IDS202 - Cartographie interactive des acteurs de l'écosystème réglementaire des Dispositifs Médicaux
Les projets ou stages publiés auxquels vous accédez sont des rapports d'étudiants et doivent être pris comme tels. Il peuvent donc comporter des imperfections ou des imprécisions que tout lecteur doit admettre et donc supporter. Il ont été réalisés pendant les périodes de formation et constituent avant-tout des travaux de compilation bibliographique, d'initiation et d'analyse sur des thématiques associées aux concepts, méthodes, outils et expériences sur les démarches qualité dans les organisations ou sur les technologies en santé.
Si, malgré nos précautions, vous avez des raisons de contester ce droit de diffusion libre, merci de nous en faire part (master@utc.fr), nous nous efforcerons d'y apporter une réponse rapide. L'objectif de la présentation des travaux sur le web est de permettre l'accès à l'information et d'augmenter ainsi la qualité des échanges professionnels.
Nous ne faisons aucun usage commercial des travaux de projet ou de stage publiés, par conséquent les citations des informations et l'emploi des outils mis à disposition sont totalement libres. Dans ce cas, nous vous demandons de respecter les règles d'éthique en citant explicitement et complètement vos sources bibliographiques.
Bonne lecture...
Auteurs





Contacts
- Maryse de PETANG : marysedepetang@gmail.com
- Oumayma ES-SIFER : oumaymaessifer@gmail.com
- Charline VIGUIER : charline.viguier01@gmail.com
- Safae HASSANI : safae.hassani@outlook.fr
- Hamza EL-MANSOURI : hamzaelmansouri79@gmail.com
Citation
A rappeler pour tout usage : M. de PETANG, O. ES-SIFER, C. VIGUIER, S. HASSANI, H. EL-MANSOURI « Cartographie interactive des acteurs de l'écosystème réglementaire des Dispositifs Médicaux », Université de Technologie de Compiègne (France), Master Ingénierie de la Santé, Mémoire de Projet, https://travaux.master.utc.fr/, réf n° IDS202, https://doi.org/10.34746/ids202, janvier 2024, https://travaux.master.utc.fr/formations-master/ingenierie-de-la-sante/ids202/
Résumé
Plusieurs acteurs interviennent à chacune des étapes du cycle de vie d'un Dispositif Médical.
Bien qu'ayant tous des activités et des obligations différentes, des caractéristiques communes peuvent tout de même leur être trouvées. Tous agissent, en effet, en interdépendance les uns des autres afin d'assurer la qualité, la sécurité et les performances des Dispositifs Médicaux mis à disposition des patients. Et surtout : tous sont influencés par le cadre réglementaire et normatif dans lequel ils s'inscrivent.
Cet ensemble (réglementation, normes et acteurs) est appelé l'écosystème réglementaire des Dispositifs Médicaux.
Les dynamiques de cet écosystème ont connu un tournant radical lorsque le 26 mai 2021, la nouvelle réglementation autour des Dispositifs Médicaux (Règlement (UE) 2017/745 et Règlement (UE) 2017/746) est entrée en application.
Depuis, l'écosystème peut paraître comme étant très étendu, complexement structuré et trop évolutif pour un professionnel néophyte. Et pourtant, s'il souhaite que son dispositif soit commercialisé et utilisé, il devra forcément interagir avec les autres membres de sa filière.
De là émerge donc une potentielle solution : la création d'un outil interactif synthétisant la composition de ce secteur.
Abstract
Several skateholders are involved at each stage of the life cycle of a Medical Device.
Although they all have different activities and obligations, they still have certain characteristics in common. Indeed, they all are interdependent on one another to ensure the quality, safety and performance of the medical devices made available to patients. Above all, they all are influenced by the regulatory and standards framework in which they operate.
This ensemble (regulations and players) is known as the medical device regulatory ecosystem.
The dynamics of this ecosystem took a radical turn when the new regulation on Medical Devices (Regulation (UE) 2017/745 and Regulation (UE) 2017/746) came into force on the 26th May 2021.
Since then, the ecosystem may appear to be very extensive, complexly structured and too fast-moving for a novice professional. And yet, if they want their device to be marketed and used, they will inevitably have to interact with the other members of their sector.
A potential solution has therefore emerged : the creation of an interactive tool summarizing the composition of this sector.
Téléchargements



Mémoire Complet
Cartographie interactive des acteurs de l'écosystème réglementaire des Dispositifs Médicaux
Introduction
Les Dispositifs Médicaux (DM) sont des éléments incontournables dans tout parcours de soins au sein du système de santé ; à la fois pour les professionnels de santé mais aussi pour les patients. Répartis en quatre classes selon leur niveau de risque, ils bénéficient d'une définition très large car ils font appel à différentes techniques à la fois (électronique, informatique, biomatériaux...), sont sous diverses formes et peuvent présenter des finalités médicales variées. Ainsi, ils peuvent être à visée diagnostique ou thérapeutique, optimiser la qualité de prise en charge, le suivi ou l'accompagnement de leur utilisateur voire radicalement améliorer sa qualité de vie.
Il s'agit aussi de produits de santé aux nombreux risques. C'est pourquoi la réglementation et les normes que doivent respecter les différents acteurs du secteur sont particulièrement rigoureuses [1].
L’écosystème réglementaire des Dispositifs Médicaux comprend l’ensemble des parties prenantes impliquées dans la mise en place, le contrôle de l’exécution et l’application de ces outils réglementaires du secteur des DM.
Cet écosystème a récemment subi des bouleversements majeurs à la suite de différents scandales sanitaires et d'une transformation digitale importante ces dernières années, avec notamment une connectivité de plus en plus importante des dispositifs et l'émergence de l'Intelligence Artificielle dans tous les domaines (imagerie, chirurgie, médecine personnalisée...). En 2017 notamment sont entrés en vigueur les deux Règlements sur les DM et DMDIV [2] [3].
Aujourd'hui, ce secteur présente une configuration réglementaire particulièrement complexe avec des procédures différentes selon la catégorie du dispositif, une identification des documents nécessaires pour la mise en conformité à faire, des documents officiels souvent peu lisibles et longs, un planning chargé pour mettre en œuvre les nouveaux Règlements dans les délais, un cumul des exigences à la fois réglementaires mais aussi normatives...
Cet écosystème s'avère d'autant plus difficile à comprendre qu'il évolue de façon perpétuelle pour s'adapter aux changements induits par l'innovation : ainsi, la Règlementation relative aux DM a été modifiée à 6 reprises depuis sa mise en œuvre en 1998.
En parallèle, malgré ces problématiques, reste un besoin toujours aussi important d'une mise à disposition large des Dispositifs Médicaux, comme en témoignent les derniers chiffres de la Consommation en Soins et Biens Médicaux (CSBM) en France d'une valeur de 235.8 Milliards d'euros en 2022 [4].
Il semble donc important, pour toute personne ayant pour projet de fabriquer et commercialiser un Dispositif Médical, de comprendre rapidement et correctement les parties prenantes de cet écosystème avec lesquelles il sera en contact à chaque étape du cycle de vie de son innovation ; de son étude marché à sa réforme en passant par son marquage CE.
Dans ce contexte, se pose donc la problématique suivante :
"Comment apporter de la lisibilité à un secteur en tension qui garantit la disponibilité de produits de santé sûrs et performants aux patients?"
Afin de répondre à cette problématique, le mémoire suivant se constitue de trois parties.
Une première permet de poser le contexte : quelles sont les grandes catégories d’acteurs du secteur des Dispositifs Médicaux, à quelles évolutions elles sont aujourd’hui confrontées et enfin, parmi ces catégories, lesquelles sont ciblées par le projet.
Puis, une deuxième partie explicite la démarche employée par le groupe afin d’accompagner ces cibles dans la compréhension des autres acteurs de leur milieu.
Enfin, une troisième partie sert à la fois de description des résultats obtenus mais aussi de bilan de la démarche employée grâce aux retours d’expérience des professionnels sur les livrables produits.
En particulier, ce mémoire propose un outil interactif qui cartographie les acteurs de l’écosystème réglementaire des Dispositifs Médicaux présents à l’échelle française, européenne et internationale. De cette manière, l’utilisateur peut à la fois connaître le rôle de chaque acteur ainsi que ses missions, mais aussi identifier rapidement ses interlocuteurs et les documents utiles ; enfin, soit le positionner dans l’ensemble de l’écosystème soit dans le processus du marquage CE.
Partie 1 : Mise en contexte
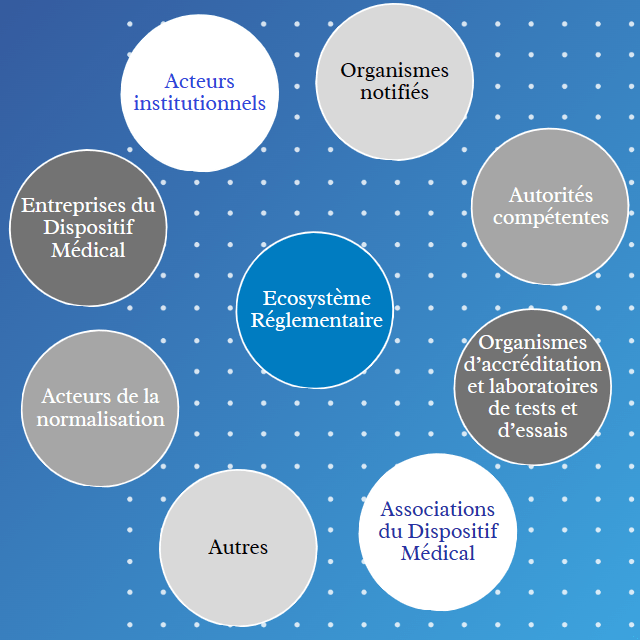
I) Présentation générale des acteurs de cet écosystème
Dans l’industrie des Dispositifs Médicaux, les Affaires Réglementaires ont pour rôle de garantir la conformité avec la réglementation en vigueur avant, pendant et après la mise sur le marché. Divers acteurs jouent aussi des rôles cruciaux pour s’assurer que les Dispositifs médicaux mis sur le marché sont sûrs et efficaces afin de protéger la santé des patients et des utilisateurs. Voici une explication du rôle de chaque catégorie d’acteurs identifiée dans l’écosystème réglementaire :
- Acteurs institutionnels
Les acteurs institutionnels dans l'écosystème réglementaire des Dispositifs Médicaux sont tous les organismes, agences et organisations gouvernementales qui jouent un rôle central dans l'élaboration, la mise en œuvre et l'application des réglementations entourant les Dispositifs Médicaux. Ces acteurs institutionnels collaborent souvent étroitement pour élaborer des politiques et des réglementations qui garantissent la sécurité et l'efficacité des Dispositifs Médicaux. Ils jouent aussi un rôle crucial dans la protection de la santé publique et la promotion de l'innovation dans le domaine des Dispositifs Médicaux.
- Entreprises du Dispositif Médical
Les entreprises du Dispositif Médical sont des entités privées ou publiques qui sont impliquées dans tout le cycle de vie du DM : la conception, la fabrication, la distribution, la vente et parfois la maintenance des Dispositifs Médicaux. Elles jouent un rôle central dans l'écosystème réglementaire des Dispositifs Médicaux en veillant à ce que les produits mis sur le marché soient sûrs, efficaces et conformes aux réglementations en vigueur.
- Acteurs de la normalisation
Un acteur de la normalisation est une entité, qu'il s'agisse d'une organisation, d'un gouvernement, d'une entreprise ou d'une association, qui participe activement au processus d'établissement de normes. Les normes sont des spécifications techniques, des lignes directrices ou des critères qui visent à réguler et à normaliser les produits, les services ou les processus dans divers secteurs [5].
- Organismes Notifiés
Un Organisme Notifié est une organisation désignée par un État membre de l’UE (ou par d’autres pays dans le cadre d’accords spécifiques) pour évaluer la conformité de certains produits avant leur mise sur le marché. Ces organismes sont habilités à effectuer des tâches liées aux procédures d’évaluation de la conformité définies dans la législation applicable lorsque l’intervention d’un tiers est requise [6].
- Autorités compétentes
Une autorité compétente est une organisation gouvernementale ou un organisme désigné par un pays ou une région spécifique pour réglementer et superviser les Dispositifs Médicaux sur son territoire. Cette autorité joue un rôle crucial dans l'application des réglementations, l'homologation, la surveillance sur le marché et l'assurance de la qualité des Dispositifs Médicaux afin de protéger la santé et la sécurité des patients et des utilisateurs. En France, l’autorité compétente est l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM). L’ANSM intervient également dans le processus de désignation et de contrôle de l’Organisme Notifié français, le GMED [7].
- Laboratoires de tests et d’essais
Un laboratoire d’essais atteste de la conformité des produits après avoir répondu à tous les essais de qualification exigés par le cahier des charges ou par des normes. Il intervient également sur les nouveaux produits en proposant ou en améliorant des méthodes de contrôle et des moyens de test.
- Organismes d’accréditation
Dans le contexte des Dispositifs Médicaux, les organismes d'accréditation sont des entités indépendantes qui évaluent et attestent que les organismes d'évaluation de la conformité disposent de la compétence technique et organisationnelle à réaliser des essais, des analyses, des activités d'inspection et de certification. Ces organismes jouent un rôle crucial dans le processus de certification et contribuent à assurer la qualité, la sécurité et la conformité réglementaire des Dispositifs Médicaux [8].
- Associations du Dispositif Médical
Les associations du Dispositif Médical sont des organisations à but non lucratif, des groupes industriels ou des regroupements professionnels qui représentent les intérêts et les préoccupations des entreprises, professionnels et parties prenantes liées à l'industrie des Dispositifs Médicaux. Elles jouent un rôle important dans l'écosystème réglementaire des Dispositifs Médicaux en agissant comme des intermédiaires entre les entreprises, les professionnels de la santé, les autorités réglementaires et autres parties prenantes pour promouvoir une industrie des Dispositifs Médicaux sûre, innovante et responsable. Elles contribuent à façonner l'écosystème réglementaire en faisant valoir les intérêts collectifs de l'industrie.
- Autres
Dans l’écosystème des Dispositifs Médicaux, cette catégorie englobe les professionnels de santé, les patients ou encore des tiers.
Les catégories d’acteurs présents dans l’écosystème réglementaire des Dispositifs Médicaux sont donc nombreuses. De plus, les dynamiques observées aujourd’hui au sein de chacun de ces groupes sont aussi très variées (parfois opposées), ce qui peut rendre la compréhension du professionnel néophyte encore plus difficile.
II) Dynamiques observables aujourd'hui au sein de cet écosystème
Dans cet écosystème en perpétuelle évolution, deux tendances opposées doivent être mises en évidence :
- D’une part, la pénurie importante traversée par les Organismes Notifiés, pourtant maillons centrales de la chaîne de mise sur le marché des DM,
- D’autre part, l’augmentation du nombre d’opérateurs économiques présents sur le territoire européen malgré un déclin consécutif à la mise en place du nouveau cadre législatif.
1) Un nombre insuffisant d'Organismes Notifiés au titre des nouveaux Règlements
L’intervention des Organismes Notifiés est nécessaire pour l’accès au marché de tous les DM qui ne sont pas de classe I : les classes I stériles, I avec fonction de mesurage ou I chirurgicaux réutilisables, IIa, IIb et III.
Toutefois, ces acteurs sont aujourd’hui en nombre insuffisant face aux nombreuses entreprises demandeuses d’une certification initiale ou d’un renouvellement de leur marquage CE [9].
Cela peut s’expliquer par le renforcement global de la réglementation.
En effet, en 2013, est arrivé le règlement d’exécution 2013/920. Celui-ci a imposé la désignation, le contrôle et la réévaluation de chaque Organisme Notifié tous les 3 ans selon un processus dit de “joint assessment” pouvant durer entre 14 et 18 mois.
Plusieurs acteurs, à l’échelle nationale et européenne, sont impliqués dans ce processus :
- L’autorité compétente nationale (l’ANSM en France qui a aussi le statut d’Autorité Responsable des Organismes Notifiés),
- La Commission Européenne,
- Le Medical Device Coordination Group (MDCG),
- Deux autres autorités compétentes européennes qui forment une équipe d’évaluation conjointe (EEC).
Dans un second temps, le Règlement 2017/745 a provoqué un renforcement important des critères de ce processus afin de remédier à une des limites de la précédente directive : le faible contrôle exercé sur les Organismes Notifiés. La conséquence a donc été une réduction drastique du nombre d’Organismes Notifiés désignés. En effet, l’entrée en vigueur du règlement d'exécution 2013/920 a provoqué une diminution de leur nombre en Europe de 87 en 2012 à 59 en 2016 [10]. Puis, en avril 2020, lors de la période de transition, sur les 59 organismes initialement notifiés au titre des trois directives, seuls 14 d’entre eux étaient en mesure d’effectuer des activités d’évaluation de la conformité des DM selon le Règlement des Dispositifs Médicaux (RDM) et 3 selon le Règlement des Dispositifs Médicaux de Diagnostic In Vitro (RDMDIV) [11].
Aujourd’hui, la situation s’est légèrement améliorée sans revenir au point de départ. D’après le système d’informations NANDO (New Approach Notified and Designated Organisations, article 42 du RDM), 40 organismes sont, en effet, notifiés au titre du RDM et 12 au titre du RDMDIV (Novembre 2023) [12].
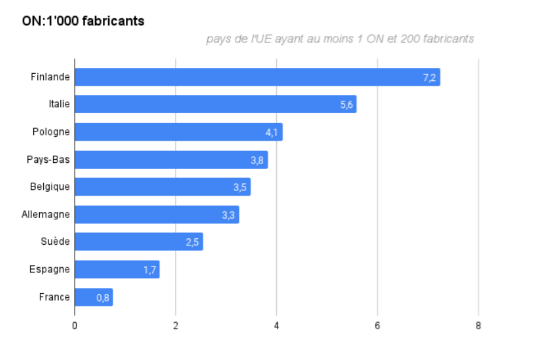
Par ailleurs, les Organismes Notifiés sont aussi répartis inégalement géographiquement. En moyenne, seuls 3.8 Organismes Notifiés sont ainsi présents dans un pays de l’UE pour 1000 fabricants [13].
La France est un exemple assez préoccupant en la matière : en effet, seul un organisme est notifié sur toute le territoire pour effectuer des contrôles sur les DM/DMIA et DMDIV, le LNE-GMED (numéro d’identification 0459), alors qu’il s’agit pourtant du deuxième marché du DM en Europe. Le pays ne se positionne ainsi qu'au neuvième rang dans l’UE (Figure 1) loin derrière l’Italie ou l’Allemagne, qui en comptent une dizaine chacun.
Figure 1 : Représentation du nombre moyen d'Organismes Notifiés présents dans un pays de l'UE pour 1000 fabricants (pays de l'UE ayant au moins un Organisme Notifié et 200 fabricants) [Source : d'après [13]]
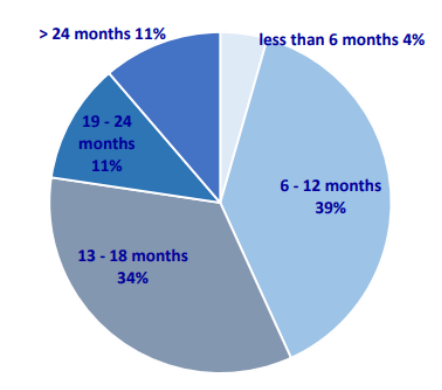
Cette pénurie d’Organismes Notifiés n’est pas sans conséquences. Synonyme de perte financière importante pour les industriels, elle entraîne notamment un retard important dans l’évaluation des dossiers. Ainsi, selon une enquête de la Team-NB (le syndicat des Organismes Notifiés avec 36 ON membres en Août 2023), les industriels doivent, pour obtenir un nouveau certificat MDR, faire face à des temps d’attente de plus de 13 mois dans 60% des cas [14] (Figure 2).
Figure 2 : Temps moyen nécessaire aux Organismes Notifiés adhérents de la Team-NB pour délivrer un nouveau certificat MDR [Source : d'après [14]]
Contrairement aux Organismes Notifiés, une autre catégorie de l’écosystème semble quant à elle connaître une pleine croissance : il s’agit de celle des opérateurs économiques aussi appelés “entreprises du DM” par le Syndicat National de l’Industrie des TEchnologies Médicales (SNITEM); une organisation professionnelle représentant les entreprises du secteur des Dispositifs Médicaux dans les instances nationales et européennes.
2) Un certain nombre d'opérateurs économiques en activité aujourd'hui...
En tout, 4 opérateurs économiques distincts sont désignés par le Règlement (UE) 2017/745 qui définit leurs responsabilités et renforce les exigences à leur égard :
- Le fabricant,
- L’importateur,
- Le mandataire,
- Le distributeur.
Tous, sauf le distributeur, sont contraints par le Règlement à s’enregistrer sur la plateforme Eudamed (acronyme pour "European DAtabank for MEdical Device") et à y contribuer. Ils obtiennent ainsi un numéro d’authentification par l’intermédiaire de l’autorité compétente nationale : le Single Registration Number (SRN) variable selon l’entité et son activité ; celui-ci permettant de conditionner l’accès de l’opérateur à différents niveaux de la base en fonction de son statut. Une revue de leurs informations peut par ailleurs être effectuée périodiquement par l’ANSM.
Eudamed est une base de données européenne de partage d’informations instaurée par l’article 33 du RDM. Elle est structurée autour de 6 modules interconnectés dans un site restreint (une partie seulement de ces informations sera rendue accessible au public) (Figure 3):
- Le module “Acteurs” disponible depuis décembre 2020,
- Le module “Dispositif/UDI”,
- Le module “Organismes notifiés, Certificats de conformité CE et Résumé des Caractéristiques de Sécurité et des Performances Cliniques ”,
- Le module “Investigations cliniques et études des performances”,
- Le module “Matériovigilance, surveillance post commercialisation et rapport périodique actualisé de sécurité (PSUR)”,
- Le module "Surveillance du marché".
Figure 3 : Architecture de la base EUDAMED [Source : d'après [15]]
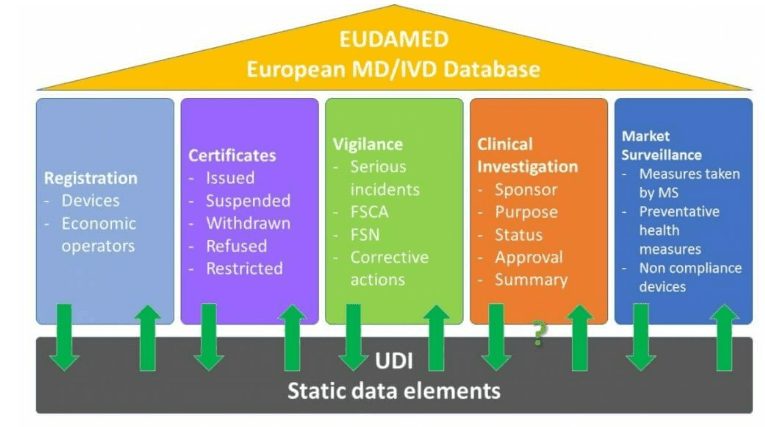
EUDAMED s’inscrit donc comme un véritable élément central de la gestion des acteurs de l’écosystème des Dispositifs Médicaux. Il est à noter que son utilisation ne sera rendue obligatoire que 6 mois après qu’elle aura été déclarée entièrement fonctionnelle suivant un audit indépendant et la publication d’une notice de la Commission dans le Journal Officiel de l’Union Européenne [16]. Son calendrier d’implémentation est donc à suivre avec attention par chaque acteur.
Bien que son utilisation ne soit donc ni obligatoire ni requise, la base est déjà utilisée volontairement par certains opérateurs. De cette manière, des chiffres ont pu être obtenus.
En effet, en octobre 2023, selon un graphique en secteurs (Figure 4) obtenu à partir des données de la base, les fabricants représentaient 70% de l'ensemble des opérateurs économiques (plus de 31 000 au total). À cette même date, près de la moitié d’entre eux provenaient de Chine (22.8%), de l’Allemagne (15.9%) et des Etats-Unis (9.4%) [13]:
Figure 4 : Répartition des opérateurs économiques enregistrés sur EUDAMED [Source : d'après [13]]
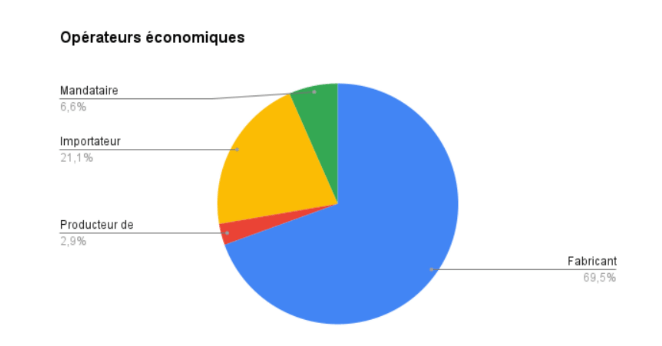
Aujourd’hui, il y a donc un certain nombre d’opérateurs économiques en activité sur le territoire de l’UE malgré un déclin qui avait été initialement observé suite à l’entrée en vigueur des deux nouveaux Règlements.
3) ...Malgré un déséquilibre engendré par l'arrivée du nouveau cadre législatif
L’arrivée du nouveau règlement a en effet non seulement impacté le nombre d’Organismes Notifiés en activité mais elle a aussi provoqué un déséquilibre au niveau du nombre d’entreprises.
C’est dans ce cadre qu’une étude nommée “Panorama et analyse qualitative de la filière industrielle des dispositifs médicaux en France en 2021” a été menée par le SNITEM et publiée le 11 juillet 2022. Cette étude est basée sur une méthode quantitative ; une enquête en ligne a été envoyée aux principales entreprises du DM.
D’après cette étude, entre 2019 et 2021, il a été constaté une réduction du nombre d'entreprises actives sur le marché. Pendant cette période, 126 entreprises ont en effet quitté le marché tandis que 63 nouvelles entreprises y sont entrées. Cela a entraîné un déséquilibre avec un nombre total d'acteurs réduit à 1 440, ce qui représente une baisse de 4,5% par rapport aux 1 502 entreprises enregistrées en 2019 [17].
Les restrictions dues aux nouveaux règlements font partie des principales causes du déséquilibre des entreprises du DM. En effet, parmi les entreprises interrogées dans le cadre de cette étude, il est dénombré (Figure 5) :
Figure 5 : Données chiffrées issues de l'étude "Panorama et analyse qualitative de la filière industrielle des DM en France en 2021" [Source : d'après [17]]
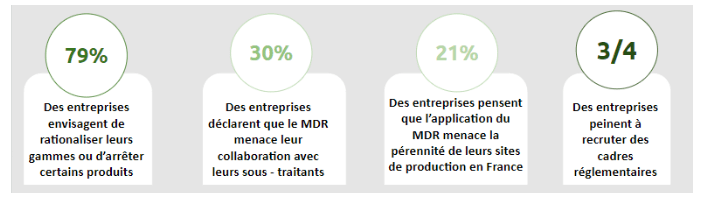
Pour répondre à cette crise, les entreprises ressentent un besoin profond de recruter des personnes qualifiées afin d'assurer la mise en conformité des DM.
Afin que ces personnes qui sont notamment de profil juniors soient moins démunies face à la complexité du secteur, une action possible est de décrypter sa composition pour le rendre plus lisible.
Pour rappel, la problématique retenue pour le MIM suivant est donc :
“Comment apporter de la lisibilité à un secteur en tension qui garantit la disponibilité de produits de santé sûrs et performants aux patients ?”
III) Objectifs du projet et détermination des cibles de l'outil
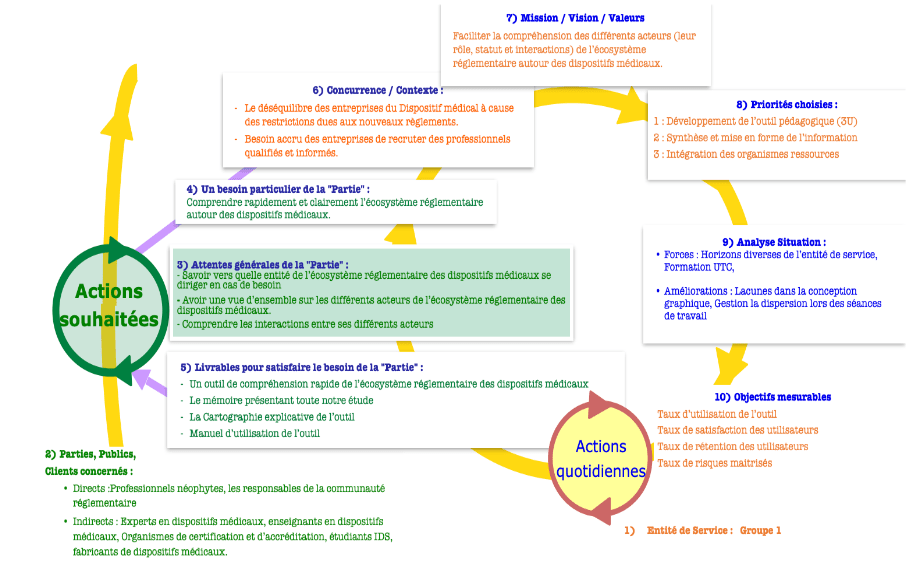
Pour répondre à cette problématique, une Planification Dynamique Stratégique (PDS) (Annexe) a été établie afin de faire ressortir les objectifs stratégiques de ce projet :
- Synthèse et Mise en Forme de l'Information : il s’agit ici d’analyser l’existant de l’écosystème réglementaire actuel des Dispositifs Médicaux pour en extraire les informations relatives aux acteurs, référentiels, afin d’organiser et présenter ces informations de manière claire, concise et accessible.
- Développement d’un outil pédagogique (3U) : qui consiste à concevoir un outil pédagogique interactif et intuitif pour faciliter la compréhension rapide de l’écosystème réglementaire des Dispositifs Médicaux tout en s'assurant de sa pertinence (Utile), de sa facilité d'utilisation (Utilisable) et de sa fréquente utilisation (Utilisé) par les parties prenantes.
- Intégration des Organismes et Ressources : c’est à dire recenser et mettre en avant des liens d’organismes, guides et ressources complémentaires qui peuvent accompagner les parties prenantes dans la compréhension de l’écosystème réglementaire des Dispositifs Médicaux.
Grâce à la Planification Dynamique Stratégique, il a aussi été possible de mettre en évidence le public concerné directement ou indirectement par cette solution, tenant compte de leurs besoins particuliers identifiés dans la littérature et le questionnaire diffusé sur Linkedin (Tableau 1).
Tableau 1 : Identification des personnes concernées par la solution proposée [Source : auteurs]
| Professionnels néophytes | Le questionnaire diffusé sur Linkedin a permis d’identifier la catégorie de professionnels néophytes par les profils ayant répondus au questionnaire, en effet, il y avait des profils de chargé qualité et Affaires réglementaires allant de 1 à 4 ans d’expérience. |
| Étudiants en formation biomédicale | D’un point de vue d’étudiants en Ingénierie de la Santé, la connaissance de cet écosystème sera très bénéfique pour la vie professionnelle future. |
| Personne Chargée de Veiller au Respect de la Réglementation | Compte tenu de ses missions visées par le Règlement européen des Dispositifs médicaux à l’Article 15, la PCVRR se doit de connaître les acteurs intervenant tout autour du Dispositif Médical. |
| Enseignants de Dispositifs Médicaux | Le questionnaire diffusé sur Linkedin a permis d’identifier la catégorie d’enseignants dans le domaine du Dispositif Médical : Questionnaire Linkedin : Est-ce que la connaissance des acteurs de l’écosystème réglementaire a déjà été un frein à la réussite de vos missions en entreprise ? Si oui, pouvez vous nous décrire un cas pratique ? “Non, mais un outil peut être utile pour de la formation interne des fonctions moins en lien avec le réglementaire” |
Fabricants de dispositifs médicaux | Les fabricants de Dispositifs Médicaux doivent impérativement connaître les acteurs de l’écosystème avec lesquels ils sont en interaction pour assurer la conformité de leurs Dispositifs Médicaux. |
Personnes étrangères aux affaires réglementaires des Dispositifs Médicaux | Le questionnaire diffusé sur Linkedin a permis d’identifier une nouvelle catégorie de partie prenante : Questionnaire Linkedin : Est-ce que la connaissance des acteurs de l’écosystème réglementaire a déjà été un frein à la réussite de vos missions en entreprise ? Si oui, pouvez vous nous décrire un cas pratique ? “Cas qui arrive rarement dans le service Affaires Réglementaires, mais il est vrai que pour une personne issue d'un autre département, l'ignorance de ces acteurs peut compliquer la conduite de certains projets…” |
Après l'établissement du contexte du projet et la définition de son public cible, la réflexion s'est naturellement orientée vers la manière de faciliter une compréhension approfondie de l'écosystème existant pour cette audience spécifique.
Partie 2 : Méthodologie
I) Démarche globale employée par le groupe projet
1) Vue d'ensemble de la démarche
À cet effet, un ensemble d’activités ont été réalisées par le groupe. Celles-ci sont récapitulées dans le logigramme suivant (Figure 6).
Figure 6 : Logigramme synthétisant la démarche employée par le groupe [Source : auteurs]
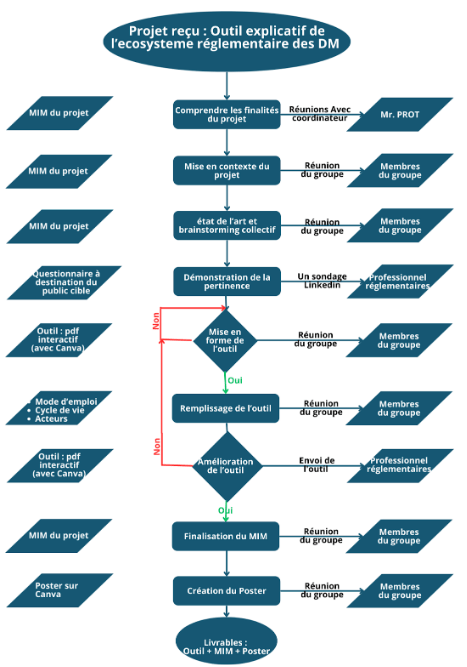
Cette démarche n’est néanmoins pas sans risques. Un diagramme de décisions lui a donc été associé en amont (Figure 7).
2) Risques associés à la démarche et alternatives envisagées
Un diagramme de décisions est une représentation visuelle utilisée pour analyser et résoudre des problèmes ; en identifiant les différentes options disponibles tout en évaluant les risques associés à chaque choix. Il offre une vue structurée des décisions à prendre, facilitant ainsi la compréhension et la gestion des scénarios complexes.
Figure 7 : Diagramme de décisions réalisé par le groupe sur le projet [Source : auteurs]
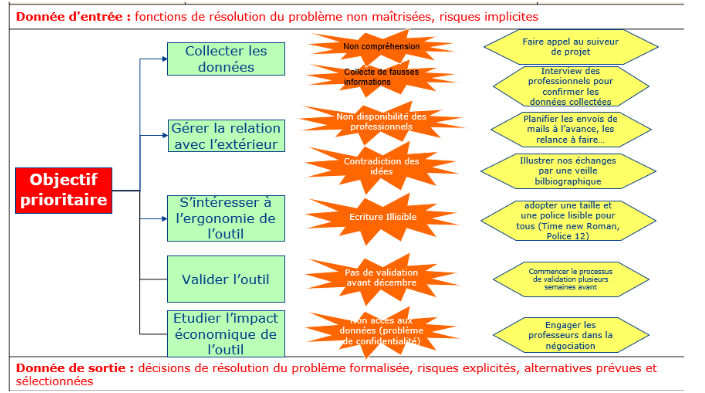
II) Zoom sur certaines actions de la démarche
1) Etat de l'art des outils similaires existants et de leurs avantages/ inconvénients
Dans la démarche globale, une des premières actions envisagées par le groupe était de faire un état de l’art des outils similaires à celui qui sera conçu.
Toutefois, cette action s’est avérée difficile à effectuer car il y a très peu d'organisations ou d'associations qui ont traité cette problématique.
Ainsi, la veille n’a permis d’identifier qu’un seul outil qui traite de l’écosystème réglementaire ; celui-ci ayant été réalisé par le SNITEM. Le travail se basera donc sur cet outil pour traiter ses avantages et inconvénients.
L’outil se présente en deux infographies sous forme de PDF téléchargeable. La première infographie tient sur deux pages et elle représente les cinq principaux acteurs des entreprises du DM : le fabricant, le mandataire, l’importateur, le distributeur, et la personne visée à l’article 22 du RDM (ou assembleur) (Figure 8).
Figure 8 : Première infographie du SNITEM [Source : d'après [18]]

La deuxième infographie représente les acteurs institutionnels nationaux et européens : l’Union Européenne, les Organismes Notifiés, l’Agence Nationale de Sécurité du Médicament et des produits de santé, la Haute Autorité de Santé, le Comité Économique des Produits de Santé, le Ministère en charge de la Santé et de la Sécurité Sociale, l’Assurance Maladie, et le Ministère en charge de l’Économie et de l’Industrie (Figure 9).
Figure 9 : Seconde infographie du SNITEM [Source : d'après [18]]

Après une analyse de ces deux infographies, leurs avantages et inconvénients ont ensuite pu être identifiés (Tableau 2).
Tableau 2 : Avantages et inconvénients des deux infographies du SNITEM [Source : auteurs]
| Avantages | Inconvénients |
| Infographie synthétique | Manque de précision sur les missions et la composition de certains acteurs |
| PDF aux couleurs attractifs et facile à lire | Manque d'interactivité |
| Traite des principaux acteurs présents à l’échelle française et européenne | Absence des acteurs présents à l’échelle internationale |
| Absence de plusieurs autres acteurs participant à l’écosystème réglementaire | |
| Manque de logigrammes représentant les relations entre les différents acteurs |
En conclusion, il n' y a pas d'outils présentant tout l'écosystème réglementaire à l'échelle internationale, européenne et française. Ce constat permet de continuer dans la démarche d’aider les professionnels à la compréhension de l’écosystème réglementaire grâce à un seul document le plus synthétique et interactif possible.
2) Revue de ressources complémentaires
Bien qu'il n'y ait aujourd'hui pas d'outil pédagogique existant qui présente tous les acteurs de l'écosystème en un seul endroit, plusieurs sites peuvent tout de même être utilisables par le public cible de l'outil pour se renseigner sur un organisme qu'il ne connaîtrait pas.
2.1) Textes législatifs et réglementaires
En effet, dans un contexte réglementaire en évolution permanente, quel que soit son métier, chaque acteur cible de l'outil doit tout d'abord surveiller régulièrement les textes législatifs et réglementaires européens et français qui pourraient avoir un impact sur les activités et le produit de l'entreprise où il exerce : identification des dernières versions applicables, amendements, révisions, suppression de textes...[19]
Cela lui permet évidemment de rester conforme aux derniers textes applicables. Mais aussi, cela peut lui fournir des informations sur les autres acteurs de son secteur avec qui il sera peut-être en interaction dans ses démarches quotidiennes telles que : la constitution de la documentation technique de son DM ou la mise à jour du Système de Management de la Qualité de sa société.
Par exemple, le Règlement 2017/745 contient à lui seul des informations sur les acteurs suivants (Tableau 3):
Tableau 3 : Liste des acteurs mentionnés par le Règlement 2017/745 avec les articles correspondants [Source : auteurs]
| Passages du Règlement | Acteurs |
| Article 10 | Le fabricant |
| Article 11 | Le mandataire |
| Article 13 | L’importateur |
| Article 14 | Le distributeur |
| Article 15 | La Personne Chargée de Veiller au Respect de la Réglementation (PCVRR) |
| Article 22 | L'assembleur de systèmes et nécessaires |
| Chapitre IV et Annexe IV | Les organismes notifiés |
| Article 101 | Les autorités compétentes |
| Article 103 | Le MDCG |
2.2) Normes, spécifications communes et guides
Appartiennent également au processus de veille :
- Toutes les normes harmonisées : il s’agit de normes élaborées par les organismes de normalisation comme le CEN et le CENELEC, à la suite d'un mandat de la Commission Européenne, et publiées au Journal Officiel de l'Union Européenne. Elles fournissent ainsi une présomption de conformité aux exigences correspondantes du Règlement et sont publiées dans des décisions d'exécution [20] ;
- Les spécifications communes (cas où il n'y a pas de normes harmonisées existantes ou qu'elles sont insuffisantes ou en cas de préoccupation de santé publique);
- Les guides, notamment ceux du MDCG (anciennement Meddev).
Ces trois points sont d'utilité cruciale car ils donnent les spécificités techniques à suivre (le "comment faire") pour démontrer la conformité avec le Règlement (le "quoi faire"). Comme les textes législatifs et réglementaires, ils sont aussi assez riches en informations sur les acteurs.
Par exemple, il est ainsi possible, entre autres, de se référer aux guides MDCG suivants pour la thématique du projet :
- Le guide MDCG 2021-5 "Guidance on standardisation for medical devices"[21]: celui-ci fournit des informations sur plusieurs acteurs de la normalisation européens et internationaux et décrit leurs relations avec les instances réglementaires. Ces acteurs, dont certains peu connus du public, sont entre autres :
1) Les organismes de normalisation européens : CEN et CENELEC,
2) L'ABHS : le conseil consultatif du CEN CENELEC sur les normes de santé,
3) Les consultants HAS (HArmonised Standards),
4) Le groupe MDCG,
5) Les acteurs internationaux de la normalisation : l'ISO, l'IEC et l'International Medical Device Regulators Forum (IMDRF).
- Le guide MDCG 2019-7 Rev.1 "Guidance on article 15 of the medical device regulation (MDR) and in vitro diagnostic device regulation (IVDR) on a person responsible for regulatory compliance (PRRC)"[22]: celui-ci permet de clarifier les exigences contenues à l'article 15 du Règlement vis-à-vis des missions et obligations du PCVRR.
2.3) Les sites officiels de chaque acteur
Enfin, le public cible de l'outil pédagogique peut se référer aux sites officiels de chaque acteur. Certains proposent même des abonnements à leurs newsletters, pour rester au courant des nouveautés [19].
Tous ces acteurs mentionnés dans les textes réglementaires peuvent ensuite être enrichis par le moyen du brainstorming collectif entre individus d’horizons variés.
3) La maîtrise de l'écosystème réglementaire facilite-t-elle l'obtention du marquage CE ?
En cherchant à simplifier la compréhension de l'écosystème réglementaire pour les parties prenantes à travers un document unique, interactif et synthétique, l'idée que cette approche pourrait faciliter l'obtention du marquage CE a été explorée.
C’est ainsi que la démarche s'est poursuivie par une séance de brainstorming collective afin de faire ressortir la totalité des acteurs présents dans cet écosystème. Celui-ci a ensuite été séparément enrichi par le biais de recherches bibliographiques et sur le web. Puis il a abouti à la réalisation d’un diagramme des affinités durant cette même phase afin de regrouper les idées similaires sous une même entête/catégorie. Ce diagramme des affinités (Figure 10) a ainsi permis la création du menu 1 de l’outil (voir Partie 3 suivante). De cette manière, étant ainsi partis de nombreuses idées plutôt diffuses, un ensemble de catégories cohérentes par rapport à la réalité actuelle du secteur a été obtenu.
Figure 10 : Diagramme d'affinités réalisé par le groupe [Source : auteurs]
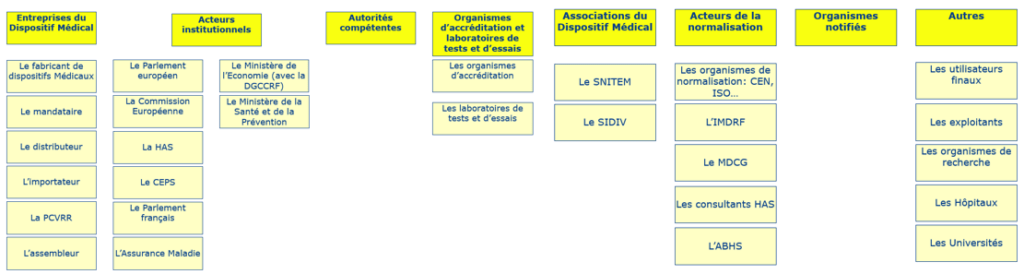
Par la suite, une investigation sur le terrain à été menée auprès de professionnels travaillant dans des entreprises du DM, permettant de récolter des informations sur leur perception de l’écosystème réglementaire et son apport quant à l’obtention du marquage CE. Les différents points évoqués ci-dessous sont issus d’une analyse du questionnaire envoyée à des responsables affaires réglementaires, des ingénieurs QARA, chargée d’affaires réglementaires et des consultants dans différentes structures biomédicales.
- Comprendre l'écosystème réglementaire
Lors de l'enquête, il a été observé que la définition de ce qu'est un écosystème réglementaire n'est pas très claire pour de nombreux professionnels. Cette lacune semble particulièrement notable parmi les néophytes travaillant au sein de start-ups et de PME. En effet, certaines réponses à la question "Pour vous, que représente la notion d'écosystème réglementaire ?" incluent des réponses telles que "Aucune idée", "Cette notion est nouvelle pour moi", "Pas de réponse", ou encore "un gros machin qui mériterait d'être remis en ordre". Parmi les 22 réponses recueillies, 6 personnes n’ont pas une compréhension claire de l'écosystème réglementaire, renforçant ainsi la nécessité de développer un outil visant à définir et clarifier cette notion.
- Besoin d'un outil interactif
Parmi les 22 réponses, 86,4% expriment le besoin d'un outil interactif résumant la composition de l'écosystème réglementaire des Dispositifs Médicaux, y compris ses acteurs, leurs missions, statuts et relations. Cette demande est particulièrement forte en raison de la transition actuelle vers l'obtention du marquage CE, conformément au MDR 2017/745. En effet, 70% des répondants veulent un classement des acteurs selon les étapes de l’obtention du marquage CE. De plus, certains professionnels, notamment ceux des start-ups, se lancent dans le processus d'obtention de leur premier marquage CE. Avec l'introduction de ce nouveau règlement, de nouveaux acteurs ont émergé, accompagnés de nouvelles exigences. Ainsi, l'idée d'un outil est de fournir à ces professionnels une vue d'ensemble des acteurs impliqués et des explications sur les exigences spécifiques de chacun.
- Impact de la connaissance des acteurs de l'écosystème réglementaire sur la réussite des missions en entreprise
7 professionnels jugent que la connaissance des acteurs de l’écosystème réglementaire n’est pas un frein à la réussite des missions. Selon un directeur affaires réglementaires travaillant dans une PME et attestant d’une dizaine d’années d’expérience : « la connaissance des acteurs de l’écosystème réglementaire n’est pas un frein à la réussite des missions en entreprise car la réglementation est applicable à tous, nous devons nous adapter quelque soit la contrainte/opportunité ». 5 autres répondants jugent que le manque de connaissance des acteurs de l’écosystème réglementaire est un frein indirect sur la réussite des missions dans la mesure où c’est un sujet de recherche. En effet, une chargée d’affaires réglementaires qui a un an d’expérience a témoigné : « Je passe beaucoup de temps au travail à chercher des informations par moi-même, je dirais donc qu'une synthèse serait probablement utile », une chargée d’AR attestant de 3 ans d’expériences a ajouté : « Le manque de connaissance des acteurs composant l’écosystème réglementaire n’est pas un frein à la réussite des missions en entreprise mais a été le sujet de recherches pour être apte à maîtriser les échanges avec des tiers », enfin un responsable qualités et affaires réglementaire dans une PME témoigne que « Les freins apparaissent quand on ne prend pas le temps de lire et de comprendre les réglementations principales. Elles font la connexion vers le reste ». Par ailleurs, 6 autres professionnels témoignent que le manque de connaissance des acteurs de l’écosystème réglementaire peut avoir un impact et devenir un frein à la réussite des missions en entreprise. Notamment, en ce qui concerne les autres services. En effet, un consultant en affaires réglementaires témoigne que : « il est vrai que pour une personne issue d'un autre département, l'ignorance de ces acteurs peut compliquer la conduite de certains projets. (Exemple : modification de la conception par la R&D --> il faut prévenir les ARSs pour enclencher les processus de change control, modifier éventuellement le dossier technique etc...) ». En résumé, bien que la majorité ne considère pas la connaissance des acteurs comme un frein majeur, une partie des répondants souligne son importance, notamment en termes de recherche d'informations et de collaborations interdépartementales.
- Utilisation courante d'outils et de guides synthétiques
La majorité des professionnels utilisent dans leur routine des outils pédagogiques ou des ressources similaires à un guide explicatif. Les travaux du SNITEM, GMED, MDCG, MEDDEV, AFNOR (COBAZ) et IMDRF ressortent le plus souvent ou encore les travaux des étudiants de l’UTC qui sont « synthétiques, gratuits et accessibles à tous ». Les points forts « sont la simplicité dans la transmission de la connaissance avec des graphes simplifiant la compréhension, l'expertise dans la manière de présenter les choses, le rapprochement à la réalité des difficultés rencontrées directement sur le terrain par les fabricants ». Les points faibles cités sont : « Ils ne sont pas vérifiés par des autorités, donc pas forcément fiables , les informations ne sont pas complètes, donc il faut toujours regarder ailleurs , ils ne sont pas toujours à jour et dépendent des activités de veille des autres, donc parfois ils ratent des informations (en particulier des actualités) » .
- Préférences en matière de formats d'apprentissage
87% des répondants privilégient des formats de PDF interactifs et d’infographies.
En conclusion, le brainstorming et l'enquête ont éclairci la démarche en accord avec les besoins actuels. La confusion des professionnels sur l'écosystème réglementaire des Dispositifs Médicaux souligne la nécessité d'un outil interactif (86,4%). Bien que la connaissance des acteurs ne soit pas considérée comme un obstacle majeur, l'utilité d'un outil synthétique pour faciliter la recherche d'informations et les collaborations interdépartementales est reconnue. La préférence générale (87%) va vers des formats tels que les PDF interactifs et les infographies.
Finalement, la démarche choisie par le groupe a donc permis de mettre en évidence trois éléments principaux à prendre en compte :
- L’outil en ligne du SNITEM : pour créer les fiches de certains acteurs de l’écosystème,
- Les passages du Règlement 2017/745 qui présentent les obligations de certains acteurs ainsi que les guides MDCG 2021-5 et 2019-7,
- Les conclusions de l’analyse du questionnaire.
Partie 3 - Résultats et Bilan de la démarche employée par le groupe
I) Structure de l'outil (manuel d'utilisation)
Ayant considéré ces trois éléments, l’outil a été créé sur l’application de conception Canva sous la forme d’un Powerpoint interactif [23], déployé sur trois grandes parties :
- Une partie 1 avec des diapositives introductives de la thématique,
- Une partie 2 qui représente un choix entre deux menus,
- Une partie 3 qui est le cœur de l'outil.
1) Partie 1 : des diapositives introductives de la thématique
Tout d’abord, après une page d'accueil, l’outil débute par une présentation de la thématique (le “Quoi ?”) qui sert de contenu : l’écosystème réglementaire des Dispositifs Médicaux. À la suite des réponses très variées obtenues lors du questionnaire, il est en effet apparu nécessaire de poser sa définition de façon claire et simple afin que celle-ci fasse consensus (Figure 11).
Figure 11 : Diapositive de l'outil qui sert à présenter sa thématique [Source : auteurs]

Ensuite, il a été choisi d’inclure dans le mode d’emploi (Figure 12) à la fois la procédure d’utilisation du document (sa structure ainsi que les fonctionnalités des boutons cliquables) mais aussi plusieurs avertissements dans l’objectif de rendre l’utilisateur le plus autonome possible. Cette même diapositive sert aussi de présentation des membres du groupe (Qui?) et inclut un lien vers le mode d’emploi sous format vidéo.
Figure 12 : Mode d'emploi de l'outil [Source : auteurs]

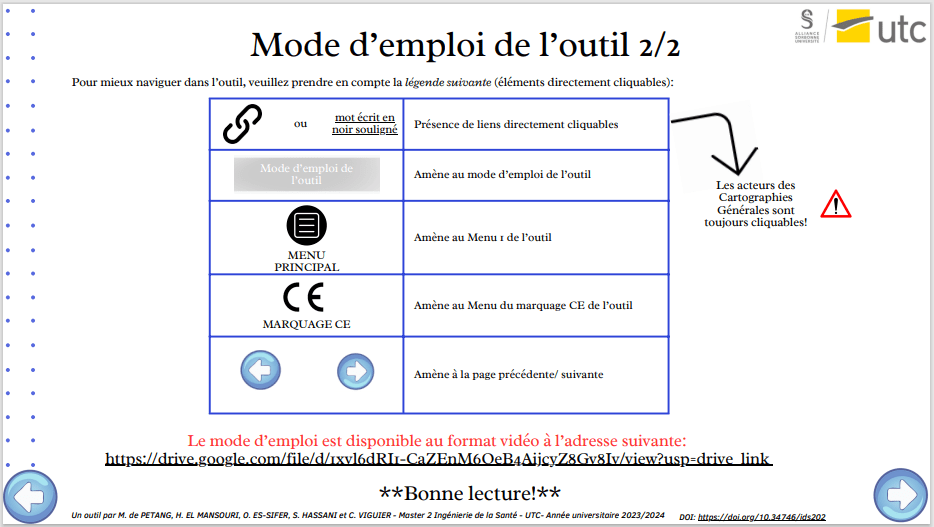
2) Partie 2 : Un choix entre deux menus
Après cette partie introductive, l’outil fait l’objet de deux menus pouvant être choisis selon l’envie et le besoin de l’utilisateur.
D’une part, le menu 1 disponible dès la page d'accueil permet de visualiser uniquement les grandes catégories d’acteurs ; 8 au total (Figure 13):
Figure 13 : Menu 1 de l'outil [Source : auteurs]

D’autre part, le menu 2 représente les étapes qui seront parcourues par tout fabricant afin d’obtenir le marquage CE de ses dispositifs (de l'étude du marché à la Surveillance Après Commercialisation); chaque organisme y étant rangé dans les étapes dans lesquelles il participe (Figure 14) :
Figure 14 : Menu 2 de l'outil [Source : auteurs]
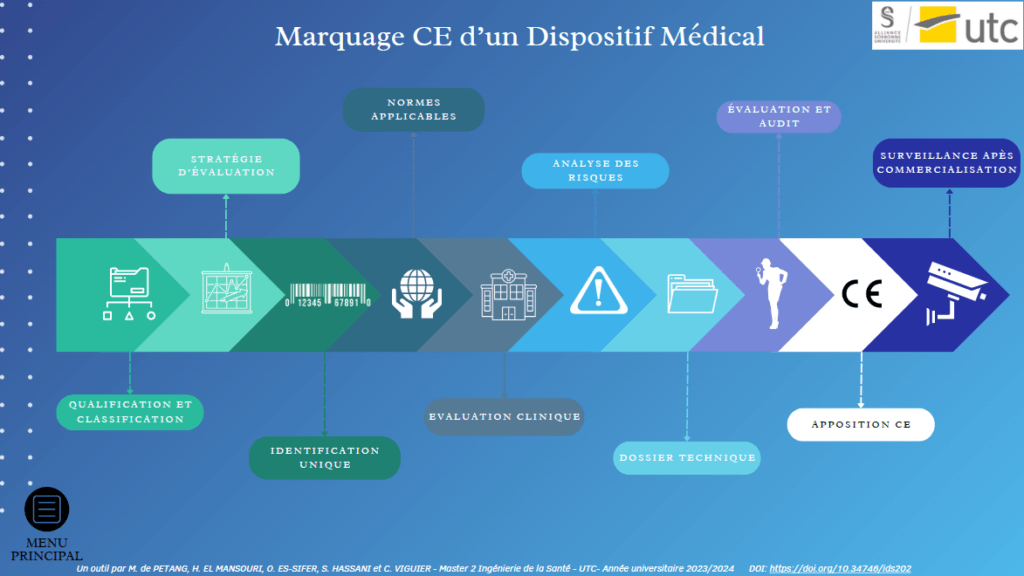
Plusieurs éléments cliquables sont par ailleurs disposés au travers de l’outil interactif afin de permettre une navigation plus fluide et plus rapide de l’utilisateur entre les pages. Il est notamment à noter la présence du bouton “MENU PRINCIPAL” qui lui permet à n’importe quel moment de revenir à l’interface de départ (menu 1) pour accéder à une nouvelle catégorie d’organismes.
3) Partie 3 : Le centre de l'outil
Ensuite, l’utilisateur rentre dans le cœur de l’outil.
Dans cette partie centrale, chaque grande catégorie débute par un avant-propos qui expose le sujet présenté par la suite avec un ensemble d’hyperliens vers des ressources complémentaires. Puis, une cartographie générale permet de représenter les relations entre les acteurs de cette catégorie donnée (Figure 15).
Figure 15 : Exemple de cartographie générale disponible dans l'outil [Source : auteurs]
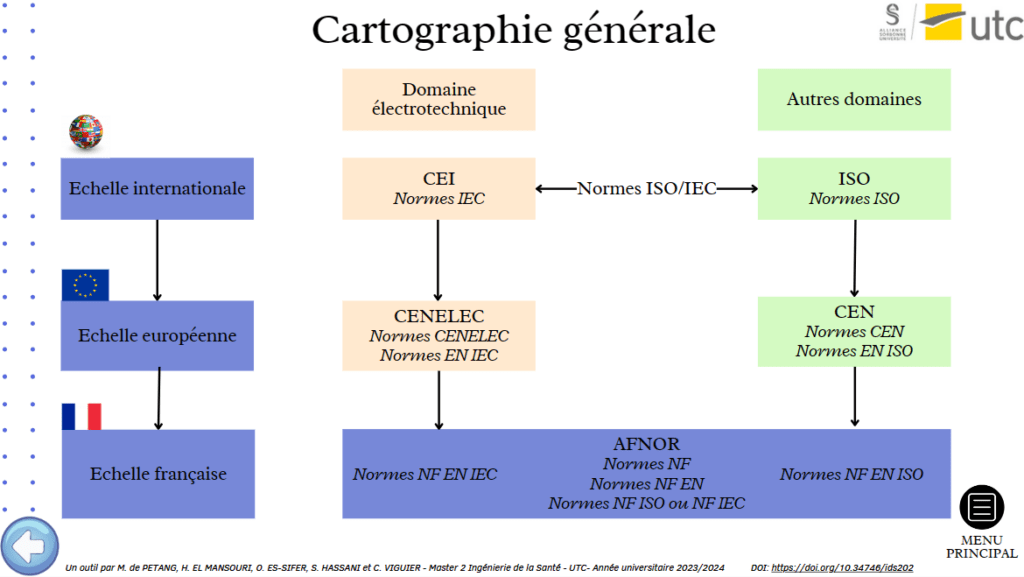
Enfin, chaque acteur de l’écosystème possède toujours sa propre fiche. Sa structure varie en fonction des cas ; cependant, il sera généralement retrouvé : une présentation de son rôle ainsi que de ses missions/obligations. Pour cette deuxième partie, il est fait référence aux passages concernés du Règlement 2017/745 quand cela est possible (=cas d’acteurs visés par le texte). Cela permet ainsi d’éviter à l’utilisateur tout le travail fastidieux de se plonger dans le Règlement. Toutes les informations indiquées pour chaque acteur sont cependant non exhaustives ; l’utilisateur devra donc à la fin de sa lecture s’assurer qu’elles sont toujours bien à jour.
À plusieurs reprises dans cette troisième grande partie, l’outil dresse par ailleurs un panorama général sur certains points de la réglementation européenne des Dispositifs Médicaux, par exemple : l’annexe Z, les normes harmonisées, la normalisation internationale…
II) Zoom sur le menu 2 de l'outil : marquage CE d'un Dispositif Médical
Le processus de mise sur le marché des Dispositifs Médicaux est strictement régulé par le marquage CE, qui signifie "Conformité Européenne". Toute commercialisation de Dispositifs Médicaux, tels que les Dispositifs Médicaux Implantables Actifs ou les Dispositifs Médicaux de Diagnostic In Vitro, exige ce marquage préalable. Le fabricant doit absolument obtenir cette certification pour chaque produit médical, suivant plusieurs étapes avant et après son obtention, comme indiqué dans la Figure 14.
Le marquage CE atteste la conformité aux normes essentielles garantissant la santé et la sécurité des utilisateurs, notamment en termes de stérilité, de compatibilité biologique et de sécurité électrique. Le Menu Marquage CE d’un Dispositif Médical offre une vue d'ensemble des principaux acteurs intervenant dans l'écosystème réglementaire des Dispositifs Médicaux, engagés dans le processus d'obtention du marquage CE d’un dispositif innovant, y compris la Surveillance Après Commercialisation. Il présente de manière concise les étapes clés de ce processus, les rôles des acteurs impliqués, les références réglementaires et normatives en vigueur et des sources supplémentaires pour obtenir des informations complémentaires.
Les étapes présentées sont les suivantes :
- Qualification et Classification du Dispositif Médical,
- Stratégie d’évaluation,
- Identification Unique du Dispositif Médical,
- Choix des normes applicables au dispositif,
- Processus d’évaluation clinique,
- Analyse des risques,
- Rédaction du dossier technique,
- Évaluation de conformité du dossier technique et Audit du Système de Management de la Qualité,
- Apposition du marquage CE,
- Surveillance Après Commercialisation.
III) Bilan de la démarche
Ayant obtenu ces résultats, la démonstration de la pertinence de la mise en œuvre choisie a constitué la dernière étape de gestion du projet.
- Méthode employée pour faire valider l’outil final et réponses obtenues
À cet effet, il a été décidé de faire soumettre l’outil final à une validation par les professionnels qui ont répondu au questionnaire et ont donc manifesté un certain intérêt pour le travail réalisé. Ceux-ci ont été laissés libres dans leurs possibilités de réponses : commentaires et suggestions leur étaient simplement demandés afin d’envisager d’éventuelles améliorations de l’outil.
Suite à cette démarche, il a donc été obtenu :
- Appréciations sur l’outil dans sa globalité,
- Remarques sur des éléments de forme,
- Remarques sur des éléments de contenu.
Tous ces retours ont ensuite été suivis d’améliorations du PDF interactif.
- Appréciations sur l'outil dans son ensemble
Concernant les appréciations, l'ensemble de l’outil a été déclaré comme étant “plutôt agréable à lire”. De plus, a été mis en valeur le travail réalisé sur la structure afin “que tout reste lisible et en même temps assez condensé”. L’aspect synthétique de l’outil a aussi été apprécié.
- Remarques sur les éléments de forme
Concernant la forme, le choix de l’interface interactive a été approuvé par les professionnels car permettant un passage plus rapide entre les pages du PDF.
- Remarques sur les éléments de contenu
En termes de contenu, les professionnels ont tous jugé positif les nombreuses références (hyperliens) présentes. Ils ont d’ailleurs recommandé l’ajout d’une page en fin de PDF reprenant l'ensemble des liens cités pour chaque catégorie ce qui a donc été fait par la suite. Ainsi, le lecteur peut désormais retrouver directement tout ce qui l'intéresse à la fin du PDF.
L’intégration de la catégorie “Associations professionnelles” dans l’outil a été appréciée car étant “un point important à citer dans le cadre du suivi des exigences réglementaires et normatives”. Dans cette même catégorie, a d’ailleurs été ajoutée l’organisation “TEAM-PRRC” créée par une ancienne étudiante de l’UTC suite à la remarque d’un professionnel.
De plus, les réponses ont également permis de mettre en évidence un point d’attention particulier lié au rôle de fabricant : celui des statuts d’Own Brand Labeller (OBL) et d’Original Equipment Manufacturer (OEM) qui ont disparu avec le Règlement 2017/745. L’OEM est aujourd’hui appelé fabricant original. L’OBL est quant à lui considéré comme fabricant ou importateur/ distributeur ; ces deux cas étant prévus à l’article 16 du Règlement :
- Soit il a conclu un accord avec le fabricant original pour prévoir que ce dernier reste mentionné en tant que tel sur l’étiquette et demeure responsable du respect des exigences applicables au fabricant : il est alors considéré comme importateur ou distributeur et applique les exigences rattachés à l’un ou l’autre des deux statuts ;
- Soit il n’a conclu aucun accord avec le fabricant original : il applique dans ce cas les obligations attachées au statut de fabricant présentes à l’article 10 du Règlement.
IV) Améliorations possibles de l'outil pédagogique
L’outil pédagogique élaboré à l’issue de ce projet peut encore faire l’objet d’évolution de ses fonctionnalités :
- L’ajout de références au Règlement 2017/746 sur les Dispositifs Médicaux de Diagnostic In Vitro,
- La mise en place de questionnaires intermédiaires pour mesurer l’état d’avancement de l’utilisateur et l’assurer ainsi de sa progression au fil de sa lecture.
Il pourrait aussi être envisagé de mettre ce projet en lien direct avec celui du groupe du Master IdS centré sur la thématique suivante : “Règlement européen en matière de dispositifs médicaux : Bilan à 6 ans d'entrée en vigueur et 2 ans d'entrée en application”(Groupe IDS203). Ainsi, plusieurs diapositives pourraient par exemple être ajoutées à l’outil afin de faire un bilan de tous les changements induits par les deux Règlements spécifiquement sur la partie des statuts et des obligations des acteurs de l’écosystème réglementaire des Dispositifs Médicaux.
Conclusion
En conclusion, l'organisation actuelle de l'écosystème réglementaire des Dispositifs Médicaux reste difficilement lisible pour les professionnels néophytes. Entre la multitude d'acteurs, les différentes échelles, les acronymes divers et variés, les regroupements de certains organismes et les évolutions permanentes, le rôle de chacun peut être confus, leurs compétences aussi et surtout leurs périmètres d'action.
Ainsi, la problématique suivante se pose : “Comment apporter de la lisibilité à un secteur en tension qui garantit la disponibilité de produits de santé sûrs et performants aux patients”?
Pour nous, trois axes sont à prendre en compte :
- Faire appel aux réglementations, normes et guides en vigueur : point de départ de la veille réglementaire, ils sont aussi très riches en informations sur tous les organismes ;
- Se rendre sur les sites officiels de chaque acteur et s'abonner à leurs newsletters peut aussi aider ;
- Enfin, consulter les outils d'aide qui existent ; ceux du SNITEM en priorité ainsi que ceux des étudiants de l'UTC !
Ainsi, le travail déployé a permis d’aboutir à un outil qui cartographie et compile, dans un support interactif, une multitude des acteurs de l'écosystème présents à l'échelle française, européenne et internationale.
Dans un premier temps, l’utilisateur peut rapidement parcourir cet outil interactif grâce à ces multiples éléments directement cliquables. Les deux menus proposés au début participent de même à son caractère interactif ; avec un qui positionne les acteurs dans des catégories et l'autre dans le processus du marquage CE.
Ensuite, les fiches synthétiques de chaque acteur lui permettent de connaître rapidement leurs rôles et obligations réglementaires. Les cartographies présentées au début de chaque catégorie lui servent de plus à prendre connaissance des parties prenantes avec lesquelles il est spécifiquement en relation.
Enfin, une fois l'outil compris, l’utilisateur peut enrichir sa compréhension des acteurs grâce à des liens vers des documents utiles dispersés dans l'outil et rassemblés dans une page à la fin.
Il est à préciser que cet outil reste néanmoins une photographie du secteur à l’instant t.
Les acteurs de l'écosystème réglementaire des Dispositifs Médicaux ont des statuts et des missions qui peuvent être amenés à changer avec le temps. C’est aussi le cas des textes réglementaires et normatifs en vigueur.
Cette évolutivité permanente impose donc au professionnel néophyte de faire preuve d’adaptabilité et de surveiller régulièrement les innovations émergentes de son secteur d’activité. Les collaborations mutuelles sont aussi particulièrement importantes, notamment entre grands groupes et petites entreprises, afin de consolider les relations existantes.
Annexe : Planification Dynamique Stratégique
Figure 16 : Planification Dynamique Stratégique établie par le groupe [Source : auteurs]