de Business Intelligence (BI) comme Power BI ou Tableau permettent de construire des tableaux de bord interactifs et d’afficher les indicateurs clés de performance (KPI) de la surveillance post-commercialisation : nombre d’événements, catégorie d’incidents, temps de traitement, CAPA ouvertes, etc. Ces visualisations en temps réel facilitent la prise de décision rapide, éclairée et factuelle. Plusieurs KPI peuvent être suivis pour assurer un suivi : fréquence d’incidents, gravité des incidents, délais de réponse, taux de réouverture des actions correctives, efficacité des mesures mises en place permettant d’évaluer les risques mais également les bénéfices en termes de diminution d’incidents. De plus, ces dashboards, intégrés à l’eQMS et à ses modules IA, garantissent la traçabilité complète du cycle de vie d’un incident : de sa déclaration à la mise en place et à la validation d’une action corrective tout en capturant les tendances émergentes. Cette traçabilité a un rôle stratégique en matière de conformité réglementaire (elle contribue au rapport PSUR sous le MDR) et améliore la transparence des mesures de vigilance post-commercialisation (inscrites dans les PMS et PSUR en conformité avec les exigences du MDR, Annexes III et VII)[37].
g) Phase de retrait ou modification du dispositif
Il s’agit de la dernière étape du cycle de vie du dispositif. Elle peut correspondre à une mise à jour du dispositif ou à son retrait du marché.
Gestion des modifications (Change Control)
L’eQMS est utilisé pour gérer efficacement les modifications apportées au dispositif grâce à des flux de travail automatisés et des processus d’approbation . Les fonctionnalités attendues de cet outil numérique sont les suivantes :
- Automatisation et gestion des processus d’acheminement, de transmission, d’escalade et d’approbation des documents, tout en facilitant la recherche et la récupération des documents lors des audits ou inspections [38].
- Unification des systèmes et des processus réglementaires au sein d’une plateforme cloud unique[38].
- Archivage des documents et conservation de l’historique des modifications .
- Gestion des changements et prise en charge des signatures électroniques [39].
Retrait du marché ou rappel produit
Les rappels de produits, souvent très coûteux pour le fabricant, peuvent être réduits grâce aux outils numériques qui améliorent les procédures de vérification, la gestion des modifications et la détection des défauts dès le début du processus [40].
- Le PLM peut être utilisé dans ce cas pour suivre les modifications et retracer les lots ou les versions d’un dispositif devant être rappelés ou retirés [40].
- L’ERP permet le contrôle de l’ensemble de la chaîne de fabrication, aussi bien ascendante que descendante, ce qui facilite l’identification des lots concernés en cas de défaut ou de non-conformité [41]
Le tableau ci-après présente une synthèse des principaux outils numériques et met en évidence, pour chacun d’entre eux, la phase du cycle de vie du dispositif médical dans laquelle il apporte le plus de valeur ainsi que son utilité réglementaire.
Tableau 3 : tableau comparatif des solutions numériques par phase. Source [source : auteur.e.s]
| Solution | Rôle principal | Phases concernées | Fonctions clés | Limites / points de vigilance |
| PLM | Gestion des données produit et du développement | Conception, Mise sur le marché, Modification/Rappel | Traçabilité , versioning, intégration UDI, design controls | Implémentation coûteuse ; dépendance à la qualité des données ; nécessite une forte structuration interne |
| eQMS | Gestion numérique de la qualité et la conformité | Conception, Fabrication, Qualité, PMS, Retrait | Gestion des CAPA, audits, NC, workflows, conformité 21 CFR Part 11 | Complexité des workflows ; rigidité si mal paramétré ; adoption difficile pour les petites équipes |
| ERP | Production, logistique, traçabilité et gestion financières | Fabrication, Mise sur le marché, Retrait | Traçabilité des lots, intégrité des données | Peu adapté à la gestion qualité fine ; intégration avec PLM/eQMS parfois limitée ; coût de maintenance élevé |
| EUDAMED | Enregistrement réglementaire UE | Mise sur le marché | UDI, SRN, transparence, traçabilité | Déploiement encore partiel ; dépend des autorités ; ne remplace pas un outil interne ; pas un système de gestion documentaire |
| IA | Automatisation intelligente | Conception, Qualité, PMS | Analyse risques, veille MDR/FDA, prédiction des incidents | Fiabilité dépend des données ; risques de biais ; conformité réglementaire encore en évolution ; nécessité de validation stricte |
2. Leviers pour accélérer l’intégration des outils
Dans cette section, nous présentons les principaux leviers permettant d’accélérer l’intégration des outils numériques dans les affaires réglementaires, en combinant formation, sensibilisation, accompagnement progressif et interopérabilité des systèmes, afin d’optimiser leur adoption tout en préservant le rôle indispensable de l’humain.
a) Formation des équipes
La formation des collaborateurs constitue un levier essentiel pour réussir l’intégration des outils numériques dans les affaires réglementaires. Il s’agit de les former à l’utilisation des systèmes ERP, PLM, eQMS, EUDAMED et de l’IA, en tenant compte de leur rôle spécifique dans le cycle de vie du dispositif médical. Cette approche permet de montrer que ces outils sont non seulement plus adaptés et flexibles que les solutions bureautiques classiques, mais qu’ils offrent également des fonctionnalités avancées qui surpassent largement les outils traditionnels (Word, Excel, gestion manuelle des documents).
La formation doit aller au-delà de l’apprentissage technique et inclure :
- L’utilisation pratique des logiciels dans des scénarios concrets du cycle de vie du dispositif médical.
- La compréhension des flux d’information entre les différents systèmes pour assurer une circulation efficace et cohérente des données.
- La maîtrise des exigences réglementaires et de conformité, afin de garantir la traçabilité documentaire et de dissiper les doutes liés à la conformité réglementaire.
Ainsi, la formation renforce non seulement la compétence technique des équipes, mais elle instaure une confiance dans l’utilisation des outils numériques, limite les erreurs et assure que les processus réglementaires sont respectés de manière fiable et efficace. De plus, elle permet aux collaborateurs de prendre pleinement conscience que l’acquisition et le déploiement de ces outils représentent un investissement pertinent, qui apporte des gains concrets en termes de productivité, de traçabilité et de conformité réglementaire.
b) Sensibilisation au rôle complémentaire des outils numériques
Il est fondamental de sensibiliser les équipes au fait que les outils numériques ne remplacent pas l’humain. Même si ERP, PLM, eQMS, EUDAMED ou l’IA automatisent certaines tâches répétitives, facilitent la gestion des documents et améliorent la traçabilité, les décisions critiques, l’analyse des risques, l’interprétation des données cliniques et la responsabilité réglementaire restent entièrement sous la supervision des professionnels qualifiés. Ce point a été confirmé par une experte en charge des affaires réglementaires dans une grande entreprise internationale, qui a digitalisé l’ensemble de son processus réglementaire de manière complète, tout en maintenant la supervision humaine pour les aspects critiques.
Cette sensibilisation joue plusieurs rôles :
- Renforcer la confiance des collaborateurs dans les outils numériques en montrant leur rôle complémentaire plutôt que substitutif.
- Réduire les craintes liées à la digitalisation, comme la peur de perdre le contrôle ou que la technologie rende certains postes obsolètes.
- Favoriser une adoption efficace, car les utilisateurs comprennent que l’outil est un support pour améliorer leur travail quotidien et non un remplacement de leur expertise.
Ainsi, une bonne sensibilisation contribue à créer un environnement où humains et technologies collaborent efficacement, garantissant la conformité et la performance des processus réglementaires
c) Accompagnement progressif
Le coût d’acquisition et de déploiement des outils numériques (ERP, PLM, eQMS, EUDAMED, IA) représente souvent un frein majeur pour les entreprises, notamment les PME. Les investissements initiaux élevés peuvent générer des réticences, même lorsque les bénéfices à long terme sont significatifs. Pour pallier ce frein, un accompagnement progressif et modulable est recommandé :
- Commencer par des fonctionnalités de base, moins coûteuses et directement utiles, afin de générer rapidement des gains visibles en termes de productivité et de conformité.
- Élargir progressivement le déploiement aux processus plus complexes, ce qui permet d’étaler les coûts dans le temps et de mieux gérer le budget.
- Favoriser l’adaptation graduelle des équipes, ce qui réduit les risques d’erreurs coûteuses et maximise le retour sur investissement.
Cette approche combine à la fois gestion du changement et optimisation financière, en rendant l’adoption des outils numériques plus accessible et moins intimidante pour l’entreprise, tout en assurant la maîtrise des processus et la conformité réglementaire.
Ce chapitre a montré comment les outils numériques peuvent optimiser chaque phase du cycle de vie du dispositif médical, en améliorant la traçabilité, la qualité des données et la conformité réglementaire. La réussite de leur intégration repose toutefois sur trois leviers essentiels : la formation des équipes, la sensibilisation au rôle complémentaire des outils par rapport à l’expertise humaine, et un accompagnement progressif permettant de gérer le coût et le changement. Ensemble, ces éléments créent les conditions nécessaires pour une adoption efficace et durable de la digitalisation dans les affaires réglementaires.
Conclusion Générale
L’ensemble de ce travail a montré que la numérisation des affaires réglementaires constitue aujourd’hui un enjeu central pour les fabricants de dispositifs médicaux, dans un contexte où le MDR/IVDR impose un niveau de conformité, de traçabilité et de réactivité sans précédent. À travers l’étude du cadre réglementaire, l’analyse des pratiques actuelles et l’exploration des outils disponibles, il apparaît clairement que les solutions numériques offrent un potentiel considérable pour améliorer l’efficacité des processus, réduire les délais de mise en conformité et renforcer la qualité des données réglementaires.
Les échanges avec les professionnels du secteur, les retours terrain et l’analyse des questionnaires ont permis d’identifier les principaux freins rencontrés par les entreprises, en particulier par les PME : le coût des outils, le manque d’interopérabilité des systèmes et l’insuffisance de formation. Ces obstacles, largement confirmés dans la littérature, soulignent que la transition numérique ne dépend pas uniquement de la technologie, mais également d’une transformation organisationnelle et humaine.
Malgré ces difficultés, les opportunités restent majeures. Les entreprises qui investissent dans des outils adaptés, structurent leurs processus et accompagnent leurs équipes constatent des bénéfices concrets : meilleure maîtrise documentaire, automatisation des tâches répétitives, plus grande cohérence des preuves réglementaires et accélération des cycles de validation. Les innovations émergentes notamment l’intelligence artificielle, les bases de données intelligentes et les environnements intégrés (ERP, PLM, eQMS) renforcent encore ces perspectives et ouvrent la voie à une digitalisation plus harmonisée et plus accessible.
Ainsi, ce rapport met en évidence que l’intégration des outils numériques dans les affaires réglementaires n’est plus une option, mais une évolution nécessaire pour garantir la conformité, la compétitivité et la fiabilité des fabricants. La compréhension des freins et des leviers présentée tout au long de ce travail constitue une base solide pour envisager des stratégies d’intégration réalistes et adaptées aux différents niveaux de maturité numérique des entreprises. La digitalisation représente donc une opportunité durable d’amélioration continue, au service d’un système réglementaire plus efficace, cohérent et sécurisé.
Références bibliographiques
[1] US Food and Drug Administration, « Part 11, Electronic Records ; Electronic Signatures - Scope and Application ». Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
[2] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. [En ligne]. Disponible sur : https://eur-lex.europa.eu/eli/reg/2017/745/oj/eng
[3] Álvaro Carpintero, Tacy Foster, Evgeniya Makarova & Vanya Telpis, « Reimagining smart quality approach | McKinsey ». Consulté le : 23 septembre 2025. [En ligne]. Disponible sur : https://www.mckinsey.com/industries/life-sciences/our-insights/smart-quality-reimagining-the-way-quality-works
[4] Spherical Insights, « Healthcare Compliance Software Market Size, Share, Growth Analysis, Report - 2033 ». Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.sphericalinsights.com/press-release/healthcare-compliance-software-market
[5] F. AHMED, M. VILLARRAGA, M. FROHBERGH, « 2023 Notified Bodies Medical Device Survey Reveals Certification Gap | Exponent », juill. 2024. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.exponent.com/article/new-medical-device-survey-reveals-eu-mdr-certification-gap
[6] Breda Kearney & Olivia McDermott, « The Challenges for Manufacturers of the Increased Clinical Evaluation in the European Medical Device Regulations : A Quantitative Study », Ther. Innov. Regul. Sci., vol. 57, no 4, p. 783‑796, mai 2023,https://pubmed.ncbi.nlm.nih.gov/37198369/
[7] R. Sriram, « Medical Device EU MDR Compliance : Challenges and Resource Crunch », The Kolabtree Blog. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.kolabtree.com/blog/medical-device-eu-mdr-compliance-challenges-and-resource-crunch/
[8] L. Wendy, « The importance of PLM, eQMS, and RIM systems for medical device manufacturers », Rimsys. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.rimsys.io/blog/plm-eqms-rim-systems-medical-device
[9] A. Corallo, M. E. Latino, M. Lazoi, S. Lettera, M. Marra, et S. Verardi, « Defining Product Lifecycle Management : A Journey across Features, Definitions, and Concepts », Int. Sch. Res. Not., vol. 2013, no 1, p. 170812, 2013, https://doi.org/10.1155/2013/170812
[10] O. Kheir, S. Smedts, A. Jacoby, et S. Verwulgen, « Efficient Quality Management in MedTech Start-Ups (Based on ISO 13485) », Med. Devices Auckl. NZ, vol. 14, p. 313‑319, oct. 2021,https://pubmed.ncbi.nlm.nih.gov/34703329/
[11] comission européenne, « Vue d’ensemble - Public Health - Commission européenne ». Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/medical-devices-eudamed/overview_fr
[12] null Amisha, P. Malik, M. Pathania, et V. K. Rathaur, « Overview of artificial intelligence in medicine », J. Fam. Med. Prim. Care, vol. 8, no 7, p. 2328‑2331, juill. 2019, https://pmc.ncbi.nlm.nih.gov/articles/PMC6691444/
[13] freyr digital, « What Is AI in Regulatory Compliance ». Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.freyrdigital.com/what-is-articles/what-is-ai-in-regulatory-compliance
[14] FDA (U.S. Food and Drug Administration), « Computer Software Assurance for Production and Quality System Software, Guidance for Industry and Food and Drug Administration Staff ». Consulté le : 16 décembre 2025. [En ligne]. Disponible sur : https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computer-software-assurance-production-and-quality-system-software
[15] WISE GUY, « rapport d’etude de marché des logiciels Erp pour la fabrication de dispositifs médicaux : tendances et opportunités 2032 ». 08 2025. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.wiseguyreports.com/fr/reports/medical-device-manufacturing-erp-software-market
[16] M. TREVINO, « 9 Strategies To Overcome Challenges In The EU Medical Device Market In 2025 And Beyond ». 17 décembre 2024. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://www.meddeviceonline.com/doc/strategies-to-overcome-challenges-in-the-eu-medical-device-market-in-and-beyond-0001
[17] « MDCG 2022-14 - Transition vers le DM et le DMIV - Capacité et disponibilité des organismes notifiés en matière de dispositifs médicaux et de DIV - Santé publique », European commission. Consulté le : 29 septembre 2025. [En ligne]. Disponible sur : https://health.ec.europa.eu/latest-updates/mdcg-2022-14-transition-mdr-and-ivdr-notified-body-capacity-and-availability-medical-devices-and-2022-08-26_en
[18] TrackMedium, « Why Excel & Spreadsheets Are Not Enough for Quality Management (and What to Use Instead) | Trackmedium eQMS ». Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.trackmedium.com/blog/why-excel-spreadsheets-are-not-enough-for-quality-management-and-what-to-use-instead/
[19] F. Landry, « Pourquoi devriez-vous interdire l’utilisation d’Excel dans un environnement réglementé ? » 7 mai 2024. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://fr.innovx.org/post/why-should-you-ban-excel-use-in-a-regulated-environment-1
[20] Access it, « Pourquoi ne pas utiliser SharePoint en GED ? » Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.access-it.fr/actualite/pourquoi-ne-pas-utiliser-sharepoint-en-tant-que-ged/
[21] « Medical Devices : Win the Speed and Quality Race », Dassault Systèmes. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.3ds.com/industries/life-sciences-healthcare/medical-devices
[22] Olivia Calder, « Dynamic Data Integrity : Why Alcoa Keeps Evolving ». septembre 2024. [En ligne]. Disponible sur : https://ispe.org/pharmaceutical-engineering/ispeak/dynamic-data-integrity-why-alcoa-keeps-evolving
[23] J. C. Mills-Baker Michalle Adkins, Hilary, « Achieving Data Integrity, Quality & Compliance in Manufacturing », Emerson Automation Experts. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.emersonautomationexperts.com/2023/industry/life-sciences-medical/achieving-data-integrity-quality-compliance-manufacturing/
[24] Sia Chong Hock, Chan Lai Wah, Vernon Tay & Vimal Sachdeva, « Pharmaceutical Data Integrity : issues, challenges and proposed solutions for manufacturers and inspectors », GaBIJ. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://gabi-journal.net/pharmaceutical-data-integrity-issues-challenges-and-proposed-solutions-for-manufacturers-and-inspectors.html
[25] « Life Sciences Product Lifecycle Management (PLM) with SAP », Crescense - SAP Partner and Value-Added Reseller. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.crescenseinc.com/insights/plm-with-sap
[26] « ISO 13485:2016 – Medical devices — Quality management systems — Requirements for regulatory purposes, International Organization for Standardization », ISO. Consulté le : 16 décembre 2025. [En ligne]. Disponible sur : https://www.iso.org/standard/59752.html
[27] csdmed, « ISO 14971 : FMEA, FTA and risk analysis for devices », CSDmed. Consulté le : 8 novembre 2025. [En ligne]. Disponible sur : https://www.csdmed.mc/en/news/medical-devices-regulation/risk-management-fmea-fta-132
[28] QBD Group, « How to Validate Your QMS : A Practical Guide for Pharma Professionals ». Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.qbdgroup.com/en/blog/validate-qms-pharma-guide
[29] « VDE - the technology organization ». Consulté le : 5 janvier 2026. [En ligne]. Disponible sur : https://www.vde.com/en
[30] O. Freyer, F. Jahed, M. Ostermann, C. Rosenzweig, P. Werner, et S. Gilbert, « Consideration of Cybersecurity Risks in the Benefit-Risk Analysis of Medical Devices : Scoping Review », J. Med. Internet Res., vol. 26, p. e65528, déc. 2024, doi : https://doi.org/10.2196/65528.
[31] UBAQ, « Simplification des processus réglementaires : 6 raisons essentielles de les digitaliser ». Consulté le : 20 septembre 2025. [En ligne]. Disponible sur : https://www.ubaq.io/fr/post/simplification-processus-reglementaires-raisons-essentielles-digitaliser
[32] « Public Health ». Consulté le : 5 janvier 2026. [En ligne]. Disponible sur : https://www.santepubliquefrance.fr/surveillance-syndromique-sursaud-R/documents/bulletin-national/2026/sante-mentale.-bulletin-mensuel-du-5-janvier-2026
[33] « How Can an eQMS Help Comply with EU MDR ? - Dot Compliance ». Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.dotcompliance.com/blog/medical-device-manufacturing/how-can-an-eqms-help-comply-with-eu-mdr/?utm
[34] « Rumb | La SAC ou PMS : surveillance après commercialisation des dispositifs médicaux ». Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://rumb.fr/la-sac-surveillance-apres-commercialisation-des-dispositifs-medicaux
[35] G. Samadrita, « Use of Artificial Intelligence in Medical Devices for Post-Market Surveillance », août 2025,Disponible sur : https://ijsrm.net/index.php/ijsrm/article/view/5630
[36] N. Merdović, L. Spahić, M. Hundur, L. G. Pokvić, et A. Badnjević, « Advancement of post-market surveillance of medical devices leveraging artificial intelligence : Infusion pumps case study », Technol. Health Care Off. J. Eur. Soc. Eng. Med., vol. 33, no 2, p. 915‑921, mars 2025, Disponible sur : https://pubmed.ncbi.nlm.nih.gov/40105162/
[37] Registrar Corp, « Surveillance post-commercialisation de l’UE : Une analyse approfondie de la conformité MDR », Registrar Corp. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.registrarcorp.com/fr/blog/medical-devices/medical-device-regulations/eu-mdr-postmarket-surveillance/
[38] MasterControl, « Medical Device eQMS - Quality Management for Life Sciences », MasterControl. Consulté le : 12 décembre 2025. [En ligne]. Disponible sur : https://www.mastercontrol.com/library/quality/medical-device-eqms
[39] Ptc, « Adoption réussie du PLM | PTC ». Consulté le : 21 septembre 2025. [En ligne]. Disponible sur : https://www.ptc.com/fr/resources/plm/ebook/successful-plm-adoption-ppc
[40] « Solution PLM pour l’industrie des dispositifs médicaux | Aras ». Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://aras.com/fr-fr/industries/medical-devices
[41] Forterro, « Traçabilité dispositifs médicaux : l’ERP comme solution clé | Proconcept ». 2025. Consulté le : 18 novembre 2025. [En ligne]. Disponible sur : https://www.proconcept.ch/fr/blog/tracabilite-dispositifs-medicaux-erp
Appendices
Appendices 1 Diagramme d'Ishikawa pour identifier les causes racines du frein 1 (coût de mise en place)
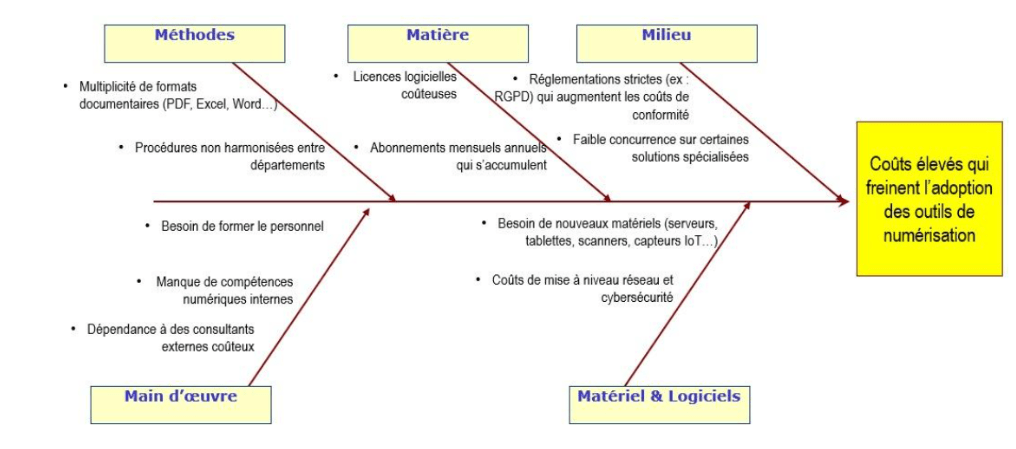
Appendices 2 Diagramme d'Ishikawa pour identifier les causes racines du frein 1 (coût de mise en place)