
IDS197 - Mise en conformité du dossier technique d'un DM de classe I aux exigences du RDM 2017/745 et internalisation d'un processus de veille réglementaire
Catégories
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteur

Steve Harison NTOMEGNE TCHIMI
Contacts
- Harison Tchimi : harisonentomegne@yahoo.fr
Citation
SH.NTOMEGNE TCHIMI « Mise en conformité du dossier technique d'un DM de classe I aux exigences du RDM 2017/745 et internalisation d'un processus de veille réglementaire », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Parcours Dispositifs médicaux et affaires réglementaires (DMAR), Mémoire d'apprentissage, https://travaux.master.utc.fr/, réf n° IDS197, juillet 2023, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids197/
Résumé
L’entrée en vigueur du RDM EU 2017/745, à renforcer les étapes à suivre pour la commercialisation des dispositifs médicaux en Europe, de nouvelles exigences ont vu le jour, notamment celles sur la traçabilité des dispositifs médicaux avec l’instauration de l’UDI, du suivi après commercialisation, etc.… poussant ainsi les fabricants de dispositifs médicaux à mettre à jour la documentation technique de leurs DM. Des questions se posent maintenant pour ceux-ci, notamment : Quelle stratégie suivre, pour mettre en conformité son dispositif médical aux exigences nouvelles du RDM et surtout, comment s’assurer de la couverture de toutes les exigences ? Dans le contexte d’une entreprise en plein renouvellement du marquage CE pour son dispositif médical, la mission a été d’accompagner le service qualité et affaires réglementaires à répondre à ce questionnement.
Ce mémoire d’apprentissage a pour but d’accompagner les fabricants de dispositifs médicaux dans la mise en conformité du dossier technique de leurs DM au RDM EU 2017/745 pour leur commercialisation en Europe. Il fournit également la voie à suivre pour construire un processus de veille réglementaire au sein d’une entreprise.
Abstract
The entry into force of MDR EU 2017/745 has strengthened the steps to be taken when marketing medical devices in Europe. New requirements have been introduced, in particular those relating to the traceability of medical devices, with the introduction of UDI, post-marketing follow-up, etc...., prompting medical device manufacturers to update the technical documentation for their medical devices. Questions now arise for manufacturers, in particular : what strategy should they follow to ensure that their medical devices comply with the new requirements of the MDR and, above all, how can they be sure that all the requirements are covered?In the context of a company in the process of renewing the CE marking for its medical device, the assignment was to help the quality and regulatory affairs department answer these questions.
The aim of this learning memorandum is to guide medical device manufacturers in ensuring that the technical file for their MDs complies with the MDR EU 2017/745 for marketing in Europe. It also provides a pathway for building a regulatory intelligence process within a company.
Téléchargements

Mémoire de fin d'étude sur la mise en conformité d'un dispositif médical de classe I aux exigences du RDM EU 2017/745 et internalisation d'un processus de veille réglementaire au sein d'une entreprise.
Outil d'évaluation de la conformité d'un dossier technique aux exigences du RDM EU 2017/745, basé sur son Annexe I.
Outil d'évaluation de la conformité d'un dossier technique aux exigences du RDM EU 2017/745 basé sur son Annexe II
Liste des abréviations
ANSM : Agence nationale de sécurité́ du médicament et des produits de santé
ATM : Articulations temporo-mandibulaires
DM : Dispositif médical
DT : Dossier technique / Documentation technique
EGSP : Exigences générales de sécurité et de performance
FDA : Food and drug administration
HAS : Haute autorité de santé
MDD : Médical devise Directive 93/42/CEE ou Directive des dispositifs médicaux 93/42/CEE
Md : Milliards
OMS : Organisation mondiale de la santé
ON : Organisme notifié
PMS : Post market surveillance/ Suivi post-marché́
PME : Petite et moyenne entreprise
QARA : Qualité et affaires réglementaires
RDM : Règlementation des dispositifs médicaux 2017/745
SMQ : Système de management de la qualité́
SNITEM : Syndicat national de l’industrie des technologies médicales
SAC : Suivi après commercialisation
TIM : Tech in Motion
TWIM : TWIN in Motion
UDI : Unique Device Identifier
UE : Union Européenne
UTC : Université́ de Technologie de Compiègne
Liste des figures
Figure 1 : Schéma de la chaine numérique en dentisterie
Figure 2 : Position des articulations temporo-mandibulaires (ATM) chez l’homme
Figure 3 : Capture d’écran du logiciel TWIn In motion
Figure 4 : Chariot opérationnel du TIM Hardware
Figure 5 : Stylet du kit patient
Figure 6 : Traqueur frontal et bandeau arrière du kit patient
Figure 7 : Fourchette du kit patient
Figure 8 : Traqueur mandibulaire du kit patient
Figure 9 : Marqueurs réflecteurs du kit patient
Figure 10 : Illustration du déroulé de l’enregistrement du mouvement mandibulaire
Figure 11 : Matrice Menaces, Opportunités, Forces et Faiblesses de MODJAW
Figure 12 : Organigramme du service qualité et affaires réglementaires de MODJAW
Figure 13 : Principaux éléments constitutifs d’un dossier technique
Figure 14 : Calendrier des dispositions transitoires du RDM UE 2017/745
Figure 15 : Extrait de la matrice de traçabilité des exigences de l’annexe 1 du RDM 2017/745
Figure 16 Cartographie de processus d’une veille réglementaire
Figure 17 : Le Check Act Plan Do de l’internalisation de la veille réglementaire
Figure 18 : Illustration du formulaire d’analyse d’écart
Liste des tableaux
Tableau 1 : Kit patient du TIM hardware
Tableau 2 : Principaux concurrents de MODJAW
Tableau 3 : Les types de veille réglementaire et normative
Tableau 4 : Mesure des indicateurs de performance du processus de veille réglementaire
Mémoire complet
Mise en conformité du dossier technique d'un DM de classe I aux exigences du RDM 2017/745 et internalisation d'un processus de veille réglementaire
Introduction
Les nombreux scandales et défaillances du système de contrôle et de commercialisation des dispositifs médicaux en Europe [1], ont conduit la commission européenne à mettre sur place en début 2017, un nouveau règlement pour les dispositifs médicaux le RDM EU 2017/745, dans le but de renforcer les exigences de la directive 93/42/CEE et garantir la sécurité des patients [2],[3].
Le marché du dispositif médical est évalué à environ 260 Md $ dans le monde, et en France il représente environ 20 Md € [4]. On estime le nombre de dispositifs médicaux utilisés en France entre 800 000 et 2 millions, et ceux dans toutes les spécialités de la médecine dont la dentisterie [4]. En effet, le dispositif médical occupe une place importante dans ce domaine car il a permis dès le début des années 1895, avec la découverte des rayons X de faciliter et d’améliorer l’exploration de la mâchoire pour diagnostiquer les pathologies dentaires. De plus, les nouvelles technologies et matériaux utilisés pour restaurer ou remplacer les dents ont permis d’améliorer les traitements [5].
Néanmoins, il reste beaucoup à faire, car selon l’OMS les pathologies dentaires représentent le 3ème fléau mondial après les maladies cardiovasculaires, on estime que près de 3,5 milliards de personnes sont touchées. De plus les couts liés aux soins dentaires sont estimés à 17 milliards d’euros [6]. Pour répondre à ces problématiques, de nombreuses innovations ont été lancées en dentisterie, notamment la numérisation des outils et méthodes de diagnostic et de conception de prothèses, afin d’améliorer et réduire les temps de traitement, pour ainsi faire baisser les dépenses de santé liées aux pathologies dentaires. C’est le cas de MODJAW qui propose un dispositif médical : le TWIN In Motion (TWIM) pour l’aide au diagnostic et au traitement des troubles de l’ATM (articulations temporo-mandibulaires) [5][7].
Dans le cadre de la mise en conformité au RDM EU 2017/745, de ce dispositif, MODJAW a lancé les projets suivants :
- Mettre en conformité la documentation technique du TWIM de classe IIa pour l’obtention du marquage CE sous Règlement EU 2017/745, avant la fin de sa période de transition initialement prévue en 2024, mais repoussé au 31 décembre 2028 tout récemment selon le règlement (UE) 2013/607 [8][9].
- Mettre en conformité la documentation technique de l’accessoire TIM Hardware de classe I pour son auto certification sous RDM 2017/745. C’est ce projet qui sera principalement détaillé dans ce mémoire.
Néanmoins, d’autres missions liées au fonctionnement du service qualité et réglementaire au quotidien y seront également présentées. Principalement celle d’internalisation complète du processus de veille réglementaire au sein du service QARA de l’entreprise, dont une partie était sous-traité, afin de conformer les produits à l’état de l’art réglementaire et normatif.
Les problématiques qui se dégagent de ces projets sont :
- Quelle stratégie à adopter pour mettre en conformité la documentation technique du TIM Hardware de classe I ?
- Comment internaliser le processus de veille réglementaire au sein de l’entreprise MODJAW anciennement sous-traité ?
Les objectifs de ce mémoire seront donc de :
- Proposer une stratégie à suivre pour la mise en conformité d’un dispositif médical de classe I aux exigences du RDM EU 2017/745.
- Proposer une méthode à suivre pour constituer un processus de veille réglementaire interne en entreprise.
I. La dentisterie une discipline transformée par les dispositifs médicaux numériques
La santé numérique est une approche de la médecine qui consiste à utiliser les outils numériques dans le processus de soin, elle comprend l’informatisation des processus et des méthodes de traitement, la numérisation des documents médicaux mais aussi l’utilisation de dispositifs tels que les logiciels en tant que dispositifs médicaux [10], [11].
La dentisterie quant à elle, est l’ensemble des connaissances et thérapies relatives aux dents et à leur environnement, elle connaît aujourd’hui de nombreuses évolutions numériques qui ont fait naitre la dentisterie numérique ou digitale [10].
1. La dentisterie numérique
La dentisterie numérique est l’utilisation d’outils numériques tels que des ordinateurs ou des équipements contrôlés par un ordinateur dans la prestation de soins dentaires, pour principalement compléter un diagnostic et/ou un examen clinique [12].
Les nouvelles techniques de dentisterie numérique ont gagné en popularité chez les dentistes depuis les dernières années car elles ont permis de réduire les temps de travail, et de remplacer les dispositifs plus anciens essentiellement mécaniques qui présentaient des risques d’erreurs. Plus globalement, elles ont permis de convertir les flux de travail en chaînes de processus numériques couvrant toutes les étapes de soin, allant de l’acquisition (avec les scanners intra-oraux) au design de prothèses (avec les logiciels de CFAO) et jusqu'à leur production, afin d’améliorer les traitements en réduisant les risques d’erreurs [10], [12] (voir figure 1).
En effet, grâce à ces technologies, les restaurations dentaires peuvent être maintenant conçues sur ordinateur, contrairement aux méthodes conventionnelles (articulateurs mécaniques) éloignés du cadre biologique réel du patient et qui utilisaient des plâtres générant de nouveaux contacts.
Figure 1 : Schéma de la chaine numérique en dentisterie
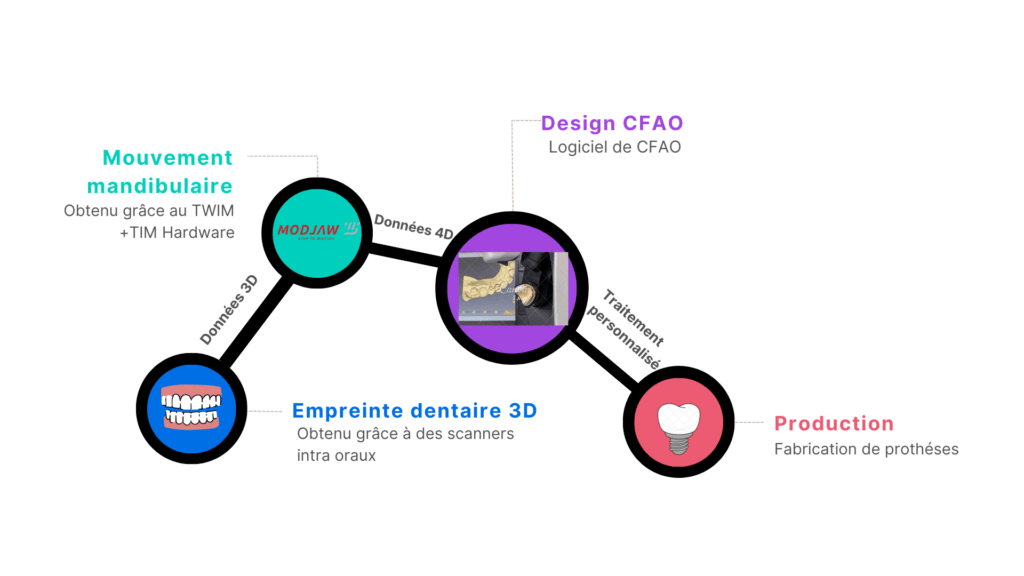
Cette chaîne numérique est issue des principales évolutions technologiques suivantes :
2. Les principales évolutions technologiques en dentisterie numérique
- Les scanners intra oraux : C’est une technologie reposant sur l’utilisation d’un faisceau de lumière (optique) en éventail projeté sur la surface des dents du patient et dont les faisceaux réfléchis sont enregistrés par une caméra CCD sous forme de signaux numériques transmis à un logiciel pour la reconstruction de l’empreinte numérique dentaire du patient, visualisable sur un écran. Cette empreinte numérique dentaire du patient sera ensuite utilisée dans toute la chaine numérique, notamment pour la simulation du mouvement mandibulaire du patient et la conception sur ordinateur de prothèses. C’est l’élément de départ de la chaine numérique [10].
- Les appareils d’enregistrement du mouvement mandibulaire : récente innovation en dentisterie, elle est née du besoin des dentistes d’avoir le mouvement réel de la mâchoire couplé aux empreintes dentaire du patient, pour obtenir un meilleur diagnostic et favoriser la création de prothèse adapté à la cinématique mandibulaire. Plus concrètement, ce sont des outils qui grâce à la capture et l’analyse de la dynamique mandibulaire, aident au diagnostic, à la caractérisation et au traitement des schémas occlusaux en travaillant sur l’empreinte dentaire du patient et en y ajoutant la notion de temps à la troisième dimension (Données 4D) [10]. C’est le cas des dispositifs médicaux de MODJAW
- Les logiciels de design CFAO : la CFAO est une technologie de conception sur ordinateur qui consiste à créer des pièces ou des équipements grâce à un logiciel informatique de dessin. En dentisterie, elle offre la possibilité aux dentistes grâce à une image de la dentition du patient obtenue soit par scanner optique ou par radiologie numérique de commander instantanément à un ordinateur la création de prothèse, au lieu de procéder à la confection d’un moule, comme il était fait précédemment [10], [12]. La CFAO permet donc des interventions plus rapides, tout en améliorant l’expérience patient. En effet, grâce à la CFAO, les restaurations dentaires peuvent être maintenant réalisées sur site le même jour de la visite du patient. De plus, il est maintenant possible d’obtenir des empreintes numériques patient qui seront ensuite utilisées par les logiciels CFAO [13].
3. L'enregistrement du mouvement mandibulaire une problématique au coeur de l'innovation Modjaw
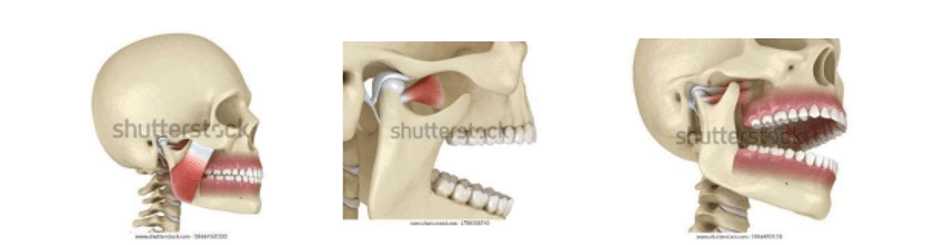
Le système masticatoire est une partie importante du complexe crânio-facial, les articulations temporo-mandibulaires (ATM), avec la dentition, les muscles masticateurs, les systèmes vasculaires et nerveux qui alimentent ces tissus constituent ses principaux composants (voir figure 2). Le mouvement mandibulaire quant à lui, est un mouvement exécuté par l’articulation temporo-mandibulaire et reflète la commande de tous les composants du système masticatoire et est essentiel pour une bonne communication orale [14].
L’enregistrement des mouvements masticatoires a été effectué par les gnathologues et a permis de comprendre la fonction normale du système stomatognathique et de diagnostiquer, puis traiter les maladies de l’ATM telles que les troubles temporo-mandibulaires (TDM) [14].
Cependant, ce système a longtemps été analysé, mais les méthodes utilisées enregistraient les points statiques ou des positions uniques de la mandibule (par exemple protrusion, excursion, etc…) alors que le système masticatoire est dynamique et sa composante principale réside dans la mandibule.
Figure 2 : Position des articulations temporo-mandibulaires (ATM) chez l'homme source : www.shutterstock.com
En effet, Pour simuler le mouvement mandibulaire de leurs patients, les dentistes ont longtemps utilisé des articulateurs mécaniques, dans le but de pouvoir diagnostiquer des malocclusions ou des altérations morphologiques de l’occlusion dentaire. Cependant ces dispositifs mécaniques étaient très éloignés du cadre biologique réel, et présentaient de nombreux problèmes :
- Les mouvements reproduits par les articulateurs mécaniques suivent les marges des structures qui le forment, ceux-ci restent invariables dans le temps et dont ne peuvent parfaitement simuler les mouvements de la mâchoire qui dépendent du schéma musculaire et la résilience des tissus mous.
- Les modèles en plâtres utilisés pour représenter la dentition du patient dans les articulateurs mécaniques ne peuvent pas simuler la mobilité dentaire. Ils sont donc incapables de reproduire les conditions dynamiques réelles d’occlusion.
- Les articulateurs mécaniques génèrent de nouveaux contacts lors de simulation du mouvement mandibulaire.
- La fiabilité de la reproduction des mouvements est faible [15].
Pour éviter toutes ces erreurs des articulateurs mécaniques, il a été développé des appareils de capture du mouvement dentaire (Jaw Motion). Ces outils permettent de capturer et de retranscrire précisément la position du modèle maxillaire, la position mandibulaire du patient au cours de son mouvement volontaire ou suggéré, ils permettent ainsi l’adaptation des morphologies dentaires lors de la conception numérique par lecture des mouvements réels du système mandibulaire du patient enregistré et non plus par simulation [16]. Parmi ces outils nous avons des :
- Dispositifs à ultrason ou optique : ils sont constitués d’une source d’ultrason ou optique, lié à la surface vestibulaire de la mandibule du patient au moyen d’un accessoire personnalisé, et d’un système de capteur logé dans un autre accessoire monté autour du patient sous forme d’un arc. Ils permettent de détecter les mouvements masticatoires du patient et de les enregistrer [16].
- Dispositifs optoélectroniques qui utilisent des caméra CCD pour enregistrer les émissions de diodes électroluminescentes positionnés au-dessus de la tête du patient et gère une image à partir des signaux [16].
- Dispositifs à caméra optique comme celui de MODJAW.
La virtualisation de l’empreinte dentaire, l’enregistrement de l'occlusion et la construction virtuelle des prothèses dentaires est aujourd’hui une réalité grâce à toutes ces évolutions de la dentisterie numérique [16].
II. MODJAW, un révolutionnaire en dentisterie numérique
1. Le dispositif médical commercialisé par MODJAW : le Twin In Motion + Tech In Motion Hardware
Le premier dispositif médical commercialisé par Modjaw était le Tech in motion : un dispositif médical sous forme de système composé d’un chariot, d’un logiciel et d’un Kit patient. Suite à de nombreuses modifications sur le logiciel du Tech in Motion pour constamment l’améliorer, et l’entrée en vigueur du RDM EU 2017/745, Modjaw décide en 2021 d’en faire un dispositif médical logiciel à part entière, le TWIN in motion accompagnée de son accessoire le Tech in motion Hardware (constitué d’un chariot opérationnel et d’un Kit patient) pour le faire fonctionner.
a) Le logiciel Twin In Motion
Le TWIN in motion est un dispositif médical de classe IIa sous le RDM, destiné à être utilisé avec son accessoire matériel le Tech in Motion Hardware pour l’enregistrement et l’analyse de la cinématique mandibulaire afin d’aider au diagnostic, à la caractérisation et à la planification des schémas d’occlusion (voir figure 3).
Ses bénéfices cliniques sont tout aussi multiples :
- Aide à générer des traitements restaurateurs et orthodontiques pertinents en se basant sur l’anatomie réel du patient
- Réduit la probabilité d’une erreur sur la restauration définitive ce qui améliore le bien-être du patient
- Assiste les spécialistes dans leurs diagnostics et leurs traitements des troubles temporo-mandibulaires
- Réduit le temps nécessaire aux traitements
Figure 3 : Capture d'écran du logiciel Twin in Motion (source : www.modjaw.com)
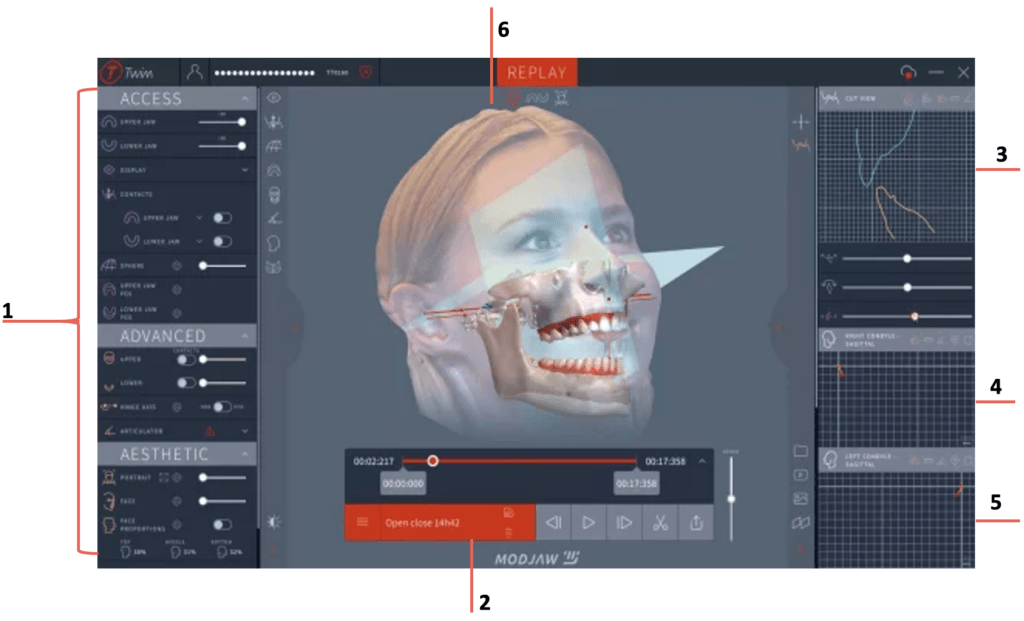
| 1 | Les différents modules du logiciel : Access : comporte les fonctionnalités basiques comme l’enregistrement de la cinématique mandibulaire Advanced : comporte des fonctionnalités avancées comme l’analyse des trajectoires Aesthetic : comporte les fonctionnalités esthétiques comme l’importation des face scans du patient |
| 2 | Lecteur de mouvement |
| 3 | Graphe de l’analyse des trajectoires mandibulaire |
| 4 | Graphe de l’analyse des trajectoires mandibulaire droits |
| 5 | Graphe de l’analyse des trajectoires mandibulaire gauches |
| 6 | Affichage du face scan du patient |
b) Le Tech In Motion Hardware (TIM Hardware): l'accessoire du dispositif médical Twin In Motion
Concernant le Tech in motion Hardware, c’est un accessoire de dispositif médical de classe I, il est composé de 2 grands éléments distincts fonctionnant en synergie pour l’enregistrement des mouvements mandibulaires :
- Un chariot opérationnel : c’est l'élément central du dispositif, il est constitué d’un Panel PC tactile porté par une potence, dans lequel est installé le TWIM et d’une caméra haute définition sur un bras articulé, pour permettre l’enregistrement, le stockage et la lecture du mouvement mandibulaire (voir figure 4).
Figure 4 : Chariot opérationnel du TIM Hardware (source : www.modjaw.com)

- Un kit patient constitué des éléments comportant des marqueurs réflecteurs suivant (voir tableau 1) :
Tableau 1 : Kit patient du TIM Hardware (source : www.modjaw.com)

c) Déroulé de l'enregistrement de la cinématique mandibulaire grace au TWIM + Tech In Motion Hardware
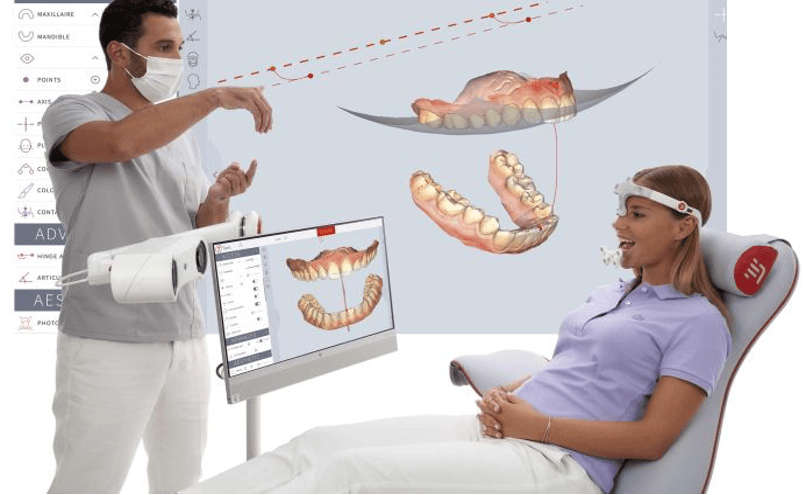
Pour procéder à l’enregistrement des mouvements mandibulaires d’un patient grâce au TWIM + Tech in Motion Hardware, il doit être préalablement positionné sur un fauteuil dentaire et le logiciel TWIM lancé sur l’ordinateur. Ensuite les étapes à suivre sont les suivantes :
- Étape 1 : Les capteurs sont positionnés sur le patient (traqueur mandibulaire équipé de ses réflecteurs devant la bouche du patient grâce à une fourchette, et le traqueur frontal positionné sur la tête du patient) et la position ATM est repérée à l’aide du stylet (voir tableau 1). Avant chaque examen, la caméra, le stylet sont calibrés pour obtenir des résultats (voir figure 10).
- Étape 2 : Une fois le patient préparé, le praticien importe le modèle 3D de la dentition du patient (obtenu grâce un scanner optique ou autre) dans le logiciel, sur lequel il identifie des points anatomiques à l’aide du stylet.
- Étape 3 : le patient est ensuite invité à effectuer une série de mouvement (ouverture, fermeture) qui sont enregistré et peuvent être rejoué selon l’envie du praticien, pour analyser les mouvements et établir son diagnostic (voir figure 10).
Figure 10 : Illustration du déroulé de l'enregistrement du mouvement mandibulaire
(source : www.modjaw.com)
2. Environnement concurrentiel de Modjaw
L’enregistrement de la cinématique mandibulaire est une problématique qui intéresse de nombreux fabricants de dispositifs médicaux. Parmis ceux-ci nous avons (voir tableau 2):
Tableau 2 : Principaux concurrents de MODJAW (source Auteur)
MODJAW est une PME située en Auvergne Rhône Alpes, fondée en 2013 par deux cousins, un dentiste et un entrepreneur. Après plusieurs années de recherche et développement, ils ont lancé leur premier dispositif médical le Tech In Motion commercialisé dés 2018 [20] (voir figure 11).
3. Historique, organisation et structure
C’est une société qui compte aujourd’hui une quarantaine de collaborateurs et qui est en plein développement en France et à l’international.
Ses principales activités sont la production et la commercialisation d’un dispositif médical de dentisterie le TWIN In Motion et son accessoire le Tech In Motion (TIM) Hardware destiné à visualiser, enregistrer et analyser directement les mouvements masticatoires du patient. Elle fournit aussi des formations aux professionnels médicaux à l’utilisation de son dispositif médical etc… [20]
Étant situé en région lyonnaise (2e pôle de développement en santé de France) [4], Modjaw jouit d’une forte attractivité et ambitionne d’ici 2024 avec la levée de nouveaux fonds de développer de nouvelles versions de son dispositif médical et de conquérir de nouveaux marchés dont le moyen orient. Sa stratégie est de continuer à faire connaître l’univers de la dentisterie digital à tous les dentistes de France et du monde entier, en les faisant basculer dans la dentisterie 4D.
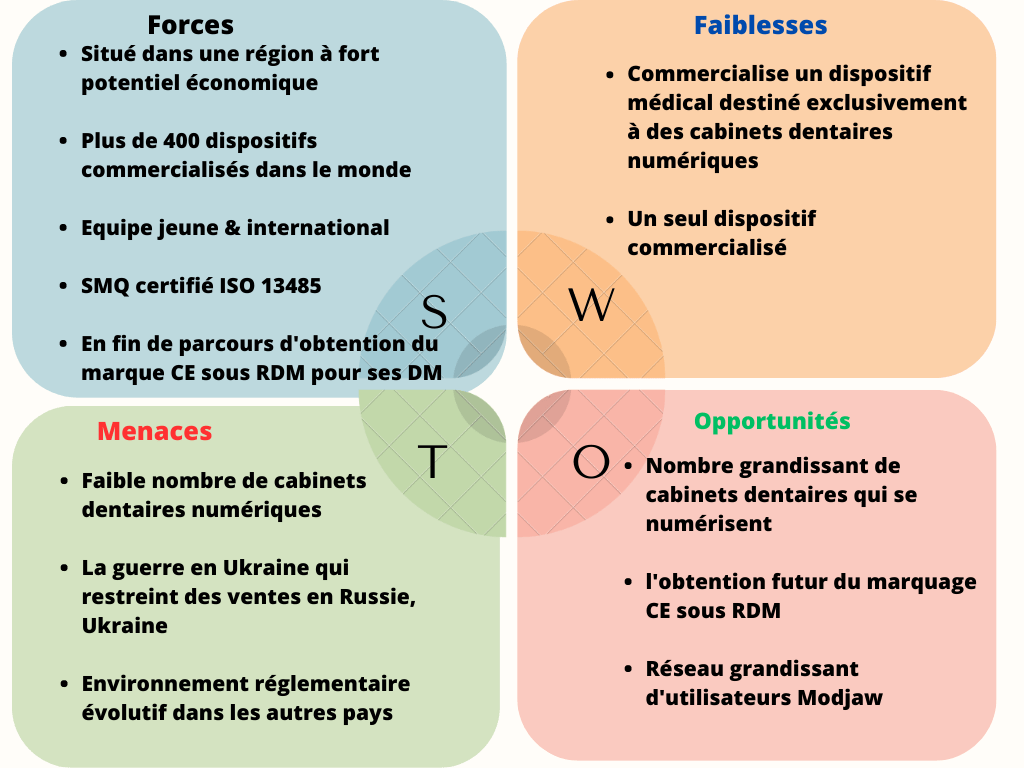
Figure 11 : Matrice Menaces,Opportunités, Forces et Faiblesses de MODJAW (source Auteur )
En termes d’organisation, Modjaw s’organise en différents services qui comprennent le service Marketing, le département clinique, le service des ventes, le département finance, le département Human ressources et le département QARA que j’ai intégré comme alternant en qualité et affaires réglementaires.
Le service Qualité et affaires réglementaire (QARA) est un service constitué de 5 personnes (voir figure 12) :
Figure 12 : Organigramme du service qualité et affaires réglementaires de MODJAW (source : Auteur)

- Un Manager d’équipe : qui est un ingénieur en qualité dont le rôle est de coordonner le fonctionnement du service, de collaborer avec les différents autres services de l’entreprise pour la bonne exécution du système de management de la qualité etc… Il est également le représentant de la direction du point de vue qualité et la personne chargée de veiller au respect de la réglementation (PCVRR).
- Un ingénieur qualité et affaires réglementaire : c’est lui, qui de façon opérationnelle déploie le système de management de la qualité en constituant les procédures, formulaires etc… et veille aussi aux respect des exigences réglementaires.
- Une chargé des affaires cliniques : c’est la personne en charge des affaires cliniques concernant le dispositif médical, elle s’occupe de la constitution des différents éléments pour l’évaluation clinique du dispositif médical.
- Une spécialiste en affaires réglementaires export : elle est chargée des activités d’enregistrement du dispositif médical à l’étranger et est le garant du respect des réglementations des pays où le DM est commercialisé.
- Un alternant en qualité et affaires réglementaires : ses missions sont d’être le support des différents personnes constituants l’équipe QARA, mais il est aussi en charge de la constitution du dossier technique du dispositif médical de classe I, de la veille réglementaire, et de l’amélioration du SMQ.
C’est un service en interaction avec tous les autres services de l’entreprise, afin d’assurer la prise en compte des exigences réglementaires et qualité dans : Le développement du produit jusqu’à sa mise en service.
III. Mise en conformité du dossier technique d'un DM de classe I aux exigences du RDM 2017/745 pour le maintien de sa commercialisation
1. Les étapes de la constitution d'un dossier technique conforme aux exigences spécifiques
La commercialisation d’un dispositif médical en Europe nécessite au préalable qu’il dispose d’un marquage CE, gage de sa conformité aux exigences du RDM 2017/745 et de sa performance, et sécurité pour les patients. L’élément essentiel pour l’obtention de ce marquage, est la constitution du dossier technique, regroupant toutes les informations sur le dispositif médical durant tout son cycle de vie. Il a pour objectif de :
- Démontrer sa conformité aux exigences du règlement EU 2017/745
- Présenter le dispositif médical, son fonctionnement etc…
- Refléter la vie du dispositif médical durant son utilisation en pratique de soin courante.
- Suivre les évolutions de l’état de l’art sur le dispositif médical et les dispositifs similaires et/ou équivalent, pour sans cesse l’améliorer (voir figure 13).
Plus globalement, c’est la fiche de vie du dispositif médical, et l'élément principale sur lequel les organismes notifiés se basent pour s’assurer de sa conformité au RDM 2017/745. En effet, les ON évaluent le dossier technique du DM et le système de management de la qualité mis en place pour le concevoir (dans le cas des DM de Classe IIa, IIb, III) afin d’attribuer le marquage CE. Et pour ceux de classe I ne nécessitant pas d'évaluation par un ON, les fabricants de dispositifs médicaux ont quand même l’obligation de disposer d’un dossier technique pour leurs DM.
C’est une exigence qui existe depuis la Directive 93/42/CEE (Annexe VII) et dont le règlement EU 2017/745 élargie et augmente les éléments à y fournir pour s’y conformer. Les éléments constitutifs d’un dossier technique sont détaillés dans l’annexe II du RDM 2017/745 : (voir figure 13)
Figure 13 : Principaux éléments constitutifs d'un dossier technique (source : Auteur)
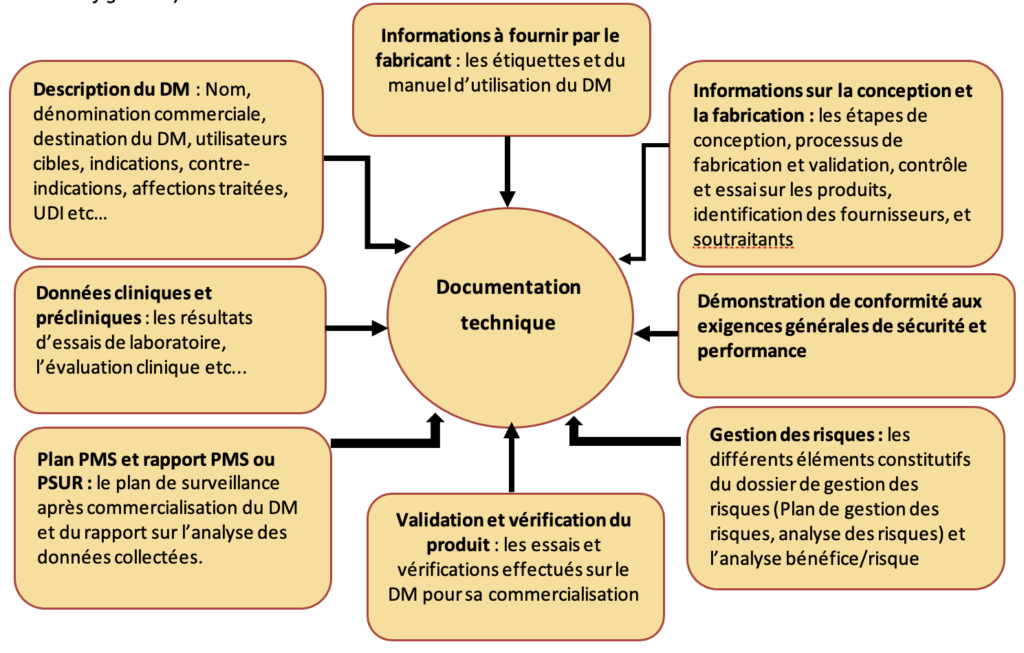
- L’UDI : Unique device identifier ou identifiant unique du dispositif en français, permet la traçabilité du dispositif médical durant tout son cycle de vie. Il est constituée d’un UDI-DI (identifiant du dispositif, intégrant l’identification du fabricant) et d’un UDI-PI (identifiant de production).
- Suivie après commercialisation ou Post Market Surveillance (PMS) en anglais : c’est le système par lequel un fabricant de dispositifs médicaux collecte et analyse les informations sur l’utilisation de son dispositif en vie réelle, pour notamment améliorer sa conception, sa sécurité et performance. Ces informations concernent toutes les réclamations clients, les questionnaires de satisfaction etc…
2. Stratégie de mise en conformité
Modjaw commercialise depuis 2018 son dispositif médical le Tech In Motion marqué CE sous la directive 93/42/CEE, qui possède déjà un dossier technique conforme aux exigences de l’annexe VII de celle-ci. Avec l’entrée en vigueur du règlement EU 2017/745 pour les dispositifs médicaux, les fabricants de DM sont contraints de mettre à jour leur documentation technique avant 2028 afin d’être autorisés à poursuivre la commercialisation.
La stratégie établie par Modjaw a d’abord été d'effectuer une analyse d’écart entre la directive 93/42/CEE notamment son annexe VII sur la documentation technique et l’annexe II du RDM. Cette analyse d’écart a permis de déterminer les exigences nouvelles à respecter pour la mise en conformité de son dossier technique, lesquelles sont les suivantes :
- Constitution d’un nouveau rationnel de classification du DM selon les nouvelles règles de classification du RDM (annexe VIII) ;
- Création des UDI pour l’identification et la traçabilité du dispositif médical ;
- Revue de la conformité des exigences générale de sécurité et de performance ;
- Évaluation clinique du dispositif médical selon (annexe XIV) ;
- Suivi après commercialisation (annexe XIII) ;
- Suivi clinique après commercialisation.
De premières actions ont été mises en œuvre pour conformer le TIM notamment la :
- Définition de l’arborescence de la documentation technique
Avant de constituer la documentation technique d’un dispositif médical, il faut définir son organisation, notamment une arborescence qui facilitera la recherche d’informations, qui regroupera les informations en fonction des exigences auxquelles elles répondent.
Pour définir l’arborescence de la documentation technique du TIM, Modjaw s'est basé sur le guide fourni par son organisme notifié, qui propose une arborescence organisée sous forme dossier, nommé selon les grandes parties de l’annexe I du RDM, facilitant ainsi la recherche et le regroupement des informations. Les éléments qui ne sont pas applicables pour MODJAW sont suivis d’un N/A (pour non applicable) (voir annexe 1).
- Reclassification du Tech in Motion (TIM) selon les règles du RDM 2017/745
La stratégie réglementaire prise pour la classification du DM, était de classer le logiciel TWIM différemment de son accessoire TIM hardware, comme le stipule la règle 3.2 de l’annexe VIII du règlement : “Si le dispositif en question est destiné́ à être utilisé en combinaison avec un autre dispositif, les règles de classification s'appliquent séparément à chacun des dispositifs. Les accessoires d'un dispositif médical et d'un produit énuméré́ à l'annexe XVI sont classés en soi, indépendamment des dispositifs avec lesquels ils sont utilisés.”
Le TWIM a donc été classé comme dispositif médical de classe IIa selon les règles 11 de l’annexe VIII et le TIM Hardware comme accessoire de DM de classe I selon la règle 1 et la règle 5 de l’annexe VIII. Suite à cela, Modjaw a décidé de créer une documentation technique pour chacun de ses dispositifs médicaux. Le Tech in motion bénéficiant de la période de transition vers le RDM, c’est à dire qu’il continue d’être commercialisé sous directive 93/42/CEE jusqu’en 2028, mais sous certaines conditions, de se conformer aux exigences en matière de vigilance, d’identification unique du DM (UDI), de surveillance après commercialisation, d’enregistrement sur la plateforme EUDAMED en suivant la chronologie de transition fixée par le règlement (voir figure 14):
Figure 14 : Calendrier des dispositions transitoires du RDM UE 2017/745 [9][3]
(source : Auteur)
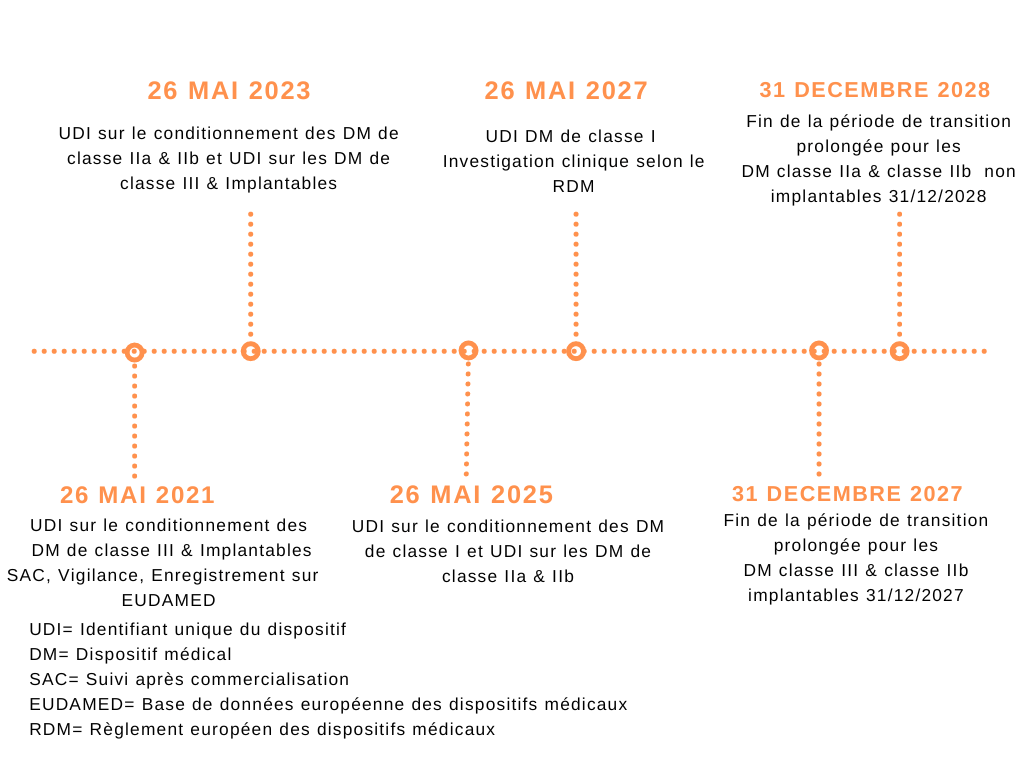
En suivant ce calendrier, la priorité a été donné à la conformité aux exigences suivantes pour le Tech in Motion :
- UDI : C’est une exigence que doivent respecter tous les dispositifs médicaux, pour s’y conformer, Modjaw a défini des UDI (UDI-DI et UDI-PI) pour les différents composants de son dispositif médical auprès de l’organisme GS1.
- Suivie après commercialisation : Pour répondre à cette exigence, Modjaw a mis en place une procédure pour la surveillance après commercialisation de son dispositif médical.
- Vigilance : c’est une autre exigence renforcée par le RDM, les fabricants de DM ont l’obligation d’assurer la matériovigilance, vigilance sur leurs DM afin de suivre les différents incidents sur celui-ci. En cas d’incident grave, ils ont l’obligation d’en faire une déclaration auprès de l’autorité compétente du pays où le DM est commercialisé dans un délais fonction du niveau de gravité de l’incident. Pour répondre à cette exigence, Modjaw a mis sur place une procédure de vigilance qui définit le traitement des cas d’incidents sur son DM, mais aussi les différentes actions à mettre en œuvre en cas d’incident grave.
- Enregistrement sur la Platform EUDAMED : EUDAMED est la base de données européenne pour les dispositifs médicaux, elle permet de suivre un dispositif médical mis sur le marché européen, de faciliter sa traçabilité, et mettre à disposition du public toutes les informations concernant le DM. Elle groupe les 6 modules suivant :
- Enregistrement des acteurs
- IUD et enregistrement des produits
- Organisme notifié et certificat
- Investigation clinique
- Vigilance et surveillance après commercialisation
- Surveillance des marchés
La platform Eudamed n’étant pas complètement opérationnel (3 modules sur 6 disponibles), Modjaw n’a commencé à se conformer qu’aux modules disponibles, notamment celui d’enregistrement des acteurs, et d’UDI et enregistrement des produits. Celui concernant les organismes notifiés et certificat est le 3e disponible mais il n’est pas applicable pour MODJAW.
Toutes ces actions n’ont permis de répondre qu’aux exigences liés à la période transitoire du RDM, et ne permettait pas d’assurer la conformité totale du dispositif médical à celui-ci. De plus, les actions menées concernaient prioritairement le TWIM, avec quelques actions pour le TIM Hardware.
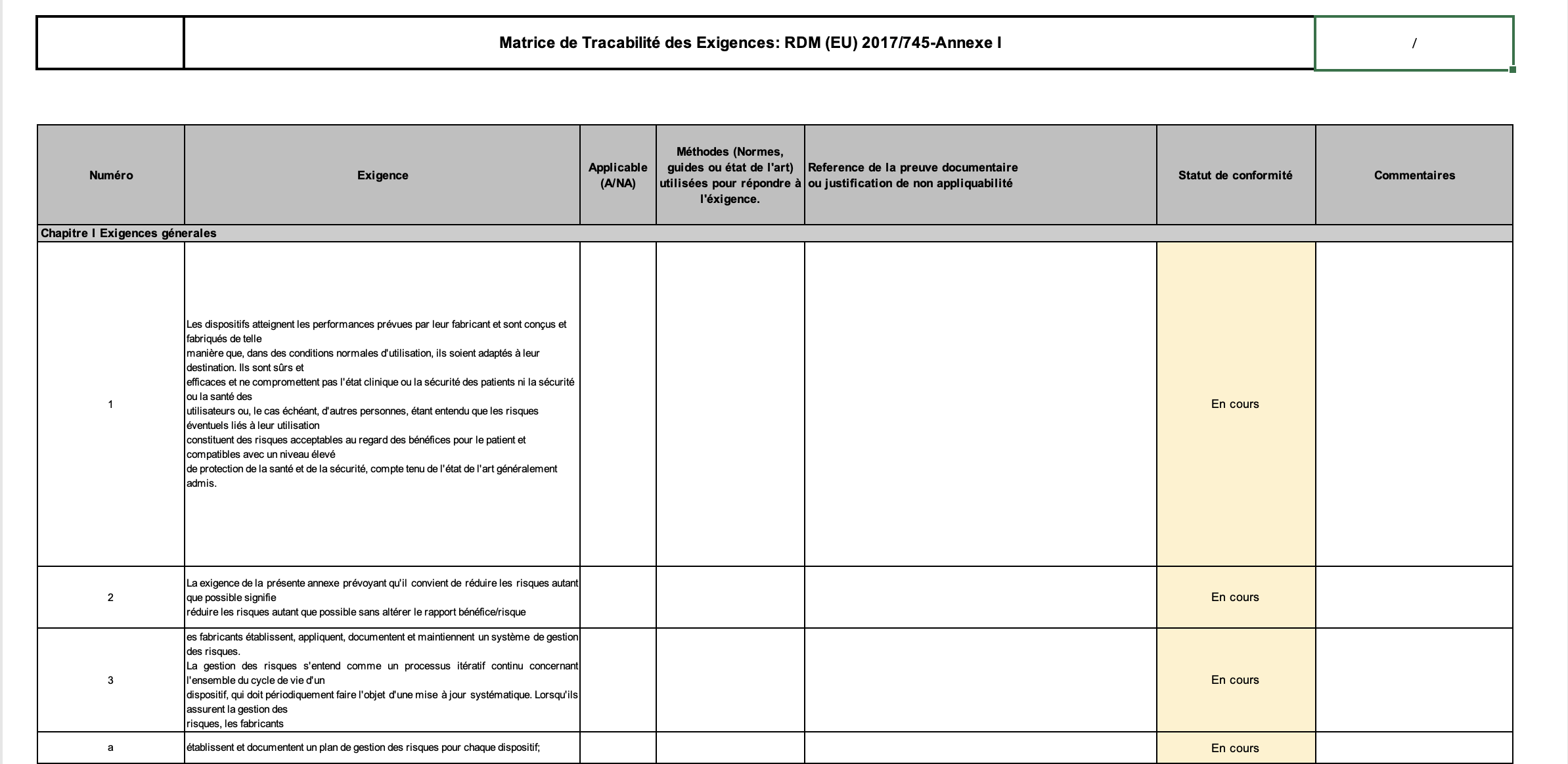
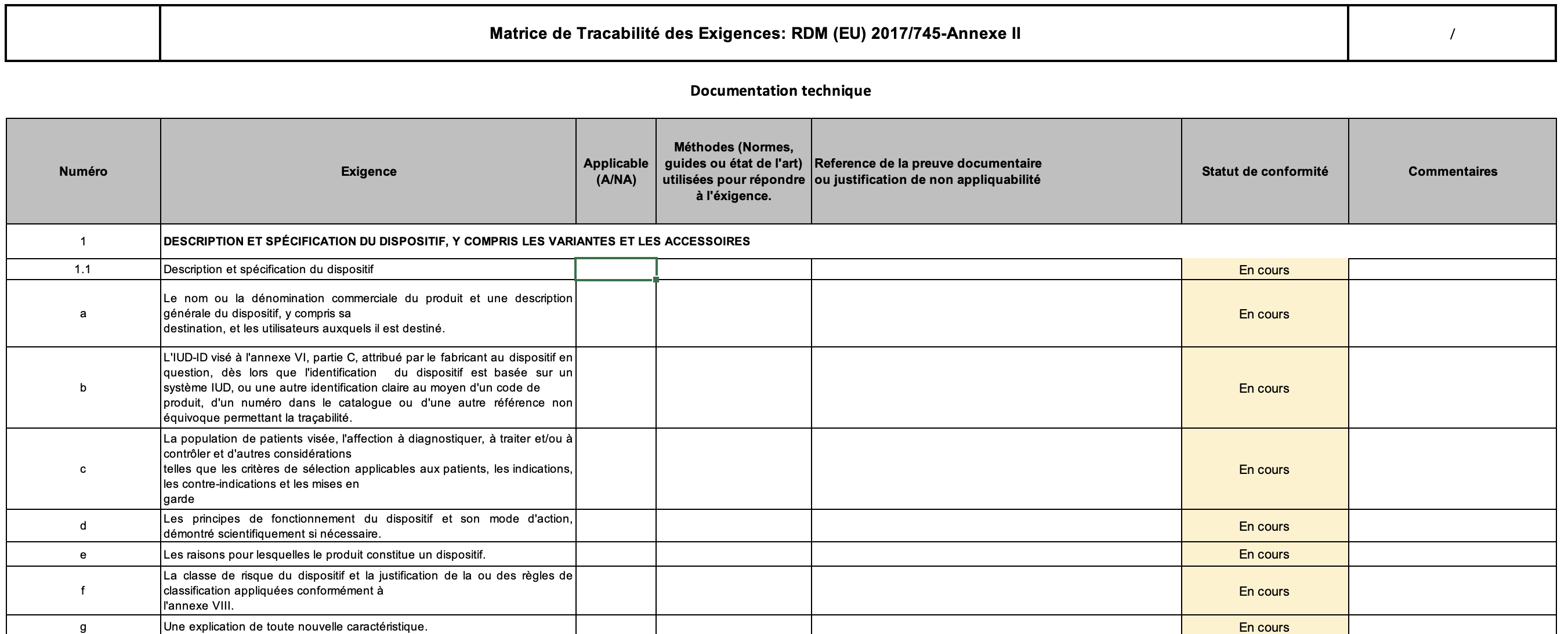
En fin de constituer le dossier technique du TIM Hardware, il a fallu évaluer l’état de sa documentation technique déjà constituée, et tracer les exigences dont la conformité avait déjà été démontrées et celles dont ça ne l’était pas. Pour cela, il a été constitué un outil de matrice de conformité des exigences du RDM (concernant l’annexe I et II).
Le choix de cet outil est basé sur sa simplicité d’utilisation et parce que c’est un outil déjà utilisé en entreprise et qui a prouvé son efficacité. Pour notre projet, la matrice de traçabilité a été adapté aux exigences de l’annexe I et II du RDM 2017/745.
3. Un outil pour l'évaluation de la conformité du dossier technique : la matrice de traçabilité des exigences du RDM 2017/745
Une matrice de traçabilité aux exigences est un outil permettant de tracer les exigences d’une norme ou d’une réglementation, avec les différentes actions mises en œuvre pour s’y conformer. Son objectif principal est de s’assurer que toutes les exigences sont respectées par des actions mises en place, afin que la conformité à la norme ou à la réglementation soit effective [21].
Comment l’utiliser ?
Une matrice de traçabilité aux exigences peut- être utilisé :
- En amont d’un projet, pour tracer toutes les exigences à respecter dans la réalisation du projet.
- En cours de réalisation du projet pour croiser les exigences aux actions en cours de réalisation.
- À la fin d’un projet pour évaluer si les exigences sont complètement respectées
Ses avantages ? :
C’est un outil qui :
- Aide à identifier les exigences à respecter dans un projet et à les faire correctement
- Aide à assurer la traçabilité des exigences de façon à enregistrer les éléments qui prouve leur respect
- Documente les liens entre les exigences et le système en construction
- En cas de départ d’un membre de l’équipe projet, elle permet d’assurer la transparence des informations, les conserve, et les rend disponible pour tous.
- Sert à confirmer la couverture des exigences à 100% via un processus de vérification approprié [21].
De part ces avantages, c’est un outil qui s’adapte bien au contexte de Modjaw, où de nombreux projets sont en cours, de plus, elle est applicable à de plusieurs projets à la fois afin de suivre les exigences à respecter.
Comment construire une matrice de traçabilité aux exigences ?
Pour constituer une matrice de traçabilité aux exigences, il faut commencer par déterminer son objectif, et le référentiel sur lequel elle se basera. Dans notre cas il s’agit de : Suivre les exigences du RDM afin de s’assurer qu'elles sont toutes respectées dans la documentation du TIM Hardware.
Ensuite, il faut choisir la forme ou le style de matrice de traçabilité, dans notre cas, nous avons opté pour un outil sous forme de tableau Excel regroupant toutes les exigences du RDM, car simple d’exploitation et de mise en œuvre. Il est organisé sous forme de colonnes, et se parcoure en se déplaçant à l’aide de sa souris entre les différentes pages du fichier.
Enfin, il faut compléter les différentes colonnes de l’outil pour suivre les exigences et les preuves de conformité associées.
Cet outil est illustré en figure 15 :
Figure 15 : Extrait de la matrice de traçabilité des exigences de l'annexe I du RDM 2017/745 (Source Auteur)
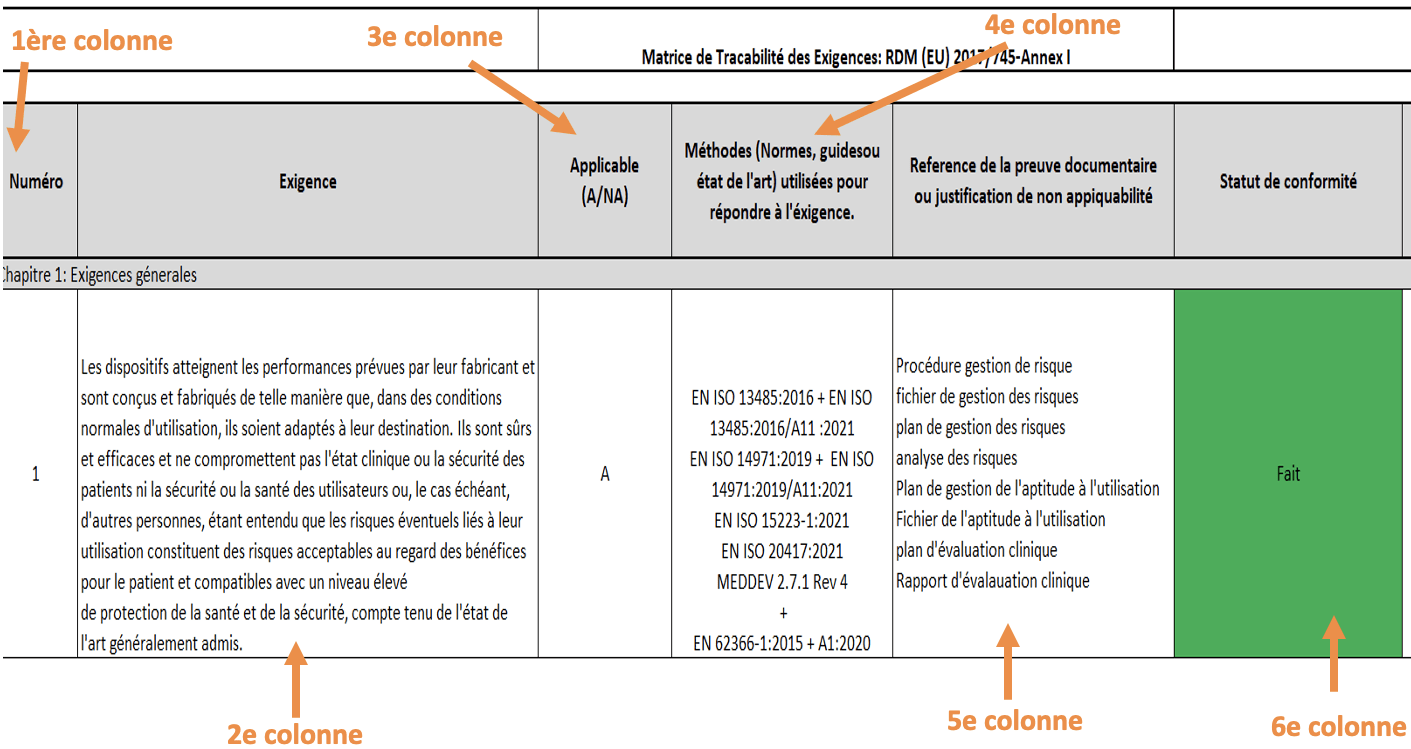
Les différentes colonnes permettent de :
- 1ère colonne : lister les numéros des exigences
- 2e colonne : lister les exigences
- 3e colonne : présenter le statut de l’exigence par rapport à Modjaw (est-elle applicable à Modjaw ou non ?)
- 4e colonne : renseigner les normes harmonisées qui aident à démontrer la conformité du produit aux exigences.
- 5e colonne : lister les éléments de preuve au respect de l'exigence
- 6e colonne : évaluer le statut du respect de l’exigence en fonction de 3 valeurs (Fait, pas fait, en cours).
Les matrices de traçabilité constituées concernent les annexes I des ESPR (environ 18 pages) et l’annexe II sur la documentation technique du RDM (environ 8 pages). Elles ont permis d'évaluer la documentation technique du TIM Hardware. Il en est ressorti que les exigences suivantes n’étaient pas respectées :
- Les informations à fournir par le fabricant (manuel d’utilisation, étiquettes)
- La description technique du dispositif médical, comprenant son usage revendiqué, sa population cible etc…
Ainsi les actions correctives qui y ont découlées pour terminer la mise en conformité du dossier technique du TIM Hardware étaient :
- Création du manuel d’utilisation du TIM Hardware
Le manuel existant au sein de l’entreprise comportait à la fois les informations du logiciel TWIM et de son accessoire le TIM Hardware. La stratégie a été de les séparer, pour constituer un manuel d’utilisation unique pour le TIM hardware. Pour cela, nous nous sommes également basés sur la norme ISO 20417 « Dispositifs médicaux — Informations à fournir par le fabricant » [22] et la norme ISO 15223 « Dispositifs médicaux — Symboles à utiliser avec les informations à fournir par le fabricant » [23] qui permettent de répondre aux exigences du RDM concernant les éléments à fournir par les fabricants de dispositifs médicaux [24]. En effet se sont les seules normes de l’état de l’art actuel qui donnent les exigences à respecter concernant les informations à fournir par les fabricants.
Plus globalement, le manuel d’utilisation a été organisé comme suite :
- Description du DM,
- Informations sur le fabricant, ses mandataires et distributeurs
- Tableaux des symboles de l’ISO 15223 utilisées pour les indications et contre-indications
- Usage revendiqué du DM
- Population cible
- Avertissements
- Contre-indications,
- Étapes à suivre pour la configuration du TIM hardware
- Fonctionnement du TIM Hardware
- Conditions et environnement d’utilisation
- Fin de vie du dispositif / Recyclage
- Création du document de description du TIM Hardware
Pareillement que pour le manuel d’utilisation, il existait un document décrivant à la fois le TWIM et le TIM Hardware. La stratégie a été de le séparer pour créer une description unique au TIM Hardware, c’est-à-dire un document présentant ses différents composants, leurs spécifications techniques, son usage revendiqué, sa population cible etc…
A la fin de la réalisation de toutes ces actions, la matrice de traçabilité des exigences du RDM a été réutilisée pour réévaluer le dossier technique et confirmer à 100% la couverture de toutes les exigences.
Ces éléments constitués ont été validés en revue de direction, et ont permis de figer le dossier technique du TIM Hardware. Il est maintenant en attente d’une déclaration EU de conformité qui sera élaboré après l’obtention du marquage CE sous RDM du TWIM, pour permettre leur commercialisation.
IV. Internalisation de la veille réglementaire chez Modjaw, pour la mise en conformité de ses processus à l'état de l'art normatif et réglementaire
Dans son Annexe II sur la documentation technique, le règlement européen 2017/745 exige aux fabricants de dispositifs médicaux de recenser dans leur documentation technique toutes les normes harmonisées, spécifications communes et/ou toutes autres solutions appliquées qui les permettent d’être conforme aux exigences générales de sécurité et de performance du RDM 2017/745 [3]. En conséquent, ils doivent mener un processus de veille réglementaire et normative qui les permettra d’identifier les textes harmonisés aux RDM EU 2017/745, qui en les appliquant valent présomption de conformité à ce règlement [25].
De façon générale toutes ces normes, guides et textes sont de bons moyens qui montrent le cheminement à suivre pour être conforme aux exigences du règlement EU 2017/745. Et dans le cas où un fabricant se base sur ceux-ci pour respecter les exigences du règlement, il a l’obligation de suivre leurs évolutions afin d’être à jour des exigences nouvelles et/ou modifiées pour mettre en œuvre des actions pour s’y conformer.
1. Definition
La veille réglementaire et normative est un processus crucial pour les fabricants de dispositifs médicaux, son fonctionnement est illustré en figure 16 [26]. Elle permet grâce à l’identification des nouveautés et changements concernant les textes réglementaires et normatif, de les analyser pour évaluer l’impact potentiel de ceux-ci sur les processus et produits de l’entreprise et de mettre en place des actions pour s’y conformer.
Figure 16 : Cartographie de processus d'une veille réglementaire (Source Auteur)
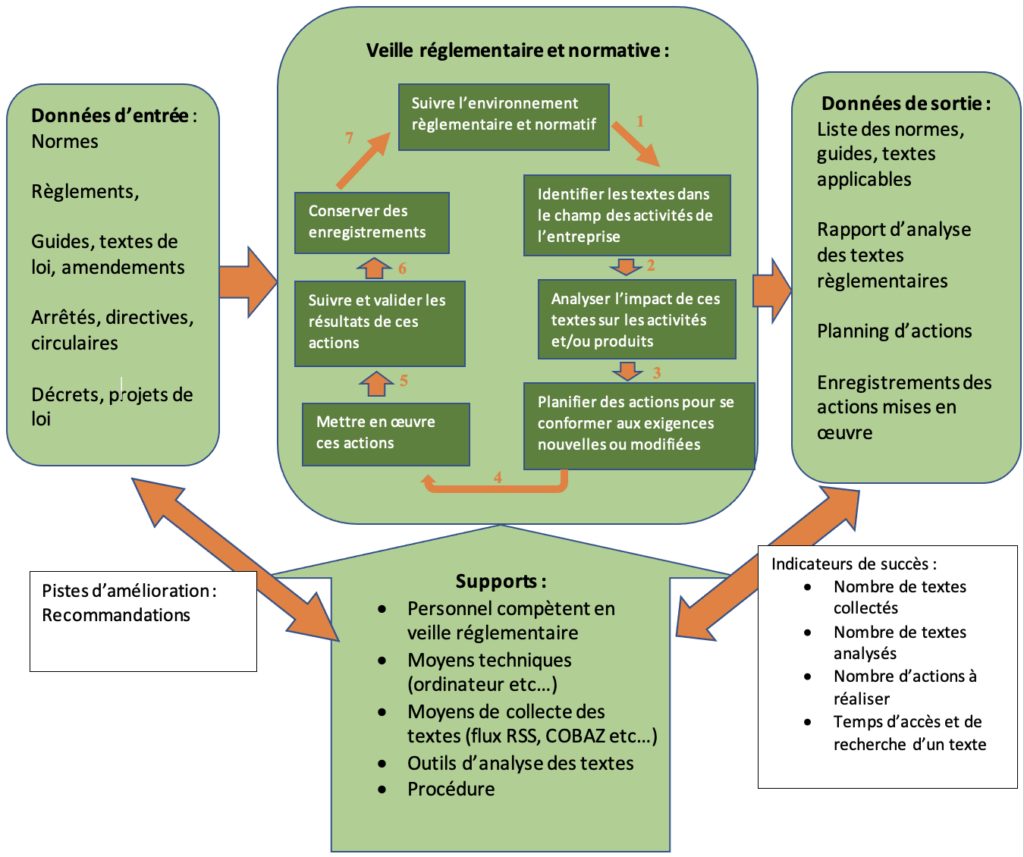
Un processus de veille réglementaire mal construit peut avoir plusieurs risques, notamment sur la conformité du produit, du SMQ et entraver l’obtention du marquage CE et par conséquent la commercialisation d’un dispositif médical. Il est donc crucial de maitriser ce processus pour avoir un dispositif médical le plus conforme possible aux exigences réglementaires et normatives [26] (voir tableau 3).
Tableau 3 : Les types de veille réglementaire et normative (Source : Auteur)
| Les types de veilles règlementaires et normatives | ||
| La veille automatisée C’est une veille qui consiste à s’inscrire à des newsletters, flux RSS sur des sites internet d’autorités compétentes (Agence nationale de sécurité des médicaments produits de santé ANSM), normatives (COBAZ) etc… pour recevoir automatiquement des informations nouvelles sur des normes, guides, règlements etc…. [25], [26] | La veille manuelle C’est une veille qui consiste à consulter des sources d’informations réglementaires, normatives manuellement tel que des sites internet comme COBAZ, RAMS, le site de la commission européenne, le site de la FDA, etc… [25], [26] | La veille sous traitée C’est une veille qui consiste à réceptionner des résumés périodiques sur des nouvelles normes, règlements, guides ou des modifications/ amendements sur ceux-ci, par une entreprise spécialisée dans la veille réglementaire. C’est cette entreprise qui s’occupe de consulter les sources d’informations, analyse et fourni les résumées des textes à l’organisme pour qui elle soustraite cette activité [25], [26]. |
2. La veille réglementaire chez Modjaw
La veille réglementaire chez Modjaw est une veille sous-traitée par un cabinet de conseil en affaires réglementaires, qui collecte et analyse les changements et nouveautés liés aux textes règlementaires et normatifs avant de les transmettre au service qualité et affaires réglementaires (QARA).
Le service QARA se charge ensuite d’enregistrer ces textes dans un document dédié, et procède à une analyse d’impact et d’écarts pour savoir si ce texte à un impact sur leur produit, processus etc.… et ensuite détermine les actions à mettre en œuvre pour y être conforme.
Ce processus de veille réglementaire présente plusieurs inconvénients et induit certains risques dont :
- Une absence de suivi régulier du processus de veille réglementaire car il n’y avait pas de personne dédiée dans le service QARA pour le faire.
- Le fichier regroupant les différents textes réglementaires n’est pas régulièrement mis à jour après la réception de la veille du sous-traitant.
- L’analyse d’impact et d’écarts n’est pas effectuée sur tous les textes issus de la veille.
Pour combler à ces lacunes, le service QARA de Modjaw souhaite aujourd’hui internaliser se processus de veille réglementaire dans l’entreprise et m’en a confié la mission.
3. Stratégie d'internalisation
Pour planifier et réaliser ce projet, le travail a été basé sur l’outil qualité le CAPD, qui permet de résoudre un problème en se basant sur 4 étapes bien définies (CHECK, ACT, PLAN, DO) et favorise l’amélioration continue.
Le CAPD est un outil qualité de résolution de problèmes, qui consiste à évaluer l’existant par rapport à ce qui devrait être fait, pour ensuite imaginer et définir des améliorations, planifier des actions à réaliser et finalement réaliser ce qui avait été prévu. Notre CAPD est est représenté dans la figure 17 ci-après :
Figure 17 : Le Check Act Plan Do de l'internalisation de la veille réglementaire (Source : Auteur)
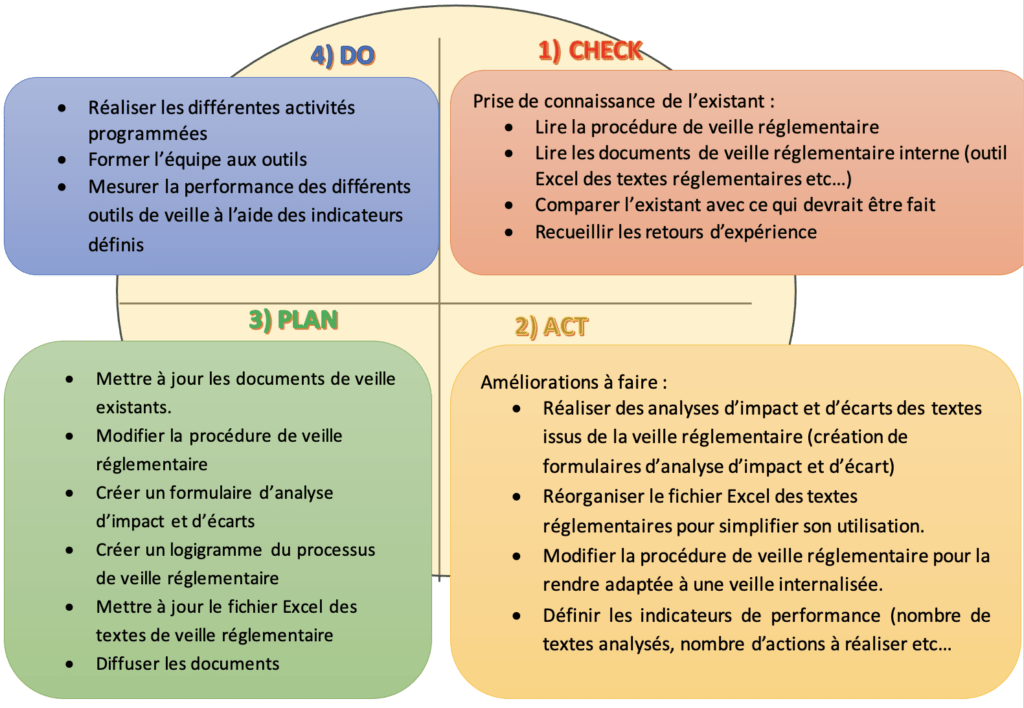
Pour comprendre la problématique à résoudre et définir les actions à mener pour sa résolution, il a fallu commencer par comprendre l’existant en termes de veille réglementaire chez Modjaw, en consultant la procédure de veille réglementaire présente dans l’entreprise, les fichiers relatifs à la veille réglementaire.
Ensuite nous avons effectués plusieurs réunions au sein du service QARA afin de déterminer le plan d’actions à suivre pour internaliser la veille réglementaire (voir figure 17).
a) Les étapes d'internalisation du processus de veille réglementaire
Pour cela, il a fallu procéder comme suite :
- Modification de la procédure de veille réglementaire :
La procédure anciennement présente chez Modjaw était basé sur une veille réglementaire sous-traité, pour la rendre adaptée à une veille réglementaire internalisée, il a fallu la modifier pour changer les responsabilités, les acteurs, mais aussi les étapes à suivre pour réaliser la veille complète. Les différentes modifications sont :
- La fréquence pour réaliser la veille : il a été défini 2 fréquences : une première fréquence mensuelle pour enregistrer tous les textes récoltés dans un fichier Excel dédié et une deuxième annuelle pour analyser les textes recensés.
- Constitution d’un logigramme expliquant le processus de veille réglementaire qui sera intégré dans la nouvelle procédure de veille réglementaire modifiée (voir Annexe 2).
- Les responsabilités : la responsabilité de la collecte et de l’analyse des textes de veille réglementaire a été attribué au service QARA de Modjaw.
- Création d’un formulaire d’analyse d’impact :
La veille réglementaire sous-traité par Modjaw n’incluait pas d’analyse d’impact des nouveaux ou modifications des textes, elle devait être faite en interne et ne l’était pas suffisamment faite ou ne l’était pas du tout.
Pour remédier à cela, il a été proposé un formulaire d’analyse d’impact afin de déterminer si les textes issus de la veille apportaient des modifications pour les produits, processus de l’entreprise (voir figure 18).
Figure 18 : Illustration du formulaire d’analyse d’écart (source Auteur)


Cette analyse d’impact est importante à réaliser, au moins une fois dans l’année pour chaque texte issu de la veille, afin de s’assurer que les processus, produits de l’entreprise sont conformes aux normes, guides revendiquées, et réglementations. Ceci permet d’éviter de produire des dispositifs qui ne sont plus conformes à l’état de l’art normatif et réglementaire et qui ne soient plus compétitifs.
- Création d’un formulaire d’analyse d’écarts :
Après l’analyse d’impact des textes issus de la veille réglementaire, l’élément à réaliser dans le cas d’une modification de normes, guides ou réglementation est l’analyse d’écarts. Son but est d’analyser les écarts qui existent entre un nouveau texte réglementaire et son ancienne version, pour ensuite déterminer les actions à mettre en œuvre pour se conformer à la nouvelle version du texte.
Cette analyse d’écart n’était pas faite chez Modjaw, pour remédier à cela il a été constitué un formulaire d’analyse d’écarts des textes réglementaires (disponible en Annexe 3).
- Mise à jour du fichier de veille réglementaire :
Un outil Excel regroupant les différents textes issus de la veille réglementaire était présent dans l’entreprise, il permettait à la fois de recenser les textes de veille et de faire une analyse d’impact. Mais son organisation, sa complexité, ne permettaient pas son utilisation optimale.
En effet, cet outil Excel était organisé en plusieurs onglets, un onglet regroupant les textes et leurs descriptions, un autre pour l’analyse d’impact, et un autre encore pour les textes réglementaires internationaux. Ces onglets ne fournissaient pas assez d’espace pour analyser les textes, et les données qui y étaient enregistrées n’étaient pas facilement visibles, ce qui a favorisé sa non utilisation.
Pour remédier à cela, il a été constitué un nouvel outil de recensement des différents textes issus de la veille en le simplifiant : en supprimant l’onglet d’analyse d’impact, pour mettre un accent uniquement sur l’enregistrement des textes et donc simplifier son utilisation.
- Constitution d’indicateur pour mesurer la performance de la veille réglementaire :
Pour s’assurer de l’efficacité du processus de veille réglementaire mis en œuvre, et trouver des pistes d’amélioration, nous avons défini les indicateurs de performance suivants :
- Nombre de textes collectés et/ou analysés selon les fréquences définies
- Nombre de non-conformité lié à la veille
- Temps d’accès et de recherche d’un texte dans l’outil Excel regroupant les textes de veille
- Nombre de modifications de processus, produits liés à la veille
- Nombre d’actions issus de la veille proportionnellement à nombre de textes analysés
Ces indicateurs ont été choisis parce qu’ils permettent d’évaluer si les outils mis en place améliorent les délais d’accès à l’information, si le nombre de textes collectés correspond bien au nombre de textes analysés, et de s’assurer que des actions sont bien issues de ce processus.
Une fois ce processus de veille mis en place, le personnel a été formé à l’utilisation des différents outils, et il a été prévu un suivi grâce aux indicateurs définis pour apporter des améliorations (voir tableau 4).
b) Suivi des résultats et perspectives d'amélioration
Après une première analyse des textes de veille réglementaire au premier trimestre 2023, les mesures des indicateurs suivants ont pu être réalisées (voir tableau 4) :
Tableau 4 : Mesure des indicateurs de performance du processus de veille réglementaire (Source Auteur)
| Textes | Règlements | Normes | Guides | Lois |
| Nombre de textes collectés | 9 | 0 | 13 | 2 |
| Nombre de texte analysés | 9 | 0 | 13 | 2 |
| Nombre d’actions à réaliser | 3 | 0 | 8 | 1 |
| Nombre total de texte : | 24 | |||
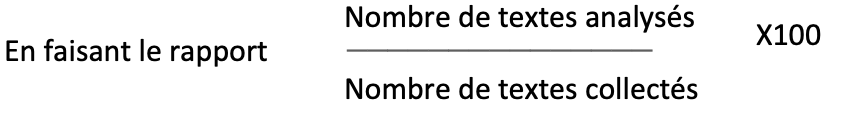
On obtient un rationnel de 1, ce qui montre que tous les textes collectés ont pu être analysé et pour 12 de ces textes, des actions y ont découlé et sont en cours de mises en œuvre au sein de l’entreprise.
De plus le temps d’accès et de recherche des textes est estimé à quelques secondes, ce qui améliore l’utilisation de l’outil regroupant les textes de veille réglementaire.
V. Bilan de mon année d'apprentissage au sein de Modjaw
Réaliser mon apprentissage au sein d’une petite et jeune entreprise comme MODJAW, m’a permis de me confronter aux réalités du monde professionnel surtout celui d’une PME en pleine évolution en France et dans le monde. Cette expérience m’a permis de développer de nombreuses compétences que voici :
1. Compétences et savoirs etre acquis
- Développer ma capacité à aller chercher de l’information et l’exploiter pour comprendre et résoudre une problématique : je sais à présent utiliser différents moyens tel que les sites internet d’organisme de règlementation (ANSM, Union européen) pour rechercher des informations sur une problématique en amont d’un projet pour mieux la comprendre et aisément proposer un plan d’action et des solutions.
- Identifier et analyser les besoins de l’entreprise pour proposer des actions pertinentes et les planifier : les différentes contraintes rencontrées en entreprise m’ont permis de mobiliser des comportements tel que le sens du dialogue et la communication.
- Conduire des audits : lors des différents audits de certifications ISO 13485, RDM et les audits internes auxquels j’ai participé, j’ai pu à la fois mobiliser des compétences d’observateur d’audit pour accompagner l’équipe d’audit avec la prise de notes, de remarques et l’écoute, mais aussi mobiliser des compétences d’auditeurs dont la capacité d’observation, d’analyse et de synthèse lors d’audit interne.
- Rédiger et améliorer des documents techniques (Procédure, formulaire, instruction) : j’ai pu grâce à cette expérience développer des stratégies pour améliorer des processus, procédures présentes dans l’entreprise pour les adapter aux évolutions réglementaires.
- Améliorer mes compétences techniques en affaires réglementaires : j’ai pu améliorer mes connaissances en gestion des risques, construction d’un système de management de la qualité selon l’ISO 13485, en réglementation internationale, en confrontant au quotidien mes connaissances déjà acquises à l’UTC aux réalités de l’entreprise.
- Construire une documentation technique et un processus de veille réglementaire : grâce à ces missions j’ai pu développer ma capacité à élaborer une stratégie pour définir un plan d’action et résoudre une problématique donnée.
- Améliorer ma communication professionnelle que ce soit lors de réunions avec les collaborateurs ou pour faire parvenir des informations sur des évolutions réglementaires. Ces améliorations concernent la communication par email professionnel, la présentation fluide de résultats de fin de projet aux collaborateurs.
- Améliorer ma capacité rédactionnelle : c’est la compétence la plus cruciale pour un ingénieur qualité, parce que savoir rédiger des procédures, des instructions et des formulaires qui seront facilement compréhensibles pour tous, est la clé pour qu’elles soient suivies et respectées. Cette expérience m’a permis de grandement développer cette compétence pour arriver à rédiger des procédures qui sont aujourd’hui suivies et utilisées dans l’entreprise.
- Développer une capacité d’adaptation à un environnement changeant : Depuis mon arrivée au sein de Modjaw, l’environnement n’a de cesse changée, avec l’arrivée de nouveaux collaborateurs et le départ de certains. Ce changement d'interlocuteur pousse à sans cesse s’adapter en prenant le temps de comprendre les méthodes de chacun, pour favoriser des collaborations fructueuses. Ce qui m’a fortement permis de développer ma capacité d'adaptation.
2. Compétences et comportements à developper
Pour continuer à m’améliorer et développer encore plus de compétences nécessaires au métier de chargé d’affaires réglementaires et qualité auquel je me destine, il faut que je m’améliore encore sur certains points notamment :
- La gestion des priorités : évoluer dans un contexte de petite entreprise, demande de travailler sur plusieurs problématiques à la fois, et souvent on peut se trouver dépassé pour terminer la réalisation des projets à temps.
- Les connaissances en réglementation internationale : j’ai développé ma connaissance de la réglementation en Europe et aux États-Unis, brésil, Australie, Japon, Canada (formation d’auditeur sur le référentiel MDSAP). Mais il me faut continuer à m’informer sur la réglementation d’autres pays tels que la chine etc…
- Ma capacité à m'auto évaluer pour gagner en performance, durant cette expérience, j’ai surtout appris à évaluer mon travail grâce à des indicateurs.
- La réalisation d’Audit : ce point est très important pour un ingénieur qualité, avec mon expérience chez Modjaw, j’ai pu encore plus développer des compétences d’auditeur. Mais concernant les audits, on peut toujours s’améliorer en participant notamment à des audits, pour apprendre en pratiquant, c’est pourquoi je continuerai à réaliser des audits pour apprendre davantage.
3. Lien avec la formation théorique en master ingénierie de la santé à l'UTC
La formation suivie en master ingénierie de la santé à l’UTC, m’a apporté un socle de connaissances suffisant pour aborder aisément mes missions en entreprise. C’est une formation riche et principalement accès sur le management de la qualité, les technologies biomédicales, la réglementation pour les dispositifs médicaux en Europe et à l’international.
Toutes ces connaissances m’ont été utiles et principalement celles concernant :
- La réglementation des dispositifs médicaux : À l’UTC nous avons appris à comprendre et décrypter le règlement européen pour les dispositifs médicaux. Grâce à ces connaissances et aux projets de cours réaliser sur la commercialisation d’un DM en Europe, il m’a été facile de réaliser mes missions de mise en conformité d’un dossier technique d'un dispositif médical aux exigences du RDM EU 2017/745.
- Le management de la qualité : la norme ISO 9001, la norme ISO 13485 sont toutes des normes qui ont été abordées à l’UTC lors de mon cursus en master ingénierie de la santé, ce qui m’a permis de facilement comprendre le système de management de la qualité selon l’ISO 13485 mis en place chez Modjaw pour l’améliorer.
- Les outils qualités tels que le PDCA, la cartographie des processus, le QQOQCP, le logigramme, le Diagramme des causes à effets sont des outils qui m’ont été utile, et que j’ai utilisé pour comprendre et résoudre les problématiques rencontré en entreprise.
- L’étude des normes ISO 14971, ISO 60601, ISO 62304, ISO 62366 vu à l’UTC, m’a également permis de facilement mobiliser ces référentiels pour répondre aux exigences du RDM EU 2017/745.
Au-delà de toutes ces connaissances techniques, la formation à l’UTC étant basé sur une pédagogie par projets, nous avons réalisé au cours de nos années d’études de nombreux projet en groupe d’étudiants. Ce qui m’a permis de développer des compétences de travail en équipe, de rigueur, de communication, qui toutes m’ont été grandement utiles pendant mon expérience en entreprise.
Conclusion
Le passage de la directive 93/42/CEE vers le règlement européen 2017/745 a renforcé la démarche d’obtention du marquage CE, dans le cas de MODJAW, il a fallu conformer la documentation technique de son dispositif médical de classe I aux exigences du règlement.
Le but de ce mémoire a été de proposer une stratégie à suivre pour conformer le dossier technique du TIM Hardware de classe I aux exigences du RDM 2017/745, mais aussi de construire un processus de veille réglementaire interne à l’entreprise.
Ce but a été atteint, en mettant en conformité le dossier technique du dispositif médical, et en proposant au sein de l’entreprise un nouveau processus de veille règlementaire complètement géré en interne et applicable à n’importe quelle organisation qui souhaite en construire un. Un outil de matrice de traçabilité a également été proposé pour faciliter la mise en conformité des dispositifs médicaux aux exigences du RDM 2017/745.
Ces deux réalisations permettront à l’entreprise d’assurer la continuité de la commercialisation de son dispositif médical, la sécurité des patients et le suivi de l’environnement réglementaire, normatif et concurrentiel.
Afin de continuer la mise en conformité du dossier technique du dispositif, il serait envisageable de croiser et d’appliquer les évolutions normatives et réglementaires à celui-ci afin de le conformer aux nouvelles exigences normatives également.
Références bibliographiques
[1] Agence nationale de securité du médicament et des produits de santé, « Les prothèses mammaires implantables PIP État des lieux », Rapport de synthèse, avr. 2013. [En ligne]. Disponible sur : https://ansm.sante.fr/uploads/2020/12/22/synthese-rapport-pip-def-01-02-12.pdf
[2] « Directive 93/42/CEE du Conseil, du 14 juin 1993, relative aux dispositifs médicaux », Journal officiel de l’Union européenne, Document 31993L0042, Journal officiel n° L 169 du 12/07/1993 p. 0001-0043. [En ligne]. Disponible sur : http://data.europa.eu/eli/dir/1993/42/oj/fra
[3] « Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE (Texte présentant de l’intérêt pour l’EEE. ) », Journal officiel de l’Union européenne, https://eur-lex.europa.eu, mai 2017. [En ligne]. Disponible sur : http://data.europa.eu/eli/reg/2017/745/oj/fra
[4] Guillaume Promé, « Dispositifs médicaux : les chiffres en France », Qualitiso. https://www.qualitiso.com/dispositifs-medicaux-chiffres-france/ (consulté le 5 juin 2023).
[5] Nicolas Bonnet, « Le système Modjaw® un nouvel outil au service de la pédagogie en occlusodontologie pour comprendre les fonctions occlusales », Thèse n°42-57-20-28, Faculté de chirurgie dentaire de Nice, Nice, 2021. Consulté le : 28 mai 2023. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-03107513
[6] « Chirurgiens-dentistes : les chiffres clés de la profession en France », Nextdentiste. https://www.nextdentiste.com/chirurgiens-dentistes-chiffres-cles-de-profession-france (consulté le 25 juin 2023).
[7] « Santé bucco-dentaire », Organisation Mondiale de la Santé. https://www.who.int/fr/news-room/fact-sheets/detail/oral-health (consulté le 14 juin 2023).
[8] LNE GMED, « La prolongation de la période de transition pour les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro est adoptée », GMED Groupe LNE, 17 février 2023. https://lne-gmed.com/fr/news/la-prolongation (consulté le 14 juin 2023).
[9] « RÈGLEMENT (UE) 2023/607 DU PARLEMENT EUROPÉEN ET DU CONSEIL du 15 mars 2023 ». https://eur-lex.europa.eu/eli/dec_impl/2022/757/oj (consulté le 25 avril 2023).
[10] Marco Tallarico, « Computerization and Digital Workflow in Medicine : Focus on Digital Dentistry », vol. 13, no 9, p. 2172, mai 2020, doi : 10.3390/ma13092172.
[11] Ivoclar Vivadent, « Dentisterie numérique et technologie dentaire : le monde dentaire en transition ». https://content.ivoclar.com/fr/dentisterie-numérique-et-technologie-dentaire-le-monde-dentaire-en-transition (consulté le 5 juin 2023).
[12] « Les évolutions technologiques dans le domaine dentaire », Santé Pratique. https://www.santepratique.fr/evolutions-technologiques-domaine-dentaire (consulté le 5 juin 2023).
[13] Syndicat National de l’Industrie des Technologies Médicales et COMIDENT, « Dispositifs médicaux & progrés en santé bucco-dentaire », SNITEM, p. 57.
[14] Royannez Marion, « Mastication et ODF », Thése en chirurgie dentaire, Faculté d’odontologie de Marseille, Marseille, 2018. Consulté le : 15 juin 2023. [En ligne]. Disponible sur : https://dumas.ccsd.cnrs.fr/dumas-01870348/document
[15] Penarrocha-Oltra David, Penarrocha-Diago Maria, Maestre-Ferrin Laura, et Romero-Millan Javier, « Virtual articulator for the analysis of dental occlusion : An update », Med Oral Patol Oral Cir Bucal, p. e160‑e163, 2012, doi : 10.4317/medoral.17147.
[16] Thibaud Casas, « Empreinte optique et flux numérique : la maturité technologique au service de l’efficience clinique », AOnews le magazine dentaire qui nous rassemble. http://www.aonews-lemag.fr/casas-numerique-empreinte-optique-aonews/ (consulté le 5 juin 2023).
[17] « SDi Matrix Diagnostic center ». https://sdimatrix.com/en/ (consulté le 24 juin 2023).
[18] « zebris Medical GmbH : Bienvenue dans le monde de la biomécanique ». https://www.zebris.de/ (consulté le 24 juin 2023).
[19] « Logiciels et scanners 3D dentaires pour la dentisterie CFAO », 3Shape. https://www.3shape.com/fr/ (consulté le 24 juin 2023).
[20] « Modjaw la dentisterie dynamique, un changement de paradigme ». https://www.modjaw.com/fr/ (consulté le 24 juin 2023).
[21] G. Cuofano Gennaro, « Qu’est-ce qu’une matrice de traçabilité des exigences ? Matrice de traçabilité des exigences en bref », FourWeekMBA, 4 janvier 2023. https://fourweekmba.com/fr/matrice-de-tra%C3%A7abilit%C3%A9-des-exigences/ (consulté le 14 juin 2023).
[22] « ISO 20417:2021 Dispositifs médicaux — Informations à fournir par le fabricant », Ed.Afnor, Paris, www.afnor.org, 5 mai 2021. Consulté le : 20 juin 2023. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/nf-en-iso-20417/FA194754?rechercheID=15536223&searchIndex=1&activeTab=all
[23] « ISO 15223-1:2021 Dispositifs médicaux — Symboles à utiliser avec les informations à fournir par le fabricant — Partie 1 : Exigences générales », Ed.Afnor, Paris, www.afnor.org, 29 septembre 2021. Consulté le : 20 juin 2023. [En ligne]. Disponible sur : https://cobaz-afnor-org.ezproxy.utc.fr/notice/norme/nf-en-iso-15223-1/FA196228?rechercheID=15536220&searchIndex=2&activeTab=all
[24] « projet de norme PR NF EN ISO 20417 - Dispositifs médicaux - Informations à fournir par le fabricant », Ed. Afnor, Paris, www.afnor.org, 13 mai 2019. [En ligne]. Disponible sur : https://sagaweb-afnor-org.ezproxy.utc.fr/fr-FR/sw/consultation/notice/1545369?recordfromsearch=True
[25] Guillaume Promé, « Faire une Veille Réglementaire », Qualitiso. https://www.qualitiso.com/veille-reglementaire-techniques/ (consulté le 2 mai 2023).
[26] S. Salmi, « VEILLE & CONSTRUCTION DE DOSSIERS TECHNIQUES DES DISPOSITIFS MEDICAUX SELON LE REGLEMENT EUROPEEN 2017/745 », Université de Technologie de Compiègne (France), Master Ingénierie de la santé, Parcours Technologies Biomédicales et Territoires de santé (TBTS) et Dispositif Médical et Affaires Réglementaires (DMAR), Mémoire de stage réf n°IDS070, juill. 2020. Consulté le : 15 juin 2023. [En ligne]. Disponible sur : https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids070/
Annexes
Annexe 1 : Arborescence du dossier technique du TIM Hardware


Annexe 2 : Logigramme du processus de veille réglementaire chez Modjaw
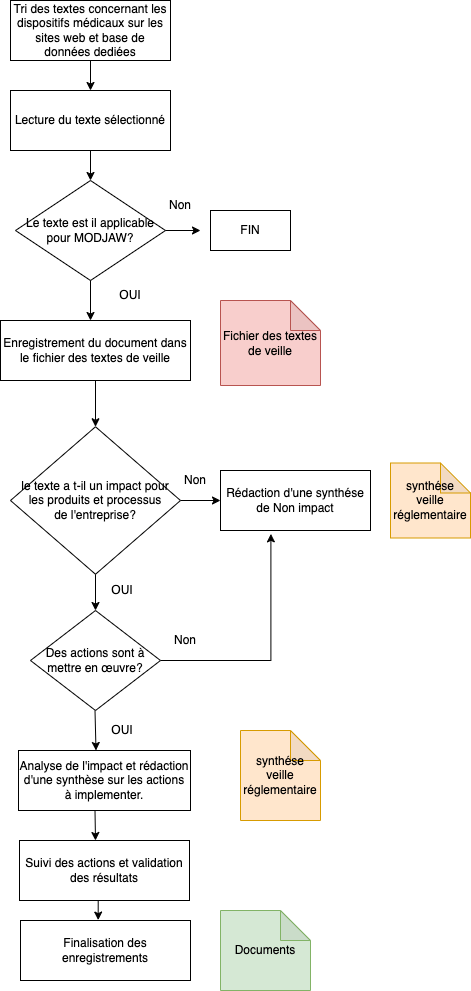
Annexe 3 : Formulaire d'analyse d'écart
